

Abstract
We have previously reported that mice with muscular dystrophy, including mdx mice, develop embryonal rhabdomyosarcoma (eRMS) with a low incidence after 1 year of age and that almost all such tumours contain cancer-associated p53 mutations. To further demonstrate the relevance of p53 inactivation, we created p53-deficient mdx mice. Here we demonstrate that loss of one or both p53 (Trp53) alleles accelerates eRMS incidence in the mdx background, such that almost all Trp53−/− mdx animals develop eRMS by 5 months of age. To ascertain whether increased tumour incidence was due to the regenerative microenvironment found in dystrophic skeletal muscles, we induced muscle regeneration in Trp53+/+ and Trp53−/− animals using cardiotoxin (Ctx). Wild-type (Trp53+/+) animals treated with Ctx, either once every 7 days or once every 14 days from 1 month of age onwards, developed no eRMS; however, all similarly Ctx-treated Trp53−/− animals developed eRMS by 5 months of age at the site of injection. Most of these tumours displayed markers of human eRMS, including over-expression of Igf2 and phosphorylated Akt. These data demonstrate that the presence of a regenerative microenvironment in skeletal muscle, coupled with Trp53 deficiency, is sufficient to robustly induce eRMS in young mice. These studies further suggest that consideration should be given to the potential of the muscle microenvironment to support tumourigenesis in regenerative therapies for myopathies.
Keywords: p53, rhabdomyosarcoma, mdx, muscular dystrophy, tumour microenvironment
Introduction
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma of childhood and adolescence [1]. The name ‘rhabdomyosarcoma’ comes from the presence of skeletal muscle cells representing various stages of muscle differentiation, from mononucleated myoblasts to multinucleated myotubes and rhabdomyoblasts, within the tumour [2]. Immunostaining for muscle proteins, particularly MyoD, myogenin and desmin, is used to aid in differentiating RMS from other types of sarcoma [3]. While RMS often emanates from skeletal muscle, it can also arise from tissues lacking skeletal muscle, such as the bladder or prostate [1,4–6]. The two major subtypes of paediatric RMS are embryonal RMS (eRMS) and alveolar RMS (aRMS). aRMS is associated with specific t(1;13)(p36;q14) or t(2;13)(q35;q14) chromosomal translocations between PAX7 or PAX3, respectively, and FKHR (Forkhead in human rhabdomyosarcoma, also known as FOXO1 ) [7–9], while eRMS is associated with particular genetic deletions (eg del11p15.5 [10,11]).
A number of mouse models for aRMS and eRMS have been made that speak to the molecular mechanisms of RMS formation and the involvement of various oncogenes in this process. Most aRMS models have been based on the forced expression of PAX3:FKHR or PAX7:FKHR fusion proteins [12–15]. By contrast, a number of eRMS models have been made implicating p53 inactivation as a primary event in oncogenesis. p53-deficient mice expressing an activated form of HER-2/neu develop eRMS of the urinary tract (and also salivary carcinomas) [16], p53-deficient mice made to express K-ras containing an oncogenic mutation develop pleomorphic RMS of the lower extremities [17] and eRMS in fish [18], p53-deficient mice with a deletion of the tumour suppressor patched 1 [19] or suppressor-of-fused [20] develop RMS and also medulloblastoma, and p53-deficient mice also deleted for c-Fos develop RMS of the facial and orbital regions [21]. A new study by Keller and colleagues defines RMS variability further by showing that the incidence of various RMS subtypes is altered depending on the types of muscle cells deleted for p53 [22]. Of particular note, deletion of p53 in differentiated myoblasts and myotubes led to exclusive formation of eRMS, while deletion of p53 in satellite cells, the predominant regenerating stem cell pool in muscle, led primarily to undifferentiated pleomorphic (or spindle cell) sarcoma [22].
We have recently described eRMS formation in two different mouse models of muscular dystrophy [23], the mdx model for Duchenne muscular dystrophy and the α-sarcoglycan-deficient (Sgca−/−) model for limb girdle muscular dystrophy 2D, after 1 year of age. Additional studies also support an association with RMS in aged mdx mice [24,25]. In the mdx and Sgca−/− animal models of muscular dystrophy, skeletal myofibres become weakened by the absence of proteins within the dystrophin-associated glycoprotein complex [26–28]. This gives rise to damage and death of skeletal myofibres. Such damage stimulates an inflammatory response, involving macrophages and T cells, and increased proliferation of satellite cells, the regenerative stem cells of adult skeletal muscles [29]. Ultimately, the differentiation and fusion of replicated satellite cells leads to the formation of new skeletal myofibres. The late onset of eRMS in these models suggests that RMS arises from the cumulative effects of chronic muscle regeneration.
As all of the eRMS tumours we previously studied in mdx mice had cancer-associated mutations or deletions in p53 (Trp53 ) [23], we have chosen here to directly engineer p53-deficiency into the mdx model. We present studies showing that Trp53 −/− mdx mice have very high incidence and early onset of muscle-derived eRMS and that induction of muscle regeneration is sufficient to induce robust eRMS formation in non-dystrophic p53-deficient mice.
Materials and methods
Materials
Antibody to dystrophin (Dys1) was purchased from Nova Castra (Newcastle upon Tyne, UK). Antibodies to Mdm2 (Sc-965), p53 (Sc-100), Rb (Sc-102), MyoD1 (Sc-760), myogenin (Sc-576) and PTEN (Sc-6817R) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibody to desmin (H-37) was purchased from Sigma (St. Louis, MO, USA). Antibodies to Akt (9275; phospho-threonine 308, 9271; phosphoserine 473, and 9272; Akt1-3 protein) were purchased from Cell Signaling Technology (Danvers, MA, USA). Antibody to Igf1 (AF791) and Igf2 (AF792) were purchased from R&D systems (Minneapolis, MN, USA). All secondary reagents were purchased from Jackson ImmunoResearch (West Grove, PA, USA).
