

Abstract
Fibrillar aggregates involved in neurodegenerative diseases have the ability to spread from one cell to another in a prion-like manner. The underlying molecular mechanisms, in particular the binding mode of the fibrils to cell membranes, are poorly understood. In this work we decipher the modality by which aggregates bind to the cellular membrane, one of the obligatory steps of the propagation cycle. By characterizing the binding properties of aggregates made of α-synuclein or huntingtin exon 1 protein displaying similar composition and structure but different lengths to mammalian cells we demonstrate that in both cases aggregates bind laterally to the cellular membrane, with aggregates extremities displaying little or no role in membrane binding. Lateral binding to artificial liposomes was also observed by transmission electron microscopy. In addition we show that although α-synuclein and huntingtin exon 1 fibrils bind both laterally to the cellular membrane, their mechanisms of interaction differ. Our findings have important implications for the development of future therapeutic tools that aim to block protein aggregates propagation in the brain.
The aggregation of proteins into fibrillar high molecular-weight species is the hallmark of human degenerative diseases, including Alzheimer’s, Parkinson’s, or Huntington’s1. During the past decade it has become clear that these fibrillar aggregates have the capacity to spread from one cell to another in a prion-like manner, inducing in the recipient cell the aggregation of like endogenous protein2,3,4,5,6,7,8. This unique property could be at the origin of the stereotypic progression of pathology inside the brain9. A better knowledge of the molecular and cellular mechanisms underlying the propagation of amyloid fibrils could allow the development of new drugs able to slow down the gradual evolution of the disease.
The docking of extra-cellular aggregates to the plasma membrane of naïve neurons is a key step of the vicious propagation-amplification cycle. Next, the aggregates are internalized and reach the cytoplasm where they can recruit the otherwise soluble like endogenous protein thus elongating/amplifying. The circle completes with the release of the amplified aggregates into the extracellular milieu, where they can target naïve cells. Very little is known about the molecular basis by which protein fibrillar aggregates bind to the plasma membrane. Different mechanisms have been proposed. The binding could be passive10, mediated by an interaction with the membrane lipids11, and/or with specific protein partners12,13,14. The efficiency of the binding seems to depend both on the aggregates characteristics, such as their charge or conformation10,15, and on the properties of the membrane, with an emphasis on the role of a specific lipid composition16,17 or of the membrane curvature18,19. Whether amyloid fibrils bind to the cellular membrane laterally or through their ends is subject to debate. Indeed, by comparing the permeabilization and toxic properties, which are related to but differ from the binding per se, of two types of β-2-microglobulin fibrils displaying different lengths, Xue and Radford proposed that the binding was mediated mainly through fibrils ends11,20. However massive lateral binding has been also observed in this model like in others11,21,22,23. Determining how amyloid fibrils interact with membranes is important owing to the role played by fibril fragmentation, both for the aggregation mechanism24,25,26 and their propagation propensity20,27. Indeed at any given monomer concentration short, fragmented fibrils display a larger number of extremities as compared to long fibrils while both short and long fibrils have the same lateral surfaces. Thus short fibrils will bind to a higher extent than long fibrils as opposed to with the same efficiency depending on whether interaction with cell membranes occurs through fibrils ends or lateral surfaces.
Here we specifically assessed this issue by studying the binding of amyloid fibrils displaying similar composition and structure but different lengths to artificial liposomes and mammalian cells. We used two different and complementary models, namely α-synuclein (αSyn) involved in Parkinson’s disease, and huntingtin protein exon 1 (HTTExon1) which aggregation is responsible of Huntington’s disease. We observed using electron microscopy massive lateral binding of αSyn and HTTExon1 fibrils to unilamellar vesicles made of brain lipids. We could demonstrate that at identical monomer concentrations, long fibrils made of either αSyn or HTTExon1 bind to the same extent or even better both to artificial liposomes and cells than their short counterparts, thus demonstrating a preeminent role of fibrils surface in the binding. We also show that although αSyn and HTTExon1 fibrils bind both laterally to the cellular membrane, their mechanisms of interaction differ.
Results
Preparation of fibrils of different lengths but identical conformation
To determine if amyloid fibrils bind to the cellular membrane laterally or through their extremities, we decided to characterize the binding of fibrils identical in everything but their length (Fig. 1). Consider the case depicted in Fig. 1: at a defined monomer concentration, short fibrils obtained by fragmenting long fibrils are in this example three times shorter than long fibrils; they are therefore three times more concentrated when considering the number of particles and possess three times more extremities but lateral surfaces that are identical in nature and area. We imagined three different scenarios for the binding of these long and short fibrils to the cellular membrane. We reasoned that if fibrils would bind to the cellular membrane exclusively or mainly through their extremities, under non-saturating conditions, short fibrils would bind to a greater extent to the cells than long fibrils, because they have more extremities (Fig. 1a). Conversely, if short fibrils do not bind to a higher extent to cells, then, under non-saturating conditions, fibrils must bind predominantly laterally to the membrane (Fig. 1b,c). Two types of lateral binding may occur: lateral binding that does not depend on the number of binding sites (Fig. 1b), in which case long and short fibrils will have equal binding capacities; and lateral binding that requires a minimal number of binding sites (Fig. 1c). In the later case short fibrils would be too short to simultaneously bind to a sufficient number of anchoring points at the membrane.
