

Abstract
Susceptibility to infection by prions is highly dependent on the amino acid sequence and host expression of the cellular prion protein (PrPC); however, cellular expression of a genetically susceptible PrPC is insufficient. As an example, it has been shown in cultured cells that permissive and resistant sublines derived from the same parental population often have similar expression levels of PrPC. Thus, additional cellular factors must influence susceptibility to prion infection. The aim of this study was to elucidate the factors associated with relative permissiveness and resistance to scrapie prions in cultured cells derived from a naturally affected species. Two closely related ovine microglia clones with different prion susceptibility, but no detectable differences in PrPC expression levels, were inoculated with either scrapie-positive or scrapie-negative sheep brainstem homogenates. Five passages post-inoculation, the transcriptional profiles of mock and infected clones were sequenced using Illumina technology. Comparative transcriptional analyses identified twenty-two differentially transcribed genes, most of which were upregulated in poorly permissive microglia. This included genes encoding for selenoprotein P, endolysosomal proteases, and proteins involved in extracellular matrix remodeling. Furthermore, in highly permissive microglia, transforming growth factor β–induced, retinoic acid receptor response 1, and phosphoserine aminotranspherase 1 gene transcripts were upregulated. Gene Set Enrichment Analysis identified proteolysis, translation, and mitosis as the most affected pathways and supported the upregulation trend of several genes encoding for intracellular proteases and ribosomal proteins in poorly permissive microglia. This study identifies new genes potentially involved in scrapie prion propagation, corroborates results from other studies, and extends those results into another cell culture model.
Introduction
Transmissible spongiform encephalopathies (TSEs, a.k.a., prion diseases) are a group of lethal and incurable neurodegenerative diseases caused by prions. TSEs include Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy in cattle, scrapie in sheep and goats, chronic wasting disease in cervids, and others [1]. Prions are primarily, if not solely, composed of PrPD (D superscript for “disease associated”), which is a misfolded isoform of the cellular prion protein (PrPC) [2]. Conversion of PrPC into PrPD is the central event in the pathogenesis of TSEs, and according to the protein-only hypothesis [2] PrPD catalyzes the conversion of PrPC into a likeness of itself by an incompletely understood mechanism of replication. Unlike PrPC, PrPD is primarily composed of β–pleated sheets; this abnormal conformational state is transmitted to newly converted molecules of PrPD and is associated with distinct biochemical features: aggregation, detergent insolubility, and partial proteinase K resistance [1].
A major determinant of a host’s susceptibility to prion infection and replication is the sequence identity between the host’s PrPC with that of the infectious PrPD. For instance, the naturally occurring Q171R and E219K variants of PrPC are known to render sheep and humans resistant to classical scrapie and CJD prions, respectively [3, 4]. Moreover, PrPC expression is required for prion propagation in cultured cells [5]. However, the inability of some PrPC expressing cell lines to propagate prions [6, 7] indicates that additional factors must play a role in susceptibility to prion infection. Identification of such factors would greatly improve the understanding of TSEs pathogenesis and enable identification of much-needed therapeutic targets.
Previous studies have identified genes potentially involved in the pathogenesis of TSEs, but have shortcomings with respect to the techniques and models employed. For instance, microarray technology has been used on ovine (natural TSE host) [8, 9] and murine (adapted TSE host) [10, 11] tissues to identify the transcriptional responses during prion infection. However, these studies could not differentiate if the transcriptional differences were directly related to prion propagation or were simply secondary to infection, nor could they assign gene transcription status to specific cell types. Additional attempts to define the factors that affect cellular permissiveness to prion infection include transcriptomic analyses through microarray technology on cultured cells [12–14]. The results of the latter studies have provided new insights into prion infection susceptibility in a pathophysiologically relevant cell type (i.e., neurons); including the role of proteins associated with extracellular matrix remodeling [13] and others that may alter trafficking of PrPC and PrPD [12] during prion propagation. Due to the difficulty in translating the biological relevance of results from cell cultures to whole organisms, and the technical limitations of microarray technology [15, 16], the aforementioned studies would be complemented by comparing the transcriptional profiles of another pathophysiologically relevant cell type using high-throughput RNA-Seq. This technique would allow for transcriptomic analysis with a broader dynamic range and a greater ability to detect low abundance novel transcripts.
We have established an immortalized ovine microglia cell culture system permissive to natural scrapie isolates (i.e., derived directly from brainstems of sheep infected with classical scrapie) [7]. Microglia are myeloid-derived monocyte cells that function in the central nervous system as resident macrophages and which have been shown to have important roles in the transport [17], accumulation [18], and degradation of prions [19] in the central nervous system and peripherally within the body. Furthermore, microglia contribute to the maintenance and degradation of the CNS extracellular matrix [20]. Similar to murine and rabbit model cell culture systems used for TSE research [6, 21], only a relatively small proportion of immortalized microglia sublines were found to be permissive to prions [7]. Also, PrPC expression levels failed to predict susceptibility to either natural scrapie isolates or culture-adapted prions across multiple ovine microglia sublines. This indicates that, in this cell line, susceptibility to scrapie prions is determined by additional factors and not only by PrPC levels. In the present study, the transcriptional profiles of highly permissive and poorly permissive ovine microglia clones were compared using RNA-Seq to test the hypothesis that a distinct transcriptional signature is associated with prion susceptibility in cultured ovine microglia. Herein, twenty-two genes with consistent differential transcription between highly permissive and poorly permissive ovine microglia cells are identified and their hypothetical roles in prion propagation at the cellular level are discussed. As such, this is the first comprehensive comparative transcriptional study that characterizes prion susceptibility in cultured cells using high throughput RNA sequencing technology.
Materials and Methods
Cell culture and inoculation with natural scrapie prions
Previously established hTERT-microglia cells [7] were used for this study. Subline H cells from a previous study [7] were cloned by limiting dilution to generate clones 438 and 439 and these were previously characterized as monocyte-derived cells by expression of CD14 [7]. Cryogenically stored clones 438 and 439 were thawed and maintained in Opti-MEM medium supplemented with 10% heat-inactivated fetal bovine serum (Atlanta Biologicals) 2 mM L-glutamine, 10 IU of penicillin, and 10 mg/ml streptomycin. Previously, in a single experiment, it was demonstrated that these clones have differential permissiveness to a scrapie isolate derived from a naturally infected sheep [7]. This phenotypic difference in scrapie-permissiveness was confirmed for the current study by challenging microglia clones with scrapie-positive and scrapie-negative brainstem homogenates. For inoculation, cells were plated at a concentration of 4 x 105 cells/well in 12-well plates and inoculated with 1% (w/v) brainstem homogenates (for additional information on inoculum preparation, inoculation protocol, and sources see [7]). Inocula made from the X124 natural scrapie isolate [22] and from a genotype-matched, scrapie-naïve lamb were respectively used as scrapie-positive and scrapie-negative controls. Inoculum was replaced with fresh culture medium seven days after inoculation and cells were kept in culture for one week prior to expansion to 25-cm2 flasks. Then, cells were split 1/5 every seven days. A total of three inoculation experiments were performed, each including three culture replicates. Permissiveness to scrapie prion infection was defined as the accumulation of nascent PrPSc (Sc superscript for scrapie) at the third and fourth passage post-inoculation. PrPSc was assessed by immunoblotting and ELISA.
Detection of PK-resistant PrP with immunoblotting
At the third passage post-inoculation, cells were collected and lysed for immunoblotting as previously described [7]. In brief, cell lysates were first normalized to total protein using the bicinchoninic acid protein assay kit (Thermo Scientific) before digestion with 50 μg/ml PK (Roche) for 1 h at 37C immediately before immunoblotting. In some experiments (S1 Fig), to increase the sensitivity of this assay, phosphotungstic acid (PTA) was used to precipitate PK-resistant PrP prior to immunoblotting, as previously described [23]. The anti-PrP monoclonal antibody 99/97.6.1 (which detects a conserved residue on the C-terminus of PrP [24]) was used for immunoblotting at a concentration of 3.5 μg/ml. Immunoblots were visually interpreted as either positive or negative. Samples that were not treated with PK were included as controls for protein extraction and PrP detection.
Detection of misfolded PrP with ELISA
At the fourth passage post-inoculation, cells were collected and lysed for detection of PrPSc using the Herd-Check CWD Ag Test (IDEXX) as previously described [23]. The corrected optical density (OD) values (OD450–OD620) of each experiment were each divided by that experiment’s kit-provided positive control to normalize for ELISA plate-to-plate variation; thus, the combined experiment results in Fig 1B are reported as “normalized units”. The kit-provided cut-off value (which is the average of three kit-provided negative controls + 0.18) was used as threshold to determine accumulation of PrPSc. In addition, the corrected OD values of lysates from scrapie-inoculated cells from clone 438 were compared to those of clone 439 using an unpaired t-test with a level of significance of P < 0.01. Also, the corrected OD values of lysates from scrapie-inoculated cells were compared to those of mock-inoculated cells from each clone using a paired t-test with a level of significance of P < 0.01. Statistical analyses were done using JMP Pro 11.2.0.