Mice
Mice deleted for p53 (Trp53−/−, B6.129s2-Trp53 < tm1Tyj>/J, stock no. 002 101) and mice deficient in dystrophin (mdx, C57Bl/10ScSn-Dmdmdx/J, stock no. 001 801) were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and bred to generate Trp53+/+, Trp53+/−, Trp53−/−, Trp53−/− mdx (mdx meaning mdx/mdx female or mdx/Y male), Trp53+/− mdx and Trp53+/+ mdx animals. Mice were bred and cared for in a clean barrier facility and all animal care and experiments were done under protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Nationwide Children’s Hospital. Only littermates from identical crosses were compared for each experiment, with the exception of wild-type (Trp53+/+) and mdx mice, where multiple generations were mixed for analysis. Mice were given free access to water and food, were housed in cages with 2–4 animals/cage and were checked daily for development of tumours.
Histology
For analysis of tumour morphology, tissue was excised after sacrifice, embedded in Optimal Cutting Temperature (OCT, Sakura Finetek; Torrance, CA, USA) and frozen in liquid nitrogen-cooled 2-methylbutane. Tumours were divided into quarters and each quarter sectioned at 6–8 μm thickness on a cryostat through multiple regions. Multiple sections of each block were stained with haematoxylin and eosin (H&E) and imaged as previously described [23,30]. Bright-field photographs were taken using a Zeiss Axioskop 40 microscope with AxioCamICc3 digital camera system. Immunohistochemical staining for myogenin, MyoD and desmin was performed by the avidin–biotin complex peroxidase method, as previously described [3,23]. Immunostaining of tumours for dystrophin, p53, Rb, Mdm2, Igf1, Igf2, phospo-Ser473-Akt, phospho-Thr308-Akt, Akt and PTEN was done much as before [23], using horseradish peroxidase species-specific secondary antibodies and species-appropriate blocking sera.
Scoring of RMS staining and histopathology
The extent of staining or the extent of histopathology was scored in a blinded fashion with respect to case by two independent investigators. Quantification was performed by capturing images with a Zeiss Axioskop 40 microscope with an AxioCamICc3 camera system microscope. Quantification of tumour cells positively stained or of histopathology indices (eg the presence of anaplasia or necrosis) was scored using Zeiss AxioVision Rel 4.7 software.
Induction of muscle regeneration with cardiotoxin
Cardiotoxin (Ctx) from Naga naga venom (Sigma C9759; St. Louis, MO) was injected at a concentration of 10 μM in a 300 μl volume into the gastrocnemius or quadriceps, or in a 100 μl volume into the tibialis anterior muscle, on the left side of Trp53+/+ or Trp53−/− animals, with the contralateral muscle used as a mock-injected control. All injections were done using a sterile 0.3 cc ultrafine insulin syringe. Mice were injected once every 7 days or once every 14 days, beginning at 4 weeks of age and continuing until tumours were evident in the injected limb.
Quantification of muscle regeneration
Gastrocnemius muscles from Trp53+/+ or Trp53−/− muscles multiply injected with cardiotoxin (from 1 to 5 months of age), but where muscles had had 2 weeks to fully regenerate prior to analysis, were frozen in liquid nitrogen-cooled isopentane, cross-sectioned at 8 μm thickness on a cryostat and stained with H&E. No tumours were present in any of the muscles used for this analysis, although Trp53−/− animals did develop muscle-derived tumours in their quadriceps (Trp53+/+ animals had no tumours). At least three images, each sized at 31 181 μm2, were captured at random and quantified for each muscle, and four animals were compared per condition. Each myofibre in each image was quantified for the presence or absence of (at least one) centrally located nuclei and its diameter measured, as before [31,32]. Errors are reported as standard deviations (SDs).
Statistics
Comparison of significance for muscle regeneration measures (myofibre diameter or percentage of myofibres with central nuclei) was done using an unpaired two-tailed Student’s t-test with Microsoft Excel software. Comparison of significance between genotypes or treatment groups for RMS-free tumour survival was done using a log-rank test with JMP 9 software.
Results
Deletion of p53 increases onset and frequency of eRMS in mdx mice
All of the mdx mice that had developed muscle-derived embryonal rhabdomyosarcoma (eRMS) in our previous study had cancer-associated, likely inactivating, p53 (Trp53) missense mutations or deletions within the tumours [23]. Therefore, we generated mdx animals with a deletion of one or two alleles of p53 to reproduce this cancer-specific molecular event. mdx (mdx/mdx or mdx/Y) and wild-type mice were bred into Trp53+/+ and Trp53−/− backgrounds to generate six genotypes of mice: Trp53+/+, Trp53+/−, Trp53−/−, Trp53+/+ mdx, Trp53+/− mdx and Trp53−/− mdx. Mice were assessed daily for evidence of tumours (Figure 1). We confirmed our previous result that about 9% of mdx animals developed muscle-derived eRMS after 1 year of age and that no wild-type animals developed eRMS. eRMS formation in mdx animals was not evident until after 1 year of age. Trp53+/− mdx mice, by contrast, developed eRMS beginning at 3 months of age, with 60% of animals having eRMS by 10 months. Less than 10% of non-mdx Trp53+/− animals developed eRMS, and these tumours only formed at 10 months of age. Even more robust were the results with Trp53−/− mdx animals. Trp53−/− mdx mice began to develop muscle-derived eRMS at 3 months of age, with 90% of animals having muscle-derived eRMS by 5 months. While non-mdx Trp53−/− animals develop many types of cancer, only 30% of these were eRMS, even at 10 months of age, with the majority of the remaining tumours being lymphoma, as previously described [33]. Only two such lymphomas were identified as palpable muscle-derived tumours in our assay. Relative to age at 5 months, the odds ratio of Trp53−/− mdx animals developing eRMS was 42 compared to Trp53−/− and 175 compared to Trp53+/− mdx. Independent of age, the odds ratio of Trp53−/− mdx animals developing eRMS was 21 compared to Trp53−/−, 37 compared to Trp53+/− mdx, 71 compared to mdx and 250 compared to Trp53+/−. Similarly, the odds ratio of Trp53+/− mdx mice developing eRMS, independent of age, was 38 relative to Trp53+/− and 11 relative to mdx. When assessed as RMS-free survival using a log-rank test of significance, mdx versus wild-type (Trp53+/+) and Trp53−/− mdx versus Trp53−/− comparisons were highly significant (p < 0.001), while the Trp53+/− mdx versus Trp53+/− comparison was very significant (p < 0.01). Thus, deletion of one or both alleles of p53 in mdx mice accelerated the age of onset and increased the frequency of eRMS formation.