Figure 1. Schematics for the different scenarios of amyloid fibrils binding to biological membranes.
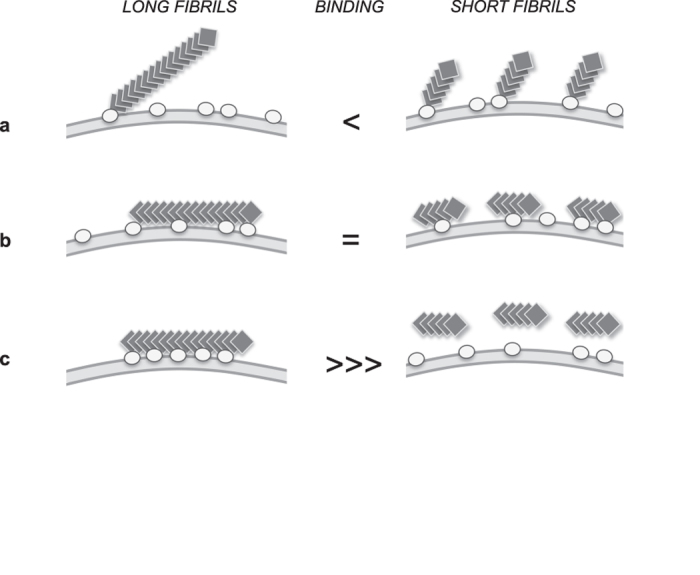
(a) Binding mainly through fibrils extremities. At identical monomer concentration and under non-saturating conditions, short fibrils will bind the cellular membrane to a higher extent than longer fibrils. (b) Lateral binding that does not depend on the number of interacting sites. Long and short fibrils will bind equally well to the cellular membrane. (c) Lateral binding with a minimal number of interacting sites required. Binding becomes dependent on fibrils length as the longer the fibrils are the higher the probability of interacting with the minimal number of interaction sites is. Fibrils of a length that do not meet with the required number of interaction sites will not bind the cellular membrane at all.
We used sonication to obtain fibrils of different lengths. We assembled αSyn and HTTExon1 into fibrils as described in the Methods section. Each sample was then divided in two: the first one was sonicated for only 10 min, to disperse the aggregates and generate individual long fibrils; the second sample was sonicated for 16 h to break the long fibrils into short ones. Representative images and the distributions of fibril lengths obtained by quantitative electron microscopy for long and short fibrils of each protein are shown in Fig. 2. For both αSyn and HTTExon1 we could obtain two distinct populations of fibrils, displaying similar morphologies but different length distributions.
Figure 2. Morphologies and length distributions of long and short αSyn and HTTExon1 fibrils.

(a,c) Representative negatively stained TEM of long (red box) and short (blue box) αSyn (a) or HTTExon1 (c) fibrils, freshly sonicated (box with solid lines) or after being incubated for 1 hour with unilamellar vesicles made from brain lipids in Dulbecco’s modified Eagle’s medium (box with dashed lines). Scale bar, 100 nm. (b,d) Length distributions of long (red) and short (blue) αSyn (b) or HTTExon1 (d) fibrils, freshly sonicated (closed circles, solid lines) or after being incubated for 1 hour with unilamellar vesicles made from brain lipids (open circles, dashed lines). The length distributions were obtained by measuring the length of at least 1000 fibrils from 3 to 10 independent experiments by quantitative negatively stained TEM.
Fibrils with different conformations are susceptible to bind differently to the membrane15. To check that sonication did not alter fibrils structure we performed several analyses (Fig. 3). Thioflavin T is an amyloid-binding dye which fluorescence intensity depends both on the amount and the conformation of proteins within fibrils28. Both αSyn and HTTExon1 long and short fibrils bind Thioflavin T to a similar extent (Fig. 3a). We previously showed that proteinase K degradation patterns can discriminate between different polymorphs of αSyn fibrils15. Long and short αSyn fibrils display identical proteinase K degradation patterns (Fig. 3b). This indicates that the protease cleavage sites are distributed similarly in both fibrils. HTTExon1 proteinase K degradation yields fragments that are too short to be resolved by SDS-PAGE, and an SDS-resistant fragment which intensity decreases with time following a kinetics that is similar for long and short fibrils (Fig. 3c). The resolubilisation of this SDS-resistant fragment in TFA:HFIP revealed the progressive disappearance of the band corresponding to full-length HTTExon1 and the progressive appearance of a band with a smaller apparent molecular weight; the pattern and kinetics of appearance and disappearance of these two bands were similar for long and short HTTExon1 fibrils (Fig. 3d).
Figure 3. Long and short fibrils have similar structures.

