

Abstract
Background and Purpose
Here, we have characterized 3‐cyclopropyl‐1‐(4‐(6‐((1,1‐dioxidothiomorpholino)methyl)‐5‐fluoropyridin‐2‐yl)benzyl)imidazolidine‐2,4‐dione hydrochloride (LEI‐101) as a novel, peripherally restricted cannabinoid CB2 receptor agonist, using both in vitro and in vivo models.
Experimental Approach
We investigated the effects of LEI‐101 on binding and functional activity. We assessed its in vitro and in vivo selectivity. Efficacy of LEI‐101 was determined in a mouse model of cisplatin‐induced nephrotoxicity.
Key Results
LEI‐101 behaved as a partial agonist at CB2 receptors using β‐arrestin and GTPγS assays and was ~100‐fold selective in CB2 /CB1 receptor‐binding assays. It did not display any activity on endocannabinoid hydrolases and nor did it react with serine hydrolases in an activity‐based protein profiling assay. In mice, LEI‐101 had excellent oral bioavailability reaching high concentrations in the kidney and liver with minimal penetration into the brain. LEI‐101 up to a dose of 60 mg·kg−1 (p.o.) did not exert any CNS‐mediated effects in the tetrad assay, in mice. LEI‐101 (p.o. or i.p.) at 3 or 10 mg·kg−1 dose‐dependently prevented kidney dysfunction and/or morphological damage induced by cisplatin in mice. These protective effects were associated with improved renal histopathology, attenuated oxidative stress and inflammation in the kidney. These effects were absent in CB2 receptor knockout mice.
Conclusion and Implications
These results indicate that LEI‐101 is a selective, largely peripherally restricted, orally available CB2 receptor agonist with therapeutic potential in diseases that are associated with inflammation and/or oxidative stress, including kidney disease.
Abbreviations
- 4‐HNE
4‐hydroxy‐2‐nonenal
- Δ9‐THC
Δ9‐tetrahydrocannabinol
- ABPP
activity‐based protein profiling
- BUN
blood urea nitrogen
- DAGL
diacylglycerol lipase
- hFAAH
human fatty acid amide hydrolase
- LEI‐101
3‐cyclopropyl‐1‐(4‐(6‐((1,1‐dioxidothiomorpholino)methyl)‐5‐fluoropyridin‐2‐yl)benzyl)imidazolidine‐2,4‐dione hydrochloride
- MAGL
monoacylglycerol lipase
- NAPE‐PLD
N‐acylphosphatidylethanolamine‐specific phospholipase D
- Vss
volume of distribution in steady state
Tables of Links
Introduction
In recent years, drug discovery efforts have focused on the design and synthesis of selective cannabinoid CB2 receptor agonists (Han et al., 2013; Dhopeshwarkar and Mackie, 2014). Selective activation of this receptor has been associated with anti‐inflammatory and tissue protective effects without the CNS‐mediated side effects, which are associated with stimulation of psychoactive CB1 receptors in the CNS (Hanus et al., 1999; Malan et al., 2001).
Although numerous putative CB2 receptor agonists from different chemical series have been characterized in vitro, only a few have been used in inflammatory (Hanus et al., 1999) and neuropathic pain models (van der Stelt et al., 2011) as well as in experimental paradigms of ischemia‐reperfusion injury (Batkai et al., 2007; Pacher et al., 2007), neurodegeneration (Zhang et al., 2009) and gastrointestinal disorders (Storr et al., 2009) and other diseases (Teixeira‐Clerc et al., 2010; Louvet et al., 2011).
Furthermore, very limited information is available on in vivo selectivity and oral availability of these compounds (Pacher and Mechoulam, 2011; Pacher and Kunos, 2013), and limited selectivity for CB2 over CB1 receptors and/or bioavailability may have contributed to the failure of clinical trials with peripherally restricted mixed CB1 /CB2 receptor agonists for pain and to recent controversies related to CB2 receptor function in the brain (Pacher and Kunos, 2013).
Recently, we reported the identification of 3‐cyclopropyl‐1‐(4‐(6‐((1,1‐dioxidothiomorpholino)methyl)‐5‐fluoropyridin‐2‐yl)benzyl)imidazolidine‐2,4‐dione hydrochloride (LEI‐101) as a novel, peripherally restricted CB2 receptor agonist (van der Stelt et al., 2011). In the present study, we have further characterized the activity and selectivity of LEI‐101 in both in vitro and in vivo models, using various pharmacological tools and CB2 receptor knockout mice. LEI‐101 is ~100‐fold more potent in binding to CB2 receptors than to CB1 receptors. It was a partial agonist in the β‐arrestin recruitment and GTPγS assays and did not display any activity on the other proteins of the endocannabinoid system. LEI‐101 up to a dose of 60 mg·kg−1 (p.o.) did not exert any CNS‐mediated side effects in the mouse tetrad assay of cannabimimetic activity.
In addition, LEI‐101 dose‐dependently ameliorated kidney dysfunction and morphological damage in the cisplatin‐induced nephrotoxicity model. At 3 or 10 mg·kg−1 (p.o.), LEI‐101 largely prevented cisplatin‐induced increases in serum creatinine and blood urea nitrogen levels, improved renal histopathological injury and attenuated oxidative/nitrative stress and inflammation in the kidney. These protective effects were absent in CB2 receptor knockout mice, which is indicative of a CB2 receptor‐mediated effect. Taken together, our findings give strength to the potential of LEI‐101 for the prevention or treatment of oxidative stress and inflammation‐induced tissue damage and establish LEI‐101 as an excellent tool to study CB2 receptor biology.
Methods
Cell culture
We used the CNR1 PathHunter® CHO‐K1 CNR1 β‐Arrestin Cell Line (catalogue number 93‐0959C2) and the CNR2 PathHunter® CHO‐K1 CNR2 β‐Arrestin Cell Line (catalogue number 93‐0706C2) stably expressing human CB1 receptors (CHOK1hCB1R_bgal) or human CB2 receptors (CHOK1hCB2R_bgal) respectively, obtained from DiscoveRx Corporation (Fremont, CA, USA). CHOK1hCB1R_bgal and CHOK1hCB2R_bgal cells were cultured in Ham's F12 Nutrient Mixture supplemented with 10% fetal calf serum, 1 mM glutamine, 50 μg·mL−1 penicillin, 50 μg·mL−1 streptomycin, 300 mg·mL−1 hygromycin and 800 μg·mL−1 geneticin in a humidified atmosphere at 37°C and 5% CO2. Cells were subcultured twice a week at a ratio of 1:20 on 10cm diameter plates by trypsinization. For membrane preparation, the cells were subcultured 1:10 and transferred to large 15cm diameter plates. Membrane fractions were prepared exactly as described before (Zweemer et al., 2013).
[3H]CP55940 displacement assays
[3H]CP55940 displacement assays to determine CB2 receptor and CB1 receptor binding affinity were performed as reported previously (Bonger et al., 2007), with the following changes:
Membrane aliquots containing 5 μg (CHOK1hCB1R_bgal) or 1 μg (CHOK1hCB2R_bgal) of membrane protein in 100 μL assay buffer (50 mM Tris–HCl, 5 mM Mgl2, 0.1% BSA, pH 7.4) were incubated at 30°C for 1 h. Non‐specific binding was determined in the presence of 10 μM AM630. Filtration was performed on GF/C filters, presoaked for 30 min with 0.25% PolyEthyleneImine (PEI), using a Brandel harvester. Filter‐bound radioactivity was determined in a β‐counter.