Figure 1.
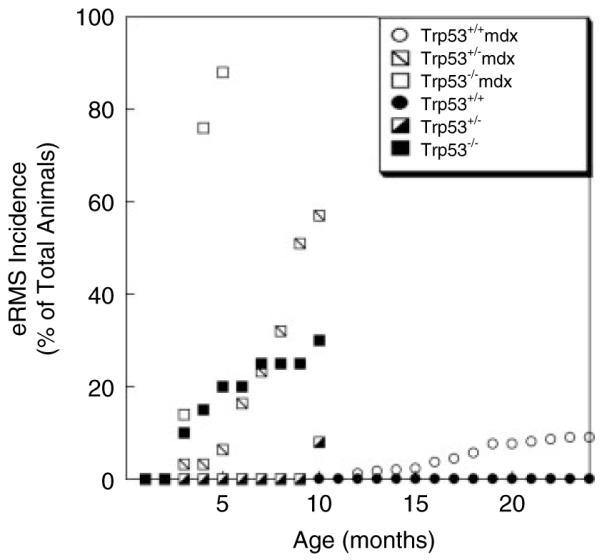
Increased embryonal rhabdomyosarcoma (eRMS) incidence and decreased age of onset in p53-deficient mdx mice. Mice defective in expression of p53 (Trp53), mice defective in the expression of dystrophin (mdx), and mice partially or completely deficient in p53 and dystrophin (Trp53+/− mdx or Trp53−/− mdx) were assessed for development of rhabdomyosarcoma at different ages. All muscle-derived RMS identified were embryonal RMS (eRMS). Data are reported as a percentage of the total animals for each individual genotype. n = 450 Trp53+/+ (wild-type), 26 Trp53+/−, 22 Trp53−/−, 360 Trp53+/+ mdx, 56 Trp53+/− mdx and 22 Trp53−/− mdx animals.
eRMS tumours in Trp53+/− mdx and Trp53−/− mdx animals were derived from skeletal muscles throughout the body plan and grew very rapidly (see Supporting information, Table S1). In addition, 6 of 34 Trp53+/− mdx animals and 10 of 20 Trp53−/− mdx animals showed multiple eRMS tumours, sometimes in distant locations along the rostral–caudal axis (see Supporting information, Table S1). All muscle-derived tumours were identified as eRMS by blinded assessment of tumour histopathology by at least two independent investigators. Such analysis showed all muscle-derived tumours in Trp53+/− mdx and Trp53−/− mdx mice to be eRMS (Figure 2). Common features in all Trp53+/− mdx and Trp53−/− mdx tumours were hypercellularity, with spindle-shaped or round cell morphology (or both) and the presence of rhabdomyoblasts (Figure 2). In addition, aspects of muscle differentiation, eg skeletal myofibres, were seen in all eRMS tumours. Where sections contained tumour borders, these were always juxtaposed with regions of dystrophic skeletal muscle (Figure 2B). Anaplastic cells were a feature of 48% of Trp53+/− mdx and 64% of Trp53−/− mdx tumours, while necrosis was a feature of 56% of Trp53+/− mdx and 75% of Trp53−/− mdx tumours (see Supporting information, Table S1). In Trp53−/− animals, we occasionally identified lymphomas (Figure 2F). These were confirmed by lack of immunostaining for MyoD and myogenin and by positive immunostaining for B220, CD3 and/or LCA (not shown).
Figure 2.

H&E staining of muscle-derived tumours from Trp53+/− mdx, Trp53−/− mdx and Trp53−/− mice. H&E staining of sections taken from muscle-derived tumours in Trp53+/− mdx (A–C), Trp53−/− mdx (D, E) and Trp53−/− (F) mice. Tumours from Trp53+/− mdx (A–C) and Trp53−/− mdx (D, E) show histopathology consistent with embryonal rhabdomyosarcoma (eRMS). Rare intramuscular tumours were identified as lymphoma in Trp53−/− mice (F). Features of eRMS tumours included hypercellularity (A–E) with spindle-shaped (A) or round (D) cell morphology. Tumours always emanated from areas containing regenerating skeletal muscle (B) and always contained rhabdomyoblasts and/or intratumoural skeletal myofibres (arrow in C). Bar = 100 μm and represents all panels.
