

Abstract
Drug-loaded nanoparticles (NPs) can improve infection treatment by ensuring drug concentration at the right place within the therapeutic window. Poly(lactic-co-glycolic acid) (PLGA) NPs are able to enhance drug localization in target site and to sustainably release the entrapped molecule, reducing the secondary effects caused by systemic antibiotic administration. We have loaded auranofin, a gold compound traditionally used for treatment of rheumatoid arthritis, into PLGA NPs and their efficiency as antibacterial agent against two Gram-positive pathogens, Streptococcus pneumoniae and Streptococcus pyogenes was evaluated. Auranofin-PLGA NPs showed a strong bactericidal effect as cultures of multiresistant pneumococcal strains were practically sterilized after 6 h of treatment with such auranofin-NPs at 0.25 μM. Moreover, this potent bactericidal effect was also observed in S. pneumoniae and S. pyogenes biofilms, where the same concentration of auranofin-NPs was capable of decreasing the bacterial population about 4 logs more than free auranofin. These results were validated using a zebrafish embryo model demonstrating that treatment with auranofin loaded into NPs achieved a noticeable survival against pneumococcal infections. All these approaches displayed a clear superiority of loaded auranofin PLGA nanocarriers compared to free administration of the drug, which supports their potential application for the treatment of streptococcal infections.
Bacterial infections are responsible for significant morbidity and mortality in clinical settings and represent a global health threat and a burden to healthcare systems1. Streptococcus pneumoniae, the pneumococcus, is a Gram-positive pathogen and a leading cause of diseases such as otitis media, bacteremia and meningitis in young children, the elderly, and persons with chronic medical conditions. Its clinical burden is about 2 million deaths per year from invasive disease (defined as isolation of S. pneumoniae from a normally sterile site such as blood or cerebrospinal fluid), half of these in children under 5 years of age, but is likely to cause many more due to non-bacteremic pneumonia and other respiratory diseases2. Thus, for every case of bacteremic pneumococcal pneumonia in adults, it has been estimated that there are, at least, 3 additional cases of non-bacteremic pneumococcal pneumonia3. The classical treatment to fight pneumococcal infections has been the use of antibiotics, but the effectiveness of this therapy has been compromised by the progressive selection for resistance against major drug classes, and treatment failures are widely reported4,5. In addition, a smaller, but growing, number of pneumococcal isolates are resistant to multiple antibiotics, leaving vancomycin as a drug of last choice6. Streptococcus pyogenes is also an important human pathogen being the bacterium most frequently isolated from patients with pharyngitis, although it causes more severe invasive infections, including necrotizing fasciitis, sepsis, and toxic shock syndrome. Antibiotic treatment failures in cases of streptococcal pharyngitis have been reported, mainly due to the biofilm formation7. Consequently, in a recent report, the US Centers for Disease Control and Prevention (CDC) has called for an aggressive and immediate action to halt the spread of drug-resistant pathogens8.
It is well established that drug discovery and development is nowadays a very expensive, time-consuming, and risky process. The so-called drug ‘repurposing’ (or ‘reprofiling’) is an alternative and promising strategy to speed up this drug discovery process, with a concomitant reduction of failure rates and associated costs9,10. In this sense, auranofin is a mixed ligand gold compound approved by the U.S. Food and Drug Administration (FDA) in 1985, commercialized under the brand name of Ridaura, and recommended for the treatment of severe rheumatoid arthritis11. Several years ago, new attractive pharmaceutical activities were disclosed for auranofin including anticancer, antiviral and against pathogenic protozoa, like Plasmodium falciparum, Entamoeba histolytica, and Giardia lamblia12,13. Although the antibacterial activity of auranofin has been less explored, a promising effect on certain Gram-positive pathogens as Clostridium difficile14, Staphylococcus aureus15 or Mycobacterium tuberculosis16 has been reported in the recent years.
One of the most successful strategies for overcoming microbial resistance is the use of nanoparticles (NPs) as drug delivery systems, since they can achieve a predictable and desired therapeutic effect in the human body17. This effect is reached when the drug plasma concentration at the relevant site is within the therapeutic window, that is, below the toxic level but above the effective level. Thus, sustained release of antibiotics from NPs could potentially improve the treatment efficacy. Additionally, NPs have been used to target antimicrobial agents to the site of infection, so that higher doses of drug can be given at the infected site, thereby increasing bactericidal activity with fewer adverse side effects18. Poly(lactic-co-glycolic acid) (PLGA) is one of the most successfully employed biodegradable polymers because the monomers produced after its hydrolysis, lactic acid and glycolic acid, are endogenous and efficiently metabolized by the body. In fact, from all the possible biomaterials established for the production of NPs, PLGA has engaged the interest of the drug delivery community because of its attractive properties. Among them, it is worth to mention: a) the biodegradability and biocompatibility; b) its capability for protecting the drug molecules from biodegradation; and c) the potential development of sustained release systems19,20,21. PLGA NPs have been employed for vaccine delivery22, cancer treatment23, treatment of inflammatory diseases24,25, on regenerative medicine26,27 and even for the treatment of some cardiovascular diseases28. Regarding infection treatments, there are some initial studies aimed at developing antibiotic-PLGA NPs to improve the treatment of certain bacterial infections. Different antibiotics have been encapsulated into PLGA NPs, such as rifampicin and azithromycin29, gentamicin30,31,32, nafcillin33, and sparfloxacin34. After several preclinical evaluations, PLGA NPs have demonstrated their antiinfective potential based upon two principles: the NPs are usually taken up by endocytosis, so antibiotic-PLGA NPs are promising delivery systems for targeting intracellular infections; and NPs can achieve sustained release, so antibiotic-PLGA NPs can be employed for the treatment or prevention of infections.
In this work we have explored the use of PLGA NPs loaded with auranofin to demonstrate, as a proof of concept, their efficiency as antibacterial agent against two important streptococcal pathogens, i.e., S. pneumoniae and S. pyogenes. Promising results have been obtained in in vitro assays using multiresistant pneumococcal strains and a biofilm model of S. pneumoniae and S. pyogenes, as well as in vivo using a zebrafish embryo infection model.
Results
Morphology of the NPs
PLGA NPs, both unloaded and auranofin-loaded, were produced following the nanoprecipitation method described in Methods section. Auranofin was successfully loaded into the PLGA nanocarrier, as digestion experiments confirmed. The morphology of NPs was observed by scanning electron microscopy (SEM) (Fig. 1a), showing the expected spherical shape of the produced NPs. Loading auranofin during the nanoprecipitation stage did not affect the morphology of the particles (data not shown).
Figure 1. Auranofin and PLGA NPs.