PathHunter® β‐arrestin recruitment assay
The assay was performed using the PathHunter hCB1R_bgal CHOK1 or hCB2R_bgal CHOK1 β‐arrestin recruitment assay kit (DiscoveRx Corporation, Fremont, CA, USA), according to the manufacturer's protocol.
[35S]GTPγS binding assay
The assay was performed as reported previously (Zweemer et al., 2013), with the following changes: 5 μg of homogenized CHOK1hCB2R_bgal membranes, pretreated with 5 μg saponin and 1 μM GDP, were used in 100 μL assay buffer per assay point (50 mM Tris–HCl buffer (pH 7.4), 5 mM MgCl2, 150 mM NaCl, 1 mM EDTA, 0.05% BSA and 1 mM DTT, freshly prepared every day). The reference agonist CP55940 (10 μM) was added to each filter for 100% stimulation.
Off‐target profiling
The enzymatic activity of anandamide and 2‐arachidonoylglycerol metabolic enzymes was assayed in human THP‐1 macrophages and was expressed as pmol of product released per min per mg of protein. Fatty acid amide hydrolase (FAAH) activity was assayed in cell extracts (100 μg of protein/test) at 37°C for 30 min by measuring the release of ethanolamine‐1‐3H from 10 μM [ethanolamine‐1‐3H]‐anandamide (Gattinoni et al., 2010). The synthesis of anandamide through the activity of N‐acylphosphatidylethanolamine‐specific phospholipase D (NAPE‐PLD) was assayed in cell homogenates (100 μg protein/test) at 37°C for 30 min by measuring the release of [3H]‐anandamide from 100 μM [3H]N‐arachidonoyl‐phosphatidylethanolamine (Tsuboi et al., 2011). The activity of diacylglycerol lipase (DAGL) was assayed in cell extracts (200 μg protein/test) with 100 μM of sn‐1‐stearoyl‐2‐[14C]arachidonoyl‐glycerol at 37°C for 30 min (Bisogno et al., 2003). Monoacylglycerol lipase (MAGL) activity was assayed (100 μg protein/test) with 10 μM [3H]2‐oleoylglycerol at 37°C for 30 min (Dinh et al., 2002).
Activity based protein profiling (ABPP) selectivity assay was performed as described previously (Baggelaar et al., 2013). Briefly, mouse brain proteome (2.0 mg·mL−1, 20 μL) was preincubated for 30 min with vehicle (0.5 μL DMSO) or ligand in 0.5 μL DMSO at room temperature and subsequently treated with 250 nM (final concentration) MB064 1 or 500 nM (final concentration) TAMRA‐FP (from Thermo Fischer Scientific) for 15 min at room temperature . The reactions were quenched with 10 μL standard 3 × SDS page sample buffer. The samples were directly loaded and resolved on SDS page gel (10% acrylamide).
Animals
All animal care and experimental procedures conformed to National Institutes of Health (NIH) guidelines and were approved by the Institutional Animal Care and Use Committee of the National Institute on Alcohol Abuse and Alcoholism (Bethesda, MD, USA). Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 206 animals were used in the experiments described here.
Six‐ to 8‐week‐old male C57BL/6 J mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). CB2 receptor knockout mice (CB2R−/−) and their wild‐type littermates (CB2R+/+) were developed as described previously and had been backcrossed to a C57BL/6 J background. (Batkai et al., 2007). All animals were kept in a temperature‐controlled environment with a 12 h light–dark cycle and were allowed free access to food and water at all times.
Pharmacokinetics of LEI‐101in mice
LEI‐101 was given i.p. or by oral gavage (p.o.). The drug was dissolved in a vehicle of DMSO: Tween 80: saline, 1:1:18. After administration of LE‐101, mice were killed by cervical dislocation under deep anaesthesia with 5% isoflurane, for collection of blood and tissue samples at the time described in the figure. Both tissues and plasma from the blood were stored at −70°C for further processing for mass spectrometry analyses.
Plasma and tissues were extracted and levels of LEI‐101 were quantified by LC‐MS/MS, using multiple reactions monitoring under conditions described previously (Godlewski et al., 2010) Briefly, the mass spectrometer was set for electrospray ionization operated in the positive ion mode. The molecular ion and fragments for each compound measured were as follows:
m/z 473.2 ➔ 200.1 and 473.2 ➔148 for LEI‐101 (CID energy 28 and 44 V, respectively.
The amounts of LEI‐101 in the samples were determined from standard curves using linear regression. Values were expressed as ng·ml−1 for serum or ng·g−1 for tissues.
Tetrad assay
ICR mice (counterbalanced Latin square within subject design), from The Jackson Laboratory, Bar Harbor, ME) were housed individually overnight. The behavioural testing was conducted in the following order: bar test (catalepsy), tail withdrawal test, rectal temperature and locomotor activity. Testing was performed according to previously described procedures (Long et al., 2009; Schlosburg et al., 2010; Ignatowska‐Jankowska et al., 2014). Mice received LEI‐101 or vehicle (DMSO,Tween 80 and saline, in 1:1:18 ratio) p.o. (via gavage) at dose of 60 mg·kg−1 of each drug, 2 h before testing in the tetrad assay.
Catalepsy was assessed on a bar 0.7 cm in diameter placed 4.5 cm off of the ground. The mouse was placed with its front paws on the bar and a timer (Timer no. 1) was started. A second timer (Timer no. 2) was turned on only when the mouse was immobile on the bar, with the exception of respiratory movements. If the mouse moved off the bar, it was placed back on in the original position. The assay was stopped when either Timer no. 1 reached 60 s, or after the fourth time the mouse moved off the bar, and the cataleptic time was scored as the amount of time on Timer no. 2. Nociception was then assessed in the tail immersion assay. The mouse was placed head first into a small bag fabricated from absorbent under pads (VWR Scientific Products; 4 cm diameter, 11 cm length) with the tail out of the bag. Each mouse was hand‐held and 1 cm of the tail was submerged into a 52°C water bath. The latency for the mouse to withdraw its tail within a 10 s cut off time was scored. Rectal temperature was assessed by inserting a thermocouple probe 2 cm into the rectum, and temperature was determined by telethermometer (BAT‐10 Multipurpose Thermometer, Clifton, NJ, USA). Locomotor activity was assessed 120 min after treatment, for a 5 min period in a Plexiglas cage (42.7 × 21.0 × 20.4 cm) and Anymaze (Stoelting, Wood Dale, Illinois, USA) software was used to determine the percentage of time spent immobile and distance travelled.
Nephropathy model
Mice (CB2R ‐/‐ and CB2R+/+) were killed (as described above) 72 h after a single injection of cisplatin (cis‐diammineplatinum(II) dichloride 20 mg·kg−1 i.p.; Sigma). LEI‐101 was dissolved as described previously and was used at 0.3, 3.0 and 10 mg·kg−1, i.p. and p.o. every day, starting 1.5 h before the cisplatin exposure.
