

Abstract
Background
Copy number variation (CNV) is an important source of genetic variability associated with phenotypic variation and disease susceptibility. Comprehensive genome-wide CNV maps provide valuable information for genetic and functional studies. To identify CNV in Japanese Black cattle, we performed a genome-wide autosomal screen using genomic data from 1,481 animals analyzed with the Illumina Bovine High-Density (HD) BeadChip Array (735,293 single-nucleotide polymorphisms (SNPs) with an average marker interval of 3.4 kb on the autosomes).
Results
We identified a total of 861 CNV regions (CNVRs) across all autosomes, which covered 43.65 Mb of the UMD3.1 genome assembly and corresponded to 1.74 % of the 29 bovine autosomes. Overall, 35 % of the CNVRs were present at a frequency of > 1 % in 1,481 animals. The estimated lengths of CNVRs ranged from 1.1 kb to 1.4 Mb, with an average of 50.7 kb. The average number of CNVR events per animal was 35. Comparisons with previously reported cattle CNV showed that 72 % of the CNVR calls detected in this study were within or overlapped with known CNVRs. Experimentally, three CNVRs were validated using quantitative PCR, and one CNVR was validated using PCR with flanking primers for the deleted region. Out of the 861 CNVRs, 390 contained 717 Ensembl-annotated genes significantly enriched for stimulus response, cellular defense response, and immune response in the Gene Ontology (GO) database. To associate genes contained in CNVRs with phenotypes, we converted 560 bovine Ensembl gene IDs to their 438 orthologous associated mouse gene IDs, and 195 of these mouse orthologous genes were categorized into 1,627 phenotypes in the Mouse Genome Informatics (MGI) database.
Conclusions
We identified 861 CNVRs in 1,481 Japanese Black cattle using the Illumina BovineHD BeadChip Array. The genes contained in CNVRs were characterized using GO analysis and the mouse orthologous genes were characterized using the MGI database. The comprehensive genome-wide CNVRs map will facilitate identification of genetic variation and disease-susceptibility alleles in Japanese Black cattle.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-016-0335-z) contains supplementary material, which is available to authorized users.
Keywords: Copy number variation (CNV), BovineHD BeadChip Array, Japanese Black cattle
Background
Copy number variation (CNV) is defined as deletions or duplications of genome segments ranging from 1 kb to several Mb [1]. Several studies have identified CNVs in cattle using three platforms: single-nucleotide polymorphism (SNP) arrays [2–9], comparative genomic hybridization (CGH) [10, 11], and next-generation sequencing (NGS) [12–15] (Additional file 1: Table S1). Previous results show that CNVs comprise a large fraction of the bovine genome, ranging from 3.29 to 498 Mb [2, 3, 5–15] (Additional file 1: Table S1).
CNVs can influence phenotypic variation or result in disease via mechanisms such as gene dosage modification and gene structure disturbance, either directly by exposing recessive alleles or indirectly by disturbing the regulatory regions of genes (for review, see [16]). Stranger et al. estimated that CNVs were responsible for 17.7 % of genetic variation in gene expression of human lymphoblastoid cell lines [17], and two studies on rodents showed that CNVs result in genome-wide expression changes in various tissues [18, 19], suggesting that CNVs alter gene dosage and are associated with phenotypic variance and disease susceptibility. In fact, several studies in cattle have demonstrated that CNVs contribute to phenotypic diversity in coat color [20] and milk production [21, 22], and also to diseases such as female fertility failure [21], nephritis [23], anhidrotic ectodermal dysplasia [24], myopathy [25], and osteopetrosis [26]. In addition, CNVs have been shown to contribute to phenotypic diversity and disease susceptibility in other species (for review, see [16, 27]).
Recently, substantial genotyping data using SNP arrays have been produced from genome-wide association studies [28] and genomic selection [29] in cattle, which can be directly exploited for CNV analysis. In particular, the Illumina Bovine High-Density (HD) BeadChip Array, with a total of 777,692 SNPs and an average marker interval of 3.4 kb, is 15-fold denser than the Illumina Bovine50K BeadChip Array [30], and provides higher resolution and a convenient screen for high-throughput CNV detection in the cattle genome [5, 8, 9].
Japanese Black cattle are highly rated owing to the abundant marbling of meat caused by intramuscular fat deposition [31]. Strict selection for marbling under a closed breeding system in Japan [32] has made the Japanese Black cattle genetically distinct from other cattle breeds [33]. A genome-wide map of CNVs has not been developed for Japanese Black cattle. Thus, to identify genotypic variability and disease-susceptibility alleles in the population, genome-wide CNV screens must be applied to this breed.
Therefore, this study aimed to investigate CNVs in Japanese Black cattle by performing a genome-wide screen of autosomes using genomic data from 1,481 animals analyzed with the Illumina BovineHD BeadChip Array.
Results and discussion
Genome-wide autosomal detection of CNVRs in 1,481 Japanese Black cattle
To identify CNVs in Japanese Black cattle, we used the Illumina BovineHD BeadChip Array, which contains 735,293 SNPs on autosomes with an average marker interval of 3.416 kb [30]. SNPs on sex chromosomes (X and Y) as well as on unknown chromosomes were excluded because of the lack of accurate information on their position in the bovine genome. In this study, a potential CNV was determined if it contained three or more consecutive SNPs [3, 5, 6, 9, 34] (Additional file 1: Table S1). To date, most SNP array-based CNV studies in cattle (Additional file 1: Table S1) and other animals (pig [35], chicken [36], sheep [37], and dog [38]) have used the hidden Markov model (HMM) approach-based PennCNV software [39, 40]. To identify CNVs and to compare our results with previous findings (Additional file 1: Table S1), we chose to use PennCNV software as well. We detected 55,593 CNV calls in 1,481 Japanese Black cattle. Of these, 1,099 singleton CNVs were identified, and 861 CNV regions (CNVRs) with overlapping CNVs [41] were detected in at least two animals. Since singleton CNVs were only detected in one animal, they were considered false positives compared to CNVRs [41] and excluded from subsequent analyses.