(a) SEM micrograph of unloaded PLGA NPs and size distribution of PLGA NPs measured by DLS (inset). (b) In vitro release of auranofin from PLGA NPs and kinetic fitting (dashed line) of the release kinetics according to a near-zero order. Chemical structure of auranofin is inserted in the panel. Ac, acetyl; Et, ethyl.
Average diameter and zeta potential
Size of NPs was evaluated using a Dynamic Light Scattering (DLS) apparatus. The mean diameter of both unloaded and auranofin-PLGA NPs was observed to be ca. 60 nm in both cases (Fig. 1a). The surface characteristics were examined through zeta potential, and all NPs were negatively charged with values ca. −30 mV, as expected.
In vitro release kinetics
The employed poly(lactic) acid to poly(glycolic) acid ratio together with their molecular weights were selected to follow a certain degradation pattern which controls the release kinetics so a constant release of auranofin was achieved in the first 6 h of the experiment. The release kinetics from PLGA NPs is mainly based on PLGA degradation via hydrolysis of its ester linkages in the presence of water. In that case, the typical release profile from PLGA NPs should consist on a zero order phase. However, there are other effects influencing the release kinetics, such as surface diffusion, bulk diffusion, polymer molecular weight variation and erosion of the NPs. All of them contribute to a complex process that is very difficult to reproduce in an individual equation. In our particular case, the auranofin release data showed in Fig. 1b can be fitted to a near-zero order kinetic models following the equation:
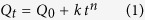 |
Being Qt the percentage of auranofin released at time t, Q0 the initial amount of auranofin in the solution (normally Q0 = 0), k is the release constant, and n is the factor that is 1 in pure zero-order release (degradation of polymeric matrix) and 0.5 in Higuchi-type release (diffusion of drug molecules).
The parameters of the kinetic fitting, shown in Fig. 1b, indicate that our release process is not a pure zero release (degradation) neither a pure Higuchi release (diffusion), but somewhere in the middle (n = 0.81), a near-zero order kinetic release.
In vitro bactericidal activity of auranofin-PLGA NPs
To compare the bactericidal activity of auranofin alone or auranofin-loaded NPs, we initially chose the noncapsulated strain S. pneumoniae R6. It is important to notice that pneumococcal strains show autolysis after several hours of stationary phase of growth. Thus, the incubation time was adjusted to take into account the release kinetics of auranofin and bacterial autolysis in the conditions tested. The compounds were added to a bacterial suspension at an OD550 of 0.6 at three different concentrations (0.1, 0.25 and 0.5 μM), and cell optical density was followed throughout the growth curve. Viable cell counting demonstrated that auranofin alone was effective as an anti-pneumococcal agent since 6 h of incubation at 0.5 μM decreased bacterial population for about three log units (Fig. 2a), whereas cultures treated with auranofin-NPs were virtually sterilized at concentrations of 0.25 or 0.5 μM (Fig. 2b). Afterwards, the bactericidal activity of auranofin-loaded NPs was also tested using encapsulated pneumococcal isolates, namely, strains D39, 48 and 69 (Table 1). As in the previous assay, free auranofin caused a decrease on viability of almost three logs on the multiresistant 48 strain at 0.5 μM (Fig. 2c), whereas auranofin-NPs also led to total killing of the culture even at 0.25 μM (Fig. 2d). Comparable results were obtained with the D39 strain (serotype 2) and the multiresistant 69 strain (serotype 19F) (Table 2). To confirm that the bactericidal effect was produced by the auranofin released from NPs rather than from the NPs themselves, unloaded PLGA NPs were always tested in all the experiments, demonstrating that the nanocarrier did not alter the bacterial population.
Figure 2. Bactericidal effects of auranofin and auranofin-PLGA NPs against pneumococcal strains.

Exponentially growing cultures of R6 strain (a,b) and 48 strain (c,d) were incubated in the absence or in the presence of auranofin alone (a,c) or auranofin-NPs (b,d). Controls with PBS buffer, DMSO or PLGA without auranofin were included. Viable cells were determined on blood agar plates after 6 h of incubation at 37 °C. Data are means of four independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared to the control in the absence of auranofin (one-way ANOVA with a post hoc Dunnet test; *P < 0.01; **P < 0.001).
Table 1. Bacterial strains used in this study.
| Strain | Relevant description | Source or reference |
|---|---|---|
| S. pneumoniae | ||
| R6 | D39 derivative; non-encapsulated | 49 |
| D39 | Serotype 2 | 52 |
| 48 | Serotype 23F; penicillin MICa = 16 mg L−1; erythromycin MICa > 128 mg L−1; ciprofloxacin MICa = 1 mg L−1; levofloxacin MICa = 1 mg L−1; chloramphenicol MICa = 4 mg L−1; tetracycline MICa > 64 mg L−1 | 53 |
| 69 | Serotype 19F; penicillin MICa = 2 mg L−1; erythromycin MICa = 16 mg L−1; levofloxacin MICa = 1 mg L−1; chloramphenicol MICa = 4 mg L−1; tetracycline MICa = 64 mg L−1 | 53 |
| P046 | lytAlytC mutant | 48 |
| S. pyogenes | ||
| Type strain | CECTb 985 | |
| SF370 | Serotype M1; biofilm-forming strain | CECTb 5109 |
aMIC, Minimum Inhibitory Concentration.
bCECT, Colección Española de Cultivos Tipo.
Table 2. Bacterial viability after treatment with auranofin or auranofin-PLGA NPsa.
| Organism (serotype) | Viability after 6 h of treatment withb: |
|||||||
|---|---|---|---|---|---|---|---|---|
| Auranofin (μM) |
PLGA-auranofin (μM) |
|||||||
| DMSO | 0.1 | 0.25 | 0.5 | PLGA | 0.1 | 0.25 | 0.5 | |
| S. pneumoniae | ||||||||
| R6 (none) | − | + | + | +++ | − | ++++ | * | * |
| D39 (2) | − | ++ | ++ | +++ | − | ++++ | ++++ | * |
| 48 (23F) | − | + | + | +++ | − | ++++ | * | * |
| 69 (19F) | − | ++ | ++ | +++ | − | ++++ | ++++ | * |
| S. pyogenes | ||||||||
| Type strain | − | − | − | − | − | − | − | + |
| SF370 (M1) | − | − | − | + | − | + | ++ | ++ |
aBacteria were incubated at 37 °C in PBS (OD550 ≈ 0.6) with the indicated compound at 0.1, 0.25 or 0.5 μM. Viability was determined after 6 h of incubation.
b−, no effect; +, decrease of 1 log in viable cells;++, decrease of 2 logs in viable cells; +++, decrease of 3 logs in viable cells; ++++, decrease of ≥4 logs in viable cells; *, <10 CFU mL−1.