Fig 1. Characterization of differential prion susceptibility in ovine microglia clones.
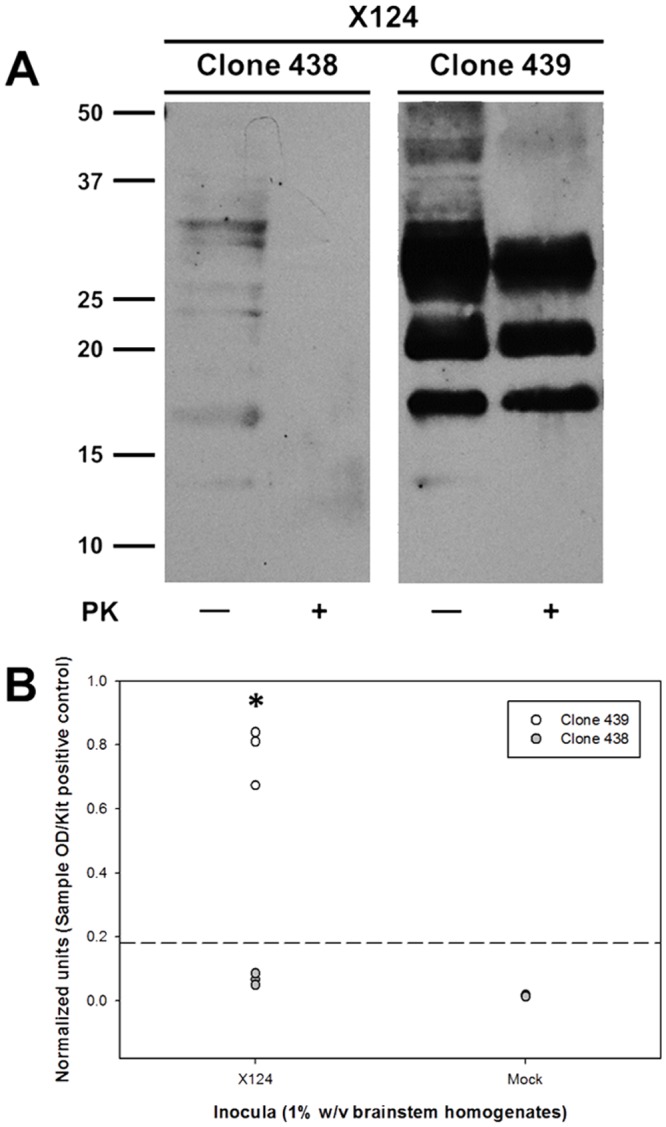
Microglia clones were inoculated with 1% (w/v) brainstem homogenates from either scrapie-positive (“X124”) or scrapie-naïve (“Mock”) sheep. Inoculated cells were passaged on a weekly basis and then tested for the accumulation of nascent PrPSc at passage three by immunoblotting (A) and at passage four by ELISA (B). Immunoblot picture (A) depicts results from one culture replicate inoculated with scrapie-positive brain homogenates and is representative of three independent experiments. In graph (B), each circle represents the mean of three culture replicates from each of three independent experiments (i.e., total of six circles per treatment, three for 439 and three for 438), and the dashed line indicates the assay cut-off threshold for detection of PrPSc. Values of normalized units in the X124 group for clone 439 are significantly higher than those of in clone 438 (*: P = 0.0048, unpaired t-test). Values of normalized units between X124 and mock groups are statistically significantly different for the clone 439 (P = 0.0044, paired t-test) and clone 438 (P = 0.0065, paired t-test); however, the values for clone 438 fail to rise above the cut-off threshold and are negative by immunoblotting ([A] and S1 Fig).
PrPC quantification
The levels of cell-associated PrPC of each uninoculated ovine microglia clone were determined using a commercial ELISA (TeSeE™ SAP Detection Kit, Bio-Rad) following manufacturer’s instructions, as previously described [7]. This kit uses an immobilized anti-PrP antibody for plate capture, but enzymatic digestion using PK was not utilized (i.e., the TeSeE™ SAP Purification Kit was not used) so that total PrP (i.e., PrPC only in uninoculated cells) content could be measured. Briefly, cell lysates were normalized to total protein with the bicinchoninic acid protein assay kit (Thermo Scientific) and the appropriate dilutions of cell lysates were added to the ELISA plate. A PrPC standard curve was prepared using half-log dilutions of uninoculated sheep PrPC expressing RK-13 (Rov) cell lysates to convert corrected OD values to relative amounts of PrPC, as previously described [7]. Lysates from three technical replicates, each with three culture replicates, of each ovine microglia clone were analyzed. The kit-provided positive and negative controls were included in all the assays. The levels of PrPC between highly permissive and poorly permissive clones were compared with an unpaired t-test with a level of significance of P < 0.05 using JMP Pro 11.2.0.
RNA isolation and sequencing
At the fifth passage after inoculation, total RNA was extracted from three culture replicates of each ovine microglia clone. TRIzol (Invitrogen) was used to lyse cells in culture flasks following manufacturer’s instructions. Total RNA was analyzed with the 2100 Bioanalyzer (Agilent Technologies) and enriched poly(A) RNA (selected with the PrepX PolyA mRNA isolation kit [Wafergen biosystems]) was used to prepare cDNA libraries. Samples were sequenced using Illumina HiSeq 2000 with 100 bp reads and all were run on a single lane. The raw sequence data have been deposited in the NCBI Sequence Read Archive (accession number: PRJNA257519).
Comparative transcriptional analysis
The CLC Genomics Workbench (CLC Bio.) was used to process RNA-Seq data. Mapping parameters were adjusted to map a maximum number of reads to the reference Ovis aries Oar_v3.1 [25]. The distribution of the expression values for all samples was analyzed and compared. Normalization by quantiles was applied to adjust the distributions for further comparison, as previously described [26]. Fold changes with respect to RPKM (Reads Per Kilobase per Million mapped reads) values were calculated. Baggerley’s statistical test on proportions was applied to evaluate significance of fold changes [27]. Bonferroni correction was used to minimize the occurrence of false positives. Comparisons of replicates were performed in order to account for variation of transcription values within each experimental group. Genes with Bonferroni corrected P values below 0.05 were selected for further evaluation.
Evaluation of genes encoding for hypothetical proteins
The available translated sequences of genes encoding hypothetical proteins were analyzed for conserved domains using the NCBI’s conserved domain database [28]. In a few cases, translated sequences were generated with the Translate tool of the ExPASy Bioinformatics Resource Portal [29] prior to domain identification. Domains with the lowest Expect value (E-value) were considered for further evaluation.
K—means clustering
K—means clustering was used to assign the mean transcriptional value of each gene to the cluster whose center is nearest, as previously described [26]. Euclidean distance was used as the distance metric and five partitions were used to generate the clusters. The mean gene expression value over all input samples was subtracted from all genes. Previously normalized expression values were used for clustering.
Gene Set Enrichment Analysis
In order to evaluate different GO (Gene Ontology) biological pathways, the Gene Set Enrichment Analysis (GSEA) test [30] was used as previously described [26]. The test calculates and uses ANOVA statistic for multiple group experiments for each feature, as measures of association. The O. aries genome was annotated using the Bos taurus GO annotations [31]. Briefly, feature IDs in the O. aries genome were matched to synonyms and gene products from IDs of the Bos taurus data base. Additionally, this was complemented with manual curation of the annotation file, including genes reported in the Kyoto Encyclopedia of Gene and Genomes (KEGG) [32].
Quantitative RT-PCR
RT-qPCR was used to confirm RNA-Seq results of selected genes employing the SsoAdvanced™ Universal SYBR Green Supermix (Bio-Rad). In brief, total RNA was collected from cells inoculated with scrapie-positive homogenates (nine culture replicates total per each microglia clone) at the sixth passage post-inoculation using the RNeasy Mini Kit (QIAGEN) following manufacturer’s instructions. Approximately 1 μg of total RNA was reverse-transcribed using the SuperScript® III First-Strand Synthesis SuperMix for RT-qPCR (Life Technologies) and 2 μl of cDNA were used for qPCR in a 20-μl reaction. Nine genes with differential transcription based on RNA-Seq analysis were analyzed by RT-qPCR, and these genes were selected based on their consistent up- or down-regulation across pair-wise comparisons. Reaction conditions for RT-qPCR were 95C for 30 s, 35 cycles of denaturation at 95C for 15 s and annealing at 60C for 10 s followed immediately by a melt curve. Negative controls for RT-qPCR included no-template controls. A standard curve was used for each run to calculate the amplification efficiency of each run and all calculations of relative expression were based on experiments with ≥ 95% efficiency. The constitutively expressed GAPDH gene was used for normalization. Also, the PRNP gene was analyzed to support ELISA results regarding PrPC quantification. Primers used in this study are shown in Table 1 and all but those for PRNP [33] and GAPDH [34] were designed using the primer-BLAST tool [35] based on the reference O. aries Oar_v3.1 [25]. Each primer set was assessed through gradient PCR to determine the optimum annealing temperature and PCR products were analyzed through gel electrophoresis to confirm the size was of the expected molecular weight. The ratios of relative expression were calculated through the REST software [36], which uses a mathematical model that includes the efficiency correction of individual transcripts and tests the expression ratio results of the investigated transcripts for significance by a randomization test.