A total of 861 identified CNVRs covered 43.65 Mb of the UMD3.1 genome assembly, corresponded to 1.74 % of the 29 bovine autosomes, and consisted of 404 loss, 257 gain, and 200 loss plus gain (loss and gain within the same CNVR) events (Fig. 1, Additional file 1: Table S2). Overall, 35 % of the CNVRs were present at a frequency of >1 % in 1,481 animals (Additional file 1: Table S2). CNVs at a frequency of >1 % were characterized as copy number polymorphisms (CNPs) with potential involvement in the genetic basis of common phenotypes and diseases [1].
Fig. 1.
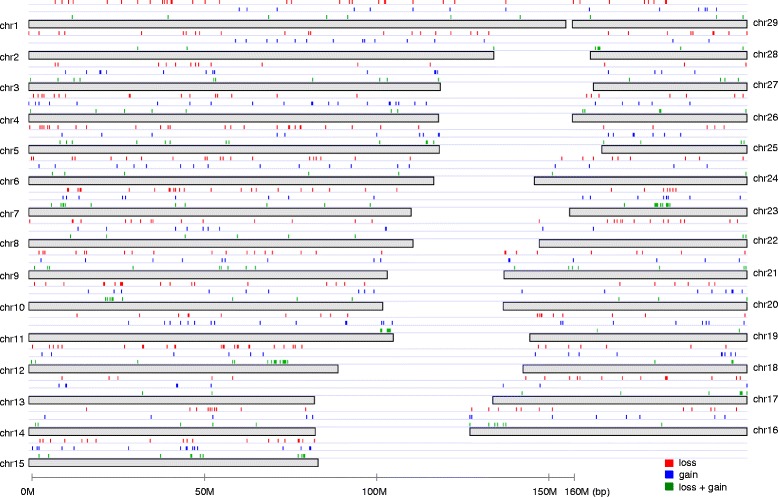
Distribution and status of CNVRs in the bovine genome. CNVRs (861 events, 43.65 Mb) in 1,481 Japanese Black cattle genotyped with the Illumina BovineHD BeadChip Array are shown on the autosomes in red (loss), blue (gain), and green (loss + gain). CNVR positions on the x-axis are based on the UMD3.1 assembly of the bovine genome
The estimated lengths of CNVRs ranged from 1.1 kb to 1.4 Mb with an average of 50.7 kb (Fig. 2a). Approximately 83 % of CNVRs were shorter than the average marker interval of the Illumina Bovine50K BeadChip Array in autosomes (68.3 kb) (Fig. 2a), indicating that a large number of small CNVRs were not detected in our population using the Illumina Bovine50K BeadChip Array. CNVRs were detected with an average of 35 events per animal (Fig. 2b). Detailed information of each CNVR is presented in Additional file 1: Table S2. In addition, we present the characteristics of CNVRs compared to previous studies in Additional file 1: Table S1.
Fig. 2.
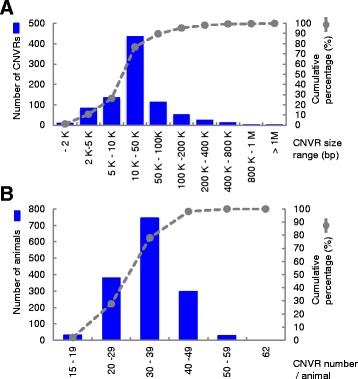
CNVR size distribution and CNVR number per animals. a The bar (blue) represents the CNVR size distribution in each size range; the line plot (gray) represents the cumulative percentage of CNVR number. b The bar (blue) represents the CNVR number per animal; the line plot (gray) represents the cumulative percentage of CNVR number
Comparison of current results with other cattle CNV studies
To characterize these CNVRs in more detail, we compared current results with three previously published cattle autosomal CNVRs using Illumina BovineHD BeadChip Arrays [5, 8, 9]. In addition, for a more comprehensive comparison, we also used the Database of Genomic Variants archive (DGVa) [42], including cattle CNV datasets from Illumina Bovine50K BeadChip Array [3, 4], CGH [11], and NGS [14], which were mapped on the UMD3.1 assembly of the bovine genome using dbVar [43]. The comparisons revealed that 72 % of the CNVR calls (620 CVNRs) detected in this study were within or overlapped with all combined non-redundant datasets (Table 2, detailed information for each comparison is presented in Additional file 1: Tables S2, S3, and S4). Comparisons with three previously reported CNV studies using the Illumina BovineHD BeadChip Array showed that CNVRs detected in this study overlapped 65.7 % of the CNVR calls reported by Hou et al. [5], 13.4 % of the CNVR calls reported by Jiang et al. [8], and 10.5 % of the CNVR calls reported by Zhang et al. [9] (Table 2 and Additional file 1: Table S2). The results were likely due to the different criteria used for the determination of CNV and the number of samples and breeds used in each study. CNV was defined as containing three or more consecutive SNPs in Hou et al. [5], in Zhang et al. [9], and in this study, whereas it was defined to contain 10 or more consecutive SNPs in Jiang et al. [8] (Additional file 1: Table S1). In the initial CNV discovery, we used three SNP windows, which is a less strict criterion than that in the study by Jiang et al. [8] (Additional file 1: Table S1). Therefore, we also inferred CNV with 5, 10, and 15 consecutive SNP windows, respectively (Additional file 1: Tables S1 and S2, Additional file 2). Of 861 CNVRs, 581 (67 %), 287 (33 %), and 181 (21 %) were also detected with 5, 10, and 15 consecutive SNP windows (Additional file 1: Table S2; Additional file 2), respectively. The average length of CNVRs in 3, 5, 10, and 15 consecutive SNP windows were 50.7, 65, 104, and 132 kb, respectively, indicating that there was an inverse correlation between the detection number of CNVRs and CNVR length, depending upon the size of SNP windows (Additional file 2). The increased number of consecutive SNPs may infer reliable CNV detection compared to a smaller number of SNPs, although the increased number of consecutive SNPs cannot capture small CNVRs. Thus, the results from this stepwise analysis of SNP windows will provide valuable information for further analyses, including experimental validation of CNVRs. Detailed SNP window information for each CNVR is presented in Additional file 1: Table S2.