Renal function monitoring
Blood was collected under deep anaesthesia with 5% isoflurane, by retro‐orbital bleeding and serum levels of creatinine and blood urea nitrogen (BUN) were measured using a Prochem‐V chemical analyser (TX, USA) or Idexx VetTest 8008 (Idexx Laboratories, Westbrook, ME, USA).
Histological examination for tubular damage
After fixation of the kidneys with 10% formalin, renal tissues were sectioned (5μm) and stained with periodic acid–Schiff (PAS) reagents for histological examination. Tubular damage in PAS‐stained sections was examined under the microscope (200× magnification) as described (Pan et al., 2010).
Nitrotyrosine staining of tissue samples
Paraffin‐embedded sections (5μm) of kidney were cut, deparaffinized and hydrated in descending gradations of ethanol, followed by microwave treatment. Next, sections were incubated in 0.3% H2O2 in PBS to block endogenous peroxidase activity. The sections were then incubated with anti‐nitrotyrosine (1:200 dilution; Cayman Chemical, Ann Arbor, MI, USA) overnight at 4°C in a moist chamber. Biotinylated secondary antibodies and ABC reagent were added according to the kit instructions (Vector Laboratories, Burlingame, CA, USA). Colour development was induced by incubation with a DAB kit (Vector Laboratories) for 3–5 min, and specific staining was visualized by light microscopy as described previously (Mukhopadhyay et al., 2010b; Mukhopadhyay et al., 2010a).
Lipid peroxidation/ROS production
4‐Hydroxynonenal (4‐HNE) in the renal tissues was determined using a kit from Cell Biolabs (San Diego, CA, USA). Renal tissue (50‐80 mg) was homogenized in 1 mL of T‐PER Reagent (Fisher Scientific) in a Precellys Ceramic Bead (1.4 mm) homogenizer and centrifuged at 10,000 g for 20 min. Supernatants were used as tissue lysate and renal tissue lysates (protein concentration, 10 μg mL‐1) were adsorbed onto a 96‐well plate for 12 h at 4°C. 4‐HNE adducts present in the sample or standard were probed with anti‐4‐HNE antibody, followed by a horseradish peroxidase‐conjugated secondary antibody. 4‐HNE‐protein adduct content in an unknown sample was determined by comparison with a standard curve as described in the manufacturer's protocol (Mukhopadhyay et al., 2012).
Detection of apoptosis by renal DNA fragmentation and caspase 3/7 activity assays
Renal tissue (50‐100 mg) was homogenized in 1 ml of TRIZOL® Reagent(Life technologies) in a Precellys Ceramic Bead (1.4 mm) homogenizer and centrifuged at 10,000 g for 20 min. Supernatants were used as tissue lysate. Caspase 3/7 activity in tissue lysates was measured using the Apo‐One Homogenous Caspase‐3/7 assay kit (Promega Corp., Madison, WI, USA). An aliquot of caspase reagent was added to each well and mixed on a plate shaker for 1 h at room temperature shielded from light, and the fluorescence was measured.
The DNA fragmentation assay was based on measuring the amount of mono‐ and oligonucleosomes in the cytoplasmic fraction of tissue extracts using a commercially available kit (Roche Diagnostics, Basel, Switzerland) according to the manufacturer's instructions as described by Mukhopadhyay et al. (2012).
Real‐time PCR analyses
Total RNA was isolated from kidney homogenate (50‐100 mg of renal tissue were homogenized in 1 ml of TRIZOL® Reagent (Life Technologies) in Precellys Ceramic Bead(1.4 mm) homogenizer), using Trizol reagents (Invitrogen; ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer's instructions. The isolated RNA was treated with RNase‐free DNase (Ambion, Austin, TX, USA) to remove traces of genomic DNA contamination. One microgram of total RNA was reverse‐transcribed to cDNA using SuperScript II (Invitrogen). The target gene expression was quantified with Power Sybr green PCR master mix using an ABI 7500 real‐time PCR instrument. Each amplified sample was analysed for homogeneity using dissociation curve analysis. After denaturation at 95°C for 2 min, 40 cycles were performed at 95°C for 10 s and 60°C for 30 s. Relative quantification was calculated using the comparative Ct method (2−ΔΔCt: ΔΔCt = ΔCt sample – ΔCt reference). Lower ΔCt values and lower ΔΔCt reflect a relatively higher amount of gene transcript. Statistical analyses were carried out for at least 6 to 15 replicate experimental samples in each set.
The following primers were used: TNF‐α, 5′‐AAGCCTGTAGCCCACGTCGTA‐3′ and 5′‐AGGTACAACCCATCGGCTGG‐3′; IL‐1β, 5′‐AAAAAAGCCTCGTGCTGTCG‐3′ and 5′‐GTCGTTGCTTGGTTCTCCTTG‐3′; gp91phox, 5′‐GACCATTGCAAGTGAACACCC‐3′ and 5′‐AAATGAAGTGGACTCCACGCG‐3′; NOX4, 5′‐TCATTTGGCTGTCCCTAAACG‐3′ and 5′‐AAGGATGAGGCTGCAGTTGAG‐3′; and actin, 5′‐TGCACCACCAACTGCTTAG‐3′ and 5′‐GGATGCAGGGATGATGTTC‐3′.
Data analysis
Results are reported as means ± SEM. Statistical significance between two measurements was determined by the two‐tailed unpaired Student t‐test (and among groups it was determined by anova followed by post hoc Student–Newman–Keuls) using GraphPad Prism 4.3 or 5.0 software (San Diego, CA, USA). Probability values of P < 0.05 were considered significant.
Materials
LEI‐101, 3‐cyclopropyl‐1‐(4‐(6‐((1,1‐dioxidothiomorpholino)methyl)‐5‐fluoropyridin‐2‐yl)benzyl)imidazolidine‐2,4‐dione hydrochloride was synthesized as described previously (van der Stelt et al. 2011). For in vivo studies, LEI‐101 was dissolved in DMSO/Tween80/saline (1/1/18 v/v/v). [3H]CP55940 (specific activity 141.2 Ci mmol‐1) and GTPγS (specific activity 1250 Ci mmol‐1) were purchased from Perkin Elmer (Waltham, MA, USA). The other labelled compounds ‐ [ethanolamine‐1‐3H]‐anandamide (60 Ci mmol‐1); [3H]N‐arachidonoyl‐phosphatidylethanolamine (200 Ci mmol‐1); sn‐1‐stearoyl‐2‐[14C]arachidonoyl‐glycerol (55 mCi mmol‐1); [3H]2‐oleoylglycerol (20 Ci mmol‐1) ‐ were from ARC (St. Louis, MO). Bicinchoninic acid (BCA) and BCA protein assay reagent were obtained from Pierce Chemical Company (Rochford, IL, USA). All other materials were obtained from standard commercial sources and were of analytical grade.
Results
LEI‐101 is a selective CB2 receptor partial agonist
As shown in Table 1, LEI‐101 behaves as a CB2 receptor partial agonist in the β‐arrestin recruitment and GTPγS assays, compared with the reference mixed CB1/ CB2 receptor agonist CP‐55940. LEI‐101 had more than 100‐fold selectivity for CB2 receptors over CB1 receptors in the binding assay. The compound does not interact with other proteins of the endocannabinoid system: human FAAH (hFAAH), MAGL, NAPE‐PLD and DAGL. LEI‐101 did not inhibit serine hydrolases as determined in an ABPP with mouse membrane proteome and MB064 and TAMRA‐FP as fluorescent probes (data not shown).