Next, we tested the efficacy of auranofin, alone or loaded in NPs, against two strains of S. pyogenes, the type strain and SF370, the latter being a typical biofilm-forming strain. As shown in Table 2, the bactericidal effect in these strains did not reach the same levels than those found against pneumococcal strains. In fact, free auranofin did not cause any effect on the survival of the S. pyogenes type strain, even at the maximum concentration tested (0.5 μM) but provoked a 90%-drop in the viability of the SF370 strain. It is worth noting, however, that auranofin-NPs effectively killed both S. pyogenes strains, with a decrease of 1- and 2-logs on type strain and SF370 strain, respectively, confirming the increased efficiency of auranofin when was loaded into NPs, compared with auranofin alone.
One of the reasons to explain the greater efficacy of auranofin loaded into PLGA NPs against the streptococcal pathogens could be due to a better stability of the encapsulated drug and, thus, to the protection against a rapid enzymatic and/or hydrolytic degradation26. To investigate this issue we performed bactericidal assays against S. pneumoniae R6 adding auranofin pulses each hour to simulate the slow release that takes place from the auranofin-NPs. Adjustment to a final concentration of 0.5 μM of auranofin by 6 pulses decreased bacterial population 6 logs after 6 h of treatment, while the same concentration of auranofin added at once at the beginning of the experiment caused a viability drop of almost 4 logs (Fig. 3). These results strongly suggest that treatments with repeated doses of auranofin could be more effective against pneumococcal infections than a high single dose. In other words, encapsulation of auranofin in PLGA NPs allows a good delivery system since a sustained release of the drug increases the efficiency of the treatment.
Figure 3. Bactericidal effects of auranofin pulses against S. pneumoniae R6.
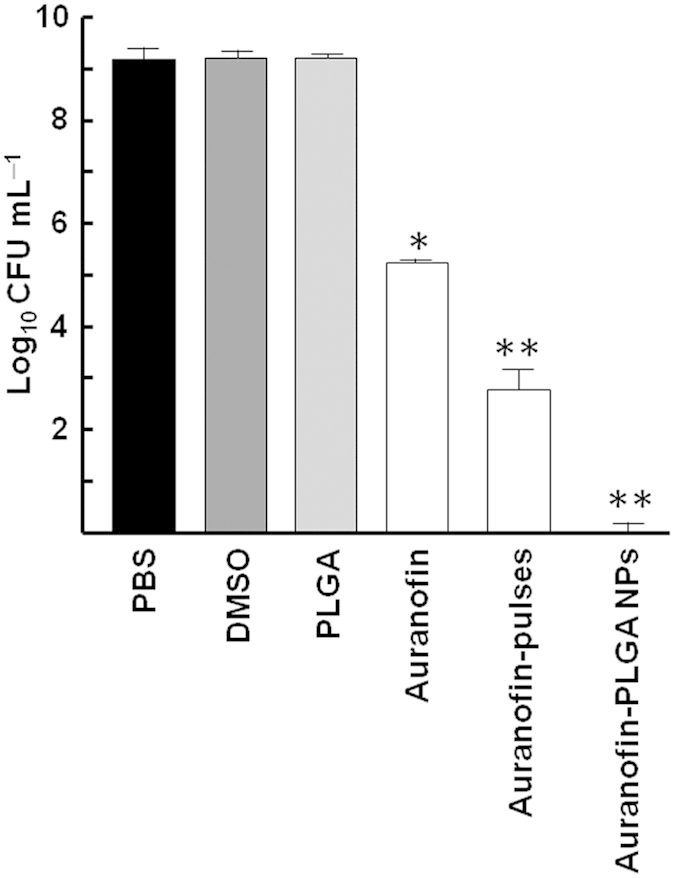
Exponentially growing cultures were incubated in the presence of a final concentration of 0.5 μM of auranofin added in three different conditions: as a single dose at the beginning of the experiment (auranofin), by 1 h pulses for 6 h (auranofin-pulses), or loaded into PLGA NPs (auranofin-PLGA NPs). Controls with PBS buffer, DMSO or PLGA without auranofin were also included. Viable cells were determined on blood agar plates after 6 h of incubation at 37 °C. Data are means of three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared to the control in the absence of auranofin (one-way ANOVA with a post hoc Dunnet test; *P < 0.01; **P < 0.001).
Lethal activity of auranofin-PLGA NPs on bacterial biofilm model
Free auranofin and auranofin-loaded in NPs were firstly assayed on S. pneumoniae P046 strain at concentrations of 0.25, 0.5 and 1 μM. This biofilm-prone strain does not autolyse after long periods of incubation either at 30 °C or 37 °C, due to its deficiency in LytA and LytC autolysins. A positive effect of any drug against bacterial biofilm is evidenced by the decrease of biofilm formation due to the dispersion of the cells and, most importantly, by the lethal action of the drug on the remaining cells. Consequently, the bactericidal effect on the P046 biofilm was checked after 6 h of the corresponding treatment and results clearly demonstrated that the drug charged in NPs killed more efficiently S. pneumoniae biofilm-grown cells than free auranofin. For instance, auranofin-NPs killed about 4 logs of the bacterial population at 0.25 μM (more than 99.9% mortality), whereas auranofin alone was virtually ineffective at the same concentration, being necessary at least 1 μM of compound to achieve 90% mortality (Fig. 4a). In addition, we checked the same treatments on S. pyogenes SF370 strain, grown as biofilm, at the same concentrations used before. The results in this case were very similar than the preceding experiment, as auranofin-NPs at a concentration of 0.25 μM were capable to kill the same 4 logs of bacterial cells. It is worth noting a dramatically different behavior in lethal activity between free and encapsulated auranofin when compared planktonic and biofilm-grown cultures of both pathogens, S. pneumoniae and S. pyogenes. As shown in Fig. 4 and Table 2, free auranofin displayed higher bactericidal effect on planktonic than on biofilm cells of S. pyogenes SF370 strain (about 1 and 0.5 log decrease, respectively, at 0.5 μM). In contrast, auranofin-loaded PLGA NPs were clearly more lethal against biofilm of both bacteria than the same cells grown planktonically (>4 and 2 log decrease, respectively, at 0.5 μM for S. pyogenes).
Figure 4. Streptococcal biofilms treated with auranofin or auranofin-PLGA NPs.