Table 2.
Gene Ontology (GO) categories significantly overrepresented in CNVRs
| GO categoriesa | gene number in CNVR | expected gene numberb | P valuec | |
|---|---|---|---|---|
| Biological | response to stimulus | 124 | 61.18 | 8.14E-12 |
| Process | cellular defense response | 51 | 15.71 | 1.25E-10 |
| immune system process | 120 | 61.91 | 4.61E-10 | |
| hemopoiesis | 23 | 5.57 | 5.45E-06 | |
| immune response | 51 | 22.36 | 1.83E-05 | |
| B cell mediated immunity | 28 | 8.8 | 3.71E-05 | |
| extracellular transport | 18 | 4.94 | 1.01E-03 | |
| response to interferon-gamma | 14 | 3.1 | 1.14E-03 | |
| Cellular | immunoglobulin complex | 20 | 0.98 | 4.32E-18 |
| Component | protein complex | 48 | 17.26 | 2.21E-08 |
| macromolecular complex | 50 | 21.76 | 4.03E-06 | |
| heterotrimeric G-protein complex | 8 | 1.24 | 2.23E-03 | |
| MHC protein complex | 10 | 2.03 | 2.53E-03 | |
| Molecular | antigen binding | 20 | 0.98 | 1.35E-17 |
| Function | ATPase activity, coupled to transmembrane movement of substances | 9 | 1.46 | 3.40E-03 |
aList of GO categories associated with overrepresentation of genes in the CNVRs (P < 0.05)
bEnsembl gene list of 19,799 Bos taurus genes compared to the list of 717 genes in the CNVRs using PANTHER 9.0
cBonferroni-corrected P value
In addition to CNV definition, the inconsistency noted among studies could also be due to differences in sample size and cattle breeds. Our study used 1,481 animals of a single cattle breed, while Hou et al. [5] used 674 animals of 27 different cattle breeds, and Jiang et al. [8] and Zhang et al. [9] used 96 and 6 animals of a single cattle breed (Additional file 1: Table S1), respectively. The present study used a larger number of animals than the studies by Jiang et al. [8] and Zhang et al. [9] from a single breed; therefore, the current CNV screen may be more effective than previous intrabreed studies [8, 9].
In this study, we did not calculate the proportion of unique CNVRs in Japanese Black cattle directly; however, 28 % of identified CNVRs have not been previously reported in non-redundant CNV datasets (Table 1) and might be unique in Japanese Black cattle. As reported in dogs [44, 45] and horses [46], integration of current CNV data with the results from different cattle breeds will facilitate the identification of unique genotypic variability and disease-susceptibility alleles in Japanese Black cattle.
Table 1.
Comparison of 861 CNVRs detected in this study with results from three other CNV studies used the Illumina BovineHD BeadChip Array and structural variants deposited in the DGVa
Experimental validation of CNVRs by quantitative PCR and CNVR_27 by PCR with flanking primers
Quantitative PCR (qPCR) was performed to verify CNVR calls using the SNP array as an independent experimental validation. Three loss-type CNVRs, CNVR_285, CNVR_437, and CNVR_631, were selected. The Basic transcription factor 3 gene (BTF3), which served as an internal qPCR standard for both copies at a locus (2n) [2], was co-amplified with the primers. The copy number estimated by qPCR was approximately one (Fig. 3), which was in agreement with the expected copy number estimated by the PennCNV analysis using the Illumina BovineHD BeadChip Array platform.
Fig. 3.
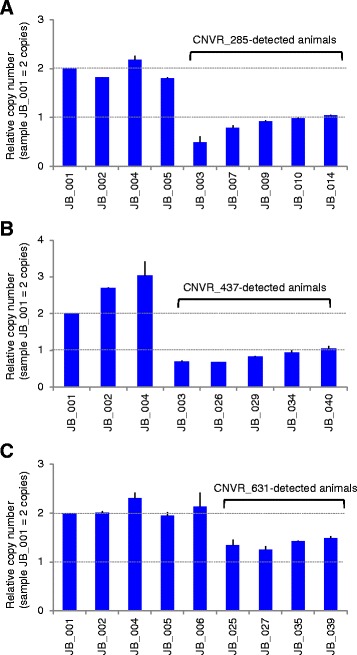
Quantitative PCR (qPCR) validation of CNVR_285 (a), CNVR_437 (b), and CNVR_631 (c). The left-most bar in each panel represents a calibrator animal (JB_001). The calibrator animal is assumed to contain two copies of the DNA segment detected from the PennCNV analysis. The Basic transcription factor 3 gene (BTF3), which served as an internal qPCR standard for both copies at a locus (2n), was co-amplified with the primers. The x-axis represents the animals. The brackets represent CNVR-detected animals using the Illumina BovineHD BeadChip Array. Error bars represent ± Standard Error of Mean (SEM) obtained from triplicate experiments
To further test the CNV calls derived from the PennCNV analysis with Illumina BovineHD BeadChip Array, we examined a region of Claudin 16 gene (CLDN16) on chromosome 1. Hirano et al. reported that the first four exons of CLDN16 were deleted in Japanese Black cattle with autosomal recessive nephritis [23]. We found that CNVR_27 overlapped with CLDN16 (Fig. 4a, b, Additional file 1: Tables S2, S5). The mean Log R ratio of 22 SNPs, which were consecutively located within a 36,382 bp window between BovineHD0100022316 (77,469,795 bp) and BovineHD0100022336 (77,506,177 bp) on chromosome 1, was decreased in 116 animals (Fig. 4a). To further confirm whether animals with CNVR_27 have the CLDN16 deletion, we performed PCR with flanking primers designed to amplify the CLDN16 deletion region in CNVR_27-detected animals. A CLDN16-deletion allele was detected in CNVR_27-detected animals, whereas this deletion was not detected in non-CNVR_27-detected animals (Fig. 4c). These results also indicated that the PennCNV analysis with Illumina BovineHD BeadChip Array inferred reliable CNVs in this study.