In addition to showing classic eRMS morphology, all muscle-derived Trp53+/− mdx and Trp53−/− mdx tumours were positive for immunostaining with MyoD, myogenin, and desmin (Figure 3A). In all such cases, staining with secondary antibody alone showed no positive immunostaining. For Trp53+/− mdx and Trp53−/− mdx tumours, MyoD and myogenin staining was predominantly in the nucleus, consistent with the fact that these proteins are transcription factors. Positive nuclear staining for MyoD and myogenin was present in a significant fraction of the eRMS tumour cells, in the range 10–50%, while staining for desmin was present in almost all tumour cells. While there are other tumours, for example malignant peripheral nerve sheath (or triton) tumour, that can show the RMS-like features described here and stain for muscle proteins [34], the histochemical findings, in aggregate, strongly supported a diagnosis of eRMS in all instances. Likewise, the very abundant presence of differentiating myotubes and rhabdomyoblasts within the tumours did not support a diagnosis of spindle cell or undifferentiated pleomorphic sarcoma in any instance.
Figure 3.

MyoD, myogenin and desmin immunostaining of muscle-derived tumours from p53-deficient muscles. (A) All Trp53+/− mdx and Trp53−/− mdx muscle-derived eRMS tumours showed positive immunostaining for MyoD, myogenin and desmin. Example is a tumour fromTrp53−/− mdx. Panel marked ‘2nd Only’ was stained with secondary antibody but no primary antibody. All sections were counterstained with haematoxylin. (B) Staining for MyoD, myogenin and desmin were similarly positive in eRMS tumours taken from muscles of cardiotoxin (Ctx)-treated Trp53−/− mice. Bar = 50 μm for all panels in (A, B).
Induction of muscle regeneration is sufficient to induce robust early onset eRMS in Trp53−/− mice
One explanation as to why p53 deficiency led to an earlier onset and a higher frequency of eRMS in mdx mice would be that the regenerative microenvironment present in mdx skeletal muscles is permissive for eRMS formation. Conversely, the loss of dystrophin in mdx animals could be directly responsible for increased tumour formation. Indeed, dystrophin is absent in most paediatric RMS [23]. To test the role of muscle regeneration directly, we induced regeneration in specific skeletal muscles of Trp53+/+ and Trp53−/− mice using cardiotoxin. Cardiotoxin (Ctx) is a cobra venom protein that induces death via the elevation of intracellular free calcium levels in skeletal myofibres. This, in turn, induces immune cell recruitment and clearance of necrotic myofibres by macrophages, while at the same time inducing the division of satellite cells, the intramuscular stem cell pool [35]. Dividing satellite cells ultimately repopulate the region of muscle damage and fuse together to make new skeletal myofibres, thereby completing the regeneration cycle. The molecular and cellular processes induced by Ctx are very similar to those that are chronically present in dystrophic skeletal muscles, such as those that occur in the mdx mouse [36]. While there are other methods to induce muscle damage (eg barium loading, muscle freezing, physical injury or forced ambulation), cardiotoxin injection is a relatively standard method to induce a very focal muscle injury from which the animal quickly recovers with minimal discomfort. Unlike some of these other methods, the use of cardiotoxin also allows for the study of regeneration within a region of an otherwise normal muscle.
Wild-type (Trp53+/+) and Trp53−/− mice were treated with cardiotoxin (Ctx) once every 7 days or once every 14 days from 4 weeks of age onwards. Skeletal muscles (gastrocnemius, quadriceps and tibialis anterior) on the left side of the animal were injected with Ctx, while the same muscles on the contralateral (right) side were mock-injected with vehicle alone. Once a tumour was identified in the lower hindlimb, or at the end of the experiment for animals with no tumours, mice were allowed to recover from their final Ctx treatment for 2 weeks prior to sacrifice. This is roughly the amount of time required for muscle regeneration to be completed in response to a single Ctx injection [36]. All Ctx-treated Trp53−/− animals, regardless of injection schedule, developed muscle-derived tumours in their left lower hindlimb muscles by 5 months of age (Figure 4). By contrast, no Ctx-treated Trp53+/+ animals showed any muscle-derived tumours. We could identify muscle-derived tumours emanating from the gastrocnemius, quadriceps or tibialis anterior muscle of Ctx-injected Trp53−/− hindlimbs, and Ctx-treated Trp53−/− animals only showed tumours in the injected muscles (see Supporting information, Table S1). Several untreated Trp53−/− animals also developed RMS tumours over the time course of this experiment (Figure 4). This level of tumour incidence (20%), however, was far lower than the incidence (100%) in Ctx-treated Trp53−/− animals. When assessed as RMS-free survival using a log-rank test of significance, Ctx-treated Trp53−/− animals showed a highly significant decrease in RMS-free survival compared to untreated Trp53−/− mice or Ctx-treated Trp53+/+ mice (p < 0.001 for both comparisons).
Figure 4.
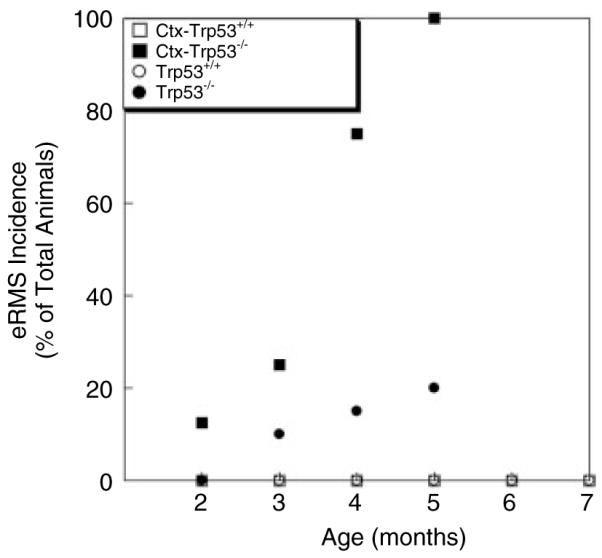
Increased eRMS incidence and decreased age of onset in cardiotoxin-treated p53-deficient mice. Muscle regeneration was induced by treatment of wild-type (Trp53+/+) and p53-deficient (Trp53−/−) mice with cardiotoxin (Ctx). Ctx-treated Trp53−/− mice showed robust induction of embryonal rhabdomyosarcoma (eRMS) by 5 months of age. n = 50 Trp53+/+ (wild-type), 12 Ctx-Trp53+/+ (cardiotoxin-injected), 22 Trp53−/−, 10 Ctx-Trp53−/− (cardiotoxin-injected).