(a) Cells of S. pneumoniae P046 strain, grown as biofilm, were treated with 0.25, 0.5 and 1 μM of auranofin and auranofin-PLGA NPs for 6 h at 37 °C. (b) Cells of S. pyogenes SF370 strain, grown as biofilm, were treated with 0.25, 0.5 and 1 μM of auranofin and auranofin-PLGA NPs for 6 h at 37 °C. Controls included biofilms incubated for 6 h in PBS buffer, DMSO or PLGA NPs. Viable cells were determined on blood agar plates. Data represent the mean of three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared to the control in the absence of auranofin (one-way ANOVA with a post hoc Dunnet test; *P < 0.01; **P < 0.001).
Once demonstrated the powerful killing effect of auranofin-NPs against two streptococcal bacteria grown as biofilm, we analyzed by confocal laser scanning microscopy (CLSM) the fine structure of the typical mesh that characterizes this bacterial growth to detect the cell dispersion capacity due to the activity of auranofin-NPs. This analysis was made on P046 pneumococcal strain and CLSM images of the biofilm in PBS and the corresponding to a control containing unloaded PLGA NPs presented similar cell density and percentage of living bacteria (Fig. 5a,b). In contrast, the biofilm treated with auranofin-NPs evidenced the great loss of remaining pneumococcal cells (Fig. 5c), which fully confirmed the lethal action of auranofin when is loaded in PLGA NPs.
Figure 5. CLSM of pneumococcal biofilms treated with auranofin-PLGA NPs.

Cells of S. pneumoniae P046 strain, grown as biofilm, were incubated with PBS (a), or with1 μM of PLGA NPs (b), or treated with1 μM of auranofin-PLGA NPs (c) for 6 h at 37 °C. Afterwards, cells in the biofilms were stained with the BacLight LIVE/DEAD kit to reveal viable (green fluorescence) and non-viable (red fluorescence) bacteria. Horizontal three-dimensional reconstructions (x–y plane) are shown. Bars, 25 μm.
Antibacterial activity in an animal model
To validate in an animal model of infection the in vitro bactericidal results of auranofin-NPs, we employed a zebrafish embryo model, which has been recently set up for S. pneumoniae and S. pyogenes35. Control experiments using the pneumococcal strain D39 showed that a bacterial challenge with about 2.5 × 108 CFU mL−1 killed 50% of the embryos in 4−5 days, when administered by immersion in E3 medium. Therefore, at 48 h post fecundation, zebrafish embryos were brought in contact with the pathogen. Eight hours after bacterial challenge, embryos were extensively washed with the same medium and treated with a single dose of free auranofin or auranofin-NPs at concentrations ranging from 0.1 to 0.5 μM per embryo. Heat-killed (10 min at 65 °C) D39 strain cells were used as negative control. Rescue of the embryos infected and treated with auranofin-NPs varied depending on the concentration used, i.e., 100% survival (46/46) when treated with 0.5 μM, 93.5% (43/46) with 0.25 μM, and 89.1% (41/46) with 0.1 μM. These results indicated the efficiency of auranofin to protect embryos from pneumococcal infection and also that treatment with the auranofin-NPs resulted in about 15% greater protection (P < 0.01) than with the drug alone, e.g. 73.9% of embryos (34/46) survived when treated with free auranofin and 89.1% with auranofin-NPs, both at the same concentration (0.1 μM) (Fig. 6). The effectiveness of auranofin on embryo rescue was compared with that of a conventional antibiotic like ampicillin, which belongs to the β-lactam family. When both drugs were added alone at 0.5 μM the zebrafish embryo survival was almost the same after 5 days post infection (84.6%) (Fig. 6a). However, treatments with the same concentration of ampicillin loaded into PLGA NPs rescued a similar rate of embryos (38/46 that means 82.6%), whereas auranofin-NPs achieved 100% of survival at the same concentration of 0.5 μM (Fig. 6b). To visualize the protection effect of auranofin-NPs, embryos from all groups of experiments were monitored over time for any morphological change. Embryos were examined using a stereomicroscope and it was found that embryos infected with S. pneumoniae D39 had a much shorter and curved tail and several deformities, mainly in pericardial cavity and yolk sac (Fig. 7b) compared to the corresponding uninfected controls (Fig. 7a). However, infected embryos and treated with 0.5 μM of auranofin-NPs were fully protected and did not show the typical deformities provoked by pneumococcal infection (Fig. 7c).
Figure 6. Rescue of zebrafish embryos from pneumococcal infection by auranofin or auranofin-PLGA NPs.

Survival of embryos infected with S. pneumoniae D39 strain and treated (or not) with different concentrations of auranofin or auranofin-PLGA NPs (n= 24 to 36 embryos per condition) is shown. Controls with DMSO, auranofin, ampicillin alone and loaded into PLGA NPs were also included. Embryos were monitored for survival over a period of 5 days and results were plotted as Kaplan–Meier survival curves. Survival curves were compared with the log-rank (Mantel–Cox) and Gehan–Breslow–Wilcoxon tests (*P < 0.01; **P < 0.001).
Figure 7. Microscopic images of zebrafish embryos infected with pneumococci and treated with auranofin-PLGA NPs.

(a) Uninfected embryo group. (b) Embryo group infected with S. pneumoniae D39 strain without treatment. (c) Infected embryo group and treated with 0.5 μM auranofin-PLGA NPs. Images were taken after 5 days post infection. Abbreviations: pe, pericardial edema; tm: tail malformation, and yse, yolk sac edema. Bars, 500 μm.
Discussion
Infections caused by multiresistant bacteria are of important concern in terms of global health as they often promote lethal pathologies. It is becoming clear the particular relevance of biofilm-derived infections since after treatment with high concentrations of antibiotics most of the bacteria survived in such biofilms, even when dead bacterial cells covered the surface7. Disease treatments have been hampered mainly due to the lack of effective antibiotics; hence, the identification of new drugs with novel mechanisms of action has become an international priority36,37. Auranofin is a drug with a gold(I) center coordinated to a thiosugar and triethylphosphine residues, which has been recently repurposed as an efficient drug not only against pathogenic protozoa12,13 but also against several Gram-positive and Gram-negative bacteria, although the latter group was less susceptible to the drug than the former. In fact, MIC values for the most susceptible tested Gram-positive bacteria ranged between 0.12 and 0.5 μg mL−1, whereas these MICs increased to >16 μg mL−1 for some Gram-negative bacteria, like Klebsiella pneumoniae, Pseudomonas aeruginosa or Acinetobacter baumannii15,16. Although the detailed mechanism of the bactericidal effect of auranofin has not been elucidated, recent data strongly suggested that the thiol-redox homeostasis is the bacterial target in M. tuberculosis16. It was demonstrated that auranofin inhibits the bacterial thioredoxin reductase, a key protein in many Gram-positive bacteria for maintaining the thiol-redox balance and protecting against reactive oxidative species15.