Table 1.
Molecular pharmacology profile of LEI‐101
| Assay | Parameter | LEI‐101 | CP‐55.940 |
|---|---|---|---|
| Binding | hCB2R pKi ± SEM | 7.5 ± 0.1 | 8.4 ± 0.1 |
| hCB1R pKi ± SEM | <5 | 9.6 ± 0.1 | |
| cAMP inhibitiona | hCB2R pEC50 ± SEM | 8.0 ± 0.1 | N.D.b |
| hCB1R pEC50 ± SEM | <5 | N.D. | |
| β‐Arrestin recruitment | hCB2R pEC50 ± SEM (Emax: % ± SEM) | 7.0 ± 0.3 (41 ± 6) | 8.5 ± 0.1 (100 ± 1) |
| hCB1R pEC50 ± SEM (Emax: % ± SEM) | <5 (−1% @ 10 μM) | 8.3 ± 0.2 (99 ± 1) | |
| GTPγS binding | hCB2R pEC50 ± SEM (Emax: % ± SEM) | 6.6 ± 0.2 (65 ± 8) | 8.3 ± 0.2 (96 ± 3) |
| Substrate hydrolysisc | hFAAH1/2 pKi ± SEM | <5 | N.D. |
| MAGL pKi ± SEM | <5 | N.D. | |
| DAGL pKi ± SEM | <5 | N.D. | |
| NAPE‐PLD pKi ± SEM | <5 | N.D. | |
| ABPP | Serine hydrolases | No off targets detected | No off targets detected |
Data from van der Stelt et al. 2011; bN.D., not determined;cResidual activity values with reference inhibitors were as follows (vs. controls, set to 100%): 24% for FAAH (with 0.1 nM URB597), 27% for MAGL (with 0.1 nM JZL184) and 38% for DAGL (with 1 μM tetrahydrolipstatin). At present, inhibitors for NAPE‐PLD are not commercially available.
LEI‐101 has excellent oral bioavailability in mice
Previously, we have determined the pharmacokinetic profile of LEI‐101 in rats (van der Stelt et al., 2011). Because the efficacy models presented in this study are in mice, we determined the plasma concentrations of LEI‐101 after 10 mg·kg−1 doses, given p.o. or i.p. to mice using DMSO/Tween80/Saline (1/1/18 v/v/v) as a vehicle. As depicted in Figure 1A–C, plasma levels were found to peak after 2 h of either administration route. In view of its low plasma protein binding (fraction unbound = 0.44), these plasma concentrations should be more than sufficient to activate CB2 receptors. Of note, the compound seemed to accumulate in the kidney (Figure 1). Lowest levels were found in the brain, thereby confirming the peripherally restricted nature of LEI‐101.
Figure 1.
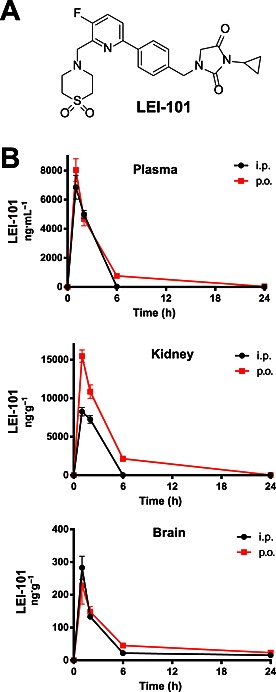
The CB2 receptor agonist LEI‐101 is an orally bioavailable and a peripherally restricted CB2 receptor agonist with high kidney exposure. Chemical Structure of LEI‐101 (A) and time‐course of tissue distribution (B) in plasma, kidney and brain after 10 mg·kg−1 p.o. and i.p. administration of LEI‐101 in mice. Results are means ± SEM; n = 4 per group.
LEI‐101 does not induce cannabimimetic effects in vivo
To determine whether the compound maintains its selectivity for CB2 receptors over CB1 receptors in vivo, we tested LEI‐101 in the mouse tetrad assay for CB1 receptor activity. This is a very sensitive assay in which four consecutive behavioural tests, related to anti‐nociception, hypothermia, catalepsy and spontaneous activity, are performed 120 min after administration of the compound. A positive result in all four tests indicates that the compound or one of its metabolites activates CB1 receptors in the CNS. LEI‐101 (60 mg·kg−1, p.o.) did not produce any effects in the tetrad assay as compared with vehicle. There were no effects on nociceptive behaviour assessed in tail withdrawal test (Figure 2A) (P = 0.2 in both cases) and no changes in body temperature (Figure 2B) (P = 0.4 and P = 0.8 for each respective drug). No effects on locomotor behaviour (Figure 2C) were found in case of distance travelled (P = 0.6 and P = 1.0, respectively), time spent mobile (P = 0.8 and P = 0.5, respectively), or running speed of mice (P = 0.3 and P = 0.9, respectively). No catalepsy was observed following administration of LEI‐101. The results indicate that LEI‐101 at high doses administered orally did not produce CB1 receptor‐mediated CNS‐side effects up to 60 mg·kg−1 (p.o.). Overall, we can conclude that LEI‐101 maintains its selectivity in vivo.
Figure 2.
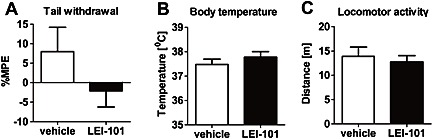
The CB2 receptor agonist LEI‐101 does not induce cannabimimetic effects in vivo. LEI‐101 (60 mg·kg−1, p.o.) did not affect nociceptive behaviour assessed in tail withdrawal test (A) and did not change body temperature (B), as compared with mice receiving vehicle. No effects on locomotor behaviour (C) were found. Results are means ± SEM; n = 8 per group.
LEI‐101 attenuates biomarkers and histological damage of cisplatin‐induced nephropathy
The known CB2 receptor agonists HU‐308 (Hanus et al., 1999; Mukhopadhyay et al., 2010a,b) and the natural product β‐carophyllene (Horvath et al., 2012) ameliorate cisplatin‐induced nephrotoxicity after i.p. administration. Therefore we sought to test whether our novel synthetic CB2 receptor partial agonist, which endowed with a different chemical scaffold, could also protect animals against cisplatin‐induced nephropathy after oral gavage. For assessment of cisplatin‐induced nephrotoxicity, the serum creatinine and total blood urea levels, both biomarkers of renal dysfunction, were measured. 72 h following the cisplatin injection (20 mg·kg−1, i.p.), a marked increase of these biomarkers was observed in C57Bl6/J mice, compared with vehicle‐treated control animals. LEI‐101 dose‐dependently attenuated these increases in creatinine and blood urea nitrogen levels in cisplatin‐treated animals (Figure 3). At 3.0 or 10 mg·kg−1 (p.o. and i.p.), LEI‐101 largely prevented cisplatin‐induced increases in serum creatinine and BUN levels. This attenuation was not observed in CB2R‐/‐ mice (Figure 4). Of note, the CB2R‐/‐ mice had an increased susceptibility to cisplatin nephrotoxicity (Figure 4).