Fig. 4.
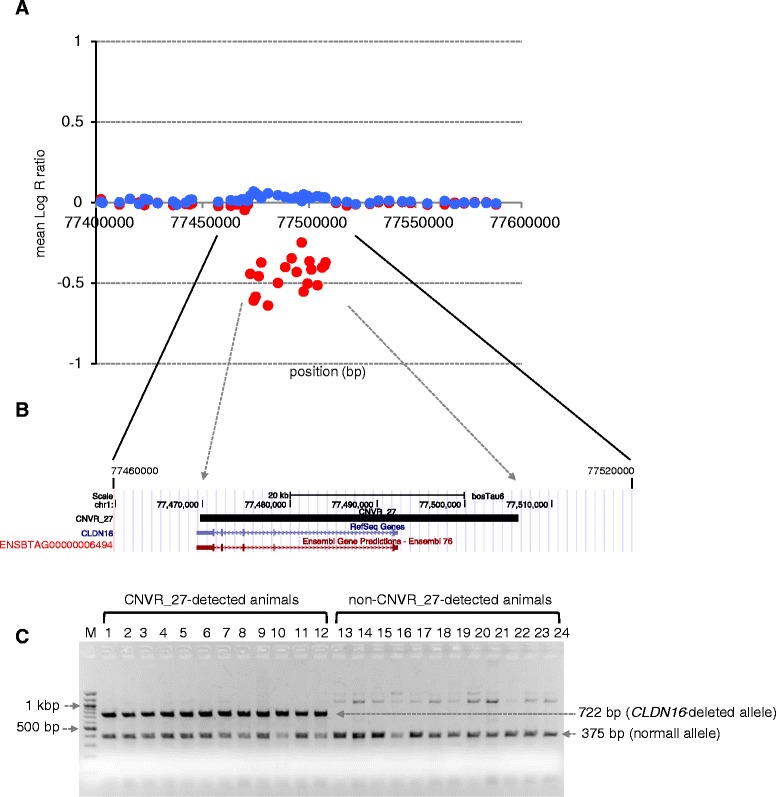
CNVR_27 overlapping with CLDN16 gene region on chromosome 1. a Regional SNP plot of CNVR_27. The mean log R ratio of CNVR_27 animals (red) and the mean log R ratio of non-CNVR_27 animals (blue). The mean log R ratio was calculated from 116 animals. SNP positions were based on the UMD3.1 assembly of the bovine genome. b CNVR_27 was visualized using the UCSC Genome Browser [62]. The RefSeq gene symbol and Ensembl IDs of CLDN16 were labeled. c Detection of CLDN16-deletion allele in CNVR_27-detected animals using PCR with flanking primers for the deleted region. CNVR_27-detected animals (lane 1 to 12) and non-CNVR_27-detected animals (lane 13 to 24), which were inferred using PennCNV analysis with the Illumina BovineHD BeadChip Array. The PCR product size of the CLDN16-deletion allele is 722 bp and that of the wild type allele is 375 bp. M, 100 bp ladder marker
Gene content of CNVRs
Of 861 CNVRs, 390 were within or overlapped with 717 Ensembl genes, including 651 protein-coding genes, 17 small nuclear RNAs, 5 small nucleolar RNAs, 1 ribosomal RNA, 15 microRNAs, 23 unprocessed pseudogenes, 3 processed pseudogenes (a processed pseudogene is a pseudogene that lacks introns and is thought to arise from reverse transcription of mRNA followed by reinsertion of DNA into the genome [47]), and two miscellaneous RNAs (Additional file 1: Tables S2 and S5). Out of 390 CNVRs, 334 contained exons, and the remaining 56 were intronic CNVRs (Additional file 1: Table S2). Of 334 CNVRs, 125 contained exons derived from multiple protein-coding genes, ranging from 2 to 21 Ensembl genes, suggesting that these CNVRs influence the function of multiple genes.
Although the 717 Ensembl-annotated genes had a wide spectrum of molecular functions (Additional file 1: Table S5), gene ontology (GO) analysis using the PANTHER classification system [48, 49] showed that the most over-represented biological processes were stimulus response, cellular defense response, and immune response (Table 2 and Additional file 1: Table S6). Of these, 63 olfactory receptors function as smell sensors (Additional file 1: Table S6), and have been frequently reported CNVs in cattle (for review, see [16]). In addition, the immune-related gene enrichment within CNVs was in agreement with previously reported bovine CNV studies (for review, see [16]). In particular, 6 CNVRs were clustered within a 3.17 Mb window from 25,325,230 bp to 28,494,393 bp on BTA23 (Fig. 1), including the major histocompatibility complex (MHC) gene family members (Additional file 1: Table S6). In addition, 12 CNVRs were clustered within a 2.97 Mb window from 22,208,187 bp to 25,173,443 bp on BTA10 (Fig. 1), including T-cell receptors (Additional file 1: Table S6), which are generated by gene rearrangement in somatic cells.
