

Abstract
Infectious diseases have plagued humankind throughout history and have posed serious public health problems. Yet vaccines have eradicated smallpox and antibiotics have drastically decreased the mortality rate of many infectious agents. These remarkable successes in the control of infections came from knowing the causative agents of the diseases, followed by serendipitous discoveries of attenuated viruses and antibiotics. The discovery of DNA as genetic material and the understanding of how this information translates into specific phenotypes have changed the paradigm for developing new vaccines, drugs, and diagnostic tests. Knowledge of the mechanisms of immunity and mechanisms of action of drugs has led to new vaccines and new antimicrobial agents. The key to the acquisition of the knowledge of these mechanisms has been identifying the elemental causes (i.e., genes and their products) that mediate immunity and drug resistance. The identification of these genes is made possible by being able to transfer the genes or mutated forms of the genes into causative agents or surrogate hosts. Such an approach was limited in Mycobacterium tuberculosis by the difficulty of transferring genes or alleles into M. tuberculosis or a suitable surrogate mycobacterial host. The construction of shuttle phasmids—chimeric molecules that replicate in Escherichia coli as plasmids and in mycobacteria as mycobacteriophages—was instrumental in developing gene transfer systems for M. tuberculosis. This review will discuss M. tuberculosis genetic systems and their impact on tuberculosis research.
“I had to know my enemy in order to prevail against him.”
Nelson Mandela
Infectious diseases have plagued humankind throughout history and have posed serious public health problems. Yet vaccines have eradicated smallpox and antibiotics have drastically decreased the mortality rate of many infectious agents (1). Although the precise viral agents had not yet been characterized, the smallpox vaccine work of Edward Jenner was critical in demonstrating that inoculation with pus from cowpox lesions could protect from a subsequent challenge with smallpox. These pioneering transfer experiments laid the ground-work for the eventual eradication of smallpox, as announced by the World Health Organization in 1979. The discovery of DNA as genetic material and the understanding of how this information translates into specific phenotypes have changed the paradigm for developing new vaccines, drugs, and diagnostic tests. Knowledge of the mechanisms of immunity and mechanisms of action of drugs has led to new vaccines and new antimicrobial agents. For example, the discovery of the Australia antigen (HBsAg) led to the subsequent engineering of the first recombinant vaccine, whose remarkable efficacy offers hope that eradication of hepatitis B in humans is not an unreasonable expectation (1, 2). Similarly, HIV infections, which not so long ago were uniformly fatal, are now controlled with drugs that were developed by understanding the HIV genome and gene products required for the HIV life cycle. The key to the acquisition of the knowledge of these mechanisms has been identifying the elemental causes (i.e., genes and their products) that mediate immunity and drug resistance. The identification of these genes is made possible by being able to transfer the genes or mutated forms of the genes into causative agents or surrogate hosts. Such an approach was limited in Mycobacterium tuberculosis by the difficulty of transferring genes or alleles into M. tuberculosis or a suitable surrogate mycobacterial host. The construction of shuttle phasmids, chimeric molecules that replicate in Escherichia coli as plasmids and in mycobacteria as mycobacteriophages, was instrumental in developing gene transfer systems for M. tuberculosis. This review will discuss M. tuberculosis genetic systems and their impact on tuberculosis (TB) research.
OVERVIEW OF KEY MUTATIONS THAT FACILITATED GENE TRANSFER IN M. TUBERCULOSIS
I distinctly remember sitting in my first bacterial genetics course, fascinated with the isolation of mutant bacteria. We had just read Francois Jacob and Jacques Monod’s (3) model of the lactose operon—an elegant hypothesis in which they postulated that Escherichia coli could selectively regulate the transcription of a set of genes required to degrade the disaccharide, lactose. They imagined the existence of a repressor protein that prevented the expression of the genes encoding lactose degradation when lactose was not available to the cell. The model was conceptualized and validated with the isolation and characterization of mutants that either had lost their ability to utilize lactose or lost their ability to regulate the degradation of lactose. Certainty of the causality of a phenotype by a mutation in a single gene was proven with gene transfer experiments (3). Key to this conceptualization was the isolation of the mutants with mutations in the specific genes. The lactose operon was a novel paradigm for gene regulation that arose from the study of bacterial mutants.
If we were to make similar advances in understanding how M. tuberculosis so successfully infects humans, we would need new tools to manipulate this once genetically intractable pathogen. I consider the following the key developments.
1. TM4::pHC79—introducing foreign DNA into mycobacteria
The shuttle phasmid phAE1 contained the cosmid pHC79 inserted in the nonessential region of the phage TM4 (4). I had hypothesized that I should be able to stably clone the entire TM4 genome into an E. coli cosmid and it would replicate in E. coli as a plasmid but replicate in mycobacteria as a phage. Furthermore, this chimeric molecule would be packageable in either bacteriophage lambda particles or TM4 mycobacteriophage particles. Although this first-generation shuttle phasmid was not useful for additional cloning experiments, it was a proof of principle that creation of shuttle phasmids was possible.
2. attLL5::L5::kan::attRL5—stable integration into mycobacterial chromosomes
This genetic alteration of mc26, in which an L5 shuttle phasmid had integrated a kanamycin resistance gene stably into the chromosome of Mycobacterium smegmatis, provided the first proof of principle that it was possible to use kanamycin and the aphgene as a selection system for mycobacteria (5). This insertion results when the attP site of the mycobacteriophage L5 integrates into the attB site of M. smegmatis or M. tuberculosis. The combination of DNA sequence analyses and genetic engineering of this region by Graham Hatfull’s lab led to the development of integration proficient vectors with which small or large fragments of foreign DNA could be stably integrated into the genomes of many different mycobacteria (6). These integration proficient vectors provided a means to stably integrate foreign antigens into bacille Calmette-Guérin (BCG) (7) or a means to screen cosmid-size fragments of the M. tuberculosis chromosome for virulence functions in mice by virtue of its stability and ease of use (8–10).
3. ept-1—plasmid transformation of mycobacteria
An efficient plasmid transformation (ept) phenotype allowed for the first plasmid transformation system of mycobacteria using pAL5000 plasmids and ept mutants of M. smegmatis (5, 11). As mentioned earlier, 25 years later we’ve now discovered that the ept phenotype results from a single point mutation causing a loss of function that normally prevents replication of pAL5000 plasmids. Despite not knowing the mechanism, the availability of a plasmid transformation system for M. smegmatis mc2155 provided a gene transfer system to facilitate the analysis of mycobacterial genes. It allowed for the development of plasmid expression vectors (12) and a system to analyze the functions required for pAL5000 replication (13–17). Importantly, it provided a surrogate host for the study of genes from the slow-growing pathogenic mycobacteria in a fast-growing, nonpathogenic mycobacterium which, unlike E. coli, had similar metabolic pathways allowing for the analyses of genes encoding complex carbohydrates (18) and previously undiscovered genes associated with resistance to isoniazid (INH) (19, 20), ethionamide (ETH) (19, 21, 22), and ethambutol (23, 24). Plasmid transformation was indispensable for identifying the activator of the prodrug pyrazinamide (25) and the common target for isoxyl and thiacetazone (26, 27). There are hundreds of papers that have used mc2155 as a system to study the biology of mycobacteria and their phages. In addition, it is noteworthy that bedaquiline, the first new TB drug to be FDA approved in 40 years, was identified using M. smegmatis mc2155, which was subsequently used to identify and validate the target (28).
4. leuD::IS1096—auxotrophic mutants of M. tuberculosis
Airborne pathogens present particular challenges in that they require biosafety level III (BSL3) containment. We reasoned that it might be possible to generate M. tuberculosis mutants that we could work with in a BSL2 laboratory if we introduced mutations that prevented the mutant from growing in mammals. This was the strategy that was used to biologically contain E. coli. We set out to delete the genes required to make diaminopimelic acid (Dap), an essential metabolite that is not synthesized by mammals. Moreover, Dap auxotrophs of E. coli undergo Dap-less death when exponentially growing cells are transferred from media with Dap to media without Dap. For E. coli, or Salmonella, the deletion of either of the genes encoding aspartokinase or aspartate semialdehyde dehydrogenase can be obtained in the presence of methionine, threonine, lysine, and Dap, but we were unable to obtain these mutants in M. smegmatis (29). In the process of testing whether there was a problem with recombination in this region of the M. smegmatis chromosome, we set up a screen to measure rare recombination events using the loss of the lacZ gene, encoding beta-galactosidase, and we serendipitously discovered a novel insertion element, IS1096, that was present in the M. smegmatis chromosome (30). This insertion element transposed with a simple cut-and-paste mechanism and was used to make an efficient tool for transposon mutagenesis that we used to generate mutants of BCG. We screened the library of insertions for auxotrophic mutants and found a leucine auxotrophic mutant in which the transposon had jumped into the leuD gene (31). What makes this auxotrophic mutant particularly noteworthy is that unlike the parental BCG strain, this mutant had lost its ability to replicate in mice. This was surprising, as Legionella, another intracellular pathogen, is naturally auxotrophic for several amino acids including leucine (32, 33), suggesting that leucine and other amino acids are not limited in the mouse. We tested a number of additional auxotrophic mutants of BCG and found that they lost their ability to replicate not only in immunocompetent mice, but also in immunocompromised mice lacking T and B cells, and yet still provided protection comparable to BCG against virulent M. tuberculosis challenge (34). The inability of these auxotrophic mutants of BCG to grow in mice suggested that they lived in a niche that differs from Legionella pneumophila, even though both are known to live in phagocytic vacuoles. Since it had been hypothesized that M. tuberculosis might escape into the macrophage cytoplasm, and BCG could not (35), it was unclear whether auxotrophic mutants of M. tuberculosis would be able to grow in mice. Leucine auxotrophs of M. tuberculosis failed to grow in mice as well (36). In fact, we discovered that M. tuberculosis mutants defective in making the amino acid lysine (37) or the vitamin pantothenate also were highly attenuated (38). After constructing double auxotroph of mutants of M. tuberculosis, we demonstrated that they are very safe and have the ability to generate protection comparable to BCG (39–42).
5. PH101—a conditionally replicating mutant of TM4
While shuttle phasmids provided a way to deliver foreign DNA into M. tuberculosis and other mycobacteria, since TM4 is a lytic phage, the recipient cell will be lysed, so we needed a way to prevent this cell death. We reasoned that we could isolate mutants of TM4 that could replicate at 30°C, but not at 37°C. To achieve this goal, we mutagenized TM4 phage and then screened plaques for those that replicated at 30°C, but not at 37°C. While nearly 100 plaques were identified that had this phenotype, all but one likely had a single mutation causing temperature-sensitive growth and thus reverted at frequencies of approximately 10−4 to 10−5. However, we identified one mutant that we named PH101 (ref), which reverted at a frequency of less than 10−8. We reasoned that this mutant phage must have contained at least two different point mutations that caused this phenotype. It is gratifying that reversion analyses and sequencing of the PH101 parent and revertants have demonstrated that point mutations in gp49 and gp66 are necessary and sufficient for this temperature-sensitive phenotype with the low reversion rate. While the function of the two proteins encoded by these two genes is yet unknown, this mutant phage has been a necessary component of efficient transposon mutagenesis, enhanced reporter phages, and specialized transduction.
TB TRANSMISSION ESTABLISHED TB CAUSALITY
In 1868, the cause of TB was unknown. Controversy followed the conclusion of Jean Antoine Villemin, first stated in his book Etudes sur la Tuberculose (43), that TB was caused by a virus. Most scientists at that time believed that TB was a hereditary disease that resulted in cancerous lesions. As a young physician in the French Army, Dr. Villemin had observed that many new recruits developed consumption (as TB used to be called) after living in confined quarters. He hypothesized that consumption was an infectious disease, a disease that was transmissible from one human to another by a virulent biological agent. He further postulated that these lesions were not cancerous, based on the observation that many of the lesions were “tubercules that looked like small, pearl-shaped globules or ‘matière caséeuse’”—a cheese-like matter (43). To test if these lesions contained a putative virus that caused consumption, Villemin isolated the tuberculous lesions (now known as granulomas) from patients that had died of consumption, cut the granulomas into pieces, and then transferred them to rabbits via subcutaneous injection. Invariably, these inoculated rabbits developed consumption-like disease, forming the tubercles he had seen in the consumptive human patients. Moreover, the transfer of tuberculous granulomas among rabbits resulted in the transmission of consumptive disease to the uninfected recipients. In contrast, the transfer of cancerous cells from deceased soldiers that had died of various neoplasms did not yield tuberculous lesions or disease.
Thus Dr. Villemin provided compelling evidence that TB was a transmissible infection, the first such published report of a communicable disease in humans. He concluded that TB was caused by a virus—a poorly defined entity at that time. The innovation of this work was the successful and reproducible transfer of infectious material from humans to animals. (Since this magnificent 600-page book is only available in French, we have relied on a translation from Dr. Catherine Vilchèze, who was impressed by the genuine passionate enthusiasm and excitement conveyed by Dr. Villemin for the scores of experiments he had performed to repeat the successful transfer.) Despite his conviction in his prescient conclusion, this work was not readily accepted by his peers.
Dr. Villemin’s transfer studies were noticed, reproduced, and extended by Dr. Robert Koch as described in his landmark presentation of “The Etiology of Tuberculosis” on March 24, 1882. Not only did Robert Koch reproduce the granuloma transfer experiments, but he was also able to develop (i) a staining procedure to visualize the tubercle bacilli in infected granulomas and (ii) a solid growth medium (bovine serum that was repeatedly heated and cooled) that allowed him to culture colonies of the tubercle bacilli from human tissue. By transferring the organisms that had been grown in pure culture to numerous types of animals, Dr. Koch established that TB was caused by tubercle bacilli and therefore unequivocally established that TB was an infectious disease (44).