Figure 3.
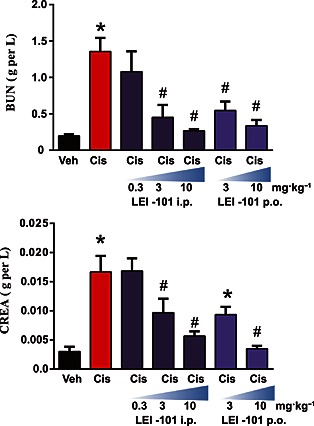
The CB2 receptor agonist LEI‐101 attenuates cisplatin‐induced renal dysfunction. Cisplatin (Cis) induced profound renal dysfunction 72 h after administration to mice as shown by increased levels of blood urea nitrogen (BUN) and creatinine (CREA), which were dose‐dependently attenuated by the CB2 receptor agonist LEI‐101 administered i.p. or by gavage (p.o.). Results are means ± SEM; n = 6 per group. * P < 0.05, significantly different from vehicle; # P < 0.05, significantly different from cisplatin.
Figure 4.
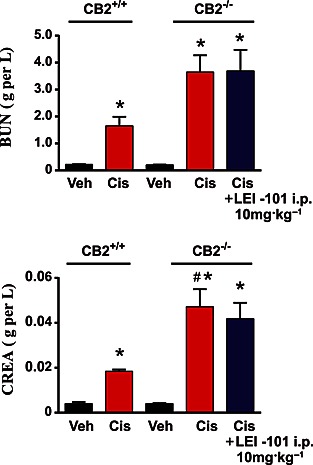
The protective effects of The CB2 receptor agonist LEI‐101 against cisplatin (Cis)‐induced nephrotoxicity are absent in CB2R‐/‐ mice. The renal dysfunction was aggravated in CB2R−/− mice compared with CB2R+/+ littermates. LEI‐101 alone had no effect on blood urea nitrogen (BUN) and creatinine (CREA) levels compared with the vehicle (Veh)‐treated group and had no protective effect in CB2R−/−mice against cisplatin‐induced nephrotoxicity. Results are means ± SEM; n = 6 per group. * P < 0.05, significantly different from vehicle; # P < 0.05, significantly different from cisplatin.
Renal dysfunction was accompanied by morphological damage to the kidney (Figure 5). Histological examination revealed necrosis, protein casts, vacuolization and desquamation of epithelial cells in the renal tubules. LEI‐101 at 10 mg·kg−1 decreased tubular damage as determined by PAS staining of the kidney (Figure 5). This effect was not observed in the CB2R‐/‐ mice, thereby indicating that the protective effect of LEI‐101 was mediated through CB2 receptors.
Figure 5.
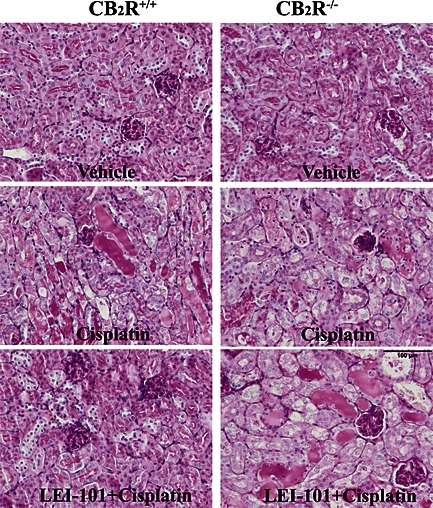
The CB2 receptor agonist LEI‐101 attenuates cisplatin‐induced renal histopathological injury in wild type, but not in CB2R‐/‐ mice. Representative images of PAS staining show that cisplatin induced profound tubular injury 72 h after administration to mice, as evidenced by protein cast, vacuolation and desquamation of epithelial cells in the renal tubules of CB2R+/+ and CB2R−/− mice. Note the decreased histopathological injury by LEI‐101 (10 mg·kg−1 i.p.) in cisplatin‐treated CB2R+/+, but not in CB2R−/− mice, and enhanced injury in CB2R−/− mice compared with their wild‐types.
LEI‐101 reduces renal oxidative and nitrative stress
Reactive oxygen species play an important role in the pathology of cisplatin‐induced tubular damage (Mukhopadhyay et al., 2010 and 2012). Consistent with previous reports (Mukhopadhyay et al., 2010, 2012), we also observed a marked increase in staining of 4‐HNE, a marker of lipid peroxidation, and 3‐nitrotyrosine, a marker of protein nitration, in the tubular cells of kidneys of cisplatin‐treated animals (Figures 6 and 7). The mRNA levels of NADPH oxidase isoform 2 (NOX2) and 4 (NOX4), enzymes known to generate ROS, were also significantly upregulated 3 days after cisplatin injection (Figure 7). These cisplatin‐induced pathological alterations were significantly reduced by LEI‐101 (10 mg·kg−1, i.p. or p.o.) pretreatment (Figures 5 and 8). Conversely, LEI‐101 did not affect the oxidative and nitrative stress markers in CB2R‐/‐ mice treated with cisplatin.
Figure 6.
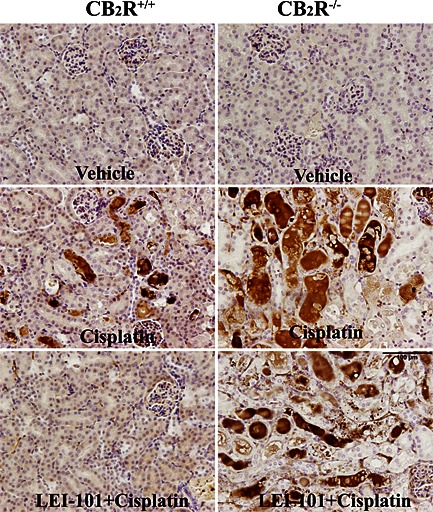
The CB2 receptor agonist LEI‐101 attenuates cisplatin‐induced renal 3‐nitrotyrosine (3‐NT) formation in wild type, but not in CB2R‐/‐ mice. Representative images show marked accumulation of 3‐NT (marker of protein nitration) predominantly in damaged renal proximal tubules (dark brown staining) 72 h after cisplatin administration to mice. Note the decreased renal 3‐NT staining by LEI‐101 (10 mg·kg−1 i.p.) in cisplatin‐treated CB2R+/+, but not in CB2R−/− mice, and enhanced staining in CB2R−/− mice compared with their wild‐types.
Figure 7.
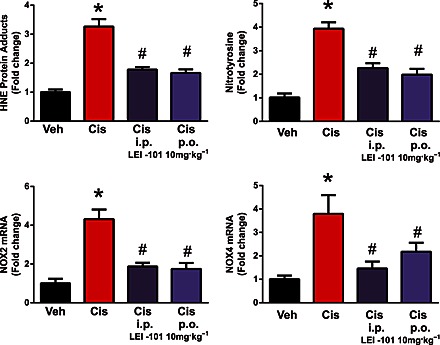
The CB2 receptor agonist LEI‐101 attenuates cisplatin‐induced renal lipid peroxidation (as HNE adducts) and nitrotyrosine generation (upper panel). The lower panel shows overexpression of superoxide‐generating enzymes NOX2 (gp91phox) and NOX4 (RENOX) after cisplatin. Cisplatin (Cis) induced marked increases in lipid peroxidation and ROS generation and in mRNA expression of NOX2 and NOX4 in the kidneys 72 h after administration to mice. These effects were attenuated by LE101 at 10 mg·kg−1 both upon i.p. and p.o. treatment (starting from 1.5 h before cisplatin injection). Results are means ± SEM; n = 6 per group; * P < 0.05, significantly different from vehicle; # P < 0.05, significantly different from cisplatin alone.