(a) ThT fluorescence spectra following binding to fibrils. (b) Proteinase-K degradation patterns of αSyn long (red) and short (blue) fibrils at identical monomeric protein concentrations. (c) Proteinase-K degradation kinetics of SDS-resistant long (red) and short (blue) HTTExon1 fibrils at identical monomeric protein concentrations. Insets, representative SYPRO-stained SDS-PAGE showing the disappearance of fibrillar HTTExon1 trapped within the gel well as a function of time. (d) Proteinase-K degradation pattern of TFA-HFIP solubilized long (red) and short (blue) HTTExon1 fibrils as a function of time. SDS-PAGE were SYPRO-stained. In panels a and c means and standard errors are calculated from 3 independent experiments.
We conclude from these physico-chemical assessments that we generated αSyn and HTTExon1 fibrils of distinct lengths but identical structures.
Binding of long and short fibrils to unilamellar vesicles made of brain lipids
We used unilamellar vesicles composed of total brain lipids to image the binding of short and long αSyn and HTTExon1 fibrils to lipid membranes. αSyn and HTTExon1 long and short fibrils (50 μM) were incubated for 1 hour at room temperature in Dulbecco’s modified Eagle’s medium containing unilamellar vesicles made of brain lipids. Representative images obtained by electron microscopy are displayed in Fig. 4. As shown in Fig. 4a, liposomes associate to the flank of long αSyn fibrils. The resulting structures look like rosaries, with “beads”, the liposomes, aligned along the “string”, the fibrils. We almost never observed liposomes bound to the end of the fibrils. Short αSyn fibrils also bind to the lipid vesicles (Fig. 4b). As illustrated in Fig. 4b, liposomes bind predominantly laterally to the fibrils. Long HTTExon1 fibrils also bind to liposomes, and as for long and short αSyn fibrils the binding was mostly lateral (Fig. 4c). In a sharp contrast, short HTTExon1 fibrils were almost never associated to liposomes (Fig. 4d). The fibrils being evenly distributed on the grid, the superposition seen between some short HTTExon1 fibrils and liposomes in the 2D-plane of the grid is most probably casual.
Figure 4. Binding of long and short αSyn and HTTExon1 fibrils to unilamellar vesicles made from brain lipids.

Binding of 50 μM αSyn long (a) and short (b) fibrils and of 50 μM HTTEx1 long (c) and short (d) fibrils to a 5× mass excess of artificial liposomes made from total brain lipids assessed by TEM. Scale bar, 100 nm.
We conclude from these experiments that both αSyn and HTTExon1 fibrils bind laterally to artificial liposomes.
Correlation between fibril length and their cell binding and permeabilization abilities
Artificial liposomes cannot recapitulate the properties of cellular membranes. We therefore assessed the binding of short and long αSyn and HTTExon1 fibrils to cultured cells.
We first controlled that neither the length nor the structure of short and long αSyn and HTTExon1 fibrils were modified upon incubation in a cellular environment. αSyn and HTTExon1 long and short fibrils (50 μM) were incubated in Dulbecco’s modified Eagle’s medium at room temperature with unilamellar vesicles made of brain lipids. Electron microscopy demonstrated that each type of fibrils retain their morphology (Fig. 2a,c) and length upon incubation (Fig. 2b,d). We determined by a filter trap assay followed by western blotting that the amount of fibrillar protein was unchanged (Figure S1a). Finally proteinase K digestion demonstrated that the structure of αSyn (compare Fig. 3b and Figure S1b) or HTTExon1 (compare Fig. 3c and Figure S1c) long and short fibrils was not affected by the experimental conditions (e.g. incubation time, cell growth media and lipid vesicles).
Next we compared the affinity of long and short αSyn and HTTExon1 fibrils for the cellular membrane. αSyn and HTTExon1 fibrils were labelled with Atto 488 before sonication, to ensure that long and short fibrils have identical monomer concentration and fluorescence intensity. We controlled that the labelling altered neither the morphology nor the lengths of the fibrils.
As demonstrated previously15,29, the long αSyn fibrils bound rapidly and efficiently to the plasma membrane of Neuro 2A cells (Figs 5a,b and S2), as assessed by flow cytometry. We measured an observed KD for the binding of long αSyn fibrils to Neuro 2A cells of 0.8 ± 0.2 μM (Fig. 5a). We demonstrated that the binding to the cellular membrane was not affected by labelling the protein. Indeed, unlabelled αSyn fibrils competed in a dose-dependent way with the binding of labelled αSyn fibrils to Neuro 2A cells (Fig. 5d–f). The binding of long αSyn fibrils induced a progressive increase in the intracellular free Ca2+ level over time as assessed by the increase in the fluorescence of Neuro 2A cells loaded with Fluo-4-AM (Fig. 6a,b,d), similarly to what we reported in earlier studies15,29. This increase in intracellular free Ca2+ level was not due to Ca2+ release from intracellular compartment, as no increase in Ca2+ level was recorded for cells loaded with Fluo-4-AM upon binding of long αSyn fibrils in the presence of 5 mM EGTA (Fig. 6d). We conclude from these experiments that αSyn long fibrils bind efficiently to the cellular membrane and subsequently induce a permeabilization of the membrane. Remarkably, the binding of short αSyn fibrils was almost identical to that of long fibrils (Figs 5a,c and S2), both under non-saturating and saturating concentrations. We measured an observed KD for the binding of short αSyn fibrils to Neuro 2A cells of 0.9 ± 0.2 μM (Fig. 5a), similar to that we determined for long fibrils. In perfect agreement with binding, short αSyn fibrils led to an increase in the intracellular free Ca2+ level over time in Neuro 2A cells to a similar extent than long αSyn fibrils (Fig. 6a,b,d). In a manner similar to what we observed for long fibrils, no increase in the intracellular free Ca2+ level was recorded when the measurements were performed in the presence of EGTA (Fig. 6d). These observations suggest that the cellular cytoplasmic membrane integrity is compromised upon binding to short αSyn fibrils.