Phenotypic information of mouse orthologous genes in CNVRs
The use of mouse models has proven to be one of the most powerful approaches to understand in vivo gene functions [47, 50]. The Mammalian Phenotype (MP) Ontology in Mouse Genome Informatics (MGI) is the most comprehensive phenotypic database that enables the annotation of phenotypes in a genetic context [51–53]. Thus, in addition to GO term analysis, to associate bovine genes contained in CNVRs with phenotypes, we converted 560 bovine Ensembl gene IDs to their 438 orthologous associated mouse gene IDs (MGI IDs) (Additional file 1: Table S7) using BioMart in both Ensembl and MGI [53, 54]. Several different bovine Ensembl gene IDs were connected with a single MGI ID, such as T cell receptor family and olfactory receptor family (Additional file 1: Table S7), and several bovine Ensembl gene IDs did not connect with a MGI ID (Additional file 1: Table S7); therefore, the number of converted mouse orthologous genes was reduced. Out of 438 MGI IDs, 195 were assigned to 1,627 phenotypic categories in MP IDs (Additional file 1: Table S8). This list will provide useful information for understanding phenotypic implications of CNV events in vivo. In particular, knockout mice for bovine orthologous genes in loss-type CNVRs may provide important information on phenotypic expression of CNV events in cattle. For instance, CNVR_27 is a loss-type CNVR exons of CLDN16 (Fig. 4, Additional file 1: Tables S2 and S5), and the mouse orthologous gene (MGI:2148742) was assigned to 9 phenotypic categories in MP IDs (Additional file 1: Table S8), such as abnormal renal reabsorbtion and abnormal renal calcium reabsorbtion. These symptoms are consistent with those of null deletions of CLDN16 in cattle [55, 56].
Conclusions
In this study, we identified 861 CNVRs in 1,481 Japanese Black cattle using the Illumina BovineHD BeadChip Array. Of these, 72 % of CNVR calls were within or overlapped with previously reported cattle CNVs. Experimentally, three CNVRs were validated using quantitative PCR, and one CNVR was validated using PCR with flanking primers specific to the deleted region. These results suggest that the current analysis inferred reliable CNV calls. Out of 861 CNVRs, 390 contained 717 Ensembl-annotated genes, which are significantly enriched for stimulus response, cellular defense response, and immune response in the Gene Ontology (GO). In addition to the GO analysis, we characterized the mouse orthologous genes using the MGI database to associate bovine genes contained in CNVRs with phenotypes. This list will provide useful information for understanding their implication in the CNV events in vivo. The comprehensive genome-wide CNVRs map generated by this study will facilitate the identification of genotypic variability and disease-susceptibility allelles in Japanese Black cattle.
Methods
Ethics statement
All animal experiments were performed according to the Guidelines for Care and Use of Laboratory Animals of Shirakawa Institute of Animal Genetics, and this research project was approved by the Shirakawa Institute of Animal Genetics Committee on Animal Research (H21-2). We have obtained the written agreement from the cattle owners to use samples and data.
Sample collection and genotyping
Genomic DNA from Japanese black cattle was isolated from blood samples of 787 cows, adipose tissue samples of 591 steers, and semen samples of 103 bulls.
Genomic DNA of all samples was genotyped using the Illumina BovineHD BeadChip Array (Illumina, cat#WG-450-1002), which contains 735,293 autosomal SNPs [30] (for SNP intervals, see Additional file 3), according to the manufacturer’s instructions. SNP clustering and genotype calling were performed using GenomeStudio version 2011 (Illumina, version 1.9.4), and all markers passed quality control (call rate > 98 %). The UMD3.1 assembly was used to map SNP positions [57].
Identification of CNVs
The ratio of observed normalized intensity of the experimental sample to the expected intensity of each locus (log R ratio: LRR) and the allelic intensity ratio (B allele frequency: BAF) of samples were reported using GenomeStudio. The population frequency of B allele (PFB) was generated based on the BAF of each SNP in the population. To identify CNVs, we used PennCNV software (version June 2011) [39, 40], which incorporates factors including LRR, BAF, marker distance, and PFB into a hidden Markov model.
In this study, only autosomes were used for the detection of CNVs. Genomic waves were adjusted for the GC content of the 1 Mb genomic region (500 kb each side) surrounding each SNP. Samples with a standard deviation of logR ratio > 0.3, BAF drift > 0.01, and wave factor > 0.05 were excluded from the analysis. We also removed CNV calls with a confidence score < 10, calls in the 15-kb centromeric and telomeric regions (Additional file 1: Table S9) [58], and calls in the immunogloblin region [59] (Additional file 1: Table S10). T-cell receptor family members were not excluded from the analysis due to mapping uncertainty. In this study, a potential CNV was determined if it contained three or more consecutive SNPs [3, 5, 6, 9, 34]. The union region of overlapping CNVs detected in at least two animals was defined as a CNV region (CNVR) [41].
Comparison of current results with other cattle CNV studies
We compared CNVRs detected in this study with results from three other CNV studies that used the Illumina BovineHD BeadChip Array [5, 8, 9]. We also compared CNVRs with structural variants deposited in the DGVa [57] and dbVar [43] databases.
Quantitative PCR validation of CNVR
Real-time qPCR was performed for CNVR validation using the 7900HT Real-Time PCR system (Applied Biosystems). Primers and probes were designed for three CNVRs (Additional file 1: Table S11). Amplification reactions (20 μl⋅well-1) were carried out in triplicate with 20 ng of genomic DNA, 1× Absolute QPCR ROX Mix (Thermo Scientific, cat#AB-1138/B), 400 nM of each primer, and 200 nM of each probe. The BTF3, which served as an internal qPCR standard for both copies at a locus (2n) [2], was co-amplified with the primers (Additional file 1: Table S12). Three replicate reactions were performed for each primer pair, and a comparative CT method was used to calculate the copy number [2]. ∆ CT was calculated by subtracting the BTF3 CT value from the sample CT value for each replicate. The average ∆ CT value for the three replicates was calculated. To determine the ∆∆ CT, the average ∆ CT of a calibrator animal, which had two copies of the DNA segment, was used. Finally, the copy number was given using the formula 2 × 2 -∆∆ CT.