Figure 8.
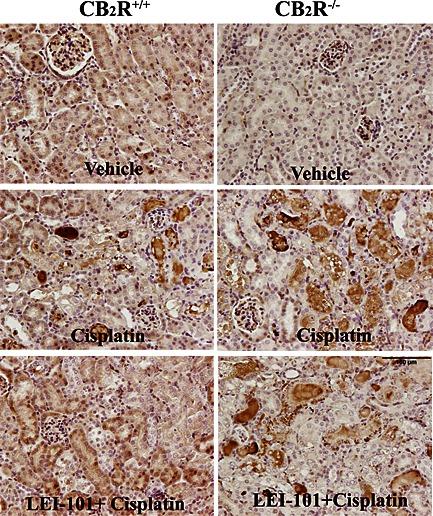
The CB2 receptor agonist LEI‐101attenuates cisplatin (Cis)‐induced renal HNE adducts formation in wild type, but not in CB2R‐/‐ mice. Representative images show marked accumulation of HNE adducts predominantly in damaged renal proximal tubules (dark brown staining) 72 h after cisplatin administration to mice. Note the decreased renal HNE staining by LEI‐101 (10 mg·kg−1 i.p.) in cisplatin‐treated CB2R+/+, but not in CB2R−/− mice, and enhanced staining in CB2R−/− mice compared with their wild‐types.
LEI‐101 attenuates renal inflammatory response and apoptotic cell death
Next, we tested whether LEI‐101 was able to limit cisplatin‐induced inflammation in the kidney. As observed in Figure 9, cisplatin increased the expression of mRNA for the proinflammatory cytokines, TNF‐α and IL‐1β. Such an increase in proinflammatory markers was reversed by LEI‐101 (10 mg·kg−1, i.p. or p.o.).
Figure 9.
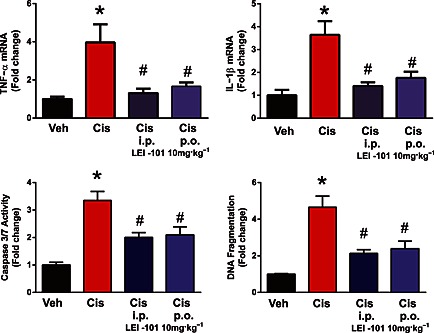
The CB2 receptor agonist LEI‐101attenuates cisplatin‐induced renal inflammation and cell death. Top panel: cisplatin (Cis) induced increases in renal TNFα and IL‐1β mRNA expression, which were attenuated by LE‐101 at 10 mg·kg−1, both i.p. and p.o. treatment. Bottom panel: cell death in the kidneys was evaluated by caspase 3/7 activity and DNA fragmentation assay. Both markers of cisplatin‐induced cell death in the kidneys were attenuated by LEI‐101 treatment starting at 1.5 h before cisplatin injection. Results are means ± SEM; n = 5–8 per group; *P < 0.05, significantly different from vehicle; # P < 0.05, significantly different from cisplatin alone.
Apoptotic cell death in the kidney following cisplatin‐induced injury was evaluated by monitoring caspase 3/7 activity and DNA fragmentation (Figure 9). All these markers were significantly increased 3 days after cisplatin injection and largely attenuated by treatment with LEI‐101 (10 mg·kg−1, i.p. or p.o.).
Discussion
In this study, we have comprehensively characterized a novel CB2 receptor ligand that may be used as a tool to interrogate CB2 receptor biology in more detail. We have shown that LEI‐101 is an orally bioavailable, largely peripherally restricted, selective CB2 receptor partial agonist that does not induce cannabimimetic effects in the mouse tetrad at high doses (up to 60 mg·kg−1 p.o.). This indicates that LEI‐101 and its metabolites did not activate CB1 receptors in vivo either directly, neither indirectly by raising endocannabinoid levels. LEI‐101 significantly reversed cisplatin‐induced nephropathy and attenuated inflammation and oxidative stress already at a dose of 3 mg·kg−1 p.o. and i.p. Importantly, this protective effect was mediated via CB2 receptors, because it was not observed in CB2R‐/‐ animals. Currently, at least three CB2 receptor agonists (GW81466, cannabinor and S‐777469) have been tested in phase 2 clinical trials for pain and atopic dermatitis, albeit with a poor outcome (Dhopeshwarkar and Mackie, 2014). Peripherally restricted, mixed CB1/ CB2 receptor agonists induced severe CB1 receptor‐mediated cardiovascular and metabolic side effects leading to termination of the clinical trials (Pacher and Kunos, 2013). At this moment, the reasons for the failure to demonstrate clinical efficacy, for these CB2 receptor agonists, are not exactly clear. One of them could be that the CB2 receptor has not been properly validated for the disease under investigation, or that the clinical candidates could have been tested at the wrong stage of the disease. For example, once inflammation‐induced tissue injury has progressed too far, CB2 receptor agonists could be ineffective (Pacher and Mechoulam, 2011). In addition, the clinical candidates may not have reached the site of action. In fact, in chronic pain, the site of action of CB2 receptor agonists as well as its mechanism of analgesic action is still under debate. At the moment, there is a lack of suitable biomarkers to correlate target engagement and modulation of CB2 receptors with efficacy in chronic pain. It has been suggested that very low receptor occupancy of CB1 receptors could be responsible, at least in part, for mediating the effect of commonly used ‘selective’ CB2 receptor agonists in preclinical inflammatory pain models. Most previous in vivo studies of inflammation and tissue injury relied on the use of only very few CB2 receptor agonists predominantly after i.p. administration, without characterization of their pharmacokinetics (Pacher and Mechoulam, 2011). In addition, most of these studies either used a single dose of CB2 receptor agonist or could not establish a clear dose‐dependence, because of the biphasic response. Given the emerging evidence that supports the pro‐inflammatory role of peripheral CB1 receptors, such a biphasic response could be attributed (at least in part) to activation of CB1 receptors at higher doses (Mukhopadhyay et al., 2010; Jourdan et al., 2014). Surprising, in vivo evidence for the latter has also been presented at a recent meeting on (endo)cannabinoids (Wiley et al., 2014).
In this study, we demonstrate that LEI‐101 dose‐dependently attenuated cisplatin‐induced kidney injury from 3 mg·kg−1 both p.o. or i.p., without inducing cannabimimetic effects in the mouse tetrad or drug discrimination paradigm up to 60 mg·kg−1 p.o. Thus, the compound and its metabolites did not activate CB1 receptors in vivo, even at high concentrations. There is accumulating evidence on the opposing role of CB1 and CB2 receptors in kidney injury. It appears that while activation of CB1 receptors promotes kidney injury, CB2 receptor activation attenuates kidney inflammation and injury in rodent models of diabetic and tubular nephropathy (Barutta et al., 2010, 2011 and 2014; Mukhopadhyay et al., 2010; Jourdan et al., 2014).