Koch elegantly wrote, “To prove that tuberculosis is a parasitic disease, that it is caused by the invasion of bacilli and that it is conditioned primarily by the growth and multiplication of the bacilli, it was necessary: 1) to isolate the bacilli from the body; 2) to grow them in pure culture until they were freed from any disease product of the animal organism which might adhere to them; and, 3) to administer the isolated bacilli to animals to reproduce the same morbid condition which, as known, is obtained by inoculation with spontaneously developed tuberculous material.” We call this philosophical argument Koch’s postulate, whose fulfillment can directly link a pathogen as the causative agent to an infectious disease. (Many textbooks call this Koch’s postulates, but it should be the singular form postulate. The postulate or “deduced truth” is that the bacilli cause the specific disease if three conditions are met.) Since its publication, this has been the paradigm that establishes the causative agents of infectious diseases. The innovations of Koch’s work were in his development of bacteriological methods—staining and bacterial culture—that allowed him to clone the tubercle bacilli. Moreover, he extended the transfer experiments of Villemin by transferring isolated bacilli to allow for the unequivocal conclusion that TB is caused by tubercle bacilli. This knowledge meant that controlling TB involved not cancer therapy, but rather killing tubercle bacilli.
GENE TRANSMISSION ESTABLISHED PHENOTYPE CAUSALITY
In 1941, the molecular basis of genes was unknown. In fact, we did not know that a specific gene encoded a single polypeptide. The discovery of this important fact came from a gene transfer experiment. George Beadle and Edward Tatum established that a single enzyme was likely encoded by a single gene. They achieved this success by isolating mutants of Neurospora crassa that required specific vitamins (45). This accomplishment began with the hypothesis that it might be possible to isolate mutants of this fungus generated by X rays that would be defective for making a specific nutrient. By treating spores with X-ray radiation and plating for isolated clones, they were able to discover three mutants that required specific vitamins to grow. By performing a genetic cross with a wild-type strain, they were able to establish that the enzymatic defect was due to amutation in a single gene, allowing them to conclude that a single gene encoded a single polypeptide. Joshua Lederberg collaborated with Edward Tatum to isolate amino acid– and vitamin-requiring mutants of E. coli. By isolating strains of E. coli that contained at least two different auxotrophic mutations, it was possible to demonstrate that these bacteria could mediate genetic recombination (46). Gene transfer confirmed that these specific enzymatic defects were due to precise mutations in particular genes.
In 1928, Frederick Griffith, an epidemiologist and bacteriologist, made the serendipitous discovery of transformation, the first identified process of gene transfer in bacteria (47). He had been studying the virulence of the pneumococcal bacterium, now known as Streptococcus pneumoniae. He was investigating the basis of virulence with two colonial morphotypes of S. pneumoniae, a smooth (S) virulent strain and a rough (R) avirulent strain. He had made the observations that the injection of either the R strain or the heat-killed S strain into mice was not lethal. Surprisingly, if he mixed the heat-killed virulent strain with the rough avirulent strain and injected this mixture into a mouse, mortality was caused, but not by either individual component alone. An even greater surprise was that when he plated out the bacteria from the dead mice, the resulting strain no longer looked like the viable rough strain, but rather had been transformed to resemble the smooth strain. Griffith also showed that bacilli could revert from one phenotype back to the other. He concluded that some sort of “transforming” event had occurred that changed the avirulent strain into a virulent strain.
Dr. Oswald Avery, a leading pneumococcal researcher at the Rockefeller Institute for Medical Research, now Rockefeller University, was very skeptical of the studies and was convinced there must be some sort of contamination. However, researchers in his lab had confirmed the transformation process. This led to Dr. Avery’s extending the work to demonstrate that the transformation event can be selected for by treatment with an antibody in vitro. In their 1944 paper, Avery, MacLeod, and McCarty concluded from their data that the transforming principle was DNA, stating, “the active fraction contains no demonstrable protein, unbound lipid, or serologically reactive polysaccharide and consists principally, if not solely, of a highly polymerized, viscous form of deoxyribonucleic acid” (48). Acceptance that DNA, not protein, was genetic material would have to wait for the Hershey and Chase experiment using radiolabeled DNA, which revealed that the transfer of nucleic acid correlated with the transfer of genetic material (49). Again, the theme common to these seminal experiments was the transfer of the causative entity (much like in the experiments conducted by Koch that led to the establishment of the Koch’s postulate), which in this case was DNA.
The transforming principle transfer experiment of Griffith for S. pneumoniae parallels the Villemin transfer experiment in that both researchers discovered that specific phenotypes were the cause of a particular disease. The Avery, McLeod, and McCarty transfer experiment parallels the Koch transfer experiment because in both cases the fundamental cause of the disease or a virulent phenotype had been purified and transferred. This knowledge of causality has served to focus modern biology to build on this basic principle and elucidate the mechanisms by which DNA functions as genetic material that replicates and encodes RNA and protein products. By focusing on elucidating the structure of DNA, James Watson and Frances Crick (50, 51) provided an explanation for how genetic material was faithfully replicated. Moreover, this knowledge led to the central dogma of biology in which DNA is transcribed into RNA, which is translated into proteins (reviewed in reference 52).
As a graduate student, I was taught by Roy Curtiss III that the Avery, MacLeod, and McCarty experiment was a fulfillment of the molecular Koch’s postulate, which was brilliantly described by Stanley Falkow (53). In other words, it is the way in which we know if a phenotype is caused by a genotype. To paraphrase Robert Koch, to prove that a phenotype is caused by a genotype, it is necessary to (i) identify a mutant with an altered phenotype, (ii) clone the genotype, and (iii) transfer the mutant genotype into the wild-type strain and demonstrate that the recipient strain acquires the mutant phenotype. The gene transfer was observed in S. pneumonia because the bacteria had a natural transformation system. Although this transformation study established that DNA is genetic material, linkage analyses or DNA sequencing would be required to prove that a specific gene is required to make an enzyme to make a complex polysaccharide. Linkage analyses—conducted by measuring frequencies of recombination mediated by either conjugation of cells to cells (46) or DNA transfer using bacterial viruses (i.e., transduction)—first described in Salmonella by Norton Zinder (54) provided a means to identify specific genes and to transfer them. Thus, the three transfer systems in bacteria, of transformation, conjugation, or transduction, allowed for a rapid accumulation of basic biological knowledge by fulfilling the molecular Koch’s postulate.
ELUCIDATING THE MECHANISMS OF DRUG KILLING: THE STREPTOMYCIN EXAMPLE
Gene transfer, in combination with the ability of a bactericidal drug to select for independent mutants, constitutes an essential system for identifying drug targets, a critical first step in characterizing the mechanism of drug action. Knowledge of the mechanism of action of a drug provides the basis for optimizing the efficacy of the agent being studied. Equally as important is discovering the mechanism of resistance of the organism. Both mechanisms lead to the identification of new and improved chemotherapeutic agents and the development of rapid susceptibility testing. This knowledge can be acquired by the isolation and characterization of mutants that are resistant to the death-inducing action of drugs. Mutations causing drug resistance can be used to identify at least four classes of genes including those encoding (i) drug activators, (ii) drug targets, (ii) modulators of drug action, and (iv) drug-degrading enzymes. Isolates whose resistance to a specific agent is due to a single mutation provide isogenic sets of strains that can be rigorously compared in various biochemical, physiological, and cellular assays.
One of the questions that Robert Koch addressed was whether different bacteria cause different diseases or whether one or a limited number of bacteria cause many different diseases. Implicit within Koch’s postulate is the premise that if the three conditions are met, the conclusion is that the particular infectious disease under investigation is caused by the pathogen being examined. In parallel reasoning, the cloning of a particular drug resistance–conferring genotype or an allele by gene transfer experiments allows for the conclusion that the cloned gene is the basis for the specific resistance. Once a bacterium or virus has been identified and proven to be the causative agent for an infectious disease by fulfilling the Koch’s postulate, experiments aimed at comparing virulence, the pathological reaction, and immune responses elicited in the host among various specific pathogens are possible. Analogously, once an allele of a gene has been demonstrated to be the cause of a drug resistance phenotype by fulfilling the molecular Koch’s postulate, studies can be conducted comparing the biochemistry, molecular biology, and physiology of cells treated with the agent with those of others.
Streptomycin is a superb example of a drug in which genetic analysis led to an understanding of its mechanism of action of killing a bacterial cell and subsequently the cell’s mechanisms of resistance. This drug, discovered by David Schatz and Salman Waksman (55), was the first antimicrobial agent found to be highly active against the tubercle bacilli. It is also effective against Gram-negative and Gram-positive organisms (56, 57). In contrast to the modern approach to drug development, which is largely based on targeting essential gene products, streptomycin was identified by empirically screening bacteria from soil for antimicrobial activities. As a result of this method of discovery, neither its mechanism of action nor its target was known. Streptomycin was first used as a selective tool to probe for the development of specific mutations and hence to measure mutation rates under defined experimental conditions, similar to studies that used lytic phages (58). Elucidation of the target of streptomycin was not mentioned in these early papers, but the unambiguous nature of the high-level streptomycin resistance phenotype provided an attractive lead for further genetic studies. Interestingly, two types of mutations were identified when streptomycin- resistant mutants of E. coli were isolated (58). One class conferred high-level streptomycin resistance and the other, streptomycin dependence. Gene transfer experiments using either conjugation or transduction in E. coli were able to demonstrate that the three phenotypes (streptomycin susceptibility, streptomycin resistance, and streptomycin dependence) all mapped to a single gene, designated strA (58–61). Results of the gene transfer studies fulfilled the molecular Koch’s postulate, because linkage analysis provided evidence that the specific allele transferred via conjugation or transduction was responsible for the observed phenotype.
Spotts and Stanier grasped the significance of these gene transfer experiments because the streptomycin-resistant, -susceptible, or -dependent phenotypes of E. coli that were generated resulted from different alleles of a single gene (62). Based on biochemical analysis of these isogenic strains, they hypothesized that this gene likely produced a polypeptide or regulated the expression of a protein that was part of the E. coli ribosome and that mutations in specific domains of this target resulted in distinct structures that either bound or failed to bind streptomycin. This idea of a specific target was a revolutionary and unifying hypothesis. In support of this hypothesis, they cited the data of Erdos and Ullman (63, 64) that suggested that streptomycin inhibited protein synthesis in a sensitive strain but failed to do so in a resistant isolate. Erdos and Ullmann had used Mycobacterium friburgenesis for their study, a species for which there were no genetic tools, and thus they could not conclude that their resistance mutation mapped to a single specific gene and hence a specific target.
When Julian Davies isolated ribosomes from isogenic streptomycin-resistant or -susceptible strains that differed by a single allele, he was able to demonstrate that protein synthesis was inhibited by streptomycin treatment of the ribosomes from the susceptible but not from the resistant strain (65). Moreover, he was able to map the resistance or susceptibility phenotype to the 30S subunit of the ribosome. Five years later, Ozaki, Mizushima, and Nomura were able to isolate the specific protein (which had been encoded by strA) of the 30S ribosome subunit that mediated resistance to streptomycin (66). It would be another 21 years before the elucidation of the structure of the ribosome allowed for direct visualization of the streptomycin bound to the ribosome (67). In M. tuberculosis, streptomycin resistance also maps to the gene encoding the 16S rRNA (68), a result that not unexpectedly was not observed in E. coli, which has seven copies of the rRNA genes. In addition, the crystal structure of streptomycin bound to the ribosome revealed how specific mutations in the 16S rRNA would mediate resistance (68).
The mechanism of action of streptomycin is one of the most definitive in the history of the study of drugs aimed at defining the basis for the observed antimicrobial effect, because it was verified by genetic, biochemical, and X-ray crystallographic analysis. Clearly, the use of mutant isolation and the fulfillment of the molecular Koch’s postulate using the transfer systems of E. coli were seminal in these efforts.
TRANSFER OF GENES FROM PATHOGENIC MYCOBACTERIA INTO THE SURROGATE HOST E. COLI: GREAT PROMISE WITH LIMITATIONS
Transfer of any gene became relatively easy with the advent of recombinant DNA technologies (69–71) and the development of plasmid (72–76) and phage (77–80) cloning vectors for E. coli. The combination of recombinant DNA technologies with DNA sequencing technologies (81, 82) opened up entirely new approaches to the study of any organism, facilitating an explosion in new biological knowledge. While cloning technology offered great promise, it also represented potential new hazards. Would the addition of virulence genes from other pathogens into E. coli render this bacterium hazardous? This issue was discussed at a conference at Asilomar in 1973, which generated a set of guidelines and proposals to move forward in a safe and thoughtful way with experiments involving recombinant DNA technologies (83). The guidelines concerned safe practices for experimentation both with recombinant DNA and with physical and biological containment issues. The latter included engineering a bacterium that would be unable to replicate in mammalian hosts. Thus, the prototype strain χ1776 was genetically engineered to be unable to replicate outside the laboratory due to auxotrophic requirements for diaminopimelic acid, which is absent from mammalian hosts, as well as a mutation in thymidylate synthase that induced bacterial death when the organism was starved for thymidine (84). Moreover, the strain was found to be sensitive to killing by complement in human serum (85). Based on the safety experiences with E. coli of researchers throughout the world, guidelines were relaxed over the years. The concepts of generating strains of bacteria that would be unable to replicate in mammalian hosts were influential in developing strains of M. tuberculosis that could safely be used in a BSL2 laboratory (see below).