All tumours were dissected and analysed by histological staining to define the form of cancer present. H&E staining of Ctx-injected Trp53−/− tumours (Figure 5) showed characteristics of eRMS tumours previously seen in mdx [23], Trp53+/− mdx, and Trp53−/− mdx mice (Figure 2). These included hypercellularity with round or spindle cell morphology and the presence of rhabdomyoblasts. Diffuse or focal anaplasia was present in 45% of tumours analysed and 63% of tumours showed some evidence of necrosis (see Supporting information, Table S1). By contrast, Ctx-treated Trp53+/+ muscles showed only normal regenerated skeletal muscle (Figure 5D). Muscle regeneration was evidenced by the presence of centrally located myofibre nuclei in almost 100% of Ctx-treated skeletal muscles. Central nuclei are an almost indelible indicator of regenerated rodent muscle, including muscles in mdx mice [31]. By contrast, centrally localized nuclei are typically found in 1–2% (and not more than 5%) of wild-type skeletal myofibres [31]. To verify that p53 deletion had not altered muscle regeneration, which in turn would alter the muscle microenvironment, we quantified the percentage of myofibres with central nuclei and average myofibre diameter in Ctx-treated Trp53−/− muscles and Ctx-treated Trp53+/+ muscles (Figure 6). Here, we chose only Ctx-treated gastrocnemius muscles where no tumours were present for analysis. There was no significant change in either of these measures (p = 0.67 for central nuclei, p = 0.98 for myofibre diameter) between Ctx-treated Trp53−/− and Trp53+/+ muscles. Thus, the extent of muscle regeneration and the extent of muscle growth within those regenerating myofibres was no different between Trp53−/− and Trp53+/+ animals, much as has been previously observed [37].
Figure 5.

H&E staining of muscle-derived tumours from cardiotoxin-treated Trp53−/− mice and skeletal muscle from cardiotoxin-treated Trp53+/+ mice. (A–C) H&E staining of sections taken from muscle-derived tumours in p53-deficient mice injected with cardiotoxin (Ctx-Trp53−/−) show histopathology consistent with embryonal rhabdomyosarcoma (eRMS). Arrow in (B) shows tumour cell with anaplasia. (D) H&E staining of skeletal muscle from wild-type mice similarly injected with cardiotoxin (Ctx-Trp53+/+) showed only normal regenerating skeletal muscle. Bar = 50 μm for all panels.
Figure 6.
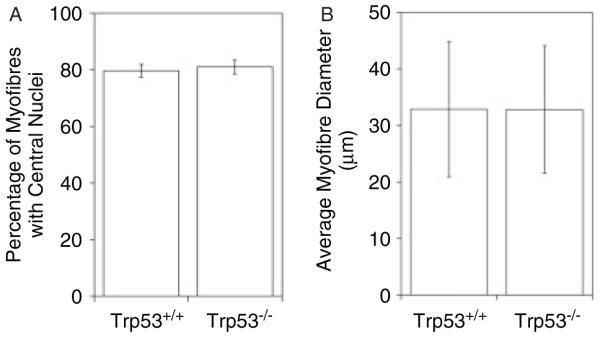
Quantification of muscle regeneration in cardiotoxin (Ctx)-treated Trp53+/+ and Trp53−/− muscle. Trp53+/+ and Trp53−/− mice were similarly treated with weekly injections of cardiotoxin (Ctx). Two weeks prior to analysis, Ctx injections were stopped and muscles were allowed to regenerate. The percentage of myofibres with centrally located myofibre nuclei, an indicator of at least one cycle of muscle degeneration and regeneration, was quantified (A), as was the average myofibre diameter (B). Errors are standard deviations for n = 4 animals per condition.
All Ctx-treated Trp53−/− tumours were positive for immunostaining of tumour cells with antibodies to MyoD, myogenin and desmin (Figure 3B), much as we had observed for Trp53+/− mdx and Trp53−/− mdx tumours (Figure 3A). Because regenerating satellite cells have increased expression of myogenic factors such as MyoD, we were careful to analyse tumour regions well removed from the border of regenerating muscle. The positive immunostaining of some differentiating myofibres in these sections, particularly for desmin, reflects the normal presence of immature and differentiating muscle within eRMS tumours (Figure 3B). These data, coupled with the presence of abundant intratumoural regions of regenerating muscle and/or rhabdomyoblasts (Figure 5), confirmed a diagnosis of eRMS in all instances.