Figure 5. Binding of long and short αSyn and HTTExon1 fibrils to Neuro 2A cells.

(a) Binding of αSyn-ATTO488 long (red) and short (blue) fibrils at increasing concentrations (0.1–5 μM) to Neuro 2A cells assessed by flow cytometry. (b,c) Representative traces of the binding of 1 μM αSyn-ATTO488 long (b) and short (c) fibrils to Neuro 2A cells. (d) Binding of 1 μM αSyn-ATTO488 long fibrils to Neuro 2A cells in the presence of increasing concentrations (0–100 μM) of unlabelled long αSyn fibrils. (e,f) Representative traces of the binding to Neuro 2A cells of αSyn-ATTO488 long fibrils (1 μM) in the presence of 10 (e) or 100 (f) μM unlabelled long αSyn fibrils. (g) Binding of HTTExon1-ATTO488 long and short fibrils at increasing concentrations (0.5–10 μM) to Neuro 2A cells. (h,i) Representative traces of the binding to Neuro 2A cells of HTTExon1-ATTO488 long fibrils (h, 5 μM), and HTTExon1-ATTO488 short fibrils (i, 5 μM). The means percentage of cells with bound fibrils and the associated standard error values, calculated from 3 to 6 independent experiments, are represented in (a,d,g). The horizontal line represents the limit between non fluorescent and fibrils-bound, fluorescent cells in (b,c,e,f,h,i).
Figure 6. Assessment of Neuro 2A cells permeabilization by long and short fibrils by imaging intracellular free Ca2+ alterations.

(a–c) Cells loaded with Fluo4-AM were imaged by epifluorescence microscopy after exposure to PBS or PBS + 5 mM EGTA (a), long and short αSyn fibrils (10 μM; b), and long and short HTTExon1 fibrils (10 μM; c). Scale bars, 20 μm. (d) Quantification of the intracellular free Ca2+ increase over time, expressed as a fraction of the maximal Ca2+ increase observed after addition of ionomycin (10 μM) to the cells. Data are means and associated standard error values calculated from 6 to 10 independent experiments. For the sake of clarity the standard errors are not represented for αSyn and HTTExon1 long and short fibrils in the presence of 5 mM EGTA. For each experiment, fluorescence increase is a mean calculated over ≈50–100 individual cells. The increase in fluorescence measured after addition of PBS + 5 mM EGTA was subtracted for each trace.
We observed significant differences with fibrillar HTTExon1. As demonstrated previously10,30 the long HTTExon1 fibrils bound efficiently to the plasma membrane of Neuro 2A cells (Figs 5g,h and S3). We measured an observed KD for the binding of long HTTExon1 fibrils to Neuro 2A cells of 3 ± 1 μM (Fig. 5g). This binding was accompanied by a progressive permeabilization of the cellular membrane causing an increase in the intracellular free Ca2+ concentration over time (Fig. 6a,c,d), in agreement with previous results29. However and contrarily to what was seen with αSyn, we found that even at concentrations that are saturating for the binding of long HTTExon1 fibrils, short HTTExon1 fibrils were neither able to bind to the cellular membrane (Figs 5g,i and S3), nor to permeabilize it (Fig. 6a,c,d).
We conclude from these observations that neither αSyn nor HTTExon1 fibrils bind to cells principally through their ends as otherwise increased binding to cells of short fibrils as compared to long fibrils would have been observed. The short and long αSyn fibrils bind to the membrane with similar affinities and permeabilize it to the same extent. This indicates that αSyn fibrils bind predominantly laterally to the cellular membrane (Fig. 1b). When HTTExon1 fibrils were fragmented by sonication, binding was lost. This cannot be accounted for by fibrils binding through their ends. We therefore conclude that HTTExon1 fibrils also bind laterally to the cellular membrane. However the lateral binding of αSyn and HTTExon1 fibrils differs. While the binding of αSyn to the cellular membrane appears not to depend tightly on the number of interaction sites (Fig. 1b), that of HTTExon1 does given that binding is lost when the fibrils are fragmented. Thus, HTTExon1 fibrils binding appear to depend on a minimal number of interaction sites, a requirement that is fulfilled for long fibrils but not for short ones (Fig. 1c).