Table 1. Primers used for RT-qPCR.
| Target gene | Forward primer sequence | Reverse primer sequence | Amplicon size (bp) |
|---|---|---|---|
| CTSB | TTGGAAGGCTGGACACAACT | TCCCTGGTCTCGGATCTCTT | 190 |
| DCN | GCTGGCCGACCATAAGTACA | TGGGTTGCTGAAAAGGCTCA | 139 |
| DPT | GCTGGTGGGAGGAGATCAAC | GACTCGAAGTAGCGGCTCTG | 97 |
| MMP14 | CGCTATGCCATCCAGGGACT | CTCCCACACTCGGAATGCCT | 126 |
| PRNP [33] | CCGTTACCCCAACCAAGTGT | CGCTCCATTATCTTGATGTCAGT | 159 |
| PTN | GCAGACTCCACAGTACCTGC | ACACACACTCCACTGCCATT | 163 |
| RARRES1 | GGCAGCTCTTACGTGATGTG | CCAGACCAAGTGAATACGGCA | 177 |
| SEPP1 | ACCGTGGTTGCTCTTCTTCAA | TCTCCAGTTTTACTCGCAGGTC | 85 |
| SQSTM1 | TTGTACCCACATCTGCCACC | AGCCGCCTTCATCAGAGAAC | 91 |
| TGFBI | TGGGCGGCAAGAAACTGAGA | GCGATTGTCCCCCTTCAGGA | 170 |
| GAPDH [34] | GGCGTGAACCACGAGAAGTATAA | CCCTCCACGATGCCAAAGT | 120 |
Identification of genes enriched in ramified and amoeboid microglia
The RNA-Seq database generated from the above experiments was used to further characterize the cell type. Given the lack of specific information of such genes in domestic sheep, a database from rats [37] was used as reference for genes enriched in ramified (42 genes) and amoeboid (43 genes) microglia. The ≥1 RPKM criterion proposed by Hebenstreit et al [38] was used as the cut-off for genes being actively transcribed (i.e., “present”).
Results
Prion permissiveness in ovine microglia clones
To limit the extent of phenotypic and genetic heterogeneity, two first-generation clones (clones 438 and 439 [7]), which were derived from the same subline (subline H [7]) but have differential prion permissiveness, were selected. Cells were inoculated with scrapie-positive and scrapie-negative brain homogenates, and were tested for the accumulation of nascent PrPSc at passage three post-inoculation by immunoblotting and at passage four post-inoculation by ELISA. In all experiments, only scrapie-inoculated clone 439 accumulated PrPSc as determined by the detection of cell-associated PK-resistant PrP by immunoblotting (Fig 1A) and high β–sheet PrP by ELISA (Fig 1B, 439/X124 vs. 439/mock [P = 0.0044, paired t-test]). While there was a statistical significance between 438/X124 and 438/mock (P = 0.0065 for clone 438, paired t-test), the levels failed to be higher than the manufacturer’s cut-off value and no PrPSc was detected in the 438/X124 samples by routine immunoblotting (Fig 1A, left panel) or after concentration by PTA precipitation (S1 Fig). Finally, the statistical analysis of normalized units from ELISA experiments revealed a significant difference (P = 0.0048, unpaired t-test) between clones 438 and 439 after inoculation with scrapie-positive brain homogenates. Therefore, these results confirm the divergent prion permissiveness phenotypes between these ovine microglia clones and for brevity these clones will be henceforth referred to as highly permissive (clone 439) and poorly permissive (clone 438) microglia.
PrPC expression levels in ovine microglia clones
Expression of PrPC in cultured cells is essential for the ex vivo propagation of PrPSc [5]. Thus, we investigated if the poorly permissive phenotype of ovine microglia clone 438 was due to markedly reduced expression of PrPC as compared to highly permissive microglia clone 439. To determine levels of PrPC, lysates of uninoculated cells of each microglia clone were analyzed by ELISA using a standard curve. No difference in PrPC levels was detected between the highly permissive and poorly permissive ovine microglia clones (P = 0.573, paired t-test, Fig 2). This finding suggests that this level of PrPC is insufficient by itself to confer prion permissiveness in different ovine microglia clones.
Fig 2. PrPC expression in ovine microglia clones.
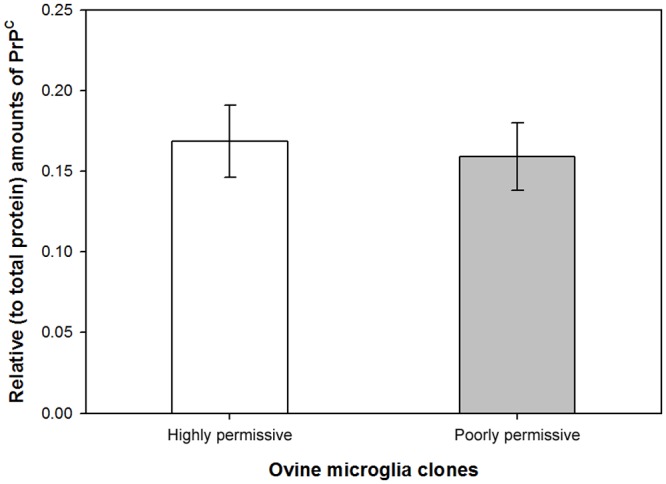
Cell lysates of uninoculated cells were normalized to total protein and used to quantitatively analyze expression levels of endogenous PrPC by ELISA. No significant difference in PrPC levels was found (P = 0.573, paired t-test).
Comparative transcriptional analysis
RNA-Seq was used to compare the transcriptional profiles of highly permissive and poorly permissive ovine microglia clones after inoculation with either scrapie-positive or scrapie-negative (i.e., mock) brainstem homogenates. The number of reads per library ranged from 25.8 to 33.7 million, and the number of reads mapped to the O. aries genome [25] ranged from 17.2 to 22.1 million (Table 2). Mapped reads were normalized using RPKM values and normalized values were used for pair-wise comparisons between microglia clones under the same inoculation conditions (i.e., scrapie or mock). Fold changes in transcription were considered significant when Bonferroni-corrected P values (Baggerley’s test) were less than 0.05. K—means clustering was used to identify genes with similar transcriptional patterns. Most of the genes with altered transcription in the comparison of mock-inoculated microglia fell into clusters 3 and 4; these genes were only slightly up- or downregulated (Fig 3A). Clusters 1, 2, and 5 from the same comparison showed higher magnitude of transcription than clusters 3 and 4; thus, the genes within the former clusters were selected for further analysis. Likewise, based on the same criteria mentioned above, the genes with altered transcription in the scrapie-inoculated microglia comparison that fell into clusters 2, 4, and 5 were further analyzed (Fig 3B).
Table 2. Reads mapped to O. aries genome.
| Experiment group (microglia clone/inoculum) | Total reads mapped to O. aries genome | % of reads mapped to O. aries genome | |
|---|---|---|---|
| Highly permissive/Mock | 30,150,424 | 19,341,429 | 64.15 |
| 33,750,532 | 21,783,758 | 64.54 | |
| 29,777,612 | 17,288,403 | 58.06 | |
| Highly permissive/Scrapie | 29,322,920 | 19,676,625 | 67.1 |
| 29,179,988 | 17,751,264 | 60.83 | |
| 30,184,738 | 21,223,906 | 70.31 | |
| Poorly permissive/Mock | 29,795,238 | 18,879,979 | 63.37 |
| 28,227,946 | 19,430,079 | 68.71 | |
| 30,521,914 | 21,390,586 | 70.08 | |
| Poorly permissive/Scrapie | 29,309,184 | 18,221,641 | 62.17 |
| 33,028,046 | 22,097,393 | 66.9 | |
| 25,814,978 | 17,952,849 | 69.54 |
Fig 3. K-means clustering of genes with differential transcription in comparisons of highly permissive against poorly permissive microglia.

The five clusters generated in the comparisons of mock-inoculated (A) and scrapie-inoculated (B) microglia clones are depicted. Each color line represents a single gene included in each cluster. The y—axis shows normalized expression values (RPKM), and microglia clones are on the x—axis.