Despite the availability of recombinant DNA technologies, research on leprosy bacillus and the tubercle bacillus were limited until the second half of the 1980s. First, both of these mycobacteria are BSL3 pathogens, whose manipulation mandates a tedious biocontainment environment that significantly curtails efficiency. More limiting was the fact that the leprosy bacillus cannot be grown in any artificial media. It was not until 1960 that Charles Shepard demonstrated that the leprosy bacillus procured from humans could be grown in mouse footpads (86). In this seminal work, Shepard showed an increase from 1,000 acid-fast bacilli to 1 million in 6 months’ time. Despite this advance, this system, which required growth monitoring by microscopic inspection of tissue samples, is limited by its inefficiency in terms of time and bacterial yield, the latter significantly hampering efforts to generate genomic libraries. Subsequently, it is debated whether the Shepard experiments fulfilled Koch’s postulate for the leprosy bacillus since the growth in the mouse does not reproduce any neurological symptoms typically associated with this pathogen. Nevertheless, the Shepard model provided a means to culture various M. leprae strains and to test drugs and vaccines (87).
The next significant advance for leprosy research was in 1971, when Wilemar Kirchheimer and Eleanor Storrs demonstrated that the leprosy bacillus replicated in nine-banded armadillos (88). In contrast to the mouse footpad model, infection of the nine-banded armadillo reproduces much of the clinical pathology associated with leprosy. Charles Shepard not only reproduced this work, but also showed it was possible to isolate 1010 to 1011 leprosy bacilli from the armadillo liver following a two-year infection. Under the supervision of my comentors Josephine E. Clark-Curtiss and Roy Curtiss III, I had generated cosmid genomic libraries of M. leprae. These reagents enabled attempts to characterize specific biosynthetic pathways of M. leprae by complementation studies using auxotrophic strains of E. coli (89, 90). I failed to obtain complementation with these cosmids and was only later successful when I used a promoter that was highly active in E. coli, allowing me to complement a mutation in the citrate synthase gene (91). The conclusions from these studies posited that promoters of mycobacterial genes did not function well in E. coli. Numerous other groups had found that genes from organisms with a high guanine plus cytosine content failed to express well in E. coli. To overcome this limitation, Richard Young and Ron Davis developed an elegant strategy to express mycobacterial genes in E. coli using a highly active promoter from λgt11, a bacteriophage lambda vector (92), to efficiently express proteins in E. coli. This bacteriophage-based system enables identification of major protein antigens of M. leprae (93) and M. tuberculosis (94) that elicit human humoral immune response by screening of the expression library using sera from infected individuals. The identification of these antigens provided novel opportunities to study the immune responses in leprosy and TB. However, identification of the genes encoding the targets of specific antibiotics used for the treatment of TB and leprosy—or elucidation of mycobacterial virulence factors—would still need the development of a gene transfer system in mycobacteria.
INTRODUCTION OF FOREIGN DNA INTO MYCOBACTERIA: SHUTTLE PHASMIDS AND THE DEVELOPMENT OF A PLASMID TRANSFORMATION SYSTEM FOR M. SMEGMATIS
Mycobacteria are substantially different from E. coli, and there were numerous questions that were unanswerable using E. coli as a surrogate host. For example, in 1987, we did not know (i) the targets of the TB-specific drugs INH, ETH, ethambutol, thioacetazones, and pyrazinamide; (ii) the genetic basis of acid-fast staining; (iii) if it was possible to isolate auxotrophic mutants of M. tuberculosis; or (iv) the basis for attenuation of BCG. This lack of knowledge was due to the inability to fulfill the third condition of the molecular Koch’s postulate, because at the time, an effective DNA transfer system for M. tuberculosis did not exist. For example, while it was feasible to isolate INH-resistant mutants of M. tuberculosis by plating a large number of wild-type bacilli on INH-containing plates, the lack of a DNA transfer system for the tubercle bacillus at the time precluded the mapping of the resistance phenotype to a specific gene. Although generalized transduction (95) and conjugation (96, 97) for fast-growing M. smegmatis had been described, the inability to transfer genes from M. tuberculosis or M. leprae into M. smegmatis limited the use of these tools for analyzing these pathogens. Even if conjugation or generalized transduction had been discovered for M. tuberculosis or BCG, the slow replication time of these strains of 16 to 24 h would have made genetic analysis difficult. An effective genetic system that enables transfer of genes into the slow-growing M. tuberculosis was needed for study of this global pathogen.
The cloning of genes into mycobacterial plasmids offered the simplest approach to identify drug targets or virulence factors. Although mycobacterial plasmids were discovered in 1979 (98) and 1985 (99), no successful transformation of any mycobacterial cell had been achieved when I started in the laboratory of Barry Bloom in 1985. Barry had wanted to develop the TB vaccine strain, BCG, into a recombinant vaccine vector, and I wanted to develop a system by which the genotype-phenotype causality can be proven according to the molecular Koch’s postulate. These shared goals both required the development of gene transfer for M. tuberculosis. Plasmid transformation was the obvious choice, but numerous individuals had told me they had been unsuccessful. While transformation in M. tuberculosis was the ultimate goal, the process was not straightforward. First, studies of M. tuberculosis require BSL3 containment. Second, M. tuberculosis has a generation time of 16 to 24 h, requiring 3 to 4 weeks to form colonies on a plate. This slow growth was enough of a deterrent to spawn the consideration of the use of a more rapidly growing non-BSL3 mycobacterium. For these reasons, M. smegmatis was an attractive surrogate host for the study of genes from M. tuberculosis and M. leprae. It is fast-growing with a generation time of 2.5 to 4 h and thus yields colonies from single cells in 3 to 4 days; it is nonpathogenic, and the ATCC607 strain is an excellent host for mycobacteriophage isolation. Dr. Wilbur Jones from the CDC shared this strain with me as well as the mycobacteriophages D29 and 33D.
The simplest vector to imagine would have been to construct an E. coli–mycobacterial shuttle plasmid that could transform E. coli and mycobacteria including M. smegmatis. While the specific DNA fragment required for replication of a plasmid in these host cells was not known, the construction of a random insertion of an E. coli plasmid into a mycobacterial plasmid was easily done, but this did not yield transformants into M. smegmatis. We reasoned that the failures of others could have been due to four possible limitations: (i) the inability of naked DNA to enter the mycobacterial cell, (ii) degradation by a restriction system of the E. coli DNA seen as foreign, (iii) failure to have a functional selection system (appropriate concentrations of antibiotic and sufficiently high levels of expression of the selectable marker gene), and/or (iv) the inability of the mycobacterial plasmid to replicate and segregate into M. smegmatis cells. By making my own bacteriophage lambda in vitro packaging mixes in the Curtiss laboratory, I had come to appreciate the utility and power of phage vectors because phages had developed highly efficient means to deliver DNA into a cell. I therefore decided to focus on developing recombinant DNA phage vectors for the genetic manipulation of mycobacteria. Using the protoplast methodologies developed by Mervyn Bibb and David Hopwood (100), I focused on optimizing DNA transfer into M. smegmatis by assaying infectious centers formed after transfecting naked D29 phage DNA (4). The advantage of this choice was that phage plaques could be observed in 24 h, allowing me a rapid readout for optimizing protoplast generation protocols and to conduct polyethylene glycol (PEG) lot evaluations. I routinely obtained greater than 1,000 plaque forming units (PFU) per microgram of DNA (4). As I was optimizing my protoplast regeneration methods from the culture of M. smegmatis ATCC607 (which I had stocked as mc21; I had decided to name my mycobacterial culture collection the mc2 collection in honor of the Einstein equation), I observed three colonial morphotypes that I cloned and stocked, the predominant one being orange rough (mc26), with the other two being white rough (mc221) and orange smooth (mc222). I chose to use the predominant mc26 for subsequent experiments.
Having established an efficient transfection assay, I decided to focus on the TM4 mycobacteriophage (101) that had been isolated from Mycobacterium avium in the laboratory of Patrick Brennan and was likely a temperate phage. I reasoned that a temperate phage vector could be used to introduce a stable selectable marker gene into the mycobacterial genome. By analyzing the restriction digests of TM4 phage DNA, I had discovered the phage was very similar to 33D, had a genome size of approximately 54 kb, and possessed cohesive ends. To construct a mycobacteriophage cloning vector patterned after what had been done with bacteriophage lambda, a unique restriction enzyme site would have to be introduced in a deleted nonessential region of the TM4 genome. Since I had discovered in my doctoral work that mycobacterial DNA did not express well in E. coli, I reasoned I could clone an entire mycobacteriophage genome into an E. coli plasmid and it would not kill the E. coli cell. Moreover, I reasoned that if I included a bacteriophage lambda cos site, I would have a shuttle phasmid—a chimeric molecule that had four important attributes that would allow it to (i) be packaged into bacteriophage lambda particles, (ii) be packaged into mycobacteriophage particles, (iii) replicate in E. coli as a plasmid, and (iv) replicate in mycobacteria as a phage (Fig. 1).
FIGURE 1.

Specialized transduction is outlined as follows: the center plasmid represents the shuttle phasmid phA159, which contains 90% TM4 phage DNA and 10% plasmid DNA. The stars mark the sites of the mutations in the TM4 genome. The nonessential genes that are deleted to create the shuttle phasmid are noted in the picture, flanked by PacI sites. This site can be replaced with one of three things: (i) a reporter gene such as green fluorescent protein (GFP), (ii) an allelic exchange substrate (AES) that contains an antibiotic resistance marker, or (iii) a transposase gene to facilitate transposon mutagenesis. Going counterclockwise in this schematic, the recombinant cosmid can be packaged into phage heads using an in vitro packaging mix, and the subsequent phages can be used to transduce E. coli to create E. coli transductant colonies. Going clockwise from the shuttle phasmid, one can transfect M. smegmatis mc2155 at 30°C to yield plaques on an M. smegmatis lawn, resulting from lysis of the cells. The plaques can then be purified and amplified to obtain a high-titer phage lysate that can subsequently be used to transduce any mycobacterial species. doi:10.1128/microbiolspec.MGM2-0037-2013.f1
To achieve this goal, TM4 phage DNA was ligated to form long concatemers, partially digested with a frequent-cutting DNA restriction enzyme, ligated to a bacteriophage lambda–based cosmid, in vitro packaged, and transduced into E. coli. Restriction analysis of individual E. coli cosmid clones showed I had generated a library of cosmid constructs containing 45-kb fragments of the TM4 genome. From the TM4 viewpoint, I had inserted cosmids at random sites around its genome with concomitant small deletions. I hypothesized that shuttle phasmids could be identified by transfecting the cosmid library into M. smegmatis protoplasts and assaying for plaques because I reasoned that the only molecules that could yield a plaque would be those in which the cosmid had inserted in the nonessential region of TM4. To my astonishment, the restriction fragment analysis of the DNA isolated from these plaques revealed the presence of the parental TM4 DNA with no cosmid. After reflecting on this result, I deduced that the wild-type TM4 phage had been generated by two or more TM4-cosmid hybrid molecules that underwent recombination to generate wild-type TM4 DNA. (It is worth noting that this high frequency of recombination yielding wild-type TM4 plaques is likely a result of a recombination function within TM4, as we have not observed this phenomenon with D29, L5, or DS6A. The possibility of this recombination function prompted us to develop specialized transduction, described in a later section, with the hope that the TM4 recombination functions could promote homologous recombination events.) Plaque hybridizations revealed that 4% of the plaques hybridized with the cosmid DNA, and these recombinant molecules were bona fide shuttle phasmids that could replicate in E. coli as a plasmid and in a mycobacterium as a phage (4). The mycobacteriophage-packaged cosmids readily infected BCG and M. tuberculosis (4).
These TM4 shuttle phasmid studies yielded important new knowledge. First, they provided the means to introduce foreign DNA into M. smegmatis, BCG, and M. tuberculosis for the first time. Second, they demonstrated not only that transfection of M. smegmatis protoplasts was highly reproducible, but also that DNA molecules propagated in E. coli were not being degraded by a restriction modification system of M. smegmatis.
Using the first-generation TM4 shuttle phasmids, a gene encoding kanamycin resistance was cloned into the shuttle phasmid in E. coli. This construct, however, failed to yield stable kanamycin-resistant transductants in M. smegmatis. We concluded that the TM4 shuttle phasmid was not able to lysogenize, and I started screening for mycobacteriophages that could site-specifically integrate into mycobacterial chromosomes. Numerous groups had identified mycobacteriophages that formed turbid plaques on M. smegmatis, so I obtained many such phages, characterized their genomes and screened them for the ability to site-specifically integrate into mc26. The mycobacteriophages L1 and L5 had been well characterized by Margaret Sellers (7). I demonstrated by performing a Southern blot on an L5 lysogen of mc26 that this phage clearly integrated site-specifically into M. smegmatis (5). It was because of this result that Graham Hatfull decided to determine the DNA sequence of this particular phage (102). Scott Snapper and I generated shuttle phasmids from L5 and demonstrated that these could transfer a kanamycin resistance gene stably into the chromosome of mc26 (5). This result allowed us to establish the concentration of kanamycin to be used to select for the presence of a newly introduced kanamycin resistance gene. We had then overcome three of the four limitations we proposed to be possible reasons for the difficulty in obtaining M. smegmatis transformants.