Discussion
It is now well established that fibrillar protein aggregates involved in neurodegenerative diseases spread from one neuron to the other2,3,4,5,6,7,8. This propagation cycle comprises different key steps the first of which being the binding of the aggregates to the cellular membrane, followed by their internalization, the recruitment of the endogenous and otherwise soluble cellular proteins into newly born aggregates, and the release of the protein aggregates. Little is known about the molecular mechanisms underlying each of these steps. It has also appeared difficult to study each step independently, many studies focusing on the global cytotoxicity resulting from aggregates internalization16,20,27,31. The binding step can be studied in vitro using artificial liposomes11,22,32,33,34, but this strategy neglects the prominent role of membrane proteins and the extracellular matrix in fibrils binding10,13,17,35. In this work we demonstrate, by measuring the binding of fibrils displaying identical structures and physico-chemical properties but different lengths to lipid vesicles and cultured cells, that fibril ends are not a prime determinant of the binding process of αSyn and HTTExon1 assemblies.
Lateral binding of fibrils to artificial liposomes was observed by electron microscopy in other models21,22,23. These results together with those we report here using artificial liposomes and cells are different from that obtained with β-2-microglobulin fibrils, which were shown to bind and permeabilize membranes mainly through their ends, although lateral binding was also observed11,20. While our results do not exclude that αSyn and HTTExon1 fibrils also bind to the cellular membrane through their extremities, they do demonstrate that fibrils ends have an affinity for the membranes that is at most similar to fibrils flanks.
Even if we demonstrate that αSyn and HTTExon1 fibrils bind both laterally to the cellular membrane, the way they interact with the membranes appears to differ significantly. While the binding of αSyn to the cellular membrane seems not to depend on the number of interaction sites (Fig. 1b), that of HTTExon1 does given that binding is lost when the fibrils are fragmented. Thus, HTTExon1 fibrils binding appears to depend on a minimal number of interaction sites, a requirement that is fulfilled for long fibrils but not for short ones (Fig. 1c). We cannot exclude that αSyn fibrils shorter than those we used in this work would exhibit a behaviour similar to that of HTTExon1 short fibrils.
Many fibrils characteristics determine their binding capacities: their net charge10, aggregation state10,33, conformation15 or length (this work). Even small sequence modifications can have dramatic effects10,32,36. The next step will be to map fibrils surfaces that binds to the cellular membrane and dissect at the molecular level the binding reaction37,38,39,40. The elucidation of this “binding code” is of fundamental importance for the development of new therapeutic tools. For example it has been proposed that fibril stabilization could be valuable to decrease the number of fibrils ends and thus their binding propensity to cells11,24. While this may impact fibrils uptake because long fibrils could be taken up with lesser efficiency than their short counterparts, our results clearly demonstrate that this strategy is not justified by the somewhat generic ability of amyloid fibrils to bind to cellular membranes through their ends. We therefore favour an alternative approach where fibrils surfaces are modified by fibrils binders that coat their surfaces compromising the crucial step where they interact with cell membranes41,42.
Methods
Expression and purification of αSyn and HTTExon1
Recombinant human wild-type αSyn was expressed in E. coli strain BL21(DE3) (Stratagene, La Jolla, CA, USA) and purified as described43. Recombinant C-terminally hexa His-tagged MBP-TEV-HTTExon1-His with a polyQ stretch of 48 glutamine residues was expressed in E. coli strain BL21(DE3) (Stratagene, La Jolla, CA, USA) and purified as described44. HTTExon1-His was obtained from MBP-TEV-HTTExon1-His as described44. Briefly, MBP-TEV protease was incubated with the purified MBP-TEV-HTTExon1-His until 100% cleavage was achieved. HTTExon1-His was purified from the resulting mixture using a Talon metal affinity resin column (Clontech, Saint-Germain-en-Laye, France), immediately filtered through a 0.22 μM filter, aliquoted, flash frozen in liquid nitrogen and stored at −80 °C until use.
Assembly into fibrils, labelling, and preparation of short and long fibrils
For fibril formation, αSyn was incubated in assembly buffer A (50 mM Tris-HCl, pH 7.5, 150 mM KCl) at 37 °C under continuous shaking in an Eppendorf Thermomixer set at 600 rpm; HTTExon1 was incubated in assembly buffer B (20 mM Tris-HCl, pH 7.5, 150 mM KCl, 10% glycerol) at 37 °C without agitation. Long and short fibrils were obtained by sonicating the fibrils with an ultrasond sonicator (Hielsher) set with an amplitude of 75 and a cycle of 0.5 s for 10 min or 16 h, respectively.
For cell sorting experiments, αSyn and HTTExon1 fibrils were first washed in PBS and then labelled by the addition of 2 molar excess of the aminoreactive fluorescent dye Atto 488 N-hydroxysuccinimide ester (ATTO-Tec GmbH) before the sonication step. Labelling was performed following the manufacturer’s recommendation. Unreacted dye was removed by two cycles of sedimentation and suspension of the fibrils in PBS.