Initially, we focused this study on the bactericidal effect of auranofin against two clinically relevant pathogens, S. pneumoniae and S. pyogenes. A summary of viability data, shown in Table 2, demonstrated that this drug has a potent bactericidal effect against all the pneumococcal strains tested, including the multiresistant ones, whereas it was rather inefficient against the S. pyogenes strains assayed. The value of the lethal concentration for pneumococci, 0.5 μM corresponds to 0.34 μg mL−1, is in the range of the MIC values reported for other Gram-positive bacteria as S. aureus and Streptococcus epidermidis16,38. After these promising results with the reprofiled auranofin against pneumococci, we looked for a method to improve and extend the bacteriolytic action of the drug, even in biofilm assays where bacterial population is more tolerant, or resistant, to antibiotic therapies. A typical solution to achieve this goal in the formulation of active compounds is loading of the desired drug in an appropriate vehicle or NP.
Polymers have been employed by the pharmaceutical industry for more than 40 years, evolving from resorbable sutures and orthopaedic implants to multifunctional NPs able of targeting and controlled release of therapeutics. In this sense, the use of polymeric NPs for developing safer and more effective medicines, so-called nanomedicines, has substantially influenced the pharmaceutical and biotechnological industries. Among others, biodegradable PLGA NPs have been widely employed for a variety of biological applications thanks to their reliable methods of preparation, the biocompatibility (FDA-approved), biodegradability39,40, the capacity to encapsulate and protect drugs from degradation21,41. Additionally, active molecule entrapping (or encapsulating) within a delivery system provides a greater control of the pharmacokinetic behavior of the desired drug. This more efficient use of pharmaceutical compounds may diminish some of the drawbacks and supplies the basis for shortening the current time required by classical treatments. This might be of capital importance when dealing with antibiotics, because it could help to avoid the resistance of bacteria to antibiotics, which is one of the main concerns in the treatment of infections nowadays. There are many examples in the literature where the encapsulation of antibiotics into PLGA NPs increases their efficiency. For example, azithromycin loaded PLGA NPs were more active than azithromycin solution against Salmonella typhi42. More recently it has been described that clarithromicine-PLGA NPs are more effective than untreated clarithromicine against E. coli, H. influenzae, S. aureus and S. pneumoniae43. Given the advantages showed by PLGA NPs and taking into account the improved efficacy of antibiotics when loaded in this delivery system, we prepared auranofin-PLGA NPs with a protocol that allowed the reproducible formation of NPs exhibiting diameters below 100 nm and low polydispersity indexes, which is indicative of a homogeneous size distribution.
Once the loading and sustained release of auranofin from PLGA NPs was confirmed, their potential benefits for infection treatments were evaluated. Specifically, preparation of auranofin-PLGA allowed the comparison of its lethal activity against S. pneumoniae and S. pyogenes with that of auranofin alone using three kinds of experiments as model systems: i) in vitro study using several pneumococcal strains, including relevant multiresistant ones; ii) biofilm assay on appropriate strains, like S. pneumoniae P046 and S. pyogenes SF370; iii) in vivo animal model, like zebrafish embryos infected with S. pneumoniae. All these approaches confirmed the improved killing activity of auranofin-NPs over the free auranofin with particular emphasis on the biofilm approach, since the potency of auranofin loaded into PLGA to disperse and kill bacterial population of S. pneumoniae and S. pyogenes biofilms exceeded about 4 logs the lethality found with the drug alone against both pathogens. This promising result could be especially relevant in foreseeing a future treatment of biofilm-based infections that are particularly recalcitrant to classical antibiotics44. An additional argument to count on clinical application of this kind of NPs comes from the successful validation of in vitro assays with the protection to infected zebrafish embryos, which highlights the benefits of NPs as drug delivery system to combat streptococcal infections.
Methods
NP preparation
The nanoprecipitation method for the formation of auranofin-encapsulated PLGA NPs was carried out according to a previously described procedure45. Briefly, 10−15 mg of auranofin [Sigma-Aldrich, >98% High Performance Liquid Chromatography (HPLC)] were dissolved in 20 mL of a mixture of dichloromethane:acetone (0.5:19.5), and then 200 mg of PLGA (Aldrich, 50:50, Mw 7,000–17,000) were added and subsequently sonicated for 2 min (solution 1). Separately, 56 mg of Pluronic F-68 (Sigma) were dissolved in 40 mL of water (0.14% w v−1) in a 100 mL round bottom flask under moderate magnetic stirring (solution 2). Solution 1 was placed on a syringe dispenser to be then transferred dropwise (0.25 mL min−1) over solution 2 under moderate magnetic stirring. The NPs were stirred for 2 h, and the remaining organic solvent was removed in a rotary evaporator at reduced pressure for 2 h. The NPs were then centrifuged at 6,600 rpm in a 7700 RCF rotor at 4 °C for 15 min, washed twice with distilled water and centrifuged again. Then, the NPs were suspended in a small amount of water, flash frozen in liquid nitrogen and freeze-dried. The same process was followed without auranofin for the synthesis of unloaded PLGA NPs.
Morphology of the produced NPs
The spherical morphology of the produced NPs was evaluated using SEM on a JEOL JSM 6335F (Electron Microscopy Centre, UCM). The samples underwent Au metallization prior to observation.
Determination of particle sizes and polydispersities
The average hydrodynamic size and the polydispersity index were measured with a DLS apparatus (Zetasizer NanoZS, Malvern Instruments) equipped with a 633 nm laser at 25 °C. Samples from the prepared suspensions with ultra-purified water (about 0.1 mg mL−1) were placed in the measurement cell.
Drug loading and encapsulation efficiency
Auranofin was quantified measuring Au in an acid-digested sample by Inductively Coupled Plasma Atomic Emission Spectroscopy (ICP-AES, Varian Vista AX Pro) through the Au emission line at 267.6 nm. Briefly, 10 mg of auranofin-loaded NPs were precisely weighed and placed in a reactor Teflon-jacketed steel case. Then, 10–20 mL of concentrated hydrofluoric/nitric acid (1:1) (Panreac) were gently added and moderately heated until complete digestion for 48 h. Finally, the clear yellow solution was taken to 10 mL volumetric flask and measured. The achieved drug loading was 1.8–6.2 mg of auranofin per gram of NPs in all synthesis.