Last, we stained Ctx-treated Trp53−/− tumours to determine whether molecular changes often found in human eRMS would be present in this new tumour model (Figure 7). We immunostained tumours with antibodies to insulin-like growth factor 2 (Igf2), insulin-like growth factor 1 (Igf1), phosphorylated-Akt kinase (pSer473 and pThr308), phosphatase and tensin homologue (PTEN), retinoblastoma (Rb), murine double minute 2 (Mdm2) or p53. As in human eRMS [38,39], we found high expression of Igf2 in tumour cells. Expression of phosphorylated (presumably activated) Akt, a downstream signal of Igf2, was also very high. Highly phosphorylated Akt was evidenced by staining with phospho-serine 473-specific and phospho-threonine 308-specific Akt antibodies in 7 of 10 tumours analysed. In each instance, phosphoserine 473 or phospho-threonine 308 Akt was high in almost all tumour cells, while Igf2 expression was high in about half of all tumour cells. PTEN, a phosphatase that can remove phosphorylation from Akt, was expressed at very low levels, as was Igf1. p53 was absent, as expected in Trp53−/− mice, and this was no different than staining in the presence of secondary antibody alone (‘2nd Only’ in the figures). Expression of dystrophin in these non-mdx muscles was by and large absent from eRMS cells, much as we had previously seen in human eRMS [23]; however, dystrophin was highly expressed in small regenerating myofibres that were very abundant within the tumours (not shown). This is consistent with the presence of muscle regeneration in eRMS. Rb expression was variable. Some small regions showed high nuclear expression, while others did not (not shown). Mdm2 expression was apparent in most tumours (not shown). We had previously shown that eRMS tumours in mdx mice do not bear transcripts consistent with Pax3/7:Fkhr chromosomal translocations found in aRMS [23] and we also found no evidence that this was the case here (not shown). Thus, eRMS tumours derived from Ctx-treated Trp53−/− muscles have high expression of certain markers of human eRMS, particularly Igf2 and phosphorylated Akt.
Figure 7.

Immunostaining of eRMS tumours from Ctx-treated Trp53−/− mice for markers of human eRMS. eRMS tumours from Ctx-treated Trp53−/− mice showed high expression of Igf2, activated Akt kinase (both phospho-serine 473 and phospho-threonine 308) and low expression of PTEN and Igf1. ‘2nd Only’ is staining in the absence of primary antibody for identical exposure time. All slides were counterstained with haematoxylin. Bar = 50 μm for all panels.
Discussion
The experiments presented here demonstrate that the regenerative microenvironment normally found in the skeletal muscles of mice with muscular dystrophy is sufficient to induce development of embryonal rhabdomyosarcoma (eRMS) in the absence of p53 (Trp53). These data strongly suggest a role for muscle inflammation, satellite cell division or myoblast differentiation in the development of eRMS. The division and differentiation of intramuscular satellite cells, the regenerative stem cell pool in skeletal muscles, and the invasion of immune cells, primarily macrophages, to clear necrotic myofibres are processes of primary importance to the regeneration of skeletal muscle. The two methods we have used here to study the regenerative microenvironment, cardiotoxin-induced muscle injury and dystrophin-deficiency, lead to similar molecular changes that are focused on muscle inflammatory factors and factors that control satellite cell division and differentiation [36,40–42]. The fact that all Trp53−/− mice develop eRMS in response to cardiotoxin-induced muscle injury and the fact that almost all Trp53−/− mdx mice develop eRMS by 5 months of age suggest that the processes involved in muscle regeneration give rise to a permissive environment for eRMS development and that dystrophin is not required for this to occur.
While the biology of p53 in human cancers is complex, p53 deficiency in Li–Fraumeni syndrome is sufficient to induce RMS [43]. What is less well understood is the knowledge of which elements cooperate with p53 to drive the preferential formation of RMS in skeletal muscle. Secondary factors, including neuregulin [16], Ras [17,18], patched [19,22], suppressor-of-fused [20] and Fos [21] can all increase p53-dependent RMS formation. It is likely that additional elements involved in regeneration can be identified that augment p53 deficiency to induce more robust RMS formation in skeletal muscle. The Trp53−/− mdx and Ctx-Trp53−/− mouse models of eRMS formation should facilitate the search for such factors. The Trp53−/− mdx and Ctx-Trp53−/− models may also be used to investigate the cell or cells of origin for eRMS development. Such ‘seed versus soil’ questions still remain in RMS biology. Evidence exists for mesenchymal stem cells, satellite cells and even skeletal myofibres as being the cell of origin for RMS formation [5,13,15]. Clearly, RMS can develop within tissues that lack skeletal muscle, eg bladder or prostate, so the model developed here is specific for only a subset of the potential oncogenic mechanisms involved in RMS development [4–6].
What triggers the transition of such cells to cancer cells in eRMS remains an open question. Muscle regeneration in dystrophic muscles and in Ctx-treated muscles may be coincident with elevated levels of reactive oxygen species [44] that may, in turn, stimulate increased DNA mutagenesis. If so, the microenvironment present in regenerating skeletal muscle may merely provide increased mutagenic potential to already rapidly dividing satellite cells or to other types of intramuscular stem cells. It is also possible that the increased cell division of satellite cells and/or other stem cells in regenerating muscle, or their maturation into myoblasts and/or myotubes, may be sufficient to stimulate eRMS formation, or that such a transition occurs only in the presence of increased inflammatory cytokines. In this regard, it is interesting to consider the recent study by Keller and colleagues, which showed that deletion of Trp53 in late skeletal myoblasts and skeletal myofibres using Myf6-driven gene deletion led to development of eRMS in 100% of tumours analysed, while deletion of Trp53 in only satellite cells using Pax7-driven gene deletion led predominantly to the formation of undifferentiated pleomorphic (or spindle cell) sarcoma [22]. Perhaps the high enrichment of maturing myoblasts and developing myotubes in Ctx-treated muscles here has biased the cell populations, such that Trp53 deletion leads to eRMS.