Cisplatin is a cornerstone in the treatment for many different types of cancer, but its use is dose‐limited by the occurrence of nephropathy as a major adverse side effect. The exact mechanisms of cisplatin's renal toxicity are still not completely understood, and currently no effective medication is available to prevent this problem (Pabla and Dong, 2008). Cisplatin accumulates in renal tubular cells because of its metabolism, reaching toxic levels (Pabla and Dong, 2008). Consistent with our recent results, enhanced superoxide production by mitochondria and various cellular NADPH oxidases, suhc as NOX2 and NOX4 (Mukhopadhyay et al., 2010a,b, 2012), overproduction of NO via activation of inducible nitric oxide synthase, and increased peroxynitrite generation formed from diffusion limited reaction of nitric oxide with superoxide (Pacher et al., 2007), and consequent lipid peroxidation and protein nitration have all been implicated in renal tubular cell death induced by cisplatin. Recent studies have also suggested a central role for the nuclear enzyme poly(ADP‐ribose)polymerase 1 (PARP‐1), which is activated by oxidative DNA injury and leads to depletion of cellular NAD+ and ATP stores, not only in cisplatin‐induced tubular cell death but also in the pathological kidney inflammatory response (Mukhopadhyay et al., 2011). In addition to damaging renal proximal tubular cells, cisplatin also injures renal endothelial cells leading to endothelial dysfunction and secondary functional hypoxia, amplifying the tubular cell death. These pathological events also trigger secondary inflammatory response in the kidneys, largely orchestrated by infiltrating immune cells. The pro‐inflammatory cytokine TNF‐α is produced by various cell types, including infiltrating immune cells, activated endothelium and renal parenchymal cells and appears to play a central role in further fuelling the inflammatory response and consequent tissue injury (Pabla and Dong, 2008). CB2 receptor agonists appear to protect against cisplatin‐induced kidney injury by attenuating endothelial cell activation and pro‐inflammatory response, as well as by limiting inflammatory cell chemotaxis, adhesion to activated endothelium, transmigration to parenchyma and activation at the site of injury (Mukhopadhyay et al., 2010a; Pacher and Mechoulam, 2011; Horvath et al., 2012). In experimental models of diabetic nephropathy, in which initially the glomerular injury dominates the pathology, activation of CB2 receptors protected glomerular podocytes, which wrap around the capillaries of the glomerulus and play critical role in maintaining the normal kidney function and filtration (Barutta et al., 2011, 2014). In addition, CB2 receptor agonists may attenuate monocyte chemokine signalling, which is also similar in the cisplatin‐induced nephropathy model (Barutta et al., 2011, 2014). Consistent with the importance of CB2 receptors in kidney disease in this study, we demonstrate marked dose‐dependent attenuation of kidney inflammation, oxidative stress and tubular damage induced by cisplatin in mice by LEI‐101.
In summary, we have demonstrated, in this study, that LEI‐101 is a selective, largely peripherally restricted, orally available agonist at CB2 receptors and could be an excellent tool to study the role of these receptors in inflammatory and other immunity‐related diseases (Chiurchiù et al., 2015). Of note, LEI‐101 behaved as a partial CB2 receptor agonist in the GTPγS‐assay and β‐arrestin recruitment assay, but as a full agonist in the cAMP‐assay, a more downstream signalling pathway. It is not uncommon that some ligands behave as partial agonists at the G‐protein activation while fully activating downstream signalling pathways. Whether a partial or full agonism was required in case of LEI‐101 to observe in vivo efficacy in the cisplatin‐induced nephrotoxicity model cannot be concluded at this moment, but would be very interesting to explore in future studies.
Our results also indicate that selective CB2 receptor (partial) agonists could be excellent targets for treating kidney disease associated with oxidative stress and inflammation. This observation, coupled with the recent demonstration of the protective effects of a novel series of CB2 receptor agonists in various kidney injury/fibrosis models (Adams et al., 2014), further substantiates the strong translational potential of selective CB2 receptor agonists in inflammatory kidney disease.
Conflict of interest
None
Author contributions
P.M., M.B., K.E., Z.C., F.F., B.I‐J., J.W., N.G., T.H., M.R., M.S. designed, performed and analysed experiments: P.M., L.H., G.K., M.M., A.L., P.P., M.vd.S. designed and analysed the experiments and wrote the paper:
Acknowledgements
We thank Henk de Vries and Judy Harvey‐White for excellent technical assistance. M.S. and M.v.d.S. are supported by ECHO‐STIP grant of the Dutch Research Council Chemical Sciences (NWO‐CW). P.P. and G.K. are supported by the Intramural Program of NIAAA/NIH. M.M. is supported by Ministero dell'Istruzione, dell'Università e della Ricerca (PRIN 2010–2011 grant).
Mukhopadhyay, P. , Baggelaar, M. , Erdelyi, K. , Cao, Z. , Cinar, R. , Fezza, F. , Ignatowska‐Janlowska, B. , Wilkerson, J. , van Gils, N. , Hansen, T. , Ruben, M. , Soethoudt, M. , Heitman, L. , Kunos, G. , Maccarrone, M. , Lichtman, A. , Pacher, P. , and Van der Stelt, M. (2016) The novel, orally available and peripherally restricted selective cannabinoid CB2 receptor agonist LEI‐101 prevents cisplatin‐induced nephrotoxicity. British Journal of Pharmacology, 173: 446–458. doi: 10.1111/bph.13338.
Footnotes
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al. 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a bAlexander et al., 2013a, 2013b).