In vitro release of auranofin from PLGA NPs
In vitro release studies were carried out dispersing 10 mg of loaded PLGA NPs in 0.5 mL of fresh phosphate-buffered saline (PBS) solution (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4 [adjusted to pH 7.4]) and placed on a Transwell permeable support with a 0.4 μm (average pore size) polycarbonate membrane. The well was filled with 1.0 mL of PBS pH 7.4 and the suspension was orbitally shaken at 100 rpm at 37 °C during all the experiment. Every hour, 1 mL of sample was removed from the Transwell plate and replaced with fresh PBS. The separation and quantification was carried out by HPLC equipped with UV-Vis detector, measuring the absorbance of 2-nitro-5-thiobenzoate (NTB, λmax 409 nm). NTB is produced when thiol groups react with 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) cleaving the disulphide bond to obtain NTB in water at neutral pH. The samples containing auranofin were treated with KI to break the gold-thiol bond46. With this purpose 250 μL of sample with 50 μL KI 2.0 M were placed in an ultrasound bath at 50 °C for 20 min, then immediately ice-cooled, according to the standard protocol provided by Sigma. An HPLC (Waters Alliance 2695) separation module (Milford, Massachusetts, USA) based on a isocratic mobile phase of ACN:MeOH:Water (3:3:94, v:v:v) at a flow rate of 1 mL min−1 was used to separate and quantify the NTB signal with a 4.2 min of retention time. The system was equipped with a variable wavelength diode array detector Water 2996 at 354 nm. A Waters X-Terra RP-18 reverse phase column (4.6 × 250 mm, 5 μm) was employed at 40 °C and 10–50 μL injection volume was selected upon the case. A scheme describing the reactions, the protocol for derivatization of auranofin, and the 3D chromatogram is shown in Fig. S1.
Bactericidal assays
Bactericidal effects of the auranofin-loaded NPs were monitored by following the optical density at 550 nm (OD550) for 6 h and the viable cells at this time point. Briefly, S. pneumoniae and S. pyogenes strains (Table 1) were grown in C medium47 and Todd-Hewitt medium, respectively, supplemented with yeast extract (0.8 mg mL−1; Difco Laboratories) at 37 °C without shaking. Once the bacteria reached the exponential phase of growth (OD550 ≈0.3), the cultures were centrifuged and washed twice with PBS, and the final OD550 was adjusted to ≈0.6 with PBS. Resuspended bacteria were then transferred into plastic tubes, and different amounts of auranofin-PLGA NPs (suspended in PBS) were added to reach a final concentration of 0.1, 0.25, or 0.5 μM auranofin. Control samples with unloaded PLGA NPs or auranofin alone at the same concentrations were always run in parallel. Additional controls with PBS and dimethylsulfoxide (DMSO), used to solubilize NPs and auranofin, respectively, were also included. All samples were incubated at 37 °C for 6 h, and the OD550 and viable cells were determined at selected time points. Viable cell counting was carried out in tryptose blood agar base plates (Difco Laboratories) supplemented with 5% defibrinated sheep blood. For each sample, a 10-fold dilution series was prepared in PBS, and 10 μL of each dilution were plated. Colonies were counted after overnight incubation at 37 °C.
Biofilm assay
Pneumococcal biofilms were produced using S. pneumoniae strain P046, a double lytA lytC mutant48, a derivative from the unencapsulated laboratory strain R649. The optimal conditions for biofilm formation on polystyrene plates have been described elsewhere50. In short, all biofilms were formed in Costar 3595 96-well polystyrene microtiter plates (Corning, New York, USA). Cells were grown in C medium supplemented with yeast extract (0.8 mg mL−1) to an optical density of 0.5–0.6 at OD595, sedimented by centrifugation, resuspended in an equal volume of C medium, diluted, and portions of 200 μL containing 2.2 × 104 CFU were dispensed into each well. After 16 h of incubation at 34 °C, the planktonic cultures were removed and the resulting biofilms were washed twice with PBS and then treated for additional 6 h at 37 °C with various concentrations of auranofin or auranofin-NPs. Controls with DMSO or unloaded NPs were also assayed. Afterwards, the supernatants were removed again, washed twice with PBS and 10-fold dilution series were prepared in PBS. 10 μL of each dilution were plated in blood agar plates and colonies were counted after overnight incubation at 37 °C. The S. pyogenes biofilm was produced using SF370 strain, as previously described51. Briefly, an overnight culture of S. pyogenes was diluted 1:20 with fresh Todd-Hewitt medium supplemented with yeast extract (0.8 mg mL−1) and grown statically in polystyrene microtiter plates for 24 h at 37 °C. After incubation, the same protocol was followed as described above for the pneumococcal biofilms.
Microscopy observation of biofilms
To observe biofilms by CLSM, S. pneumoniae P046 strain was grown on glass-bottomed WillCo-dishes (WillCo Wells) for 16 h at 34 °C. Following incubation, the culture medium was removed and the biofilm rinsed with PBS to remove non-adherent bacteria and then treated for additional 6 h at 37 °C with 1 μM of auranofin-NPs. Control biofilms with PBS or unloaded PLGA NPs were also assayed. Afterwards, the supernatants were removed again and then stained with the LIVE/DEAD BacLight bacterial viability kit L-13152 (Invitrogen-Molecular Probes) for monitoring the viability of bacterial populations. Cells with a compromised membrane — considered dead or dying— stain red, whereas those with an intact membrane stain green48. The biofilms were observed at ×63 magnification using a Leica TCS-SP2-AOBS-UV CLSM. The excitation/emission maxima were around 488/500–546 nm. Images were analyzed using LCS software (Leica). Projections were obtained through the x–y plane (individual scans at 0.5-μm intervals).
Zebrafish embryo infection assay
The pneumococcal infection model was based on a method described previously35, using wild-type zebrafish embryos (Danio rerio) obtained from ZFBioLabs (Tres Cantos, Spain). Briefly, embryos were dechorionated at 24 h post fecundation by treatment with pronase (2 mg mL−1) for 1 min, distributed in 96-well plates and incubated in 100 μL of E3 medium. At 48 h post fecundation, embryos were infected with 100 μL of a 2.5 × 108 colony-forming units (CFU) mL−1 suspension of S. pneumoniae strain D39. Bacterial inoculation titers were calculated by serial dilution and plating onto tryptose blood agar plates for each experiment. Seven hours post infection, infected embryos were extensively washed with E3 medium to remove the bacteria and 100 μL of the same autoclaved fresh medium supplemented with auranofin (0.1, 0.25 or 0.5 μM) or auranofin-loaded NPs (containing the same concentration of the drug) were added. Non-infected controls without treatment or treated either with DMSO, auranofin, or unloaded PLGA NPs were washed in the same way. Embryos were incubated at 27.5 °C under sterile conditions and mortality was followed in all samples for 5 days, changing fresh E3 medium every day. Zebrafish embryos were considered dead when coagulation was observed as well as absence of heartbeat during 15 seconds observation. Each experiment was repeated at least 3 times, and 24 to 36 embryos were used per condition and experiment.