Treatment-matched pair-wise comparisons (i.e., highly permissive microglia/mock vs. poorly permissive microglia/mock and highly permissive microglia/scrapie vs. poorly permissive microglia/scrapie) revealed 82 genes with altered transcription in the comparison of mock-inoculated microglia and 40 genes in the comparison of scrapie-inoculated microglia (S1 File). Fifty-seven genes with altered transcription from the comparison of mock-inoculated microglia and 32 from the scrapie-inoculated microglia comparison had known biological function (thus, there are 25 and 8 hypothetical loci identified, respectively [Tables 3 and 4]). Of these, 7/57 and 6/32 genes were upregulated in highly permissive microglia, and 50/57 and 26/32 were upregulated in poorly permissive microglia. The transcriptional status of 22 genes with known biological function (APLP2, CTSB, CTSL1, DCN, DPT, FABP5, GPNMB, LGMN, MMP14, NREP, MRCL3, P4HB, PDIA3, PLSCR4, PSAT1, PTN, RARRES1, RPL22L1, SEPP1, SQSTM1, TGFBI, and TM4SF1) was consistently altered in both pair-wise comparisons (Fig 4). The fold change in transcription of these genes varied from 1.26 to 432.14, with SEPP1, having the most dramatic change in transcription (i.e., 339.85 fold change in the mock-inoculated comparison and 432.14 fold change in the scrapie-inoculated comparison). Treatment-mismatched comparisons (i.e., mock vs. scrapie and scrapie vs. mock) revealed similar results (S2 Fig and S1 File) as those mentioned above with the addition of genes ITM2B (up-regulated in highly permissive microglia), and CRLS1 and RPS20 (up-regulated in poorly permissive microglia). For the treatment-matched pair-wise comparisons, the 33 hypothetical genes were analyzed for conserved domains (Tables 3 and 4), but only seven were consistently altered in both comparisons. Hypothetical genes from the treatment-mismatched pair-wise comparisons were not further analyzed.
Table 3. Genes encoding for hypothetical proteins with altered transcription in mock-inoculated microglia clones.
| Feature symbol | FC | Identity | Conserved domains on | E-value |
|---|---|---|---|---|
| LOC101104567¶ | 9.36 | Envelope glycoprotein-like | Env polyprotein* | 2.18E-110 |
| LOC101122294 | 3.67 | Membrane cofactor protein-like | Complement control modules | 6.90E-12 |
| LOC101116132 | -1.46 | 40S ribosomal protein S3a-like | Ribosomal S3Ae family | 1.21E-100 |
| LOC101106384 | -1.53 | Thymosin beta-4-like | Thymosin beta-4 family | 4.19E-14 |
| LOC101110467¶ | -1.66 | Transmembrane protein 45A-like | Family of unknown function (DUF716) | 3.44E-41 |
| LOC101103097 | -1.68 | Translationally-controlled tumor protein-like | Translationally controlled tumor protein | 1.23E-60 |
| LOC101104079 | -1.81 | Cystatin C | Cystatin-like domain | 8.98E-36 |
| LOC101109246 | -1.82 | 40S ribosomal protein S4-like | KOW motif of ribosomal protein S4 | 2.10E-28 |
| LOC101105484¶ | -2.11 | Ferritin heavy chain-like | Eukaryotic ferritins | 1.10E-93 |
| LOC443512¶ | -2.16 | Collagen I pro-alpha 2 chain precursor | Fibrillar collagens C-terminal domain | 1.18E-134 |
| LOC100037666 | -2.20 | Ribosomal protein S11 | 40s ribosomal protein S11 | 5.93E-91 |
| LOC100037669 | -2.23 | Niemann-Pick disease type C2 | Niemann-Pick type C2 | 7.10E-54 |
| LOC101104961 | -2.27 | 40S ribosomal protein S24-like | 40S ribosomal protein S24 | 1.52E-43 |
| LOC101104661 | -2.34 | Growth-regulated alpha protein-like | Chemokin_CXC | 1.21E-24 |
| LOC101103639 | -2.48 | Uncharacterized LOC101103639 | None | None |
| LOC100037665 | -2.52 | Ribosomal protein s6 | Ribosomal protein S6e | 2.60E-101 |
| LOC780524 | -2.54 | Ribosomal protein S2 | Ribosomal protein S5, N-terminal domain | 3.21E-30 |
| LOC100101231¶ | -2.56 | Collagen type III alpha 1 | Fibrillar collagens C-terminal domain | 7.92E-138 |
| LOC101122112 | -2.68 | 60S ribosomal protein L36a-like | 60S ribosomal protein L36a | 1.93E-95 |
| LOC100037664 | -2.76 | Ribosomal protein L35a | Ribosomal protein L35Ae | 3.17E-53 |
| LOC101112245 | -2.77 | Adenosylhomocysteinase-like | S-adenosylhomocysteine hydrolase | 0E+00 |
| LOC100037667 | -3.29 | Ribosomal protein S12 | Ribosomal protein L7A3/L30e/S12e/Gadd45 family | 8.51E-30 |
| LOC101102096 | -3.81 | Laminin receptor 1 pseudogene | 40S ribosomal protein SA* | 9.07E-125 |
| LOC101108131¶ | -3.89 | Complement C3-like | Proteins similar to C3, C4, C5 of vertebrate complement | 1.90E-94 |
| LOC101102861¶ | -5.31 | Phospholipid scramblase 2-like | Scramblase | 1.42E-122 |
FC: fold change in transcription (relative to highly permissive microglia).
*: Protein sequence manually generated prior to identification of conserved domains.
¶: Genes with altered transcription in both treatment-matched comparisons.
Table 4. Genes encoding for hypothetical proteins with altered transcription in scrapie-inoculated microglia clones.
| Gene symbol | FC | Identity | Conserved domains | E-value |
|---|---|---|---|---|
| LOC101104567¶ | 8.31 | Envelope glycoprotein-like | Env polyprotein* | 2.18E-110 |
| LOC101105484¶ | -1.55 | Ferritin heavy chain-like | Eukaryotic ferritins | 1.10E-93 |
| LOC101110467¶ | -1.68 | Transmembrane protein 45A-like | Family of unknown function (DUF716) | 3.44E-41 |
| LOC101103238 | -2.06 | Uncharacterized LOC101103238 | Chemokine_CXC | 2.09E-23 |
| LOC443512¶ | -2.27 | Collagen I pro-alpha 2 chain precursor | Fibrillar collagens C-terminal domain | 1.18E-134 |
| LOC101108131¶ | -3.08 | Complement C3-like | Proteins similar to C3, C4, C5 of vertebrate complement | 1.90E-94 |
| LOC100101231¶ | -3.13 | Collagen type III alpha 1 | Fibrillar collagens C-terminal domain | 7.92E-138 |
| LOC101102861¶ | -6.29 | Phospholipid scramblase 2-like | Scramblase | 1.42E-122 |
FC: Fold change in transcription (relative to highly permissive microglia).
*: Protein sequence manually generated prior to identification of conserved domains.
¶: Genes with altered transcription in both treatment-matched comparisons.
Fig 4. Transcript fold-change in highly permissive microglia compared to poorly permissive microglia.
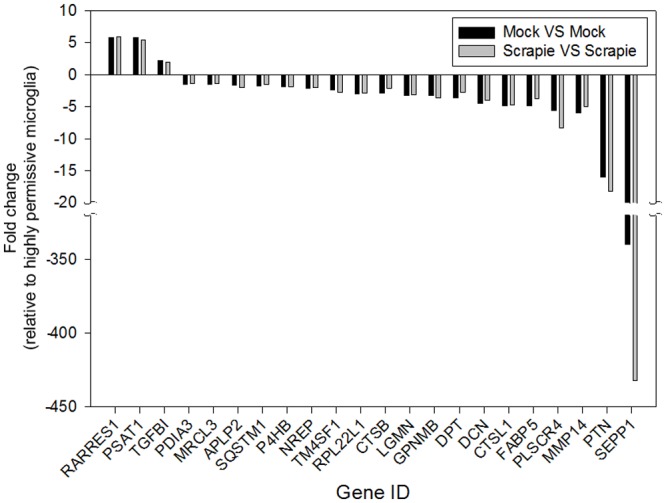
Transcriptional profiles of highly permissive and poorly permissive microglia clones under two experimental conditions (mock [black bars] or scrapie [gray bars] inoculation) were compared. All the genes with differential transcription in both comparisons (P < 0.05 [Baggerley’s test and Bonferroni correction]) and known biological function across three culture replicates are shown. Gene IDs are on the x—axis. The fold change in transcription relative to highly permissive microglia is on the y—axis. Thus, positive fold changes indicate upregulation in highly permissive microglia and negative fold changes indicate upregulation in poorly permissive microglia.
GSEA identified 59 altered biological pathways in the comparison of mock-inoculated clones and 46 altered biological pathways in the comparison of scrapie-inoculated clones (P < 0.01, S2 File). Of these, twenty-one pathways were similarly altered in both comparisons (Tables 5 and 6), and the proteolysis, positive regulation of cell migration, chromosome segregation, translation, and mitosis pathways had the lowest scores. Proteolysis, translation, and mitosis consistently included the highest number of features, suggesting significant alteration of these pathways. Genes of the Ovis aries genome assigned to biological pathways are listed in S4 File.