Tobias Kieser had made a library of insertions at random sites in the 5-kb plasmid pAL5000, which had been isolated from Mycobacterium fortuitum (99) with an E. coli plasmid bearing a kanamycin resistance gene. The use of a library was important because the DNA sequence that mediated plasmid replication was not known. Having the knowledge that we could introduce DNA into mycobacteria, not worry about restriction, and use kanamycin as a selection, Scott Snapper set up to transform M. smegmatis mc26 using this library. The one concern of using protoplasts was that regeneration could be an inefficient process. At the time, the technique of electroporation was being used to transform bacteria (103), thereby circumventing the regeneration step. The electroporation of mc26 cells with recombinant DNAs of the E. coli plasmid pAL5000 chimeric library yielded three kanamycin-resistant transformants. From these M. smegmatis transformants, plasmid DNA was isolated, revealing extrachromsomal chimeric plasmids. These plasmids yielded transformants in both E. coli and BCG (5), thus validating the first plasmid transformation system for mycobacteria. Scott Snapper and I hypothesized that the transformants contained a mutation that allowed for replication of the pAL5000 replicon. To test this hypothesis, we cured one of the original transformants (namely, mc2154) of its plasmid by propagating the strain in the absence of kanamycin. A kanamycin-sensitive clone was stocked as mc2155. Astonishingly, transformation of mc2155 yielded 104 to 106 transformants compared to mc26. A series of experiments allowed us to demonstrate that this efficient plasmid transformation phenotype was not due to DNA uptake, restriction, or selective expression of kanamycin resistance genes. We concluded that it was due to a mutation that specifically affected the replication of pAL5000 plasmids (11).
Despite not knowing the mechanism underlying the significantly enhanced transformation efficiency observed, the availability of mc2155 revolutionized the field of mycobacteria because it provided the surrogate mycobacterial host to study the genes of the pathogenic mycobacteria as well as a genetically tractable organism for studying mycobacterial biology. It is gratifying that 25 years after the discovery of mc2155, we have just recently found a single-point mutation that confers the efficient plasmid transformation phenotype. The mutation causes a loss of function of the DNA binding protein that prevents the replication of pAL5000 (M. Panas, P. Jain, and W.R. Jacobs, Jr., submitted). Shuttle phasmids showed us that M. smegmatis does not restrict E. coli DNA and enabled the first successful transfer of a selectable marker gene to M. smegmatis, leading to the development of a plasmid transformation system for mycobacteria.
SECOND GENERATION SHUTTLE PHASMIDS: REPORTER MYCOBACTERIOPHAGES, TRANSPOSON DELIVERY, AND SPECIALIZED TRANSDUCTION
Conditional Replication and PacI Excisable Cosmids
Shuttle phasmids packaged in TM4 mycobacteriophage particles facilitate the transfer of DNA into M. tuberculosis, M. smegmatis, and many other mycobacteria, because it simply requires a phage infection. One of the limitations of this system was that TM4 is a lytic phage that lyses and kills the infected cells. In retrospect, it was fortuitous the TM4 had lost its ability to be a temperate phage because it allowed us to focus on delivering DNA transiently. Sequence analysis of the Cluster K family phages has revealed that the genes encoding the integrase and repressor genes (104) are deleted from TM4, thereby making it unable to stably integrate into the mycobacterial chromosome. Conditionally replicating lytic phages, such as repressor and integrase-deleted mutants of bacteriophage lambda, have proven to be very useful for delivery of transposons to E. coli (105). We reasoned that we could isolate mutations that allowed the TM4 mycobacteriophage to replicate at 30°C but not at 37°C. Stoyan Bardarov, Jordan Kriakov, and Christian Carriere had screened a large number of TM4 mycobacteriophage mutants for those that were not able to replicate at 37°C but were able to replicate at 30°C and reverted at frequencies of less than 1 in 10 million (106, 107). One such phage mutant, named PH101, was identified out of hundreds that were temperature-sensitive for growth and reverted at frequencies of 1 in 10,000. PH101 has recently been sequenced and found to have nine independent point mutations, of which two were found by Graham Hatfull and colleagues to be necessary and sufficient for the low reversion frequency temperature-sensitive phenotype (see Fig. 1).
Since it was desirable to be able to test a variety of different transposons for mutagenesis, we developed a very simple system for replacing the cosmid that had been used to create new TM4 shuttle phasmids by flanking it with unique restriction enzyme sites. We took advantage of the fact that the restriction enzyme PacI recognizes the sequence TTAATTAA, which is rarely found in mycobacteriophages that generally (but with some notable exceptions; see reference 150) have a GC content of greater than 60%. Shuttle phasmids are constructed by partial digestion of concatemerized phage genomes with the frequent-cutting restriction enzyme Sau3A. These fragments are then ligated to a bacteriophage lambda cosmid, pYUB328, which has a unique restriction enzyme site that is compatible with Sau3A like BamHI or BglII. We engineered this cosmid to have PacI sites flanking the unique BamHI or BglII site, and as such, shuttle phasmids generated with pYUB328 allowed easy excision and replacement with other cosmids. The ability to package the shuttle phasmids into bacteriophage lambda heads makes the cloning of any cosmid with a unique PacI site a relatively easy construction. The transfection of the resulting shuttle phasmid into M. smegmatis allows for the generation of mycobacteriophage particles that can infect most other mycobacterial strains. The conditionally replicating TM4 phage provides a means to deliver virtually any DNA fragment in a way that does not kill the recipient cells. We have utilized these shuttle phasmids to deliver reporter genes, transposons, and allelic exchange substrates to develop novel diagnostic tests, transposon libraries, and specialized transductants (Fig. 1).
Reporter Mycobacteriophages for Rapid Drug Susceptibility Testing of M. tuberculosis
Increasing numbers of drug-resistant M. tuberculosis strains are compromising first-line therapies. Unfortunately, determination of drug resistances can require a minimum of 6 to 12 weeks if done by culture methodologies. The recent development of a molecular beacon-based test has made diagnosis of M. tuberculosis infection and rifampin resistance available in 90 min to 48 h, but knowledge of viability and resistance to other first-line drugs is also important for proper treatment. Shimon Ulitzer and Jonathan Kuhn had proposed the idea of introducing a bioluminescence gene to identify bacteria in any sample (108). Since the TM4 shuttle phasmids infected M. tuberculosis strains and had a cloning capacity of over 5 kb of DNA, it was possible to clone the firefly luciferase gene into a TM4 shuttle phasmid to develop luciferase reporter mycobacteriophages (109). In the presence of exogenous luciferin, we were able to detect light production from M. smegmatis or M. tuberculosis cells within 30 min following the addition of the phage. We reasoned that since the luciferase reaction required ATP for light production, luciferase reporter mycobacteriophages would be great tools for assessing drug susceptibilities. Rather than waiting a month for M. tuberculosis cells to grow, then another month to do drug susceptibility testing, we hypothesized that it would be possible to assess drug action by incubating samples of M. tuberculosis cells with or without drugs and then adding the luciferase reporter mycobacteriophage. If the drug was effective, we reasoned that no light would be emitted. If the strain was drug resistant, it would become luminescent.
This technique works very well in ideal laboratory conditions but has proven challenging in a clinical laboratory in the developing world (110–114). To circumvent some of these issues, we made a number of innovative improvements including the development of the “Bronx Box,” which used a Polaroid film–based assay (115) and then the isolation of temperature-sensitive mutant that prevented the killing of the infected cells (107). The incorporation of the gene encoding the green fluorescent protein (116, 117) with a highly expressed phage promoter (118) has provided the means to visualize individual cells and the potential to measure fractional drug resistance. We are collaborating with Dr. Alex Pym at the K-RITH laboratories in Durban, South Africa, to test the utility of these next-generation fluorophages for rapidly assessing drug susceptibilities of patient samples.
Transposon Mutagenesis
Transposons, genetic elements that can jump from one piece of DNA to another, have proven to be indispensable tools for the genetic analysis of organisms, because they generate random insertions into genes, thereby creating a mutation that often inactivates the target gene (105). Transposition was first observed in mycobacteria in the laboratory of Brigitte Gicquel using a new transposon that they had identified in M. fortuitum (119). The Gicquel laboratory went on to develop a temperature-sensitive plasmid that replicated at 30°C but not at 37°C as a system for efficient transposon mutagenesis (120). Unfortunately, these initially isolated transposons did not transpose by a simple cut-and-paste mechanism or in a random manner, limiting transposon mutagenesis as a genetic tool. Serendipitously, while trying to understand the inability to disrupt the gene encoding aspartate semialdehyde dehydrogenase, Jeffery Cirillo discovered a novel insertion element in M. smegmatis, which we named IS1096 (30). IS1096 is 2.3 kb and possesses two open reading frames (ORFs). By cloning a gene encoding kanamycin resistance into various sites within IS1096, we were able to identify which gene was responsible for transposition and also to generate a transposon library of BCG (31). Following transformation of the IS1096 transposon, the cells were plated on media with Casamino Acids, and then the resulting colonies were screened for mutants that failed to grow on minimal media. Three auxotrophic mutants were found: two were leucine auxotrophs and one was a methionine auxotroph. These transposon mutants and the illegitimate recombination mutants discussed below were the first auxotrophic mutants isolated for BCG or M. tuberculosis. The studies also established the IS1096-derived transposons as an excellent system for transposon mutagenesis.
Since phages can deliver their DNA genomes to every cell in a population of bacteria, the goal of developing a conditionally replicating phage that could deliver a transposon transiently to every cell, but not kill the infected cells, was highly desirable. As described above, we isolated PH101, a temperature-sensitive mutant of TM4 that failed to form plaques at 37°C and had a low reversion frequency. It is worth noting that, in addition to PH101, we isolated numerous independent temperature-sensitive mutants of TM4 and D29 shuttle phasmids, all with reversion frequencies of less than 1 in 100,000, that all proved unsuitable for transposon mutagenesis experiments. Two shuttle phasmids, phAE87 and phAE159, were constructed using the PacI excisable cosmid for efficient transposon mutagenesis. The shuttle phasmid phAE87 was used to make our first library of thousands of transposon mutants in M. tuberculosis with Tn5367 (an IS1096-derived transposon [106]). The shuttle phasmid was useful for generating transposon libraries in M. avium (121), Mycobacterium ulcerans (122), Mycobacterium paratuberculosis (123), and Mycobacterium abscessus (L. Kremer, in press). Michael Glickman screened a library of transposon mutants of M. tuberculosis and identified one that failed to cord, a property that has long been associated with virulence (124). Using the clever strategy of signature-tagged mutagenesis developed by the laboratory of David Holden (125), in which bar codes were incorporated into transposons to screen for underrepresented mutants, Jeffery Cox used tagged Tn5367 to identify a novel lipid secretion system for M. tuberculosis (126). Sequence analysis of a large number of transposon insertions revealed a bias in IS1096 transposition, but it was still useful in revealing a number of new genes required for virulence (127). John McKinney’s laboratory used this system to screen for M. tuberculosis counter-immune mutants (128). The selection for underrepresented clones was improved by Christopher Sassetti and Eric Rubin by cloning the Himar transposon into the conditionally replicating shuttle phasmid phAE87 and performing TraSH (transposon site hybridization) (129). TraSH has been used extensively to identify the essential genes under various growth conditions in vitro (130) and in vivo (131). Common to all the transposon studies in slow-growing mycobacteria is the use of the conditionally replicating shuttle phasmid to generate transposon libraries.
Specialized Transduction and Linkage Cotransduction
Phages were discovered to be capable of transferring DNA from one strain to another to repair auxotrophic mutations. This process of transduction was characterized as being generalized if a single phage could transfer genes to a diverse set of genes around the chromosome as for P22 of Salmonella (54) and P1 of E. coli (60). In contrast, lysates induced from bacteriophage lambda lysogens were found to be able to specifically transduce genes involved the galactose utilization (132). This was called specialized transduction. Generalized transducing phages package by headful mechanisms and occasionally package genome fragments of the bacterial chromosome. Thus, in lysates of generalized transducing phages, a small percentage of phage particles exist that contain headful packaged lengths of the bacterial chromosome. In contrast, because specialized transducing particles from lambda phage in E. coli are made from aberrant excisions of the integrated lysogenized phage DNA, these rare particles only contain a small fraction of the chromosome adjacent to the integrated phage. For generalized or specialized transduction, the DNA from the bacterial chromosome has the ability to recombine by homologous recombination into the recipient cell. We hypothesized that it should be possible to deliver homologous recombination substrates efficiently to the M. tuberculosis chromosome and select for the introduction of precise null deletions of any gene of any mycobacteria (133). We demonstrated the proof of principle by disrupting numerous genes encoding amino acid or vitamin biosynthetic functions in M. tuberculosis, BCG, and M. smegmatis (133) and the gene encoding a mycolic acid biosynthesis enzyme that is required for the cording phenotype of M. tuberculosis (124).
The robustness of specialized transduction was demonstrated with the deletion of not only a single gene, but also all 11 genes of the esx1 locus (the primary attenuating mutation of BCG) in three different M. tuberculosis strains and M. bovis (134). Ken Stover’s group had performed subtractive hybridizations and found many deletions that were absent from BCG yet present in the M. tuberculosis genome. The one deletion that was common to all the BCG strains was RD1, later named esx1 (135). Tsungda Hsu had been successful in obtaining one deletion mutant of this region in M. tuberculosis that was unmarked by a plasmid transformation. He was trying to re-create the primary attenuating mutation of BCG that had been hypothesized to be the deletion of esx1 since it is common to all BCG strains. But Drs. Calmette and Guérin had isolated this mutant from Mycobacterium bovis, so Dr. Hsu wanted to make the strain in M. bovis, not just M. tuberculosis. Dr. Hsu performed over 100 independent transformations with M. bovis over a 5-month span, and although he obtained hundreds of hygromycin-resistant colonies, he obtained no knockouts of esx1. We were unsure if specialized transduction would be successful in making such a large deletion. To our delight, the esx1 deletion was successfully constructed in three M. tuberculosis strains (H37Rv, Erdman, and CDC1551) as well as in M. bovis (Fig. 2). All four of these strains had demonstrated a marked attenuation in both immunocompetent C57BL/6 mice and immunocompromised SCID mice and could be fully complemented back to virulence with a cosmid that spanned this region (134). Interestingly, we had shared our SCID data and the complementing cosmid with Stewart Cole, and he transformed BCG and showed that it was able to restore virulence in SCID mice but not in immunocompetent mice (136). Specialized transduction allowed us to fulfill the molecular Koch’s postulate for the primary attenuating mutation of BCG.