Ethics statement
All experiments with zebrafish embryos were performed under strict accordance to the new EU Directive 2010/63/EU on the protection of animals. Signs of infection were monitored three times daily throughout the experimental time course. Moribund embryos were anesthetized by immersion in tricaine (MS222) (Sigma-Aldrich) at 200 mg mL−1, followed by immobilization by submersion in ice water (5 parts ice/1 part water, 0–4 °C) for at least 20 min to ensure death by hypoxia. Approval for these experimental protocols was granted by the Spanish Royal Decree 53/2013 laying down basic standards for the protection of animals used in animal experiments and other scientific purposes.
Imaging analysis
Live embryos (5 days post infection) were treated to ensure death as described above, mounted in 3% methylcellulose in depression slides and photographed under an Olympus SZX16 stereoscope with a QImaging MicroPublisher 5.0 RVT camera. Images were processed with NIH ImageJ. For all the experiments described, the images shown are representative of the effects observed in at least 90% of the individuals.
Statistical analysis
All in vitro results are representative of data obtained from repeated independent experiments, and each value represents the mean standard deviations for 3 to 4 replicates. Statistical analysis was performed by using the two-tailed Student’s t test (for two groups), whereas analysis of variance (ANOVA) was chosen for multiple comparisons. For all in vivo data, the log-rank (Mantel-Cox) and Gehan-Breslow-Wilcoxon tests were used to draw, analyze and compare the survival curves. GraphPad InStat version 5.0 (GraphPad Software, San Diego, CA) was used for statistical analysis.
Additional Information
How to cite this article: Díez-Martínez, R. et al. Auranofin-loaded nanoparticles as a new therapeutic tool to fight streptococcal infections. Sci. Rep. 6, 19525; doi: 10.1038/srep19525 (2016).
Supplementary Material
Acknowledgments
We thank E. García for critical reading of the manuscript. The excellent technical assistance of E. Cano is greatly appreciated. This research was funded by grants from Ministerio de Economía y Competitividad, Spain (SAF2012-39444-C02-01, MAT2012-35556 and CSO2010-11384-E, Ageing Network of Excellence). We also thank the National Electron Microscopy Center at UCM for the SEM analyses.
Footnotes
Author Contributions M.V.-R. and P.G. conceived the research. R.D.-M., E.G.-F., M.M., M.V.-R. and P.G. designed the experiments. R.D.-M., E.G.-F., M.D., A.M. and M.M. performed the experiments and analyzed the data. R.D.-M., E.G.-F., M.M., M.V.-R. and P.G. wrote the paper. All authors discussed the results, edited, and approved the manuscript.
References
- WHO. Antimicrobial resistance, global report on surveillance, < http://www.who.int/drugresistance/documents/surveillancereport/en/.> (2014) (last accessed 16/11/2015).
- O’Brien K. L. et al. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374, 893−902 (2009). [DOI] [PubMed] [Google Scholar]
- Said M. A. et al. Estimating the burden of pneumococcal pneumonia among adults: a systematic review and meta-analysis of diagnostic techniques. PloS one 8, e60273 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugman K. P. Bacteriological evidence of antibiotic failure in pneumococcal lower respiratory tract infections. Eur. Respir. J. 20 (Suppl. 36), 3s–8s (2002). [DOI] [PubMed] [Google Scholar]
- Fuller J. D. & Low D. E. A review of Streptococcus pneumoniae infection treatment failures associated with fluoroquinolone resistance. Clin. Infect. Dis. 41, 118−121 (2005). [DOI] [PubMed] [Google Scholar]
- Jernigan D. B., Cetron M. S. & Breiman R. F. Minimizing the impact of drug-resistant streptococcus pneumoniae (DRSP): A strategy from the DRSP working group. JAMA: the journal of the American Medical Association 275, 206–209 (1996). [PubMed] [Google Scholar]
- Ogawa T. et al. Biofilm formation or internalization into epithelial cells enable Streptococcus pyogenes to evade antibiotic eradication in patients with pharyngitis. Microb. Pathog. 51, 58–68 (2011). [DOI] [PubMed] [Google Scholar]
- McCarthy M. CDC calls for urgent action to combat rise of drug resistant pathogens. British Med. J . 347, f5649 (2013). [DOI] [PubMed] [Google Scholar]
- Chong C. R. & Sullivan D. J. Jr. New uses for old drugs. Nature 448, 645−646 (2007). [DOI] [PubMed] [Google Scholar]
- Aronson J. K. Old drugs – new uses. Br. J. Clin. Pharmacol. 64, 563–565 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw C. F. Gold-based therapeutic agents. Chem. Rev. 99, 2589–2600 (1999). [DOI] [PubMed] [Google Scholar]
- Madeira J. M., Gibson D. L., Kean W. F. & Klegeris A. The biological activity of auranofin: implications for novel treatment of diseases. Inflammopharmacol . 20, 297–306 (2012). [DOI] [PubMed] [Google Scholar]
- Roder C. & Thomson M. Auranofin: Repurposing an old drug for a golden new age. Drugs in R&D 15, 13–20 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson-Rosario S. et al. Auranofin disrupts selenium metabolism in Clostridium difficile by forming a stable Au-Se adduct. J. Biol. Inorg. Chem. 14, 507−519 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokai Y. et al. Auranofin and related heterometallic gold(I)-thiolates as potent inhibitors of methicillin-resistant Staphylococcus aureus bacterial strains. J. Inorg. Biochem. 138, 81−88 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbut M. B. et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 112, 4453–4458 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandi V. & Kandi S. Antimicrobial properties of nanomolecules: potential candidates as antibiotics in the era of multi-drug resistance. Epidemiol. Health 37, e2015020 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leid J. G. et al. In vitro antimicrobial studies of silver carbene complexes: activity of free and nanoparticle carbene formulations against clinical isolates of pathogenic bacteria. J. Antimicrob. Chemother. 67, 138–148 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vert M., Mauduit J. & Li S. Biodegradation of PLA/GA polymers: increasing complexity. Biomaterials 15, 1209–1213 (1994). [DOI] [PubMed] [Google Scholar]
- Prokop A. & Davidson J. M. Nanovehicular intracellular delivery systems. J. Pharm. Sci. 97, 3518–3590 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danhier F. et al. PLGA-based nanoparticles: an overview of biomedical applications. J. Control Release 161, 505–522 (2012). [DOI] [PubMed] [Google Scholar]