Preparation of unilamellar vesicles made of brain lipids and incubation with fibrils
Porcine brain lipid extract (total) was purchased from Avanti Polar Lipids (Alabaster, AL). Unilamellar vesicles were prepared as previously described29 with slight modifications. Briefly, the brain lipids in chloroform were dried in glass tubes under a gentle nitrogen stream. The lipid films were hydrated at 10 mg/ml in Dulbecco’s modified Eagle’s medium, subjected to five freeze-thaw cycles of 2 min each, subjected to sonication for 20 min and centrifuged at 10000 × g for 10 min. αSyn and HTTExon1 long and short fibrils (50 μM) were incubated in Dulbecco’s modified Eagle’s medium for 1 hour at room temperature with unilamellar vesicles at a protein to lipid mass ratio of 1:5.
Quantitative electronic microscopy
Protein assemblies were examined by transmission electron microscopy (TEM) in a Jeol 1400 transmission electron microscope (Jeol SAS, Croissy-sur-Seine, France) following adsorption onto carbon-coated 200 mesh grids and negative staining with 1% uranyl acetate. The images were recorded with a Gatan Orius CCD camera (Gatan Inc., Pleasanton, CA, USA). Length measurements were performed on individual fibrils with the software ImageJ (NIH, USA). For each fibril type (αSyn long, αSyn short, HTTExon1 long, HTTExon1 short, incubated or not with liposomes) lengths distribution were obtained by measuring the lengths of at least 1000 fibrils, using more than 20 negatively stained electron micrographs obtained from 3 to 10 independent experiments.
Thioflavin T measurements
αSyn (10 μM monomer concentration) and HTTExon1 (50 μM monomer concentration) long and short fibrils were incubated with Thioflavin T (10 μM). Thioflavin T fluorescence was recorded with a Cary Eclipse spectrofluorimeter (Varian Medical Systems Inc.) using excitation and emission wavelengths set at 440 and 480 nm, respectively.
Proteolytic digestion and analysis of the proteolytic patterns
αSyn (100 μM monomer concentration) and HTTExon1 (50 μM monomer concentration) long and short fibrils in their respective assembly buffers or incubated in Dulbecco’s modified Eagle’s medium with unilamellar vesicles from brain lipids were treated at 37 °C by Proteinase K (Roche; 3.8 μg.ml−1). Aliquots (10 μl) were removed at different time intervals following addition of the protease, immediately mixed with denaturing buffer (10 μl; 180 mM Tris-HCl, pH 6.8, 6% SDS, 15% β-mercaptoethanol, 30% glycerol and 0.01% bromophenol blue) to arrest immediately the cleavage reaction, incubated for exactly 5 min at 90 °C and frozen at −80 °C until analysis on Tris-glycine SDS-polyacrylamide (15%) gel electrophoresis (PAGE). For HTTExon1 aliquots were taken for solubilization. 40 μl of the reaction mixture were withdrawn and immediately diluted in 200 μl of TFA:HFIP 1:1. Fibril solubilization was allowed for 2 hours. The solvent was then removed under a stream of nitrogen for 30 min. After removal from the gas flow the samples were immediately resuspended in 40 μl NaOH 0.1M and then diluted in 40 μl of denaturing buffer. For the analysis of αSyn fibrils cleavage, the gels were stained by coomassie. For the analysis of HTTExon1 fibrils cleavage, the gels were stained with SYPRO Orange (Invitrogen, Paisley, UK) diluted 5000 fold in 10% acetic acid for 1h and visualized using LAS-3000 imager (Fujifilm, Tokyo, Japan).
Filter trap assay and Western blotting
The amount of long and short aSyn and HTTExon1 fibrils present before and after incubation with unilamellar vesicles from brain lipids in Dulbecco’s modified Eagle’s medium was assessed by a filter retardation assay45, where 10 μl of each reaction were withdrawn in triplicate at time zero and after 1 hour of incubation, diluted in 200 μl of sarkosyl 1% (αSyn) or SDS 2% (HTTExon1), filtered through cellulose acetate membranes (0.2 μm pore size, Millipore Corp., Bedford, MA) using a 48-slot slot-blot filtration apparatus (GE Healthcare), and washed twice with 200 μl of sarkosyl 1% (αSyn) or SDS 2% (HTTExon1). The cellulose acetate membranes were incubated with 3% skim milk, probed with a mouse monoclonal anti-αSyn antibody (BD Transduction Laboratories) or a rabbit polyclonal anti-HTTExon1 antibody we raised, and developed with the enzyme-coupled luminescence technique (ECL, Thermo Scientific) following the manufacturer recommendations.
Cell culture
Murine neuroblastoma Neuro 2a cells (ATCC) were culture at 37 °C in humidified air with 5% CO2 in Dulbecco’s modified Eagle’s medium containing 10% foetal bovin serum, 2 mM glutamine, 100 units.ml−1 penicillin and 100 μg.ml−1 streptomycin. All materials used for cell culture were from PAA Laboratories GmbH (Pasching, Austria).