Table 5. Significantly altered biological pathways in highly permissive vs. poorly permissive microglia comparison after mock inoculation.
| Category | Description | Size | Test statistic | Lower tail | Upper tail |
|---|---|---|---|---|---|
| 6412 | translation (GO_REF:0000002 [IEA]q InterPro:IPR000039|InterPro:IPR021132) | 109 | -42.079 | 0 | 1 |
| 30335 | positive regulation of cell migration (GO_REF:0000024 [ISS] UniProtKB:Q8N4T4) | 76 | -16.3188 | 0.0003 | 0.9997 |
| 7059 | chromosome segregation (GO_REF:0000019 [IEA] Ensembl:ENSP00000362702) | 34 | -17.8086 | 0.0004 | 0.9996 |
| 6508 | proteolysis (GO_REF:0000003 [IEA] EC:3.4.19.9) | 372 | -13.713 | 0.0006 | 0.9994 |
| 71285 | cellular response to lithium ion (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000020974) | 11 | -20.2306 | 0.0009 | 0.9991 |
| 70830 | tight junction assembly (GO_REF:0000019 [IEA] Ensembl:ENSP00000345731) | 13 | -18.1835 | 0.0015 | 0.9985 |
| 6270 | DNA replication initiation (GO_REF:0000002 [IEA] InterPro:IPR003874) | 12 | -16.9102 | 0.0016 | 0.9984 |
| 7067 | mitosis (GO_REF:0000019 [IEA] Ensembl:ENSP00000297596) | 97 | -13.8772 | 0.0016 | 0.9984 |
| 60766 | negative regulation of androgen receptor signaling pathway (GO_REF:0000019 [IEA] Ensembl:ENSP00000362649) | 12 | -17.9763 | 0.0018 | 0.9982 |
| 90102 | cochlea development (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000077492) | 10 | -16.9598 | 0.0025 | 0.9975 |
| 22408 | negative regulation of cell-cell adhesion (GO_REF:0000019 [IEA] Ensembl:ENSP00000354040) | 10 | -16.8262 | 0.0031 | 0.9969 |
| 3382 | epithelial cell morphogenesis (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000128056) | 10 | -15.6154 | 0.0045 | 0.9955 |
| 51301 | cell division (GO_REF:0000037 [IEA] UniProtKB-KW:KW-0132) | 44 | -12.6405 | 0.0046 | 0.9954 |
| 71560 | cellular response to transforming growth factor beta stimulus (GO_REF:0000019 [IEA] Ensembl:ENSP00000457230) | 21 | -13.6831 | 0.0046 | 0.9954 |
| 7076 | mitotic chromosome condensation (GO_REF:0000002 [IEA] InterPro:IPR027120) | 11 | -13.4107 | 0.0076 | 0.9924 |
| 21766 | hippocampus development (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000019911) | 29 | 12.99072 | 0.9939 | 0.0061 |
| 2088 | lens development in camera-type eye (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000087870) | 16 | 12.89431 | 0.9947 | 0.0053 |
| 48013 | ephrin receptor signaling pathway (GO_REF:0000019 [IEA] Ensembl:ENSP00000332118) | 31 | 14.16371 | 0.9951 | 0.0049 |
| 42733 | embryonic digit morphogenesis (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000019911) | 46 | 13.21356 | 0.9952 | 0.0048 |
| 16358 | dendrite development (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000019911) | 29 | 15.72994 | 0.9976 | 0.0024 |
| 42384 | cilium assembly (GO_REF:0000019 [IEA] Ensembl:ENSP00000424757) | 68 | 16.71427 | 0.9981 | 0.0019 |
Lower and Upper tail values show the mass in the permutation based p-value distribution below or above the value of the test statistic. P values represented as 0 are less than 10-16.
Table 6. Significantly altered biological pathways in highly permissive vs. poorly permissive microglia comparison after scrapie inoculation.
| Category | Pathway description | Size | Test statistic | Lower tail | Upper tail |
|---|---|---|---|---|---|
| 6508 | proteolysis (GO_REF:0000003 [IEA] EC:3.4.19.9) | 372 | -17.1577 | 0 | 1 |
| 30335 | positive regulation of cell migration (GO_REF:0000024 [ISS] UniProtKB:Q8N4T4) | 76 | -21.8761 | 0 | 1 |
| 7067 | mitosis (GO_REF:0000019 [IEA] Ensembl:ENSP00000297596) | 97 | -22.4604 | 0.0001 | 0.9999 |
| 7059 | chromosome segregation (GO_REF:0000019 [IEA] Ensembl:ENSP00000362702) | 34 | -21.7366 | 0.0002 | 0.9998 |
| 70830 | tight junction assembly (GO_REF:0000019 [IEA] Ensembl:ENSP00000345731) | 13 | -18.3619 | 0.0008 | 0.9992 |
| 7076 | mitotic chromosome condensation (GO_REF:0000002 [IEA] InterPro:IPR027120) | 11 | -18.4568 | 0.0014 | 0.9986 |
| 71285 | cellular response to lithium ion (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000020974) | 11 | -19.3928 | 0.0016 | 0.9984 |
| 6270 | DNA replication initiation (GO_REF:0000002 [IEA] InterPro:IPR003874) | 12 | -15.5962 | 0.0039 | 0.9961 |
| 90102 | cochlea development (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000077492) | 10 | -13.4579 | 0.0046 | 0.9954 |
| 6412 | translation (GO_REF:0000002 [IEA] InterPro:IPR000039|InterPro:IPR021132) | 109 | -12.6216 | 0.0048 | 0.9952 |
| 22408 | negative regulation of cell-cell adhesion (GO_REF:0000019 [IEA] Ensembl:ENSP00000354040) | 10 | -13.3994 | 0.0049 | 0.9951 |
| 60766 | negative regulation of androgen receptor signaling pathway (GO_REF:0000019 [IEA] Ensembl:ENSP00000362649) | 12 | -13.9031 | 0.0052 | 0.9948 |
| 71560 | cellular response to transforming growth factor beta stimulus (GO_REF:0000019 [IEA] Ensembl:ENSP00000457230) | 21 | -12.6096 | 0.0062 | 0.9938 |
| 3382 | epithelial cell morphogenesis (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000128056) | 10 | -13.3184 | 0.0066 | 0.9934 |
| 51301 | cell division (GO_REF:0000037 [IEA] UniProtKB-KW:KW-0132) | 44 | -11.682 | 0.0083 | 0.9917 |
| 2088 | lens development in camera-type eye (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000087870) | 16 | 9.050416 | 0.9912 | 0.0088 |
| 42733 | embryonic digit morphogenesis (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000019911) | 46 | 8.110665 | 0.992 | 0.008 |
| 48013 | ephrin receptor signaling pathway (GO_REF:0000019 [IEA] Ensembl:ENSP00000332118) | 31 | 8.717496 | 0.9922 | 0.0078 |
| 42384 | cilium assembly (GO_REF:0000019 [IEA] Ensembl:ENSP00000424757) | 68 | 7.913188 | 0.9932 | 0.0068 |
| 21766 | hippocampus development (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000019911) | 29 | 8.947898 | 0.9946 | 0.0054 |
| 16358 | dendrite development (GO_REF:0000019 [IEA] Ensembl:ENSMUSP00000019911) | 29 | 9.441165 | 0.9947 | 0.0053 |
Lower and Upper tail values show the mass in the permutation based p-value distribution below or above the value of the test statistic. P values represented as 0 are less than 10-16.
Validation of RNA-Seq results by RT-qPCR
To confirm the RNA-Seq results, the transcriptional status of nine genes (CTSB, DCN, DPT, MMP14, PTN, RARRES1, SEPP1, SQSTM1, and TGFBI) with different transcriptional patterns and one gene (PRNP) without change in transcription (according to the RNA-Seq data) was analyzed by RT-qPCR. Only the experimental groups inoculated with natural scrapie prions were selected for this analysis. The transcriptional status of nine genes (CTSB, DCN, DPT, MMP14, PTN, SEPP1, PRNP, and SQSTM) obtained from the RNA-Seq experiment was confirmed by RT-qPCR (Fig 5). Upregulation of TGFBI in highly permissive microglia was not confirmed.
Fig 5. Validation of RNA-Seq results by RT-qPCR.

The fold changes in transcription of 10 genes across nine culture replicates of ovine microglia clones inoculated with scrapie prions are shown. Black bars represent RNA-Seq results and gray bars represent RT-qPCR results. RNA-Seq results were confirmed by RT-qPCR in 9 of 10 cases. ¶: P < 0.05. *: P < 0.0001. Gene IDs are on the x—axis. The log10 fold change in transcription relative to highly permissive microglia is on the y—axis. Thus, positive fold changes indicate upregulation in highly permissive microglia and negative fold changes indicate upregulation in poorly permissive microglia.