- Mahapatro A. & Singh D. Biodegradable nanoparticles are excellent vehicle for site directed in-vivo delivery of drugs and vaccines. J. Nanobiotechnol . 9, 55 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J. et al. Formulation of functionalized PLGA–PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 28, 869–876 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner Y., Pellequer Y. & Lamprecht A. Nanoparticles in inflammatory bowel disease: particle targeting versus pH-sensitive delivery. Int. J. Pharm. 316, 138–143 (2006). [DOI] [PubMed] [Google Scholar]
- Meissner Y. & Lamprecht A. Alternative drug delivery approaches for the therapy of inflammatory bowel disease. J. Pharm. Sci. 97, 2878–2891 (2008). [DOI] [PubMed] [Google Scholar]
- Panyam J. & Labhasetwar V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 55, 329–347 (2003). [DOI] [PubMed] [Google Scholar]
- Ladewig K. Drug delivery in soft tissue engineering. Expert Opin. Drug Deliv. 8, 1175–1188 (2011). [DOI] [PubMed] [Google Scholar]
- Lim H. J. et al. A novel technique for loading of paclitaxel-PLGA nanoparticles onto ePTFE vascular grafts. Biotechnol. Prog. 23, 693–697 (2007). [DOI] [PubMed] [Google Scholar]
- Toti U. S. et al. Targeted delivery of antibiotics to intracellular chlamydial infections using PLGA nanoparticles. Biomaterials 32, 6606–6613 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecaroz C., Gamazo C. & Blanco-Prieto M. J. Nanocarriers with gentamicin to treat intracellular pathogens. J. Nanosci. Nanotechnol. 6, 3296–3302 (2006). [DOI] [PubMed] [Google Scholar]
- Lecaroz M. C., Blanco-Prieto M. J., Campanero M. A., Salman H. & Gamazo C. Poly(D,L-lactide-coglycolide) particles containing gentamicin: pharmacokinetics and pharmacodynamics in Brucella melitensis- infected mice. Antimicrob. Agents Chemother. 51, 1185–1190 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbuluzqueta E. et al. Novel bioactive hydrophobic gentamicin carriers for the treatment of intracellular bacterial infections. Acta Biomater. 7, 1599–1608 (2011). [DOI] [PubMed] [Google Scholar]
- Pillai R. R., Somayaji S. N., Rabinovich M., Hudson M. C. & Gonsalves K. E. Nafcillin-loaded PLGA nanoparticles for treatment of osteomyelitis. Biomed. Mater. 3, 034114 (2008). [DOI] [PubMed] [Google Scholar]
- Gupta H. et al. Sparfloxacin-loaded PLGA nanoparticles for sustained ocular drug delivery. Nanomedicine: nanotechnology, biology, and medicine 6, 324–333 (2010). [DOI] [PubMed] [Google Scholar]
- Díez-Martínez R. et al. Improving the lethal effect of Cpl-7, a pneumococcal phage lysozyme with broad bactericidal activity, by inverting the net charge of its cell wall-binding module. Antimicrob. Agents Chemother. 57, 5355–5365 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellberg B., Bartlett J., Wunderink R. & Gilbert D. N. Novel approaches are needed to develop tomorrow’s antibacterial therapies. Am. J. Respir. Crit. Care Med. 191, 135–140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadakh B. & Aerschot A. V. Renaissance in antibiotic discovery: some novel approaches for finding drugs to treat bad bugs. Curr. Med. Chem. (in press) (2015). [DOI] [PubMed] [Google Scholar]
- Cassetta M., Marzo T., Fallani S., Novelli A. & Messori L. Drug repositioning: auranofin as a prospective antimicrobial agent for the treatment of severe staphylococcal infections. Biometals: an international journal on the role of metal ions in biology, biochemistry, and medicine 27, 787–791 (2014). [DOI] [PubMed] [Google Scholar]
- Avgoustakis K. et al. PLGA–mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J. Control Release. 79, 123–135 (2002). [DOI] [PubMed] [Google Scholar]
- Makadia H. K. & Siegel S. J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers (Basel) 3, 1377–1397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. M. & Shive M. S. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug Deliv. Rev. 28, 5–24 (1997). [DOI] [PubMed] [Google Scholar]
- Mohammadi G. et al. Development of azithromycin–PLGA nanoparticles: physicochemical characterization and antibacterial effect against Salmonella typhi. Colloids Surf. B Biointerfaces . 80, 34–39 (2010). [DOI] [PubMed] [Google Scholar]
- Valizadeh H. et al. Antibacterial activity of clarithromycin loaded PLGA nanoparticles. Die Pharmazie 67, 63–68 (2012). [PubMed] [Google Scholar]
- Lebeaux D., Ghigo J.-M. & Beloin C. Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol. Mol. Biol. Rev. 78, 510–543 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca C., Simões S. & Gaspar R. Paclitaxel-loaded PLGA nanoparticles: preparation, physicochemical characterization and in vitro anti-tumoral activity. J. Control Release 83, 273–286 (2002). [DOI] [PubMed] [Google Scholar]
- Kizu R., Kaneda M., Yamauchi Y. & Miyazaki M. Determination of auranofin, a chrysotherapy agent, in urine by HPLC with a postcolumn reaction and visible detection. Chem. Pharm. Bull (Tokyo) 41, 1261–1265 (1993). [DOI] [PubMed] [Google Scholar]
- Lacks S. & Hotchkiss R. D. A study of the genetic material determining an enzyme activity in Pneumococcus. Biochim. Biophys. Acta 39, 508−518 (1960). [DOI] [PubMed] [Google Scholar]
- Moscoso M., García E. & López R. Biofilm formation by Streptococcus pneumoniae: role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J. Bacteriol. 188, 7785−7795 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoskins J. et al. Genome of the bacterium Streptococcus pneumoniae strain R6. J. Bacteriol. 183, 5709−5717 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domenech M., García E. & Moscoso M. In vitro destruction of Streptococcus pneumoniae biofilms with bacterial and phage peptidoglycan hydrolases. Antimicrob. Agents Chemother. 55, 4144–4148 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Köller T., Kreikemeyer B. & Nelson D. C. Rapid degradation of Streptococcus pyogenes biofilms by PlyC, a bacteriophage-encoded endolysin. J. Antimicrob. Chemother. 68, 1818–1824 (2013). [DOI] [PubMed] [Google Scholar]
- Lanie J. A. et al. Genome sequence of Avery’s virulent serotype 2 strain D39 of Streptococcus pneumoniae and comparison with that of unencapsulated laboratory strain R6. J. Bacteriol. 189, 38−51 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano F. et al. Breakthrough in penicillin resistance? Streptococcus pneumoniae isolates with penicillin/cefotaxime MICs of 16 mg/L and their genotypic and geographical relatedness. J. Antimicrob. Chemother. 62, 1234−1240 (2008). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.