PCR validation of Claudin 16 (CLDN16) deletion
PCR was performed as described by Hirano et al. [23]. The following forward and reverse primer pairs were used: DN-F (5′-TATGCTGTTGATGTTTATGTAG-3′)/DN-R (5′-CCCCCCCCCGCCTTTTTC-3′) to detect the wild type allele, and DA-F (5′-ATTGTATTTTTAGGAGTGACTC-3′)/DA-R (5′-CCCCCCCCCACTCTATAC-3′) to detect the CLDN16 deletion allele.
Gene annotation and Gene Ontology (GO) analysis
Gene content of CNVRs was assessed based on the gene annotation of the UMD3.1 genome assembly using Ensembl (Cow release 77) [54]. The PANTHER classification system (PANTHER 9.0) [48, 49] was used to assess the probability of overrepresented genes in CNVRs within biological process, cellular composition, and molecular function using Bonferroni correction for multiple comparisons.
Phenotype annotation
To obtain phenotypic information of genes in the CNVRs, the mouse orthologs of bovine genes in the CNVRs were obtained from the Mouse Genome Information (MGI) resource [53]. Before the analysis, bovine Ensembl gene IDs were converted to their mouse orthologous Ensembl gene IDs or MGI IDs using BioMart in both Ensembl and MGI [53, 54]. Phenotypic annotations in MGI were obtained from MGI_PhenoGenoMP.rpt.txt [60] and VOC_MammalianPhenotype.rpt.txt [61].
Availability of supporting data
The datasets supporting the results of this article are included within the article and its additional files.
Acknowledgements
We would like to thank Emiko Watanabe, Takatoshi Kojima, and other lab members for generous support and valuable suggestions. This work was supported by the Japan Racing and Livestock Promotion (K. S.), the Programme for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (S. S., K. S.), the Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries, and Food Industry (K. S.), JSPS KAKENHI Grant Number 26450384 (S. S.).
Abbreviations
- CNV
copy number variation
- CNVR
CNV region
- PCR
polymerase chain reaction
- SNP
single nucleotide polymorphism
Additional files
Characterization of cattle autosomal CNVRs based on previously published studies. Table S2 – Detailed features of 861 autosomal CNVRs identified in this study. Table S3 – Comparison of CNVRs detected in this study with the results from three other CNV studies using the Illumina BovineHD BeadChip Array. Table S4 – Comparison of CNVRs detected in this study with structural variants deposited in the DGVa. Table S5 – Detailed features of Ensembl genes in CNVRs. Table S6 – PANTHER pathway analysis of genes in CNVRs in this study. Table S7 – The corresponding MGI gene to Bovine Ensembl gene in CNVRs. Table S8 – Detailed phenotypic description of mouse orthologous genes for Bovine Ensembl genes in CNVRs. Table S9 – Detailed features of excluded centromeric and telemeric regions in this study. Table S10 – Detailed features of excluded immunoglobulin regions in this study. Table S11 – Primers and probes for CNVR validation. Table S12 – Primers and probes for the basic transcriptional factor 3. (XLSX 686 kb)
Detection number and mean size of CNVRs in 3, 5, 10, and 15 consecutive SNP windows. (PPTX 53 kb)
Autosomal SNPs intervals of Illumina BovineHD BeadChip Array. (PPTX 48 kb)
Footnotes
Competing interests
The authors declare no conflicts of interest.
Authors’ contributions
SS designed the research; SS, SN, and YS performed SNP genotyping; SS and TW performed the CNV analysis; SS performed CNV validation analyses; and SS and YS wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Shinji Sasaki, Phone: +81-248-25-5641, Email: sasakis@siag.or.jp.
Toshio Watanabe, Email: t4watanb@nlbc.go.jp.
Shota Nishimura, Email: nshota0651@yahoo.co.jp.
Yoshikazu Sugimoto, Email: kazusugi@siag.or.jp.
References
- 1.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet. 2006;7(2):85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
- 2.Bae JS, Cheong HS, Kim LH, NamGung S, Park TJ, Chun JY, et al. Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genomics. 2010;11:232. doi: 10.1186/1471-2164-11-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou Y, Liu GE, Bickhart DM, Cardone MF, Wang K, Kim ES, et al. Genomic characteristics of cattle copy number variations. BMC Genomics. 2011;12:127. doi: 10.1186/1471-2164-12-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hou Y, Liu GE, Bickhart DM, Matukumalli LK, Li C, Song J, et al. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct Integr Genomics. 2011;12(1):81–92. doi: 10.1007/s10142-011-0252-1. [DOI] [PubMed] [Google Scholar]
- 5.Hou Y, Bickhart DM, Hvinden ML, Li C, Song J, Boichard DA, et al. Fine mapping of copy number variations on two cattle genome assemblies using high density SNP array. BMC Genomics. 2012;13:376. doi: 10.1186/1471-2164-13-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang L, Jiang J, Wang J, Ding X, Liu J, Zhang Q. Genome-wide identification of copy number variations in Chinese Holstein. PLoS One. 2012;7(11):e48732. doi: 10.1371/journal.pone.0048732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cicconardi F, Chillemi G, Tramontano A, Marchitelli C, Valentini A, Ajmone-Marsan P, et al. Massive screening of copy number population-scale variation in Bos taurus genome. BMC Genomics. 2013;14:124. doi: 10.1186/1471-2164-14-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang L, Jiang J, Yang J, Liu X, Wang J, Wang H, et al. Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genomics. 2013;14:131. doi: 10.1186/1471-2164-14-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Q, Ma Y, Wang X, Zhang Y, Zhao X. Identification of copy number variations in Qinchuan cattle using BovineHD Genotyping Beadchip array. Mol Genet Genomics. 2015;290(1):319–27. doi: 10.1007/s00438-014-0923-4. [DOI] [PubMed] [Google Scholar]