Identification of genes enriched in ramified and amoeboid microglia
Microglia derive from circulating monocytes that originate from the bone marrow; during post-natal life, these cells migrate into the brain and two morphologically different populations are recognized: amoeboid and ramified microglia [39]. Given that the cells used in this study had been only characterized as monocyte-derived cells by expression of CD14 [7], we sought to determine if these microglia clones were more consistent with either ramified or amoeboid microglia by identifying the presence/absence of expression of genes enriched depending on the microglia phenotype. Due to the lack of sheep-specific amoeboid and ramified microglia gene database, a published gene database from rat microglia was used as reference [37]. Both groups of microglia clones (mock-inoculated and scrapie-inoculated, three replicates each) were considered for this analysis. Genes were considered expressed when their RPKM average values were ≥1 [38]. IDs of genes analyzed are all listed in S3 File. Out of 43 genes enriched in rat amoeboid microglia, 38 genes were mappable to the ovine microglia RNA-Seq database. Thirty-one (82%) mappable genes enriched in neonatal amoeboid microglia were expressed in poorly permissive microglia and had an average RPKM of 50.9 (mock-inoculated) and 51.9 (scrapie-scrapie-inoculated); and 29 (76%) were expressed in highly permissive microglia with an average RPKM of 51.4 (mock-inoculated) and 56.2 (scrapie-inoculated) (Table 7 and S3 File). On the other hand, out of 42 genes enriched in rat ramified microglia, 39 genes were mappable to the ovine microglia RNA-Seq database. Of these, only 15 (38%) were expressed in poorly permissive microglia and had an average RPKM of 7.3 (mock-inoculated) and 6.3 (scrapie inoculated); and 14 (36%) were in highly permissive microglia with an average RPKM of 5.9 (mock-inoculated) and 6.2 (scrapie-inoculated) (Table 7 and S3 File). These findings suggest that the genotype of the ovine microglia clones used in this study is more consistent with neonatal amoeboid microglia rather than ramified microglia.
Table 7. Enriched genes of rat amoeboid microglia present in ovine microglia.
| Gene group | Poorly permissive microglia | Highly permissive microglia | ||
|---|---|---|---|---|
| Mock | Scrapie | Mock | Scrapie | |
| Mappable amoeboid genes (n = 38) | 31 | 31 | 29 | 29 |
| expressed in microglia (%) | (82%) | (82%) | (76%) | (76%) |
| Mappable ramified genes (n = 39) | 15 | 14 | 14 | 14 |
| expressed in microglia | (38%) | (36%) | (36%) | (36%) |
Discussion
Identification of cellular factors associated with prion conversion and degradation would greatly improve the understanding of TSEs pathogenesis and enable investigation of therapeutic interventions. It is known that PrPC is essential for replication of PrPD in animal models [40] and cultured cells [5]; however, the presence of PrPC does not guarantee prion conversion, highlighting the requirement for additional cell-associated factors. In this study, an immortalized microglia ex vivo system, derived from a natural TSE host, was used to identify potential cellular factors associated with relative prion permissiveness and resistance. This was accomplished by using RNA-Seq to compare the global transcriptional profiles of two clonal populations of microglia with differential prion permissiveness.
When comparing between permissibility phenotypes, transcriptional analysis identified 40 and 82 genes with altered transcription in two pair-wise comparisons, of which only 32 and 57 genes have known biological function. Twenty-two genes with known biological functions were consistently altered in both comparisons between highly permissive and poorly permissive microglia. The relative paucity of differential transcription indicates the high transcriptomic similarity between these two clonal populations of ovine microglia. Furthermore, the transcriptome suggests that the ovine microglia cells used in this study are more likely to be amoeboid microglia. This finding is expected as amoeboid microglia predominate in developing (fetal) brains, from which these microglia were isolated.
In regards to transcriptomic differences between highly permissive and poorly permissive clones, the fold change in transcription of these genes varied from 1.26- to 432.14-fold. Thirty-three genes encoding for hypothetical proteins were identified as differentially regulated; however, the significance of these is more tenuous based on the lack of definitive biological function. When comparing between inoculation statuses, most of the transcriptional differences between highly permissive and poorly permissive microglia appeared not to be induced by prion inoculation. This indicates that altered transcription of most of these genes is likely a pre-existing condition in these microglia clones.
Most of the genes with altered transcription were upregulated in poorly permissive microglia. SEPP1 was the gene with the most dramatic fold change in transcription in this study; it encodes for selenoprotein P, an extracellular selenium transporter glycoprotein that contains most of the selenium in plasma [41]. Selenoprotein P co-localizes with amyloid-β plaques and neurofibrillary tangles in individuals with Alzheimer’s disease [42] and inhibits aggregation and neurotoxicity of amyloid-β in mouse neuroblastoma cells [43]. Similarly, in poorly permissive ovine microglia, aggregation of PrPSc may be inhibited by selenoprotein P.
Multiple genes encoding for enzymes involved in proteolysis were upregulated in poorly permissive microglia. Of these, CTSB and CTSL1 encode for the cysteine proteases cathepsin B and cathepsin L, correspondingly. These cathepsins are located within lysosomal compartments and plasma membrane [44], subcellular locations in which prion conversion is thought to occur [45–47]. In murine neuronal (GT1-1) [48] and bone marrow-derived dendritic cells [19], cathepsins B and L partially degrade prions. Thus, in ovine microglia, it is possible that cathepsins B and L degrade internalized and cell membrane-associated scrapie prions. The potential effect of these proteases in poorly permissive microglia appears to be PrPSc-specific as no quantitative (see Fig 3) and qualitative [7] differences are found in PrPC between the two ovine microglia clones.
In neuroblastoma cells, the upregulation of matrix metalloproteinases (MMPs) 2 and 9 results in decreased deposition of PrPC at the extracellular matrix and resistance to prion infection [13]. Relatedly in this study, MMP14, an important activator of MMP2 [49], was consistently upregulated in poorly permissive microglia. Furthermore, in poorly permissive cells, MMP2 and FN1 were found to be upregulated, but only when these cells were inoculated with scrapie prions (see S1 File). These findings suggest that expression of FN1 and MMP2 is a response of poorly permissive microglia to inoculation with scrapie prions and reinforce the proposal by Marbiah et al [13] that FN1 activates expression of MMPs. Also, an alternative pathway of MMP activation in ovine microglia may involve HTRA1, which was upregulated in poorly permissive cells in two comparisons (see S1 File). HTRA1 encodes for a serine protease that targets extracellular matrix components (including fibronectin) and whose degradation products increase expression of MMPs [50]. Studies to further confirm and elucidate the potential role of MMPs in cellular permissibility to prions are ongoing.
SQSTM1 encodes for sequestosome 1, a protein required for the degradation of polyubiquitin-containing bodies that has been co-localized with intraneuronal ubiquitinated protein aggregates in individuals with protein misfolding diseases [51–53]. In mouse neuronal and microglia culture systems, SQSTM1 is overexpressed after inoculation with mouse-derived prions and is associated with degradation of PrPSc [54]. Our findings regarding SQSTM1 transcription are consistent with the latter studies and indicate that sequestosome 1 may contribute to degradation of scrapie prions in cultured cells of sheep, a natural TSE host. The proteins encoded by APLP2, PTN, DCN, GPNMB, P4HB, and PDIA3, have been associated with either protein misfolding diseases or TSEs [8, 55–59] but their specific role in prion protein degradation has not been described. Also, to the authors’ knowledge, the genes LGMN, SERPINH1, DPT, NREP, PLSCR4, FABP5, RPL22L1, TM4SF1, and MRCL3 have not been associated with either TSEs or protein misfolding disorders and their speculative role in resistance to prion infection is unclear at the moment. Genes encoding for multiple characterized and putative ribosomal proteins were upregulated in poorly permissive microglia after mock inoculation, but remained without significant change in transcription after scrapie infection as compared to the corresponding highly permissive microglia. The reason and significance for this difference is unclear.
Six to seven genes were upregulated in highly permissive microglia and only RARRES1, PSAT1, and TGFBI were consistently upregulated across pair-wise comparisons. The gene TGFBI encodes for the transforming growth factor beta-induced protein, which contributes to cell-collagen interaction and has been linked to protein aggregates in individuals with corneal dystrophy [60]. Moreover, mutants of TGFBI induce ex vivo aggregation of amyloid–β [61]. Thus, the transforming growth factor beta-induced protein may favor accumulation of scrapie prions by increasing aggregation of PrPSc in highly permissive microglia; however, RT-qPCR failed to verify altered transcript levels of TGFBI. RARRES1 encodes for the membrane-associated retinoic acid receptor responder membrane protein. While there is no previous evidence of this specific protein contributing to prion disease pathogenesis, retinoic acid treatment of N2a cells did increase their prion permissibility [13].
As mentioned above, our stringent algorithm and previous studies [13] have demonstrated a potential role for extracellular matrical proteins in prion permissibility, such as FN1 and MMP2, a modulator of the matrix. Furthermore, eleven genes (LOC101103238, ATP5A1, CYTB, FN1, FSTL1, HSP90B1, ITM2B, LMO4, MDD2, ND5, and VWA5A) are differentially regulated following prion inoculation (S1 File), but are not included as differentially expressed following mock inoculation. The gene LOC101103238 (recently named CXCL5) and follistatin (FST) have been shown to be upregulated in mice inoculated with 22L prions prior to the development of clinical signs [62] and in prion susceptible neuroblastoma cells [13], correspondingly. Likewise, ND5 and VWA5A have been found to be upregulated in patients with Creutzfeldt-Jakob disease [63] and sheep infected with scrapie [8], respectively. However, the specific role of the latter genes in prion diseases and/or permissiveness to prion infection has not been characterized.