FIGURE 2.

Generation of mutants in the RD1 region of M. tuberculosis and M. bovis. (A) Schematic of M. tuberculosis H37Rv RD1 region showing predicted NcoI sites. Arrows at the top represent the genes in this region. UFSs and DFSs used to generate the knockout are indicated as filled bars above the grid line. Each increment in the grid line represents 1 kbp. The RD1 sequence deleted from M. bovis BCG is represented by an open bar spanning from Rv3871 to Rv3879c. The site of the insertion of transposon Tn5370 is also indicated. (B) Southern analysis of the NcoI-digested genomic DNA isolated from the wild type and the ΔRD1 mutants generated by using specialized transduction in M. tuberculosis and M. bovis. Lane 1, M. tuberculosis H37Rv; lane 2, M. tuberculosis H37Rv ΔRD1; lane 3, M. tuberculosis Erdman; lane 4, M. tuberculosis Erdman ΔRD1; lane 5, M. tuberculosis CDC1551; lane 6, M. tuberculosis CDC1551 ΔRD1; lane 7, M. bovis Ravenel; lane 8, M. bovis Ravenel ΔRD1. The probe used in the Southern analysis was either DFS (left), demonstrating the deletion of RD1, or IS6110-specific (right). The IS6110 probe is used to characterize the four strains. Reprinted with permission. doi:10.1128/microbiolspec.MGM2-0037-2013.f2
Specialized transduction also enabled us to transfer a specific point mutation within an essential gene by linkage cotransduction. As shown in the streptomycin section above, conjugation or generalized transduction had allowed investigators to transfer specific mutant alleles within a single defined gene by linking this allele to some other mutation. The end result was the generation of isogenic strains that differed by a single point mutation. We had identified a single point mutation in the inhA gene in M. smegmatis and had linked that mutation to a kanamycin resistance gene on a cosmid, transformed it into INH-susceptible M. smegmatis, and selected for kanamycin resistance transformants. By this method, we had linked a kanamycin resistance gene to inhA and then generated INH-susceptible or INH-resistant M. smegmatis strains. Due to inefficient allelic exchange in M. tuberculosis and the high degree of illegitimate recombination, this approach was not possible in M. tuberculosis. However, by generating a specialized transducing phage in which we had genetically engineered a hygromycin resistance gene closely linked to the inhA Ser 94 Ala allele, we were able to obtain hygromycin-resistant transductants that yielded INH-resistant or INH-susceptible phenotypes (Fig. 3). Thus, we were able to generate isogenic strains of M. tuberculosis that differed by a single point mutation in the inhA allele. Specialized transduction allowed us to prove that a single point mutation within an essential gene caused an INH-resistant phenotype, consistent with the hypothesis that InhA was the target of INH and ETH (137).
FIGURE 3.
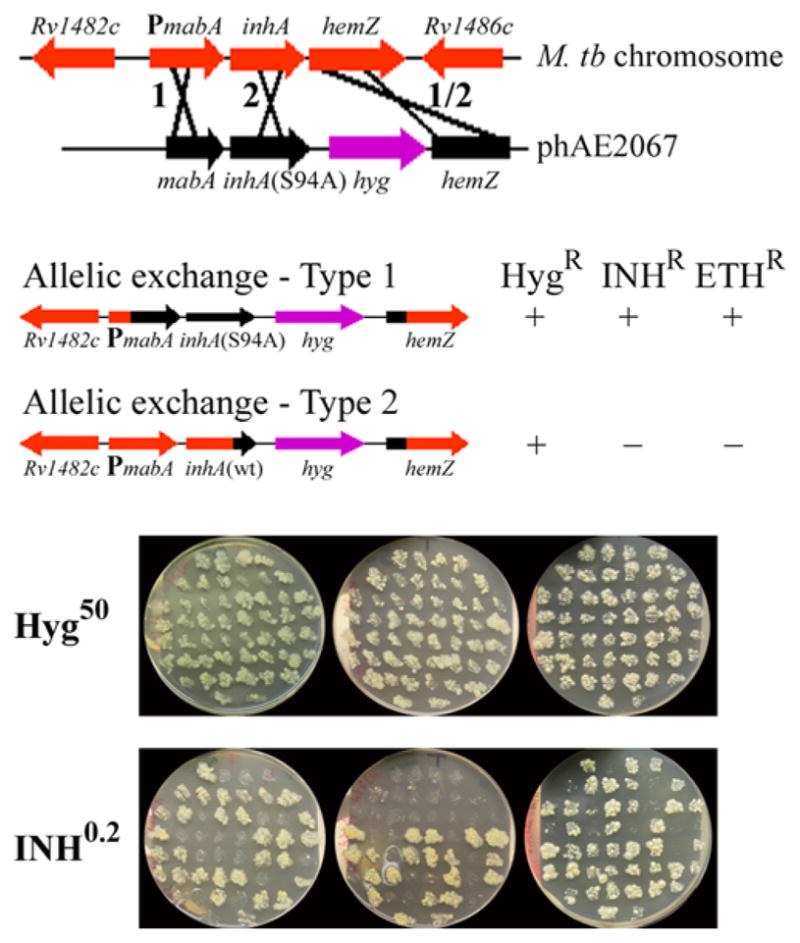
(A) Schematic representation of the specialized transducing phage. A replicating shuttle phasmid phAE2067 containing mabA, inhA carrying the S94A mutation, a hyg resistance cassette, and hemZ was used to transduce M. tuberculosis (M. tb). The two possible sites of recombination are marked 1 and 2. (B) The recombination can occur either before the point mutation (crossover type 1), resulting in an INH-resistant and ETH-resistant recombinant carrying the S94A mutation, or after the point mutation (crossover type 2; the strain contains a wild-type inhAgene). (C) Individual M. tuberculosis H37Rv inhA(S94A) transductants (n= 150) were screened by picking and patching onto plates containing either hygromycin (50 μg/ml) or INH (0.2 μg/ml). Reprinted with permission.doi:10.1128/microbiolspec.MGM2-0037-2013.f3
HIGH-THROUGHPUT SPECIALIZED TRANSDUCTION
The generation of the complete genome sequences of a bacterium, the first being Haemophilus influenzae (138), altered the way we investigated organisms. The first eukaryotic organism to have its genome fully sequenced and annotated was Saccharomyces cerevisiae (139). From such a sequence, it was possible to predict the number of ORFs and the numbers of functional RNA molecules. These predictions came from our understanding of genes, gene structures, ORFs, and operons in bacteria, primarily in E. coli. Although it was possible to assign putative functions to a subset of ORFs based on homology to related genes with proven functions, 40% of the ORFs in the H. influenzae genome had no known homology. It is important to note that all functional phenotypes are concluded to be assigned to a specific gene by the fulfillment of the molecular Koch’s postulate. Functions had been identified by isolating mutants with defined phenotypes in the organism of interest and then proving that the specific phenotype was caused by a specific genotype by performing a gene transfer.
The most direct way to determine the function of a gene from an organism is to generate a precise null deletion of an ORF from the organism with the concomitant introduction of a selectable marker gene. Such mutated alleles can be readily made in vitro using PCR (140, 141). Again, the operative theme is gene transfer, where a mutated allele is introduced into a chromosome to generate a mutant strain. S. cerevisiae was the first organism for which a complete set of precise null deletion mutants were made, primarily because it has a highly efficient allelic exchange system that enabled in vitro null deletion alleles containing a selectable marker gene to be readily introduced into the genome (142). Another property that made S. cerevisae attractive for this analysis was that the strain was a donor diploid strain. By allowing it to sporulate and form haploid spores, tetrad analyses made it possible to test whether genes were essential, depending on if it produced four viable spores. Moreover, the ability to mate the knockout mutated alleles with any knockout recipient query strain allowed for the screening of synthetic lethal alleles and provided a new way to observe previously unknown genetic interactions (147). In addition, by introducing a unique bar code into every knockout allele, screens for mutants that are underrepresented in specific growth conditions can be readily identified. Such a strategy had been first employed by David Holden’s group using bar-coded transposons for Salmonella (125).
The completion of the genomic sequence of M. tuberculosis was first achieved by Stewart Cole and colleagues on the strain H37Rv (148) and later by others on CDC1551 (149). These sequences reveal the presence of over 4000 ORFs, with over 40% of unknown functions. In a collaboration with the Genomic Foundation of Novartis, Graham Hatfull, and William Bishai, we have initiated the generation of the entire set of specialized transducing phages to make precise null deletions of every gene of M. tuberculosis. To achieve this knockout set:
We have developed a high-throughput method to construct each allelic exchange substrate (AES) for each ORF deletion mutation with the concomitant hyg selectable marker gene that possesses 500- to 1,000-bp homologous DNA fragments flanking the deletion. Included in each construct is a unique bar code that associates with each deletion mutation as well as a system to excise the hyg gene from the resulting M. tuberculosis mutant to make unmarked mutations that retain their bar codes. The AESs are generated in vitro, cloned into a PacI cosmid, sequence verified, and then stored in E. coli as plasmid transformants.
We have constructed AESs that can be readily introduced into any PacI-flanked conditionally replicating shuttle phasmid, such as phAE159, and then introduced into E. coli using the in vitro lambda packaging system.
We then electroporate the shuttle phasmid form of the specialized transducing phages into M. smegmatis, yielding plaques that then can be amplified at 30°C to generate high-titered lysates of TM4 packaged specialized transducing phages.
We can infect almost any M. tuberculosis mutant or strain with conditionally replicating specialized transducing phages. (We have screened hundreds of clinical isolates of M. tuberculosis to date with the reporter phage assay and have yet to find any strain that is not infected.)
We can store at −80°C or lyophilize the TM4 phages to retain stability.
We have shown that the yields of specialized transductants can be enhanced 50- to 400-fold with recombineering containing hosts (unpublished data), making them amenable for an HTS approach in microtiter plates.
Specialized transduction is particularly well suited to this purpose because of the high efficiency of gene transfer (Table 1). Our recent work showing significantly enhanced recombination by combining specialized transduction with recombineering plasmids convinced us that this is amenable to high throughput. We believe that this set of phages will be useful for the TB research community as the donor transfer has been for the yeast community.
TABLE 1.
Improvements for high-throughput specialized transduction
| Steps for specialized transduction | Optimization for high-throughput knockouts |
|---|---|
| Construction of plasmid with AES (500 to 1,000 bp of upstream and downstream sequence of gene targeted for deletion). AES flanks a hygromycin (hyg) and sacB cassette | Improve efficiency of four-component ligation by use of type IIP restriction enzymes that recognize symmetric palindromic sequence interrupted by a few degenerate interior base pairs |
| Cloning of AES plasmid into temperature-sensitive phage backbone to create AES phasmid | Increased cloning capacity of phasmid |
| Transfection of M. smegmatis with phasmid DNA to recover plaques | Adapted for 96-well electroporation format |
| Production of high-titer phage lysate | Adapted protocol for high-volume high-titer lysate production |
| Transduction of M. tuberculosis | Optimized transduction conditions for smaller volumes and 96-well format |
| Screening of potential M. tuberculosis knockouts | Unique barcodes in each AES allow for efficient screening |
| Removal of hygromycin cassette for unmarked knockout strain | Phage-based delivery of resolvase to efficiently unmark knockout strains |
The use of defined bar-coded deleted sets should allow for screening of genes required for growth and virulence in diverse animal models. By using such a set, we can expedite screens in animals for loss of virulence and specific immune evasion function(s). In addition, comprehensive screens looking for mutants that are defective in persisting in the presence of drugs in vitro and in vivo should be feasible.
Lastly, the generation of this complete set of specialized transducing phages will provide many new avenues for exploring the biology of M. tuberculosis because specific phenotypes can be tested with synthetic lethal screens. Synthetic lethality is the observation that two independent nonessential genes cannot be disrupted. In the process of defining interactive genetic pathways, the combination of making the whole set of mutants in a specific set of selected unmarked mutants will allow for the identification of analogous synthetic phenotypes in virulence, persistence, and survival.
IMAGINING A WORLD WITHOUT TB
Albert Einstein wrote, “Imagination is more important than knowledge because knowledge tells you what is and imagination tells you what can be.” Before we had gene transfer for M. tuberculosis, we did not know the targets of TB-specific drugs, why M. tuberculosis stains acid-fast, if it were even possible to isolate auxotrophic mutants, and why the BCG vaccine strain that had been given to half the world’s population was attenuated. By developing gene transfer for M. tuberculosis, we now know exactly how INH kills M. tuberculosis; we know what genes are involved in INH resistance and which enzymes when inactivated lead to the death of the tubercle bacillus. We now know that if the KasB enzyme is inactivated or deleted, it causes M. tuberculosis to lose its ability to be stained acid-fast and that such mutants can cause a truly latent infection in the mouse (145). We now know that auxotrophic mutants of M. tuberculosis are unable to grow in mammals, a somewhat surprising finding since other intracellular pathogens are naturally auxotrophic. Nevertheless, this knowledge has allowed us to make new vaccine candidates that are safer than BCG and precisely defined deletion mutants of M. tuberculosis that because of their safety are allowed to be used in a BSL2 lab, thereby making work on these M. tuberculosis strains much easier. We now know that the primary attenuating mutation of BCG is the loss of a specialized secretion system that is required for M. tuberculosis to exit phagosomes and in the right oxygen conditions causes M. tuberculosis to form a DNA net from an infected macrophage (146). These nets may be ideal substrates for M. tuberculosis to form a biofilm in vivo.