- 10.Fadista J, Thomsen B, Holm LE, Bendixen C. Copy number variation in the bovine genome. BMC Genomics. 2010;11:284. doi: 10.1186/1471-2164-11-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu GE, Hou Y, Zhu B, Cardone MF, Jiang L, Cellamare A, et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010;20(5):693–703. doi: 10.1101/gr.105403.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhan B, Fadista J, Thomsen B, Hedegaard J, Panitz F, Bendixen C. Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genomics. 2011;12:557. doi: 10.1186/1471-2164-12-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X, et al. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics. 2011;12:559. doi: 10.1186/1471-2164-12-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bickhart DM, Hou Y, Schroeder SG, Alkan C, Cardone MF, Matukumalli LK, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012;22(4):778–90. doi: 10.1101/gr.133967.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi JW, Lee KT, Liao X, Stothard P, An HS, Ahn S, et al. Genome-wide copy number variation in Hanwoo, Black Angus, and Holstein cattle. Mamm Genome. 2013;24(3-4):151–63. doi: 10.1007/s00335-013-9449-z. [DOI] [PubMed] [Google Scholar]
- 16.Bickhart DM, Liu GE. The challenges and importance of structural variation detection in livestock. Front Genet. 2014;5:37. doi: 10.3389/fgene.2014.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315(5813):848–53. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guryev V, Saar K, Adamovic T, Verheul M, van Heesch SA, Cook S, et al. Distribution and functional impact of DNA copy number variation in the rat. Nat Genet. 2008;40(5):538–45. doi: 10.1038/ng.141. [DOI] [PubMed] [Google Scholar]
- 19.Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, et al. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 2009;41(4):424–9. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- 20.Durkin K, Coppieters W, Drogemuller C, Ahariz N, Cambisano N, Druet T, et al. Serial translocation by means of circular intermediates underlies colour sidedness in cattle. Nature. 2012;482(7383):81–4. doi: 10.1038/nature10757. [DOI] [PubMed] [Google Scholar]
- 21.Kadri NK, Sahana G, Charlier C, Iso-Touru T, Guldbrandtsen B, Karim L, et al. A 660-kb deletion with antagonistic effects on fertility and milk production segregates at high frequency in nordic red cattle: additional evidence for the common occurrence of balancing selection in livestock. PLoS Genet. 2014;10(1):e1004049. doi: 10.1371/journal.pgen.1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu L, Cole JB, Bickhart DM, Hou Y, Song J, VanRaden PM, et al. Genome wide CNV analysis reveals additional variants associated with milk production traits in Holsteins. BMC Genomics. 2014;15:683. doi: 10.1186/1471-2164-15-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirano T, Kobayashi N, Itoh T, Takasuga A, Nakamaru T, Hirotsune S, et al. Null mutation of PCLN-1/Claudin-16 results in bovine chronic interstitial nephritis. Genome Res. 2000;10(5):659–63. doi: 10.1101/gr.10.5.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Drogemuller C, Distl O, Leeb T. Partial deletion of the bovine ED1 gene causes anhidrotic ectodermal dysplasia in cattle. Genome Res. 2001;11(10):1699–705. doi: 10.1101/gr.182501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sugimoto M, Furuoka H, Sugimoto Y. Deletion of one of the duplicated Hsp70 genes causes hereditary myopathy of diaphragmatic muscles in Holstein-Friesian cattle. Anim Genet. 2003;34(3):191–7. doi: 10.1046/j.1365-2052.2003.00990.x. [DOI] [PubMed] [Google Scholar]
- 26.Meyers SN, McDaneld TG, Swist SL, Marron BM, Steffen DJ, O’Toole D, et al. A deletion mutation in bovine SLC4A2 is associated with osteopetrosis in Red Angus cattle. BMC Genomics. 2010;11:337. doi: 10.1186/1471-2164-11-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clop A, Vidal O, Amills M. Copy number variation in the genomes of domestic animals. Anim Genet. 2012;43(5):503–17. doi: 10.1111/j.1365-2052.2012.02317.x. [DOI] [PubMed] [Google Scholar]
- 28.Goddard ME, Hayes BJ. Genome-wide association studies and linkage disequilibrium in cattle. In: Womack JE, editor. Bovine genomics. Oxford, UK: Wiley-Blackwell; 2012. pp. 192–210. [Google Scholar]
- 29.Taylor JF, McKay SD, Rolf MM, Ramey HR, Decker JE, Schnabel RD. Genomic selection in beef cattle. In: Womack JE, editor. Bovine genomics. Oxford, UK: Wiley-Blackwell; 2012. pp. 211–33. [Google Scholar]
- 30.Rincon G, Weber KL, Eenennaam AL, Golden BL, Medrano JF. Hot topic: performance of bovine high-density genotyping platforms in Holsteins and Jerseys. J Dairy Sci. 2011;94(12):6116–21. doi: 10.3168/jds.2011-4764. [DOI] [PubMed] [Google Scholar]
- 31.Cameron PJ, Zembayashi M, Lunt DK, Mitsuhashi T, Mitsumoto M, Ozawa S, et al. Relationship between Japanese beef marbling standard and intramuscular lipid in the M. longissimus thoracis of Japanese Black and American Wagyu Cattle. Meat Sci. 1994;38(2):361–4. doi: 10.1016/0309-1740(94)90125-2. [DOI] [PubMed] [Google Scholar]
- 32.Namikawa K. Japanese Beef Cattle - Historical Breeding Processes of Japanese Beef Cattle and Preservation of Genetic Resources as Economic Farm Animal (in Japanese) Kyoto: Wagyu Registry Association. Wagyu 1992.