GSEA identified several altered biological pathways with proteolysis, translation, and mitosis being the most consistently affected in both comparisons. The results of this analysis are consistent with two trends observed in poorly permissive microglia. The first corresponds to the proteolysis pathway, as discussed above. The second trend includes the alteration of translation pathway. Many genes encoding for ribosomal proteins were upregulated in poorly permissive microglia; however, the speculative impact of this pathway and these genes in prion infection resistance is unclear.
Conclusions
The global transcriptional profiles of two ovine microglia clones with differential scrapie prion permissiveness were compared using RNA-Seq, resulting in identification of 22 genes with consistently altered transcription and known biological function. The transcription of several other genes was altered but their difference in transcription was inconsistent and, in some cases, the biological function of such genes was unknown. Most of the genes with altered transcription were upregulated in poorly permissive microglia. The proteins encoded by many of these genes have known activity that may contribute to prion resistance (CTSB, CTSL1, SEPP1, and SQSTM1), possibly by degradation or neutralization of prions. The role in resistance to prion infection of other genes (APLP2, DCN, DPT, FABP5, GPNMB, MRCL3, P4HB, PDIA3, PLSCR4, PTN, RPL22L1, and TM4SF1) remains unclear. Only three genes, RARRES1, PSAT1, and TGFBI, were consistently upregulated in highly permissive microglia. Overall, the transcriptomic similarity of these clones and the overlapping results (e.g., MMP14 and FN1) with previous studies support the relevance of these findings, providing new insights in the cellular pathophysiology of TSEs and new candidate genes for therapeutic targets and markers of prion resistance and susceptibility.
Supporting Information
At passage 3 post-inoculation, cell lysates were collected, treated with PK, and incubated with PTA to increase sensitivity of immunoblotting. PK-resistant PrP was precipitated with PTA only from cells of clone 439. The results of three independent culture replicates inoculated with scrapie-positive brainstem homogenates (PrPSc +, lanes 1–3 and 5–7) and one with scrapie-negative inoculum (PrPSc–, lanes 4 and 8) of each microglia clone are shown, and are representative of three experiments.
(TIFF)
Transcriptional profiles of highly permissive and poorly permissive microglia clones under two different inoculation conditions were compared (i.e., mock VS scrapie and scrapie VS mock). Genes with differential transcription in both comparisons (P < 0.05 [Baggerley’s test and Bonferroni correction]) and known biological function across three culture replicates are shown. Gene IDs are on the x—axis and the fold change in transcription relative to highly permissive microglia is on the y—axis. Positive fold changes indicate up-regulation in highly permissive microglia and negative fold changes indicate up-regulation in poorly permissive microglia.
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Acknowledgments
The scrapie prion inoculum X124 was gently provided by Justin Greenlee and Eric Nicholson (National Animal Disease Center, United States Department of Agriculture). Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture.
Data Availability
All relevant data are within the paper and its Supporting Information files. The raw sequence data have been deposited in the NCBI Sequence Read Archive (accession number: PRJNA257519).
Funding Statement
This work was supported by the United States Department of Agriculture, Agricultural Research Service (5348-32000-030-00D).
References
- 1.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95(23):13363–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216(4542):136–44. [DOI] [PubMed] [Google Scholar]
- 3.Westaway D, Zuliani V, Cooper CM, Da Costa M, Neuman S, Jenny AL, et al. Homozygosity for prion protein alleles encoding glutamine-171 renders sheep susceptible to natural scrapie. Genes Dev. 1994;8(8):959–69. [DOI] [PubMed] [Google Scholar]
- 4.Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T. Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1998;43(6):826–8. [DOI] [PubMed] [Google Scholar]
- 5.Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, Lehmann S, et al. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci U S A. 2001;98(7):4055–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosque PJ, Prusiner SB. Cultured Cell Sublines Highly Susceptible to Prion Infection. J Virol. 2000;74(9):4377–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muñoz-Gutiérrez JF, Schneider DA, Baszler TV, Greenlee JJ, Nicholson EM, Stanton JB. hTERT-immortalized ovine microglia propagate natural scrapie isolates. Virus Res. 2015;198:35–43. 10.1016/j.virusres.2014.10.028 [DOI] [PubMed] [Google Scholar]
- 8.Filali H, Martin-Burriel I, Harders F, Varona L, Hedman C, Mediano DR, et al. Gene expression profiling of mesenteric lymph nodes from sheep with natural scrapie. BMC Genomics. 2014;15:59 10.1186/1471-2164-15-59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gossner A, Roupaka S, Foster J, Hunter N, Hopkins J. Transcriptional profiling of peripheral lymphoid tissue reveals genes and networks linked to SSBP/1 scrapie pathology in sheep. Vet Microbiol. 2011;153(3–4):218–28. 10.1016/j.vetmic.2011.05.036 [DOI] [PubMed] [Google Scholar]
- 10.Riemer C, Neidhold S, Burwinkel M, Schwarz A, Schultz J, Krätzschmar J, et al. Gene expression profiling of scrapie-infected brain tissue. Biochem Biophys Res Comm. 2004;323(2):556–64. [DOI] [PubMed] [Google Scholar]
- 11.Sorensen G, Medina S, Parchaliuk D, Phillipson C, Robertson C, Booth SA. Comprehensive transcriptional profiling of prion infection in mouse models reveals networks of responsive genes. BMC Genomics. 2008;9:114 10.1186/1471-2164-9-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown CA, Schmidt C, Poulter M, Hummerich H, Klöhn P-C, Jat P, et al. In vitro screen of prion disease susceptibility genes using the scrapie cell assay. Hum Mol Genet. 2014;23(19):5102–8. 10.1093/hmg/ddu233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marbiah MM, Harvey A, West BT, Louzolo A, Banerjee P, Alden J, et al. Identification of a gene regulatory network associated with prion replication. EMBO J 2014;33(14):1527–47. 10.15252/embj.201387150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanton JB, Knowles DP, Call DR, Mathison BA, Baszler TV. Limited transcriptional response of ovine microglia to prion accumulation. Biochem Biophys Res Comm. 2009;386(2):345–50. 10.1016/j.bbrc.2009.06.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casneuf T, Van de Peer Y, Huber W. In situ analysis of cross-hybridisation on microarrays and the inference of expression correlation. BMC Bioinformatics. 2007;8:461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okoniewski MJ, Miller CJ. Hybridization interactions between probesets in short oligo microarrays lead to spurious correlations. BMC Bioinformatics. 2006;7:276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang FP, Farquhar CF, Mabbott NA, Bruce ME, MacPherson GG. Migrating intestinal dendritic cells transport PrP(Sc) from the gut. J Gen Virol. 2002;83(Pt 1):267–71. [DOI] [PubMed] [Google Scholar]
- 18.Baker CA, Martin D, Manuelidis L. Microglia from Creutzfeldt-Jakob disease-infected brains are infectious and show specific mRNA activation profiles. J Virol. 2002;76(21):10905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luhr KM, Nordstrom EK, Low P, Ljunggren HG, Taraboulos A, Kristensson K. Scrapie protein degradation by cysteine proteases in CD11c+ dendritic cells and GT1-1 neuronal cells. J Virol. 2004;78(9):4776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakanishi H. Microglial functions and proteases. Mol Neurobiol. 2003;27(2):163–76. [DOI] [PubMed] [Google Scholar]
- 21.Raymond GJ, Olsen EA, Lee KS, Raymond LD, Bryant PK, Baron GS, et al. Inhibition of protease-resistant prion protein formation in a transformed deer cell line infected with chronic wasting disease. J Virol. 2006;80(2):596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamir AN, Richt JA, Kunkle RA, Greenlee JJ, Bulgin MS, Gregori L, et al. Characterization of a US sheep scrapie isolate with short incubation time. Vet Pathol. 2009;46(6):1205–12. 10.1354/vp.08-VP-0258-H-FL [DOI] [PubMed] [Google Scholar]
- 23.Stanton JB, Knowles DP, O'Rourke KI, Herrmann-Hoesing LM, Mathison BA, Baszler TV. Small-ruminant lentivirus enhances PrPSc accumulation in cultured sheep microglial cells. J Virol. 2008;82(20):9839–47. 10.1128/JVI.01137-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Rourke KI, Baszler TV, Besser TE, Miller JM, Cutlip RC, Wells GA, et al. Preclinical diagnosis of scrapie by immunohistochemistry of third eyelid lymphoid tissue. J Clin Microbiol. 2000;38(9):3254–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y, Xie M, Chen W, Talbot R, Maddox JF, Faraut T, et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science. 2014;344(6188):1168–73. 10.1126/science.1252806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierle SA, Dark MJ, Dahmen D, Palmer GH, Brayton KA. Comparative genomics and transcriptomics of trait-gene association. BMC Genomics. 2012;13:669 10.1186/1471-2164-13-669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baggerly KA, Deng L, Morris JS, Aldaz CM. Differential expression in SAGE: accounting for normal between-library variation. Bioinformatics. 2003;19(12):1477–83. [DOI] [PubMed] [Google Scholar]