Cell sorting experiments
To monitor the binding of αSyn long and short fibrils to the cell membranes, Neuro 2A cells lifted with EDTA were resuspended in serum-free, phenol red free-Dulbecco’s modified Eagle’s medium containing 1 μg.ml−1 of propidium iodide. At time zero, Atto 488-labelled αSyn long or short fibrils at increasing monomer concentrations (0.1–5 μM) were added to the cells (106 cells per ml) and analysis were performed using a MoFlo Astrios Cell Sorter (Beckman Coulter). The cell population was gated by forward and side scatter and at least 15,000 cells were recorded. The fraction of fluorescent cells was calculated over a time frame where the signal was stable.
As the binding of HTTExon1 fibrils to cells is a slow process30, we proceeded differently. 105 cells were platted in a 6 cm-diameter Petri dish 48 h before the experiment. They were then washed with PBS and incubated with Atto 488-labelled HTTExon1 long or short fibrils at increasing monomer concentrations (0.5–10 μM) for 2 h at 37 °C. After incubation cells were lifted with EDTA and analysed as described above.
Intracellular Ca2+ levels measurements and epifluorescence microscopy imaging
Subconfluent Neuro 2A cells cultured on ibidi-μ-Dishes (Biovalley) were loaded for 30 min at 25 °C with 2 μM of the fluorescent dye Fluo-4-acetoxymethyl ester (Fluo-4-AM) (Invitrogen) in the presence of 0.02% (w/v) Pluronic Acid F127 (Invitrogen) and 2.5 mM of the organic anion-transport inhibitor probenicid (Invitrogen). After washing the cells deesterification of the Fluo-4-AM was allowed for 10 min at 25 °C in serum-free Dulbecco’s modified Eagle’s Medium in the presence of 0.02% (w/v) Pluronic Acid F127 (Invitrogen). PBS, PBS + EGTA (5 mM), αSyn or HTTExon1 long or short fibrils (10 μM monomer concentration) with or without EGTA (5 mM) were then added to the cells. The cells were imaged over time on the Axio Observer ZI epifluorescence microscope (Zeiss) at a magnification of 20x. Images were acquired every 1 (αSyn) or 2 (HTTExon1) min for 40 (αSyn) or 60 (HTTExon1) min at 25 °C in humidified air with 5% CO2. The fluorescence was quantified for each cell of the field (≈50–100 cells/field) using the software ImageJ, after background subtraction for each image, and expressed as a fraction of the maximum fluorescence recorded upon addition of ionomycin (10 μM) to the cells.
Additional Information
How to cite this article: Monsellier, E. et al. α-Synuclein and Huntingtin exon 1 amyloid fibrils bind laterally to the cellular membrane. Sci. Rep. 6, 19180; doi: 10.1038/srep19180 (2016).
Supplementary Material
Acknowledgments
This work benefited from the imaging and cytometry facilities of Imagerie-Gif. This work was supported by the Agence Nationale de la Recherche (ANR-11-BSV8-021-01), the EC Joint Programme on Neurodegenerative Diseases (JPND-NeuTARGETs-ANR-14-JPCD-0002-02), the Centre National de la Recherche Scientifique, France Parkinson (contract 113344) and a “Coup d’Elan à la Recherche Française” award from Fondation Bettencourt-Schueller.
Footnotes
Author Contributions E.M. and R.M. designed the study; E.M. performed the experiments; L.B. contributed reagents; E.M. and R.M. wrote the manuscript. All authors reviewed the manuscript.
References
- Knowles T. P. J., Vendruscolo M. & Dobson C. M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 15, 384–396 (2014). [DOI] [PubMed] [Google Scholar]
- Ren P.-H. et al. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat. Cell Biol. 11, 219–225 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angot E. et al. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One 7, 23–27 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peelaerts W. et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522, 340–344 (2015). [DOI] [PubMed] [Google Scholar]
- Holmes B. B. et al. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. 111, E4376–E4385 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T. et al. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134 (2013). [DOI] [PubMed] [Google Scholar]
- Costanzo M. & Zurzolo C. The cell biology of prion-like spread of protein aggregates: mechanisms and implication in neurodegeneration. Biochem. J. 452, 1–17 (2013). [DOI] [PubMed] [Google Scholar]
- Costanzo M. et al. Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J. Cell Sci. 126, 3678–3685 (2013). [DOI] [PubMed] [Google Scholar]
- Brundin P., Melki R. & Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 11, 301–307 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevino R. S. et al. Fibrillar structure and charge determine the interaction of polyglutamine protein aggregates with the cell surface. J. Biol. Chem. 287, 29722–29728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi L. et al. Direct three-dimensional visualization of membrane disruption by amyloid fibrils. Proc. Natl. Acad. Sci. 109, 20455–20460 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurén J., Gimbel D. A., Nygaard H. B., Gilbert J. W. & Strittmatter S. M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 457, 1128–1132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton A., Zamponi G. W. & Ferguson S. S. G. Glutamate receptors function as scaffolds for the regulation of β-amyloid and cellular prion protein signaling complexes. Mol. Brain 8, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shrivastava A. N. et al. α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 34, 2408–2423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousset L. et al. Structural and functional characterization of two alpha-synuclein strains. Nat. Commun. 4, 2575 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelisti E. et al. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 125, 2416–2427 (2012). [DOI] [PubMed] [Google Scholar]