- 33.McKay SD, Schnabel RD, Murdoch BM, Matukumalli LK, Aerts J, Coppieters W, et al. An assessment of population structure in eight breeds of cattle using a whole genome SNP panel. BMC Genet. 2008;9:37. doi: 10.1186/1471-2156-9-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu L, Hou Y, Bickhart DM, Song J, Liu GE. Comparative analysis of CNV calling algorithms: Literature survey and a case study using bovine high-density SNP data. microarrays. 2013;2:171–85. doi: 10.3390/microarrays2030171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Wang H, Jiang J, Kang H, Feng X, Zhang Q, et al. Identification of genome-wide copy number variations among diverse pig breeds using SNP genotyping arrays. PLoS One. 2013;8(7):e68683. doi: 10.1371/journal.pone.0068683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Du ZQ, Dong JQ, Wang HX, Shi HY, Wang N, et al. Detection of genome-wide copy number variations in two chicken lines divergently selected for abdominal fat content. BMC Genomics. 2014;15:517. doi: 10.1186/1471-2164-15-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J, Zhang L, Xu L, Ren H, Lu J, Zhang X, et al. Analysis of copy number variations in the sheep genome using 50 K SNP BeadChip array. BMC Genomics. 2013;14:229. doi: 10.1186/1471-2164-14-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Molin AM, Berglund J, Webster MT, Lindblad-Toh K. Genome-wide copy number variant discovery in dogs using the CanineHD genotyping array. BMC Genomics. 2014;15:210. doi: 10.1186/1471-2164-15-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17(11):1665–74. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.PennCNV. [http://www.openbioinformatics.org/penncnv/].
- 41.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, et al. Global variation in copy number in the human genome. Nature. 2006;444(7118):444–54. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Genomic Variation Archive (DGVa). [ftp://ftp.Ensembl.org/pub/release-76/variation/gvf/bos_taurus/].
- 43.dbVar. [ftp://ftp.ncbi.nlm.nih.gov/pub/dbVar/data//Bos_taurus/by_assembly/Bos_taurus_UMD_3.1/gvf/Bos_taurus_UMD_3.1.remap.all.germline.gvf].
- 44.Alvarez CE, Akey JM. Copy number variation in the domestic dog. Mamm Genome. 2011;23(1-2):144–63. doi: 10.1007/s00335-011-9369-8. [DOI] [PubMed] [Google Scholar]
- 45.Nicholas TJ, Baker C, Eichler EE, Akey JM. A high-resolution integrated map of copy number polymorphisms within and between breeds of the modern domesticated dog. BMC Genomics. 2011;12:414. doi: 10.1186/1471-2164-12-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh S, Qu Z, Das PJ, Fang E, Juras R, Cothran EG, et al. Copy number variation in the horse genome. PLoS Genet. 2014;10(10):e1004712. doi: 10.1371/journal.pgen.1004712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schofield PN, Gkoutos GV, Gruenberger M, Sundberg JP, Hancock JM. Phenotype ontologies for mouse and man: bridging the semantic gap. Dis Model Mech. 2010;3(5-6):281–9. doi: 10.1242/dmm.002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8(8):1551–66. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.PANTHER classification system. [http://www.pantherdb.org/].
- 50.Rosenthal N, Brown S. The mouse ascending: perspectives for human-disease models. Nat Cell Biol. 2007;9(9):993–9. doi: 10.1038/ncb437. [DOI] [PubMed] [Google Scholar]
- 51.Smith CL, Goldsmith CA, Eppig JT. The mammalian phenotype ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005;6(1):R7. doi: 10.1186/gb-2004-6-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blake JA, Bult CJ, Kadin JA, Richardson JE, Eppig JT. The Mouse Genome Database (MGD): premier model organism resource for mammalian genomics and genetics. Nucleic Acids Res. 2010;39(Database issue):D842–8. doi: 10.1093/nar/gkq1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mouse Genome Informations (MGI) resources. [(http://www.informatics.jax.org/)].
- 54.Ensembl genome browser 77: Bos taurus. [ftp://ftp.Ensembl.org/pub/release-76/mysql/].
- 55.Ohba Y, Kitoh K, Nakamura H, Okuda H, Kunieda T, Sasaki Y, et al. Renal reabsorption of magnesium and calcium by cattle with renal tubular dysplasia. Vet Rec. 2002;151(13):384–7. doi: 10.1136/vr.151.13.384. [DOI] [PubMed] [Google Scholar]
- 56.Okada K, Ishikawa N, Fujimori K, Goryo M, Ikeda M, Sasaki J, et al. Abnormal development of nephrons in claudin-16-defective Japanese black cattle. J Vet Med Sci. 2005;67(2):171–8. doi: 10.1292/jvms.67.171. [DOI] [PubMed] [Google Scholar]
- 57.Center for Bioinformatics and Computational Biology at University of Maryland. [ftp.ccb.jhu.edu/pub/data/assembly/Bos_taurus/Bos_taurus_UMD_3.1/].
- 58.Miyashita N, Shiga K, Yonai M, Kaneyama K, Kobayashi S, Kojima T, et al. Remarkable differences in telomere lengths among cloned cattle derived from different cell types. Biol Reprod. 2002;66(6):1649–55. doi: 10.1095/biolreprod66.6.1649. [DOI] [PubMed] [Google Scholar]
- 59.Ekman A, Niku M, Liljavirta J, Iivanainen A. Bos taurus genome sequence reveals the assortment of immunoglobulin and surrogate light chain genes in domestic cattle. BMC Immunol. 2009;10:22. doi: 10.1186/1471-2172-10-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.MGI_PhenoGenoMP.rpt.txt. [ftp://ftp.informatics.jax.org/pub/reports/index.html].
- 61.VOC_MammalianPhenotype.rpt.txt. [ftp://ftp.informatics.jax.org/pub/reports/index.html].
- 62.UCSC Genome Browser. [http://genome.ucsc.edu/].