As eRMS most often occurs in very young children [1], it may well be that satellite cell division and fusion into skeletal myofibres, a normal part of postnatal muscle growth [45], may be present at significant enough levels to trigger the low incidence of eRMS seen in the paediatric population. If so, the experiments presented here may merely be amplifying such naturally occurring events. Given that RMS incidence in children is quite low, four to seven per million children under age 15 [1], understanding how to lessen the expression of secondary eRMS factors will be important for understanding how to lower eRMS incidence further. Use of the two models described here may help in speeding such an analysis and in testing new RMS therapies.
Supplementary Material
Acknowledgment
This work was funded by a grant to LTM from the Research Institute at Nationwide Children’s Hospital and NIH Grant No. AR049722 to PTM.
Footnotes
Author contributions
All authors designed and/or performed the research, collected, analysed and interpreted the data and were involved in the writing of the manuscript, but MB performed the majority of the research, while PTM performed the majority of the writing.
SUPPORTING INFORMATION ON THE INTERNET
The following supporting information may be found in the online version of this article:
No conflicts of interest were declared.
References
- 1.Arndt CA, Crist WM. Common musculoskeletal tumors of childhood and adolescence. N Engl J Med. 1999;341:342–352. doi: 10.1056/NEJM199907293410507. [DOI] [PubMed] [Google Scholar]
- 2.Parham DM, Ellison DA. Rhabdomyosarcomas in adults and children: an update. Arch Pathol Lab Med. 2006;130:1454–1465. doi: 10.5858/2006-130-1454-RIAACA. [DOI] [PubMed] [Google Scholar]
- 3.Morotti RA, Nicol KK, Parham DM, et al. An immunohistochemical algorithm to facilitate diagnosis and subtyping of rhabdomyosarcoma: the Children’s Oncology Group experience. Am J Surg Pathol. 2006;30:962–968. doi: 10.1097/00000478-200608000-00005. [DOI] [PubMed] [Google Scholar]
- 4.Hettmer S, Wagers AJ. Muscling in: Uncovering the origins of rhabdomyosarcoma. Nat Med. 2010;16:171–173. doi: 10.1038/nm0210-171. [DOI] [PubMed] [Google Scholar]
- 5.Charytonowicz E, Cordon-Cardo C, Matushansky I, et al. Alveolar rhabdomyosarcoma: is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009;279:126–136. doi: 10.1016/j.canlet.2008.09.039. [DOI] [PubMed] [Google Scholar]
- 6.Kikuchi K, Rubin BP, Keller C. Developmental origins of fusion-negative rhabdomyosarcomas. Curr Top Dev Biol. 2011;96:33–56. doi: 10.1016/B978-0-12-385940-2.00002-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis RJ, D’Cruz CM, Lovell MA, et al. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer Res. 1994;54:2869–2872. [PubMed] [Google Scholar]
- 8.Barr FG, Galili N, Holick J, et al. Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat Genet. 1993;3:113–117. doi: 10.1038/ng0293-113. [DOI] [PubMed] [Google Scholar]
- 9.Barr FG. Gene fusions involving PAX and FOX family members in alveolar rhabdomyosarcoma. Oncogene. 2001;20:5736–5746. doi: 10.1038/sj.onc.1204599. [DOI] [PubMed] [Google Scholar]
- 10.Koufos A, Hansen MF, Copeland NG, et al. Loss of heterozygosity in three embryonal tumours suggests a common pathogenetic mechanism. Nature. 1985;316:330–334. doi: 10.1038/316330a0. [DOI] [PubMed] [Google Scholar]
- 11.Anderson J, Gordon A, McManus A, et al. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia. 1999;1:340–348. doi: 10.1038/sj.neo.7900052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keller C, Arenkiel BR, Coffin CM, et al. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 2004;18:2614–2626. doi: 10.1101/gad.1244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keller C, Hansen MS, Coffin CM, et al. Pax3:Fkhr interferes with embryonic Pax3 and Pax7 function: implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 2004;18:2608–2613. doi: 10.1101/gad.1243904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galindo RL, Allport JA, Olson EN. A Drosophila model of the rhabdomyosarcoma initiator PAX7–FKHR. Proc Natl Acad Sci USA. 2006;103:13439–13444. doi: 10.1073/pnas.0605926103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren YX, Finckenstein FG, Abdueva DA, et al. Mouse mesenchymal stem cells expressing PAX–FKHR form alveolar rhabdomyosarcomas by cooperating with secondary mutations. Cancer Res. 2008;68:6587–6597. doi: 10.1158/0008-5472.CAN-08-0859. [DOI] [PubMed] [Google Scholar]
- 16.Nanni P, Nicoletti G, De Giovanni C, et al. Development of rhabdomyosarcoma in HER-2/neu transgenic p53 mutant mice. Cancer Res. 2003;63:2728–2732. [PubMed] [Google Scholar]
- 17.Tsumura H, Yoshida T, Saito H, et al. Cooperation of oncogenic K-ras and p53 deficiency in pleomorphic rhabdomyosarcoma development in adult mice. Oncogene. 2006;25:7673–7679. doi: 10.1038/sj.onc.1209749. [DOI] [PubMed] [Google Scholar]
- 18.Langenau DM, Keefe MD, Storer NY, et al. Effects of RAS on the genesis of embryonal rhabdomyosarcoma. Genes Dev. 2007;21:1382–1395. doi: 10.1101/gad.1545007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eichenmuller M, Bauer R, Von Schweinitz D, et al. Hedgehog-independent overexpression of transforming growth factor-β1 in rhabdomyosarcoma of Patched1 mutant mice. Int J Oncol. 2007;31:405–412. [PubMed] [Google Scholar]