References
- Adams J, Apfel CM, Bendels S, Bissantz C, Fingerle J, Formentini I et al (2014) Triazolopyrimidines – a novel class of highly potent, highly selective and in vivo active CB2 agonists, 24th Annual Symposium on the Cannabinoids, International Cannabinoid Research Society: Research Triangle Park, NC, USA, page 6567 [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The concise guide to PHARMACOLOGY 2013/14: G protein‐coupled receptors. Br J Pharmacol 170: 1459–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al (2013b). The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggelaar MP, Janssen FJ, van Esbroeck A, den Dulk H, Allara M, Hoogendoorn S et al. (2013). Development of an activity, ÄêBased probe and in silico design reveal highly selective inhibitors for diacylglycerol lipase‚alpha in brain. Angew Chem Int Ed 52: 12081–12085. [DOI] [PubMed] [Google Scholar]
- Barutta F, Corbelli A, Mastrocola R, Gambino R, Di Marzo V, Pinach S et al. (2010). Cannabinoid receptor 1 blockade ameliorates albuminuria in experimental diabetic nephropathy. Diabetes 59: 1046–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutta F, Piscitelli F, Pinach S, Bruno G, Gambino R, Rastaldi MP et al. (2011). Protective role of cannabinoid receptor type 2 in a mouse model of diabetic nephropathy. Diabetes 60: 2386–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barutta F, Grimaldi S, Franco I, Bellini S, Gambino R, Pinach S et al. (2014). Deficiency of cannabinoid receptor of type 2 worsens renal functional and structural abnormalities in streptozotocin‐induced diabetic mice. Kidney Int 86: 979–90. [DOI] [PubMed] [Google Scholar]
- Batkai S, Osei‐Hyiaman D, Pan H, El‐Assal O, Rajesh M, Mukhopadhyay P et al. (2007). Cannabinoid‐2 receptor mediates protection against hepatic ischemia/reperfusion injury. Faseb J 21: 1788–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham B, Jones PG, Uveges AJ, Kotnis S, Lu P, Smith VA et al. (2007). Species‐specific in vitro pharmacological effects of the cannabinoid receptor 2 (CB2) selective ligand AM1241 and its resolved enantiomers. Br J Pharmacol 151: 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Howell F, William G, Minassi A, Cascio MG, Ligresti A et al. (2003). Cloning of the first sn1‐DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J Cell Biol 163: 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonger KM, van den Berg RJBHN, Heitman LH, IJzerman AP, Oosterom J, Timmers CM et al. (2007). Synthesis and evaluation of homo‐bivalent GnRHR ligands. Bioorg & Med Chem 15: 4841–4856. [DOI] [PubMed] [Google Scholar]
- Chiurchiù V, Battistini L, Maccarrone M (2015). Endocannabinoid signaling in innate and adaptive immunity. Immunol 144: 352–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhopeshwarkar A, Mackie K (2014). CB2 cannabinoid receptors as a therapeutic target‐what does the future hold? Mol Pharmacol 86: 430–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL et al. (2002). Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci U S A 99: 10819–10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni S, De Simone C, Dallavalle S, Fezza F, Nannei R, Amadio D et al. (2010). Enol carbamates as inhibitors of fatty acid amide hydrolase (FAAH) endowed with high selectivity for FAAH over the other targets of the endocannabinoid system. ChemMedChem 5: 357–360. [DOI] [PubMed] [Google Scholar]
- Godlewski G1, Alapafuja SO, Bátkai S, Nikas SP, Cinar R, Offertáler L et al. (2010). Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem Biol 17: 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Thatte J, Buzard DJ, Jones RM (2013). Therapeutic utility of cannabinoid receptor type 2 (CB(2)) selective agonists. J Med Chem 56: 8224–8256. [DOI] [PubMed] [Google Scholar]
- Hanus L, Breuer A, Tchilibon S, Shiloah S, Goldenberg D, Horowitz M et al. (1999). HU‐308: a specific agonist for CB(2), a peripheral cannabinoid receptor. Proc Natl Acad Sci U S A 96: 14228–14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath B, Mukhopadhyay P, Kechrid M, Patel V, Tanchian G, Wink DA et al. (2012). beta‐Caryophyllene ameliorates cisplatin‐induced nephrotoxicity in a cannabinoid 2 receptor‐dependent manner. Free Radic Biol Med 52: 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatowska‐Jankowska B, Ghosh S, Crowe M, Kinsey S, Niphakis M, Abdullah R et al. (2014). In vivo characterization of the highly selective monoacylglycerol lipase inhibitor KML29: antinociceptive activity without cannabimimetic side effects. Br J Pharmacol 171: 1392–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdan T, Szanda G, Rosenberg AZ, Tam J, Earley BJ, Godlewski G et al. (2014). Overactive cannabinoid 1 receptor in podocytes drives type 2 diabetic nephropathy. Proc Natl Acad Sci U S A 111: 5420–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X et al. (2009). Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci U S A 106: 20270–20275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvet A, Teixeira‐Clerc F, Chobert MN, Deveaux V, Pavoine C, Zimmer A et al. (2011). Cannabinoid CB2 receptors protect against alcoholic liver disease by regulating Kupffer cell polarization in mice. Hepatology 54: 1217–1226. [DOI] [PubMed] [Google Scholar]
- Malan TP Jr, Ibrahim MM, Deng H, Liu Q, Mata HP, Vanderah T et al. (2001). CB2 cannabinoid receptor‐mediated peripheral antinociception. Pain 93: 239–245. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Pan H, Patel V, Mukhopadhyay B, Batkai S et al. (2010a). Cannabinoid‐2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic Biol Med 48: 457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Pan H, Rajesh M, Bátkai S, Patel V, Harvey‐White J et al. (2010b). CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol 160 (3): 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horváth B, Kechrid M, Tanchian G, Rajesh M, Naura AS et al. (2011). Poly(ADP‐ribose) polymerase‐1 is a key mediator of cisplatin‐induced kidney inflammation and injury. Free Radic Biol Med 51: 1774–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Horváth B, Zsengellér Z, Zielonka J, Tanchian G, Holovac E et al. (2012). Mitochondrial‐targeted antioxidants represent a promising approach for prevention of cisplatin‐induced nephropathy. Free Radic Biol Med 52: 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabla N, Dong Z (2008). Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L (2007). Nitric oxide and peroxynitrite in health and disease. Physiol Rev 87: 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Mechoulam R (2011). Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res 50: 193–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Kunos G (2013). Modulating the endocannabinoid system in human health and disease‐successes and failures. FEBS J 280: 1918–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Mukhopadhyay P, Rajesh M, Patel V, Mukhopadhyay B, Gao B et al. (2010). Cannabidiol attenuates cisplatin‐induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation, and cell death. Br J Pharmacol 160: 657–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, Davenport AP, McGrath JC, Peters JA, Southan C, Spedding M, Yu W, Harmar AJ; NC‐IUPHAR. (2014) The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 42 (Database Issue): D1098‐1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG et al. (2010). Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nat Neurosci 13: 1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storr MA, Keenan CM, Zhang H, Patel KD, Makriyannis A, Sharkey KA (2009). Activation of the cannabinoid 2 receptor (CB2) protects against experimental colitis. Inflamm Bowel Dis 15: 1678–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira‐Clerc F, Belot MP, Manin S, Deveaux V, Cadoudal T, Chobert MN et al. (2010). Beneficial paracrine effects of cannabinoid receptor 2 on liver injury and regeneration. Hepatology 52: 1046–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K, Okamoto Y, Ikematsu N, Inoue M, Shimizu Y, Uyama T et al. (2011). Enzymatic formation of N‐acylethanolamines from N‐acylethanolamine plasmalogen through N‐acylphosphatidylethanolamine‐hydrolyzing phospholipase D‐dependent and ‐independent pathways. Biochim Biophys Acta 1811: 565–577. [DOI] [PubMed] [Google Scholar]
- van der Stelt M, Cals J, Broeders Josten S, Cottney J, Avd D, Hermkens M et al. (2011). Discovery and optimization of 1‐(4‐(pyridin‐2‐yl) benzyl) imidazolidine‐2, 4‐dione derivatives as a novel class of selective Cannabinoid CB2 receptor agonists. J Med Chem 54: 7350–1062. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Thomas BF, Yang H and Mahadevan A. (2014) CB2 agonists: how selective are they? , 24th Annual Symposium on the Cannabinoids, International Cannabinoid Research Society, Research Triangle Park, NC, USA, page 65 [Google Scholar]
- Zhang M, Adler MW, Abood ME, Ganea D, Jallo J, Tuma RF (2009). CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc Res 78: 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweemer JM, Nederpelt I, Vrieling H, Hafith S, Doornbos MLJ, de Vries H et al. (2013). Multiple binding sites for small‐molecule antagonists at the CC chemokine receptor 2. Mol Pharm 84: 551–561. [DOI] [PubMed] [Google Scholar]