- 28.Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, et al. CDD: NCBI's conserved domain database. Nucleic Acids Res. 2015;43(D1):D222–D6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Artimo P, Jonnalagedda M, Arnold K, Baratin D, Csardi G, de Castro E, et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40(W1):W597–W603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian L, Greenberg SA, Kong SW, Altschuler J, Kohane IS, Park PJ. Discovering statistically significant pathways in expression profiling studies. Proc Natl Acad Sci U S A. 2005;102(38):13544–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–14. 10.1093/nar/gkr988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stanton JB, Schneider DA, Dinkel KD, Balmer BF, Baszler TV, Mathison BA, et al. Discovery of a novel, monocationic, small-molecule inhibitor of scrapie prion accumulation in cultured sheep microglia and rov cells. PLoS One. 2012;7(11):e51173 10.1371/journal.pone.0051173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Budhia S, Haring LF, McConnell I, Blacklaws BA. Quantitation of ovine cytokine mRNA by real-time RT—PCR. J Immunol Methods. 2006;309(1–2):160–72. [DOI] [PubMed] [Google Scholar]
- 35.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134 10.1186/1471-2105-13-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30(9):e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parakalan R, Jiang B, Nimmi B, Janani M, Jayapal M, Lu J, et al. Transcriptome analysis of amoeboid and ramified microglia isolated from the corpus callosum of rat brain. BMC Neurosci. 2012;13:64 10.1186/1471-2202-13-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hebenstreit D, Fang M, Gu M, Charoensawan V, van Oudenaarden A, Teichmann SA. RNA sequencing reveals two major classes of gene expression levels in metazoan cells. Mol Syst Biol. 2011;7:497 10.1038/msb.2011.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553. 10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- 40.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73(7):1339–47. [DOI] [PubMed] [Google Scholar]
- 41.Burk RF, Hill KE. Selenoprotein P—Expression, functions, and roles in mammals. Biochim Biophys Acta. 2009;1790(11):1441–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bellinger FP, He QP, Bellinger MT, Lin Y, Raman AV, White LR, et al. Association of selenoprotein p with Alzheimer's pathology in human cortex. J Alzheimers Dis. 2008;15(3):465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Du X, Zheng Y, Wang Z, Chen Y, Zhou R, Song G, et al. Inhibitory act of selenoprotein p on cu(+)/cu(2+)-induced tau aggregation and neurotoxicity. Inorg Chem. 2014;53(20):11221–30. 10.1021/ic501788v [DOI] [PubMed] [Google Scholar]
- 44.Fonović M, Turk B. Cysteine cathepsins and extracellular matrix degradation. Biochim Biophys Acta. 2014;1840(8):2560–70. 10.1016/j.bbagen.2014.03.017 [DOI] [PubMed] [Google Scholar]
- 45.Vey M, Pilkuhn S, Wille H, Nixon R, DeArmond SJ, Smart EJ, et al. Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci U S A. 1996;93(25):14945–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pimpinelli F, Lehmann S, Maridonneau-Parini I. The scrapie prion protein is present in flotillin-1-positive vesicles in central- but not peripheral-derived neuronal cell lines. Eur J Neurosci. 2005;21(8):2063–72. [DOI] [PubMed] [Google Scholar]
- 47.Goold R, McKinnon C, Rabbanian S, Collinge J, Schiavo G, Tabrizi SJ. Alternative fates of newly formed PrPSc upon prion conversion on the plasma membrane. J Cell Sci. 2013;126(16):3552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luhr KM, Nordstrom EK, Low P, Kristensson K. Cathepsin B and L are involved in degradation of prions in GT1-1 neuronal cells. Neuroreport. 2004;15(10):1663–7. [DOI] [PubMed] [Google Scholar]
- 49.Will H, Atkinson SJ, Butler GS, Smith B, Murphy G. The soluble catalytic domain of membrane type 1 matrix μetalloproteinase cleaves the propeptide of progelatinase A and initiates autoproteolytic activation. Regulation by TIMP-2 and TIMP-3. J Biol Chem. 1996;271(29):17119–23. [DOI] [PubMed] [Google Scholar]
- 50.Tiaden AN, Klawitter M, Lux V, Mirsaidi A, Bahrenberg G, Glanz S, et al. Detrimental role for human high temperature requirement serine protease A1 (HTRA1) in the pathogenesis of intervertebral disc (IVD) degeneration. J Biol Chem. 2012;287(25):21335–45. 10.1074/jbc.M112.341032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakano T, Nakaso K, Nakashima K, Ohama E. Expression of ubiquitin-binding protein p62 in ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis with dementia: analysis of five autopsy cases with broad clinicopathological spectrum. Acta Neuropathol. 2004;107(4):359–64. [DOI] [PubMed] [Google Scholar]
- 52.Nakaso K, Yoshimoto Y, Nakano T, Takeshima T, Fukuhara Y, Yasui K, et al. Transcriptional activation of p62/A170/ZIP during the formation of the aggregates: possible mechanisms and the role in Lewy body formation in Parkinson's disease. Brain Res. 2004;1012(1–2):42–51. [DOI] [PubMed] [Google Scholar]
- 53.Zatloukal K, Stumptner C, Fuchsbichler A, Heid H, Schnoelzer M, Kenner L, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. The Am J Pathol. 2002;160(1):255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Homma T, Ishibashi D, Nakagaki T, Satoh K, Sano K, Atarashi R, et al. Increased expression of p62/SQSTM1 in prion diseases and its association with pathogenic prion protein. Sci Reports. 2014;4:4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huzarewich RL, Medina S, Robertson C, Parchaliuk D, Booth SA. Transcriptional modulation in a leukocyte-depleted splenic cell population during prion disease. J Toxicol Environ Health A. 2011;74(22–24):1504–20. 10.1080/15287394.2011.618979 [DOI] [PubMed] [Google Scholar]
- 56.Tanaka H, Shimazawa M, Kimura M, Takata M, Tsuruma K, Yamada M, et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci Rep. 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tseveleki V, Rubio R, Vamvakas S-S, White J, Taoufik E, Petit E, et al. Comparative gene expression analysis in mouse models for multiple sclerosis, Alzheimer's disease and stroke for identifying commonly regulated and disease-specific gene changes. Genomics. 2010;96(2):82–91. 10.1016/j.ygeno.2010.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cappai R, Cheng F, Ciccotosto GD, Needham BE, Masters CL, Multhaup G, et al. The amyloid precursor protein (APP) of Alzheimer disease and its paralog, APLP2, modulate the Cu/Zn-Nitric Oxide-catalyzed degradation of glypican-1 heparan sulfate in vivo. J Biol Chem. 2005;280(14):13913–20. [DOI] [PubMed] [Google Scholar]
- 59.Erickson RR, Dunning LM, Olson DA, Cohen SJ, Davis AT, Wood WG, et al. In cerebrospinal fluid ER chaperones ERp57 and calreticulin bind β-amyloid. Biochem Biophys Res Comm. 2005;332(1):50–7. [DOI] [PubMed] [Google Scholar]
- 60.El Kochairi I, Letovanec I, Uffer S, Munier FL, Chaubert P, Schorderet DF. Systemic investigation of keratoepithelin deposits in TGFBI/BIGH3-related corneal dystrophy. Mol Vis. 2006;12:461–6. [PubMed] [Google Scholar]
- 61.Yam GH, Wang K, Jhanji V, Choy KW, Baum L, Pang CP. In vitro amyloid aggregate forming ability of TGFBI mutants that cause corneal dystrophies. Invest Ophthalmol Vis Sci. 2012;53(9):5890–8. 10.1167/iovs.11-9068 [DOI] [PubMed] [Google Scholar]
- 62.Carroll JA, Striebel JF, Race B, Phillips K, Chesebro B. Prion infection of mouse brain reveals multiple new upregulated genes involved in neuroinflammation or signal transduction. J Virol. 2015;89(4):2388–404 10.1128/JVI.02952-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang J, Zhang ZX, Du PC, Zhou W, Wu SD, Wang QL, et al. Analyses of the mitochondrial mutations in the Chinese patients with sporadic Creutzfeldt-Jakob disease. Eur J Hum Genet. 2015;23(1):86–91. 10.1038/ejhg.2014.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
At passage 3 post-inoculation, cell lysates were collected, treated with PK, and incubated with PTA to increase sensitivity of immunoblotting. PK-resistant PrP was precipitated with PTA only from cells of clone 439. The results of three independent culture replicates inoculated with scrapie-positive brainstem homogenates (PrPSc +, lanes 1–3 and 5–7) and one with scrapie-negative inoculum (PrPSc–, lanes 4 and 8) of each microglia clone are shown, and are representative of three experiments.
(TIFF)
Transcriptional profiles of highly permissive and poorly permissive microglia clones under two different inoculation conditions were compared (i.e., mock VS scrapie and scrapie VS mock). Genes with differential transcription in both comparisons (P < 0.05 [Baggerley’s test and Bonferroni correction]) and known biological function across three culture replicates are shown. Gene IDs are on the x—axis and the fold change in transcription relative to highly permissive microglia is on the y—axis. Positive fold changes indicate up-regulation in highly permissive microglia and negative fold changes indicate up-regulation in poorly permissive microglia.
(TIF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files. The raw sequence data have been deposited in the NCBI Sequence Read Archive (accession number: PRJNA257519).