- 20.Lee Y, Kawagoe R, Sasai K, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–6447. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- 21.Sharp R, Recio JA, Jhappan C, et al. Synergism between INK4a/ARF inactivation and aberrant HGF/SF signaling in rhabdomyosarcomagenesis. Nat Med. 2002;8:1276–1280. doi: 10.1038/nm787. [DOI] [PubMed] [Google Scholar]
- 22.Rubin BP, Nishijo K, Chen HI, et al. Evidence for an unanticipated relationship between undifferentiated pleomorphic sarcoma and embryonal rhabdomyosarcoma. Cancer Cell. 2011;19:177–191. doi: 10.1016/j.ccr.2010.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fernandez K, Serinagaoglu Y, Hammond S, et al. Mice lacking dystrophin or α-sarcoglycan spontaneously develop embryonal rhabdomyosarcoma with cancer-associated p53 mutations and alternatively spliced or mutant Mdm2 transcripts. Am J Pathol. 2010;176:416–434. doi: 10.2353/ajpath.2010.090405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chamberlain JS, Metzger J, Reyes M, et al. Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J. 2007;21:2195–2204. doi: 10.1096/fj.06-7353com. [DOI] [PubMed] [Google Scholar]
- 25.Schmidt WM, Uddin MH, Dysek S, et al. DNA damage, somatic aneuploidy, and malignant sarcoma susceptibility in muscular dystrophies. PLoS Genet. 2011;7:e1002042. doi: 10.1371/journal.pgen.1002042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sicinski P, Geng Y, Ryder-Cook AS, et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 27.Duclos F, Straub V, Moore SA, et al. Progressive muscular dystrophy in α-sarcoglycan-deficient mice. J Cell Biol. 1998;142:1461–1471. doi: 10.1083/jcb.142.6.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsumura K, Ervasti JM, Ohlendieck K, et al. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 29.De la Porte S, Morin S, Koenig J. Characteristics of skeletal muscle in mdx mutant mice. Int Rev Cytol. 1999;191:99–148. doi: 10.1016/s0074-7696(08)60158-8. [DOI] [PubMed] [Google Scholar]
- 30.Martin LT, Glass M, Dosunmu E, et al. Altered expression of natively glycosylated α-dystroglycan in pediatric solid tumors. Hum Pathol. 2007;38:1657–1668. doi: 10.1016/j.humpath.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen HH, Jayasinha V, Xia B, et al. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci USA. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu R, DeVries S, Camboni M, et al. Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of α-sarcoglycan-deficient mice. Am J Pathol. 2009;175:235–247. doi: 10.2353/ajpath.2009.080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 34.Stasik CJ, Tawfik O. Malignant peripheral nerve sheath tumor with rhabdomyosarcomatous differentiation (malignant triton tumor) Arch Pathol Lab Med. 2006;130:1878–1881. doi: 10.5858/2006-130-1878-MPNSTW. [DOI] [PubMed] [Google Scholar]
- 35.Tidball JG, Villalta SA. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1173–1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirata A, Masuda S, Tamura T, et al. Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: a role for osteopontin. Am J Pathol. 2003;163:203–215. doi: 10.1016/S0002-9440(10)63644-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.White JD, Rachel C, Vermeulen R, et al. The role of p53 in vivo during skeletal muscle post-natal development and regeneration: studies in p53 knockout mice. Int J Dev Biol. 2002;46:577–582. [PubMed] [Google Scholar]
- 38.Makawita S, Ho M, Durbin AD, et al. Expression of insulin-like growth factor pathway proteins in rhabdomyosarcoma: IGF-2 expression is associated with translocation-negative tumors. Pediatr Dev Pathol. 2009;12:127–135. doi: 10.2350/08-05-0477.1. [DOI] [PubMed] [Google Scholar]
- 39.Makawita S, Ho M, Durbin AD, et al. Expression of insulin-like growth factor pathway proteins in rhabdomyosarcoma: IGF-2 expression is associated with translocation-negative tumors. Pediat Dev Pathol Offic J Soc Pediat Pathol Paediat Pathol Soc. 2009;12:127–135. doi: 10.2350/08-05-0477.1. [DOI] [PubMed] [Google Scholar]
- 40.Bakay M, Zhao P, Chen J, et al. A web-accessible complete transcriptome of normal human and DMD muscle. Neuromusc Disord. 2002;12(suppl 1):S125–141. doi: 10.1016/s0960-8966(02)00093-7. [DOI] [PubMed] [Google Scholar]
- 41.Boer JM, de Meijer EJ, Mank EM, et al. Expression profiling in stably regenerating skeletal muscle of dystrophin-deficient mdx mice. Neuromusc Disord. 2002;12(suppl 1):S118–124. doi: 10.1016/s0960-8966(02)00092-5. [DOI] [PubMed] [Google Scholar]
- 42.Goetsch SC, Hawke TJ, Gallardo TD, et al. Transcriptional profiling and regulation of the extracellular matrix during muscle regeneration. Physiol Genom. 2003;14:261–271. doi: 10.1152/physiolgenomics.00056.2003. [DOI] [PubMed] [Google Scholar]
- 43.Li FP, Fraumeni JF., Jr. Rhabdomyosarcoma in children: epidemio-logic study and identification of a familial cancer syndrome. J Natl Cancer Inst. 1969;43:1365–1373. [PubMed] [Google Scholar]
- 44.Whitehead NP, Yeung EW, Allen DG. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol. 2006;33:657–662. doi: 10.1111/j.1440-1681.2006.04394.x. [DOI] [PubMed] [Google Scholar]
- 45.Bischoff R. In: Satellite Cells in Myology. 3rd edn Franzini-Armstrong C, Engel A, editors. McGraw-Hill; New York: 2004. pp. 66–86. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.