- Bucciantini M. et al. Toxic effects of amyloid fibrils on cell membranes: the importance of ganglioside GM1. FASEB J. 26, 818–831 (2012). [DOI] [PubMed] [Google Scholar]
- Chaibva M., Burke K. A. & Legleiter J. Curvature enhances binding and aggregation of huntingtin at lipid membranes. Biochemistry 53, 2355–2365 (2014). [DOI] [PubMed] [Google Scholar]
- Garten M. et al. Methyl-branched lipids promote the membrane adsorption of α-synuclein by enhancing shallow lipid-packing defects. Phys. Chem. Chem. Phys. 17, 15589–15597 (2015). [DOI] [PubMed] [Google Scholar]
- Xue W. F. et al. Fibril fragmentation enhances amyloid cytotoxicity. J. Biol. Chem. 284, 34272–34282 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien O. et al. Unraveling the mechanism of cell death induced by chemical fibrils. Nat. Chem. Biol. 10, 969–976 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suopanki J. et al. Interaction of huntingtin fragments with brain membranes–clues to early dysfunction in Huntington’s disease. J. Neurochem. 96, 870–84 (2006). [DOI] [PubMed] [Google Scholar]
- Engel M. F. M. et al. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 105, 6033–6038 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W. & Radford S. E. An imaging and systems modeling approach to fibril breakage enables prediction of amyloid behavior. Biophys. J. 105, 2811–2819 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J. et al. Observation of spatial propagation of amyloid assembly from single nuclei. Proc. Natl. Acad. Sci. 108, 14746–14751 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J. et al. An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537 (2009). [DOI] [PubMed] [Google Scholar]
- Wu J. W. et al. Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J. Biol. Chem. 288, 1856–1870 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancalana M. & Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieri L., Madiona K., Bousset L. & Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 102, 2894–2905 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Arlandis G., Pieri L., Bousset L. & Melki R. Binding, internalization and fate of Huntingtin Exon1 fibrillar assemblies in mitotic and non-mitotic neuroblastoma cells. Neuropathol. Appl. Neurobiol. In press (2015). [DOI] [PubMed] [Google Scholar]
- Couceiro J. et al. Sequence-dependent internalization of aggregating peptides. J. Biol. Chem. 290, 242–258 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel K. B. et al. Polyglutamine expansion in huntingtin alters its interaction with phospholipids. J. Neurochem. 110, 1585–1597 (2009). [DOI] [PubMed] [Google Scholar]
- Grey M., Linse S., Nilsson H., Brundin P. & Sparr E. Membrane interaction of α-synuclein in different aggregation states. J. Parkinsons. Dis. 1, 359–371 (2011). [DOI] [PubMed] [Google Scholar]
- Burke K. A., Yates E. A. & Legleiter J. Amyloid-forming proteins alter the local mechanical properties of lipid membranes. Biochemistry 52, 808–817 (2013). [DOI] [PubMed] [Google Scholar]
- Holmes B. B. et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. 110, E3138–E3147 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke K. A., Kauffman K. J., Umbaugh C. S., Frey S. L. & Legleiter J. The interaction of polyglutamine peptides with lipid membranes is regulated by flanking sequences associated with huntingtin. J. Biol. Chem. 288, 14993–15005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalek M., Salnikov E. S., Werten S. & Bechinger B. Membrane interactions of the amphipathic amino terminus of huntingtin. Biochemistry 52, 847–858 (2013). [DOI] [PubMed] [Google Scholar]
- Michalek M., Salnikov E. S. & Bechinger B. Structure and topology of the huntingtin 1-17 membrane anchor by a combined solution and solid-state NMR approach. Biophys. J. 105, 699–710 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco G. et al. Direct observation of the three regions in α-synuclein that determine its membrane-bound behaviour. Nat. Commun. 5, 3827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivanandam V. N. et al. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 133, 4558–4566 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton S. & Melki R. The interaction of Hsc70 protein with fibrillar alpha-Synuclein and its therapeutic potential in Parkinson’s disease. Commun Integr Biol 5, 94–95 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki R. Role of different alpha-Synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J. Parkinsons. Dis. 5, 217–227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghee M., Melki R., Michot N. & Mallet J. PA700, the regulatory complex of the 26S proteasome, interferes with alpha-synuclein assembly. FEBS J. 272, 4023–4033 (2005). [DOI] [PubMed] [Google Scholar]
- Monsellier E., Redeker V., Ruiz-Arlandis G., Bousset L. & Melki R. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J. Biol. Chem. 290, 2560–2576 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanker E. E. et al. Membrane filter assay for detection of amyloid-like polyglutamine-containing protein aggregates. Methods Enzymol. 309, 375–386 (1999). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.