During the time that gene transfer systems were being developed for M. tuberculosis, we saw a rise in the HIV epidemic—an epidemic that collided with the existing TB epidemic. The result has been a syndemic that has caused the worsening of each disease when a patient is co-infected. Unfortunately, this has led to an increased spread of TB and the emergence of strains of M. tuberculosis that are resistant to multiple or even all of the existing anti-TB drugs. Clearly, there is an unquestionable need for more research to develop new vaccines, new drugs, and new diagnostic tests for TB. While the emergence of multiple, extensively or totally drug-resistant strains may seem ominous for health care in impoverished areas of the world, the ability to discover new ways to battle TB has never been greater. Having studied TB for the last 25 years, I believe that there are a few questions that are paramount to the control and eradication of the disease. I would like to end with these questions: (i) What is the molecular basis for the slow growth phenotype of M. tuberculosis? I believe this is a strategy that M. tuberculosis has employed to resist death. (ii) What are the genes required for M. tuberculosis to enter into a persistent state? We know that if you add a bactericidal drug to an actively growing culture of M. tuberculosis you can only kill 99.9% of the cells in 4 to 5 days. The remaining 0.1% of cells are persisters that resist killing. Is it possible that resisting killing by antibiotics overlaps with resisting killing by an adaptive immune response? We will have to understand this process of persistence in M. tuberculosis, the process by which M. tuberculosis cells enter into a slowly replicating state. This might be induced by hypoxia, nutrient limitation, or growth in a biofilm. The fact is, we just don’t know, but now we have the power to find out. (iii) Is it possible to elicit a bactericidal immune response against M. tuberculosis? A number of new vaccine candidates have been developed that elicit better T-cell responses to M. tuberculosis than the BCG vaccine, yet we do not know how to engender an immune response that is able to sterilize an M. tuberculosis infection. Is this because persister cells are totally refractory to any T-cell–mediated or macrophage-mediated immune response? Is this because M. tuberculosis can persist in an extracellular biofilm in infected animals and thereby be hidden from an adaptive immune response? Do we need to understand how to make an antibody response that prevents the formation of these extracellular biofilms?
I believe that the answer to the control of TB will come from continued rational studies and serendipitous observations. The word serendipity comes from a Persian tale of the Three Princes of Serendip, who would discover new things while pursuing other quests. Louis Pasteur described this process in a different way when he said, “Chance favors the prepared mind.” The rational project to delete every gene from M. tuberculosis will undoubtedly result in a number of serendipitous observations. This endeavor along with knowledge and wisdom might lead to the eradication of TB.
References
- 1.Kaji M. Prevention of viral hepatitis—with special reference to the possibility of development of a vaccine in relation to Australia antigen. Nihon Rinsho. 1972;30:1159–1163. In Japanese. [PubMed] [Google Scholar]
- 2.Yap SF. Hepatitis B: review of development from the discovery of the “Australia Antigen” to end of the twentieth century. Malaysian J Pathol. 2004;26:1–12. [PubMed] [Google Scholar]
- 3.Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 4.Jacobs WR, Jr, Tuckman M, Bloom BR. Introduction of foreign DNA into mycobacteria using a shuttle phasmid. Nature. 1987;327:532–535. doi: 10.1038/327532a0. [DOI] [PubMed] [Google Scholar]
- 5.Snapper SB, Lugosi L, Jekkel A, Melton RE, Kieser T, Bloom BR, Jacobs WR., Jr Lysogeny and transformation in mycobacteria: stable expression of foreign genes. Proc Natl Acad Sci USA. 1988;85:6987–6991. doi: 10.1073/pnas.85.18.6987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee MH, Pascopella L, Jacobs WR, Jr, Hatfull GF. Site-specific integration of mycobacteriophage L5: integration-proficient vectors for Mycobacterium smegmatis, Mycobacterium tuberculosis, and bacille Calmette-Guerin. Proc Natl Acad Sci USA. 1991;88:3111–3115. doi: 10.1073/pnas.88.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stover CK, Delacruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Bloom BR. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 8.Collins DM, Kawakami RP, de Lisle GW, Pascopella L, Bloom BR, Jacobs WR., Jr Mutation of the principal sigma factor causes loss of virulence in a strain of the Mycobacterium tuberculosis complex. Proc Natl Acad Sci USA. 1995;92:8036–8040. doi: 10.1073/pnas.92.17.8036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pascopella L, Collins FM, Martin JM, Jacobs WR, Jr, Bloom BR. Identification of a genomic fragment of Mycobacterium tuberculosis responsible for in vivo growth advantage. Infect Agents Dis. 1993;2:282–284. [PubMed] [Google Scholar]
- 10.Pascopella L, Collins FM, Martin JM, Lee MH, Hatfull GF, Stover CK, Bloom BR, Jacobs WR., Jr Use of in vivo complementation in Mycobacterium tuberculosis to identify a genomic fragment associated with virulence. Infect Immun. 1994;62:1313–1319. doi: 10.1128/iai.62.4.1313-1319.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snapper SB, Melton RE, Mustafa S, Kieser T, Jacobs WR., Jr Isolation and characterization of efficient plasmid transformation mutants of Mycobacterium smegmatis. Mol Microbiol. 1990;4:1911–1919. doi: 10.1111/j.1365-2958.1990.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 12.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Jr, Bloom BR. New use of BCG for recombinant vaccines. Nature. 1991;351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 13.Labidi A, Mardis E, Roe BA, Wallace RJ., Jr Cloning and DNA sequence of the Mycobacterium fortuitum var fortuitum plasmid pAL5000. Plasmid. 1992;27:130–140. doi: 10.1016/0147-619x(92)90013-z. [DOI] [PubMed] [Google Scholar]
- 14.Ranes MG, Rauzier J, Lagranderie M, Gheorghiu M, Gicquel B. Functional analysis of pAL5000, a plasmid from Mycobacterium fortuitum: construction of a “mini” mycobacterium-Escherichia coli shuttle vector. J Bacteriol. 1990;172:2793–2797. doi: 10.1128/jb.172.5.2793-2797.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stolt P, Stoker NG. Protein-DNA interactions in the ori region of the Mycobacterium fortuitum plasmid pAL5000. J Bacteriol. 1996;178:6693–6700. doi: 10.1128/jb.178.23.6693-6700.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stolt P, Stoker NG. Mutational analysis of the regulatory region of the Mycobacterium plasmid pAL5000. Nucleic Acids Res. 1997;25:3840–3846. doi: 10.1093/nar/25.19.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Villar CA, Benitez J. Functional analysis of pAL5000 plasmid in Mycobacterium fortuitum. Plasmid. 1992;28:166–169. doi: 10.1016/0147-619x(92)90047-e. [DOI] [PubMed] [Google Scholar]
- 18.Belisle JT, Pascopella L, Inamine JM, Brennan PJ, Jacobs WR., Jr Isolation and expression of a gene cluster responsible for biosynthesis of the glycopeptidolipid antigens of Mycobacterium avium. J Bacteriol. 1991;173:6991–6997. doi: 10.1128/jb.173.21.6991-6997.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR., Jr inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science. 1994;263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Heym B, Allen B, Young D, Cole S. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature. 1992;358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- 21.Baulard AR, Betts JC, Engohang-Ndong J, Quan S, McAdam RA, Brennan PJ, Locht C, Besra GS. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem. 2000;275:28326–28331. doi: 10.1074/jbc.M003744200. [DOI] [PubMed] [Google Scholar]
- 22.DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE., 3rd Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 2000;97:9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belanger AE, Besra GS, Ford ME, Mikusova K, Belisle JT, Brennan PJ, Inamine JM. The embAB genes of Mycobacterium avium encode an arabinosyl transferase involved in cell wall arabinan biosynthesis that is the target for the antimycobacterial drug ethambutol. Proc Natl Acad Sci USA. 1996;93:11919–11924. doi: 10.1073/pnas.93.21.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Telenti A, Philipp WJ, Sreevatsan S, Bernasconi C, Stockbauer KE, Wieles B, Musser JM, Jacobs WR., Jr The emb operon, a gene cluster of Mycobacterium tuberculosis involved in resistance to ethambutol. Nature Med. 1997;3:567–570. doi: 10.1038/nm0597-567. [DOI] [PubMed] [Google Scholar]
- 25.Scorpio A, Zhang Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nature Med. 1996;2:662–667. doi: 10.1038/nm0696-662. [DOI] [PubMed] [Google Scholar]
- 26.Gannoun-Zaki L, Alibaud L, Kremer L. Point mutations within the fatty acid synthase type II dehydratase components HadA or HadC contribute to isoxyl resistance in Mycobacterium tuberculosis. Antimicrobial Agents Chemother. 2013;57:629–632. doi: 10.1128/AAC.01972-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grzegorzewicz AE, Kordulakova J, Jones V, Born SE, Belardinelli JM, Vaquie A, Gundi VA, Madacki J, Slama N, Laval F, Vaubourgeix J, Crew RM, Gicquel B, Daffe M, Morbidoni HR, Brennan PJ, Quemard A, McNeil MR, Jackson M. A common mechanism of inhibition of the Mycobacterium tuberculosis mycolic acid biosynthetic pathway by isoxyl and thiacetazone. J Biol Chem. 2012;287:38434–38441. doi: 10.1074/jbc.M112.400994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 29.Pavelka MS, Jr, Jacobs WR., Jr Biosynthesis of diaminopimelate, the precursor of lysine and a component of peptidoglycan, is an essential function of Mycobacterium smegmatis. J Bacteriol. 1996;178:6496–6507. doi: 10.1128/jb.178.22.6496-6507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van Kessel JC, Hatfull GF. Recombineering in Mycobacterium tuberculosis. Nat Methods. 2007;4:147–152. doi: 10.1038/nmeth996. [DOI] [PubMed] [Google Scholar]
- 31.McAdam RA, Weisbrod TR, Martin J, Scuderi JD, Brown AM, Cirillo JD, Bloom BR, Jacobs WR., Jr In vivo growth characteristics of leucine and methionine auxotrophic mutants of Mycobacterium bovis BCG generated by transposon mutagenesis. Infect Immun. 1995;63:1004–1012. doi: 10.1128/iai.63.3.1004-1012.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.George JR, Pine L, Reeves MW, Harrell WK. Amino acid requirements of Legionella pneumophila. J Clin Microbiol. 1980;11:286–291. doi: 10.1128/jcm.11.3.286-291.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tesh MJ, Miller RD. Amino acid requirements for Legionella pneumophila growth. J Clin Microbiol. 1981;13:865–869. doi: 10.1128/jcm.13.5.865-869.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guleria I, Teitelbaum R, McAdam RA, Kalpana G, Jacobs WR, Jr, Bloom BR. Auxotrophic vaccines for tuberculosis. Nat Med. 1996;2:334–337. doi: 10.1038/nm0396-334. [DOI] [PubMed] [Google Scholar]
- 35.McDonough KA, Kress Y, Bloom BR. Pathogenesis of tuberculosis: interaction of Mycobacterium tuberculosis with macrophages. Infect Immun. 1993;61:2763–2773. doi: 10.1128/iai.61.7.2763-2773.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hondalus MK, Bardarov S, Russell R, Chan J, Jacobs WR, Jr, Bloom BR. Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect Immun. 2000;68:2888–2898. doi: 10.1128/iai.68.5.2888-2898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pavelka MS, Jr, Chen B, Kelley CL, Collins FM, Jacobs WR., Jr Vaccine efficacy of a lysine auxotroph of Mycobacterium tuberculosis. Infect Immun. 2003;71:4190–4192. doi: 10.1128/IAI.71.7.4190-4192.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, Morris SL, Jacobs WR., Jr A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat Med. 2002;8:1171–1174. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- 39.Jensen K, Ranganathan UD, Van Rompay KK, Canfield DR, Khan I, Ravindran R, Luciw PA, Jacobs WR, Jr, Fennelly G, Larsen MH, Abel K. A recombinant attenuated Mycobacterium tuberculosis vaccine strain is safe in immunosuppressed simian immunodeficiency virus-infected infant macaques. Clin Vaccine Immunol. 2012;19:1170–1181. doi: 10.1128/CVI.00184-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sampson SL, Dascher CC, Sambandamurthy VK, Russell RG, Jacobs WR, Jr, Bloom BR, Hondalus MK. Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infect Immun. 2004;72:3031–3037. doi: 10.1128/IAI.72.5.3031-3037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sampson SL, Mansfield KG, Carville A, Magee DM, Quitugua T, Howerth EW, Bloom BR, Hondalus MK. Extended safety and efficacy studies of a live attenuated double leucine and pantothenate auxotroph of Mycobacterium tuberculosis as a vaccine candidate. Vaccine. 2011;29:4839–4847. doi: 10.1016/j.vaccine.2011.04.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zimmerman DM, Waters WR, Lyashchenko KP, Nonnecke BJ, Armstrong DL, Jacobs WR, Jr, Larsen MH, Egan E, Dean GA. Safety and immunogenicity of the Mycobacterium tuberculosis DeltalysA DeltapanCD vaccine in domestic cats infected with feline immunodeficiency virus. Clin Vaccine Immunol. 2009;16:427–429. doi: 10.1128/CVI.00396-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Villemin JA. Etudes sur la Tuberculose. J.-B. Baillière et Fils; Paris: 1868. [Google Scholar]
- 44.Koch R. Die Aetiologie der Tuberkulose. Berl Klin Wochenschr. 1882;19:221–230. [Google Scholar]
- 45.Beadle GW, Tatum EL. Genetic control of biochemical reactions in Neurospora. Proc Natl Acad Sci USA. 1941;27:499–506. doi: 10.1073/pnas.27.11.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lederberg J, Tatum EL. Gene recombination in Escherichia coli. Nature. 1946;158:558. doi: 10.1038/158558a0. [DOI] [PubMed] [Google Scholar]
- 47.Griffith F. The significance of pneumococcal types. J Hyg. 1928;27:113–159. doi: 10.1017/s0022172400031879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Avery OT, Macleod CM, McCarty M. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J Exp Med. 1944;79:137–158. doi: 10.1084/jem.79.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hershey AD, Chase M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J Gen Physiol. 1952;36:39–56. doi: 10.1085/jgp.36.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watson JD, Crick FH. Genetical implications of the structure of deoxyribonucleic acid. Nature. 1953;171:964–967. doi: 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]
- 51.Watson JD, Crick FH. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature. 1953;171:737–738. doi: 10.1038/171737a0. [DOI] [PubMed] [Google Scholar]
- 52.Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- 53.Falkow S. Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10(Suppl 2):S274–S276. doi: 10.1093/cid/10.supplement_2.s274. [DOI] [PubMed] [Google Scholar]
- 54.Zinder ND. Forty years ago: the discovery of bacterial transduction. Genetics. 1992;132:291–294. doi: 10.1093/genetics/132.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waksman SA, Schatz A. Strain specificity and production of antibiotic substances. Proc Natl Acad Sci USA. 1943;29:74–79. doi: 10.1073/pnas.29.2.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schatz A, Waksman SA. Strain specificity and production of antibiotic substances. IV. Variations among actionomycetes, with special reference to Actinomyces griseus. Proc Natl Acad Sci USA. 1945;31:129–137. doi: 10.1073/pnas.31.5.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waksman SA, Reilly HC, Schatz A. Strain specificity and production of antibiotic substances. V. Strain resistance of bacteria to antibiotic substances, especially to streptomycin. Proc Natl Acad Sci USA. 1945;31:157–164. doi: 10.1073/pnas.31.6.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Demerec M. Studies of the streptomycin-resistance system of mutations in E. coli. Genetics. 1951;36:585–597. doi: 10.1093/genetics/36.6.585c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hashimoto K. Streptomycin resistance in Escherichia coli analyzed by transduction. Genetics. 1960;45:49–62. doi: 10.1093/genetics/45.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lennox ES. Transduction of linked genetic characters of the host by bacteriophage P1. Virology. 1955;1:190–206. doi: 10.1016/0042-6822(55)90016-7. [DOI] [PubMed] [Google Scholar]
- 61.Newcombe HB, Nyholm MH. The inheritance of streptomycin resistance and dependence in crosses of Escherichia coli. Genetics. 1950;35:603–611. doi: 10.1093/genetics/35.6.603b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Spotts CR, Stanier RY. Mechanism of streptomycin action on bacteria: a unitary hypothesis. Nature. 1961;192:633–637. doi: 10.1038/192633a0. [DOI] [PubMed] [Google Scholar]
- 63.Erdos T, Ullmann A. Effect of streptomycin on the incorporation of amino-acids labelled with carbon-14 into ribonucleic acid and protein in a cell-free system of a mycobacterium. Nature. 1959;183:618–619. doi: 10.1038/183618a0. [DOI] [PubMed] [Google Scholar]
- 64.Erdos T, Ullmann A. Effect of streptomycin on the incorporation of tyrosine labelled with carbon-14 into protein of Mycobacterium cell fractions in vivo. Nature. 1960;185:100–101. doi: 10.1038/185100a0. [DOI] [PubMed] [Google Scholar]
- 65.Davies JE. Studies on the ribosomes of streptomycin-sensitive and resistant strains of Escherichia coli. Proc Natl Acad Sci USA. 1964;51:659–664. doi: 10.1073/pnas.51.4.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ozaki M, Mizushima S, Nomura M. Identification and functional characterization of the protein controlled by the streptomycin-resistant locus in E. coli. Nature. 1969;222:333–339. doi: 10.1038/222333a0. [DOI] [PubMed] [Google Scholar]
- 67.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Functional insights from the structure of the 30S ribosomal subunit and its interactions with antibiotics. Nature. 2000;407:340–348. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 68.Finken M, Kirschner P, Meier A, Wrede A, Bottger EC. Molecular basis of streptomycin resistance in Mycobacterium tuberculosis: alterations of the ribosomal protein S12 gene and point mutations within a functional 16S ribosomal RNA pseudoknot. Mol Microbiol. 1993;9:1239–1246. doi: 10.1111/j.1365-2958.1993.tb01253.x. [DOI] [PubMed] [Google Scholar]
- 69.Jackson DA, Symons RH, Berg P. Biochemical method for inserting new genetic information into DNA of Simian Virus 40: circular SV40 DNA molecules containing lambda phage genes and the galactose operon of Escherichia coli. Proc Natl Acad Sci USA. 1972;69:2904–2909. doi: 10.1073/pnas.69.10.2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lark C, Arber W. Host specificity of DNA produced by Escherichia coli. 13. Breakdown of cellular DNA upon growth in ethionine of strains with r plus-15, r plus-P1 or r plus-N3 restriction phenotypes. J Mol Biol. 1970;52:337–348. doi: 10.1016/0022-2836(70)90034-3. [DOI] [PubMed] [Google Scholar]
- 71.Smith HO, Wilcox KW. A restriction enzyme from Haemophilus influenzae. I. Purification and general properties. J Mol Biol. 1970;51:379–391. doi: 10.1016/0022-2836(70)90149-x. [DOI] [PubMed] [Google Scholar]
- 72.Chang AC, Lansman RA, Clayton DA, Cohen SN. Studies of mouse mitochondrial DNA in Escherichia coli: structure and function of the eucaryotic-procaryotic chimeric plasmids. Cell. 1975;6:231–244. doi: 10.1016/0092-8674(75)90014-8. [DOI] [PubMed] [Google Scholar]
- 73.Cohen SN, Chang AC, Boyer HW, Helling RB. Construction of biologically functional bacterial plasmids in vitro. Proc Natl Acad Sci USA. 1973;70:3240–3244. doi: 10.1073/pnas.70.11.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kedes LH, Chang AC, Houseman D, Cohen SN. Isolation of histone genes from unfractionated sea urchin DNA by subculture cloning in E. coli. Nature. 1975;255:533–538. doi: 10.1038/255533a0. [DOI] [PubMed] [Google Scholar]
- 75.Morrow JF, Cohen SN, Chang AC, Boyer HW, Goodman HM, Helling RB. Replication and transcription of eukaryotic DNA in Escherichia coli. Proc Natl Acad Sci USA. 1974;71:1743–1747. doi: 10.1073/pnas.71.5.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ratzkin B, Carbon J. Functional expression of cloned yeast DNA in Escherichia coli. Proc Natl Acad Sci USA. 1977;74:487–491. doi: 10.1073/pnas.74.2.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Borck K, Beggs JD, Brammar WJ, Hopkins AS, Murray NE. The construction in vitro of transducing derivatives of phage lambda. Mol Gen Genet. 1976;146:199–207. doi: 10.1007/BF00268089. [DOI] [PubMed] [Google Scholar]
- 78.Hohn B, Murray K. Packaging recombinant DNA molecules into bacteriophage particles in vitro. Proc Natl Acad Sci USA. 1977;74:3259–3263. doi: 10.1073/pnas.74.8.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murray NE, Murray K. Manipulation of restriction targets in phage lambda to form receptor chromosomes for DNA fragments. Nature. 1974;251:476–481. doi: 10.1038/251476a0. [DOI] [PubMed] [Google Scholar]
- 80.Struhl K, Cameron JR, Davis RW. Functional genetic expression of eukaryotic DNA in Escherichia coli. Proc Natl Acad Sci USA. 1976;73:1471–1475. doi: 10.1073/pnas.73.5.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Maxam AM, Gilbert W. Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 1980;65:499–560. doi: 10.1016/s0076-6879(80)65059-9. [DOI] [PubMed] [Google Scholar]
- 82.Sanger F, Donelson JE, Coulson AR, Kossel H, Fischer D. Use of DNA polymerase I primed by a synthetic oligonucleotide to determine a nucleotide sequence in phage fl DNA. Proc Natl Acad Sci USA. 1973;70:1209–1213. doi: 10.1073/pnas.70.4.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Berg P, Baltimore D, Brenner S, Roblin RO, Singer MF. Summary statement of the Asilomar conference on recombinant DNA molecules. Proc Natl Acad Sci USA. 1975;72:1981–1984. doi: 10.1073/pnas.72.6.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Curtiss R., 3rd Biological containment and cloning vector transmissibility. J Infect Dis. 1978;137:668–675. doi: 10.1093/infdis/137.5.668. [DOI] [PubMed] [Google Scholar]
- 85.Alexander WJ, Alexander LS, Curtiss R., 3rd Bactericidal activity of human serum against Escherichia coli chi1776. Infect Immun. 1980;28:837–841. doi: 10.1128/iai.28.3.837-841.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shepard CC. The experimental disease that follows the injection of human leprosy bacilli into foot-pads of mice. J Exp Med. 1960;112:445–454. doi: 10.1084/jem.112.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shepard CC. The first decade in experimental leprosy. Bull World Health Organ. 1971;44:821–827. [PMC free article] [PubMed] [Google Scholar]
- 88.Kirchheimer WF, Storrs EE. Attempts to establish the armadillo (Dasypus novemcinctus Linn.) as a model for the study of leprosy. I. Report of lepromatoid leprosy in an experimentally infected armadillo. Int J Lepr Other Mycobact Dis. 1971;39:693–702. [PubMed] [Google Scholar]
- 89.Clark-Curtiss JE, Jacobs WR, Docherty MA, Ritchie LR, Curtiss R., 3rd Molecular analysis of DNA and construction of genomic libraries of Mycobacterium leprae. J Bacteriol. 1985;161:1093–1102. doi: 10.1128/jb.161.3.1093-1102.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jacobs WR, Barrett JF, Clark-Curtiss JE, Curtiss R., 3rd In vivo repackaging of recombinant cosmid molecules for analyses of Salmonella typhimurium, Streptococcus mutans, and mycobacterial genomic libraries. Infect Immun. 1986;52:101–109. doi: 10.1128/iai.52.1.101-109.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jacobs WR, Docherty MA, Curtiss R, 3rd, Clark-Curtiss JE. Expression of Mycobacterium leprae genes from a Streptococcus mutans promoter in Escherichia coli K-12. Proc Natl Acad Sci USA. 1986;83:1926–1930. doi: 10.1073/pnas.83.6.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Young RA, Davis RW. Efficient isolation of genes by using antibody probes. Proc Natl Acad Sci USA. 1983;80:1194–1198. doi: 10.1073/pnas.80.5.1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Young RA, Mehra V, Sweetser D, Buchanan T, Clark-Curtiss J, Davis RW, Bloom BR. Genes for the major protein antigens of the leprosy parasite Mycobacterium leprae. Nature. 1985;316:450–452. doi: 10.1038/316450a0. [DOI] [PubMed] [Google Scholar]
- 94.Young RA, Bloom BR, Grosskinsky CM, Ivanyi J, Thomas D, Davis RW. Dissection of Mycobacterium tuberculosis antigens using recombinant DNA. Proc Natl Acad Sci USA. 1985;82:2583–2587. doi: 10.1073/pnas.82.9.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Raj CV, Ramakrishnan T. Transduction in Mycobacterium smegmatis. Nature. 1970;228:280–281. doi: 10.1038/228280b0. [DOI] [PubMed] [Google Scholar]
- 96.Mizuguchi Y, Tokunaga T. Genetic recombination between Mycobacterium smegmatis (strains jucho and lactocola) Igaku to Seibutsugaku. 1970;81:163–167. In Japanese. [PubMed] [Google Scholar]
- 97.Takahashi M, Tanaka M, Okudera T, Mihara K, Tokunaga M. Angiography with a new contrast medium, Isopaque. Rinsho Hoshasen. 1973;18:1001–1006. [PubMed] [Google Scholar]
- 98.Crawford JT, Bates JH. Isolation of plasmids from mycobacteria. Infect Immun. 1979;24:979–981. doi: 10.1128/iai.24.3.979-981.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Labidi A, David HL, Roulland-Dussoix D. Restriction endonuclease mapping and cloning of Mycobacterium fortuitum var. fortuitum plasmid pAL5000. Ann Inst Pasteur Microbiol. 1985;136B:209–215. doi: 10.1016/s0769-2609(85)80045-4. [DOI] [PubMed] [Google Scholar]
- 100.Bibb MJ, Ward JM, Hopwood DA. Transformation of plasmid DNA into Streptomyces at high frequency. Nature. 1978;274:398–400. doi: 10.1038/274398a0. [DOI] [PubMed] [Google Scholar]
- 101.Timme TL, Brennan PJ. Induction of bacteriophage from members of the Mycobacterium avium, Mycobacterium intracellulare, Mycobacterium scrofulaceum serocomplex. JGen Microbiol. 1984;130:2059–2066. doi: 10.1099/00221287-130-8-2059. [DOI] [PubMed] [Google Scholar]
- 102.Hatfull GF, Sarkis GJ. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol Microbiol. 1993;7:395–405. doi: 10.1111/j.1365-2958.1993.tb01131.x. [DOI] [PubMed] [Google Scholar]
- 103.Miller JF, Dower WJ, Tompkins LS. High-voltage electroporation of bacteria: genetic transformation of Campylobacter jejuni with plasmid DNA. Proc Natl Acad Sci USA. 1988;85:856–860. doi: 10.1073/pnas.85.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pope WH, Ferreira CM, Jacobs-Sera D, Benjamin RC, Davis AJ, DeJong RJ, Elgin SC, Guilfoile FR, Forsyth MH, Harris AD, Harvey SE, Hughes LE, Hynes PM, Jackson AS, Jalal MD, MacMurray EA, Manley CM, McDonough MJ, Mosier JL, Osterbann LJ, Rabinowitz HS, Rhyan CN, Russell DA, Saha MS, Shaffer CD, Simon SE, Sims EF, Tovar IG, Weisser EG, Wertz JT, Weston-Hafer KA, Williamson KE, Zhang B, Cresawn SG, Jain P, Piuri M, Jacobs WR, Jr, Hendrix RW, Hatfull GF. Cluster K mycobacteriophages: insights into the evolutionary origins of mycobacteriophage TM4. PloS One. 2011;6:e26750. doi: 10.1371/journal.pone.0026750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kleckner N, Roth J, Botstein D. Genetic engineering in vivo using translocatable drug-resistance elements. New methods in bacterial genetics. J Mol Biol. 1977;116:125–159. doi: 10.1016/0022-2836(77)90123-1. [DOI] [PubMed] [Google Scholar]
- 106.Bardarov S, Kriakov J, Carriere C, Yu S, Vaamonde C, McAdam RA, Bloom BR, Hatfull GF, Jacobs WR., Jr Conditionally replicating mycobacteriophages: a system for transposon delivery to Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 1997;94:10961–10966. doi: 10.1073/pnas.94.20.10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carriere C, Riska PF, Zimhony O, Kriakov J, Bardarov S, Burns J, Chan J, Jacobs WR., Jr Conditionally replicating luciferase reporter phages: improved sensitivity for rapid detection and assessment of drug susceptibility of Mycobacterium tuberculosis. J Clin Microbiol. 1997;35:3232–3239. doi: 10.1128/jcm.35.12.3232-3239.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ulitzur S, Kuhn J. Detection and/or identification of microorganisms in a test sample using bioluminescence or other genetically introduced marker. 4,861,709 US Patent No. 1989
- 109.Jacobs WR, Jr, Barletta RG, Udani R, Chan J, Kalkut G, Sosne G, Kieser T, Sarkis GJ, Hatfull GF, Bloom BR. Rapid assessment of drug susceptibilities of Mycobacterium tuberculosis by means of luciferase reporter phages. Science. 1993;260:819–822. doi: 10.1126/science.8484123. [DOI] [PubMed] [Google Scholar]
- 110.Banaiee N, Bobadilla-Del-Valle M, Bardarov S, Jr, Riska PF, Small PM, Ponce-De-Leon A, Jacobs WR, Jr, Hatfull GF, Sifuentes-Osornio J. Luciferase reporter mycobacteriophages for detection, identification, and antibiotic susceptibility testing of Mycobacterium tuberculosis in Mexico. J Clin Microbiol. 2001;39:3883–3888. doi: 10.1128/JCM.39.11.3883-3888.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Banaiee N, Bobadilla-del-Valle M, Riska PF, Bardarov S, Jr, Small PM, Ponce-de-Leon A, Jacobs WR, Jr, Hatfull GF, Sifuentes-Osornio J. Rapid identification and susceptibility testing of Mycobacterium tuberculosis from MGIT cultures with luciferase reporter mycobacteriophages. J Med Microbiol. 2003;52:557–561. doi: 10.1099/jmm.0.05149-0. [DOI] [PubMed] [Google Scholar]
- 112.Bardarov S, Jr, Dou H, Eisenach K, Banaiee N, Ya S, Chan J, Jacobs WR, Jr, Riska PF. Detection and drug-susceptibility testing of M. tuberculosis from sputum samples using luciferase reporter phage: comparison with the Mycobacteria Growth Indicator Tube (MGIT) system. Diagn Microbiol Infect Dis. 2003;45:53–61. doi: 10.1016/s0732-8893(02)00478-9. [DOI] [PubMed] [Google Scholar]
- 113.Riska PF, Jacobs WR., Jr The use of luciferase-reporter phage for antibiotic-susceptibility testing of mycobacteria. Methods Mol Biol. 1998;101:431–455. doi: 10.1385/0-89603-471-2:431. [DOI] [PubMed] [Google Scholar]
- 114.Riska PF, Jacobs WR, Jr, Bloom BR, McKitrick J, Chan J. Specific identification of Mycobacterium tuberculosis with the luciferase reporter mycobacteriophage: use of p-nitro-alpha-acetylamino-beta-hydroxy propiophenone. J Clin Microbiol. 1997;35:3225–3231. doi: 10.1128/jcm.35.12.3225-3231.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Riska PF, Su Y, Bardarov S, Freundlich L, Sarkis G, Hatfull G, Carriere C, Kumar V, Chan J, Jacobs WR., Jr Rapid film-based determination of antibiotic susceptibilities of Mycobacterium tuberculosis strains by using a luciferase reporter phage and the Bronx Box. J Clin Microbiol. 1999;37:1144–1149. doi: 10.1128/jcm.37.4.1144-1149.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Piuri M, Jacobs WR, Jr, Hatfull GF. Fluoromycobacteriophages for rapid, specific, and sensitive antibiotic susceptibility testing of Mycobacterium tuberculosis. PloS One. 2009;4:e4870. doi: 10.1371/journal.pone.0004870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rondon L, Piuri M, Jacobs WR, Jr, de Waard J, Hatfull GF, Takiff HE. Evaluation of fluoromycobacteriophages for detecting drug resistance in Mycobacterium tuberculosis. J Clin Microbiol. 2011;49:1838–1842. doi: 10.1128/JCM.02476-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Jain P, Hartman TE, Eisenberg N, O’Donnell MR, Kriakov J, Govender K, Makume M, Thaler DS, Hatfull GF, Sturm AW, Larsen MH, Moodley P, Jacobs WR., Jr phi(2)GFP10, a high-intensity fluorophage, enables detection and rapid drug susceptibility testing of Mycobacterium tuberculosis directly from sputum samples. J Clin Microbiol. 2012;50:1362–1369. doi: 10.1128/JCM.06192-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martin C, Timm J, Rauzier J, Gomez-Lus R, Davies J, Gicquel B. Transposition of an antibiotic resistance element in mycobacteria. Nature. 1990;345:739–743. doi: 10.1038/345739a0. [DOI] [PubMed] [Google Scholar]
- 120.Guilhot C, Otal I, Van Rompaey I, Martin C, Gicquel B. Efficient transposition in mycobacteria: construction of Mycobacterium smegmatis insertional mutant libraries. J Bacteriol. 1994;176:535–539. doi: 10.1128/jb.176.2.535-539.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Otero J, Jacobs WR, Jr, Glickman MS. Efficient allelic exchange and transposon mutagenesis in Mycobacterium avium by specialized transduction. Appl Environ Microbiol. 2003;69:5039–5044. doi: 10.1128/AEM.69.9.5039-5044.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stinear TP, Mve-Obiang A, Small PL, Frigui W, Pryor MJ, Brosch R, Jenkin GA, Johnson PD, Davies JK, Lee RE, Adusumilli S, Garnier T, Haydock SF, Leadlay PF, Cole ST. Giant plasmid-encoded polyketide synthases produce the macrolide toxin of Mycobacterium ulcerans. Proc Natl Acad Sci USA. 2004;101:1345–1349. doi: 10.1073/pnas.0305877101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Harris NB, Feng Z, Liu X, Cirillo SL, Cirillo JD, Barletta RG. Development of a transposon mutagenesis system for Mycobacterium avium subsp. paratuberculosis. FEMS Microbiol Lett. 1999;175:21–26. doi: 10.1111/j.1574-6968.1999.tb13597.x. [DOI] [PubMed] [Google Scholar]
- 124.Glickman MS, Cox JS, Jacobs WR., Jr A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–727. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 125.Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 126.Cox JS, Chen B, McNeil M, Jacobs WR., Jr Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 127.McAdam RA, Quan S, Smith DA, Bardarov S, Betts JC, Cook FC, Hooker EU, Lewis AP, Woollard P, Everett MJ, Lukey PT, Bancroft GJ, Jacobs WR, Jr, Duncan K. Characterization of a Mycobacterium tuberculosis H37Rv transposon library reveals insertions in 351 ORFs and mutants with altered virulence. Microbiology. 2002;148:2975–2986. doi: 10.1099/00221287-148-10-2975. [DOI] [PubMed] [Google Scholar]
- 128.Hisert KB, Kirksey MA, Gomez JE, Sousa AO, Cox JS, Jacobs WR, Jr, Nathan CF, McKinney JD. Identification of Mycobacterium tuberculosis counterimmune (cim) mutants in immunodeficient mice by differential screening. Infect Immun. 2004;72:5315–5321. doi: 10.1128/IAI.72.9.5315-5321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sassetti CM, Boyd DH, Rubin EJ. Comprehensive identification of conditionally essential genes in mycobacteria. Proc Natl Acad Sci USA. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Morse ML, Lederberg EM, Lederberg J. Transduction in Escherichia coli K-12. Genetics. 1956;41:142–156. doi: 10.1093/genetics/41.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bardarov S, Bardarov S, Jr, Pavelka MS, Jr, Sambandamurthy V, Larsen M, Tufariello J, Chan J, Hatfull G, Jacobs WR., Jr Specialized transduction: an efficient method for generating marked and unmarked targeted gene disruptions in Mycobacterium tuberculosis, M. bovis BCG and M. smegmatis. Microbiology. 2002;148:3007–3017. doi: 10.1099/00221287-148-10-3007. [DOI] [PubMed] [Google Scholar]
- 134.Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR., Jr The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci USA. 2003;100:12420–12425. doi: 10.1073/pnas.1635213100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mahairas GG, Sabo PJ, Hickey MJ, Singh DC, Stover CK. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J Bacteriol. 1996;178:1274–1282. doi: 10.1128/jb.178.5.1274-1282.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pym AS, Brodin P, Brosch R, Huerre M, Cole ST. Loss of RD1 contributed to the attenuation of the live tuberculosis vaccines Mycobacterium bovis BCG and Mycobacterium microti. Mol Microbiol. 2002;46:709–717. doi: 10.1046/j.1365-2958.2002.03237.x. [DOI] [PubMed] [Google Scholar]
- 137.Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, Jacobs WR., Jr Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat Med. 2006;12:1027–1029. doi: 10.1038/nm1466. [DOI] [PubMed] [Google Scholar]
- 138.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 139.Anonymous. The yeast genome directory. Nature. 1997;387:5. [PubMed] [Google Scholar]
- 140.Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp Quant Biol. 1986;51(Pt 1):263–273. doi: 10.1101/sqb.1986.051.01.032. [DOI] [PubMed] [Google Scholar]
- 141.Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 142.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Guldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kotter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 143.Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR., Jr Genetic manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol. 2007;Chapter 10(Unit 10A):12. doi: 10.1002/9780471729259.mc10a02s6. [DOI] [PubMed] [Google Scholar]
- 144.Larsen MH, Biermann K, Jacobs WR., Jr Laboratory maintenance of Mycobacterium tuberculosis. Curr Protoc Microbiol. 2007;Chapter 10(Unit 10A):11. doi: 10.1002/9780471729259.mc10a01s6. [DOI] [PubMed] [Google Scholar]
- 145.Bhatt A, Fujiwara N, Bhatt K, Gurcha SS, Kremer L, Chen B, Chan J, Porcelli SA, Kobayashi K, Besra GS, Jacobs WR., Jr Deletion of kasB in Mycobacterium tuberculosis causes loss of acid-fastness and subclinical latent tuberculosis in immunocompetent mice. Proc Natl Acad Sci USA. 2007;104:5157–5162. doi: 10.1073/pnas.0608654104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wong KW, Jacobs WR., Jr Mycobacterium tuberculosis exploits human interferon gamma to stimulate macrophage extracellular trap formation and necrosis. J Infect Dis. 2013;208:109–119. doi: 10.1093/infdis/jit097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Giaever G, Chu AM, Ni L, Connelly C, Riles L, Véronneau S, Dow S, Lucau-Danila A, Anderson K, André B, Arkin AP, Astromoff A, El-Bakkoury M, Bangham R, Benito R, Brachat S, Campanaro S, Curtiss M, Davis K, Deutschbauer A, Entian KD, Flaherty P, Foury F, Garfinkel DJ, Gerstein M, Gotte D, Güldener U, Hegemann JH, Hempel S, Herman Z, Jaramillo DF, Kelly DE, Kelly SL, Kötter P, LaBonte D, Lamb DC, Lan N, Liang H, Liao H, Liu L, Luo C, Lussier M, Mao R, Menard P, Ooi SL, Revuelta JL, Roberts CJ, Rose M, Ross-Macdonald P, Scherens B, Schimmack G, Shafer B, Shoemaker DD, Sookhai-Mahadeo S, Storms RK, Strathern JN, Valle G, Voet M, Volckaert G, Wang CY, Ward TR, Wilhelmy J, Winzeler EA, Yang Y, Yen G, Youngman E, Yu K, Bussey H, Boeke JD, Snyder M, Philippsen P, Davis RW, Johnston M. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- 148.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 149.Fleischmann RD, Alland D, Eisen JA, Carpenter L, White O, Peterson J, DeBoy R, Dodson R, Gwinn M, Haft D, Hickey E, Kolonay JF, Nelson WC, Umayam LA, Ermolaeva M, Salzberg SL, Delcher A, Utterback T, Weidman J, Khouri H, Gill J, Mikula A, Bishai W, Jacobs WR, Jr, Venter JC, Fraser CM. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol. 2002;184:5479–5490. doi: 10.1128/JB.184.19.5479-5490.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Hatfull GF. Molecular genetics of mycobacteriophages. Microbiol Spectrum. 2014;2(2) doi: 10.1128/microbiolspec.MGM2-0032-2013. MGM2-0032-2013. [DOI] [PubMed] [Google Scholar]