

Abstract
Retinal dystrophies (RDs) are degenerative diseases of the retina which have marked clinical and genetic heterogeneity. Common presentations among these disorders include night or colour blindness, tunnel vision and subsequent progression to complete blindness. The known causative disease genes have a variety of developmental and functional roles with mutations in more than 120 genes shown to be responsible for the phenotypes. In addition, mutations within the same gene have been shown to cause different disease phenotypes, even amongst affected individuals within the same family highlighting further levels of complexity. The known disease genes encode proteins involved in retinal cellular structures, phototransduction, the visual cycle, and photoreceptor structure or gene regulation. This review aims to demonstrate the high degree of genetic complexity in both the causative disease genes and their associated phenotypes, highlighting the more common clinical manifestation of retinitis pigmentosa (RP). The review also provides insight to recent advances in genomic molecular diagnosis and gene and cell-based therapies for the RDs.
Keywords: Retinal dystrophy (RD), retinitis pigmentosa (RP), massively parallel sequencing (MPS), CRISPR/Cas9
Introduction
Retinal dystrophies (RDs) are a group of conditions that have a range of clinical manifestations which are estimated to affect as many as 1 in 4,000 individuals (1). Cases may be syndromic or non-syndromic. Vision impairment may vary from poor peripheral or night vision to complete blindness, and severity usually increases with age. Cases may be familial with autosomal recessive, autosomal dominant or X-linked modes of inheritance described, with sporadic cases also observed (2-4). Due to the high genetic heterogeneity underlying these disorders, prioritisation in examining the >120 genes known to be associated with the inherited RDs is challenging (5). This has led to a lack of readily available testing in many countries for examination of all associated genes in a cost-effective and timely manner. Recent advances in genomic analysis technologies, including next generation sequencing (NGS) and chromosome microarrays, allow the prospect of genome-wide approaches to be feasible for the first time in a diagnostic setting. Studies examining copy number variation in RD are limited and have used multiplex ligation-dependant probe amplification (MLPA) techniques to target regions of interest (6). Until now, the usual diagnostic approach has been to use array-based primer extension (APEX) technology or Sanger sequencing to examine specific mutations, exons or gene targets. These techniques are reported to have a diagnostic yield of approximately 10-20% in RD patients (7). There are currently no cure or treatment options for patients with RD, with only an inevitable progression to blindness. New NGS genomic strategies and genome engineering technologies provide revolutionary opportunities in improving both diagnostic and therapeutic approaches in the RDs, emphasising the need for understanding of these conditions and applications of these new technologies.
RD can be categorized into broad groups depending on the type of photoreceptor affected and the manifestations, or degree, of atrophy within the retina. Rod and cone photoreceptors are the primary cellular units that facilitate the conversion of light energy to a neural action potential in the retina and facilitate an image to be perceived in the brain (Figure 1). RD groups can include rod-dominated diseases, cone-dominated diseases and generalised retinal degenerations involving both rod and cone photoreceptors. Syndromic forms whereby the phenotype extends to more organ systems than just retinal degeneration also exist, however are beyond the scope of this review. RD occurs due to abnormalities of retinal cellular structures including the photoreceptors as well as defects in the phototransduction and visual cycle pathways which are required to facilitate the conversion of light energy into a neuronal signal that is perceived by the brain [reviewed in (8)]. Phototransduction describes the process whereby a neural action potential is generated and propagated along the photoreceptor allowing an amplified response. In the visual cycle light sensitive pigments are generated and recycled and this involves the movement of intermediates through different cell layers of the retina. This review aims to highlight our understanding of non-syndromic RD, primarily regarding the rod and cone dominated dystrophies while also making reference to generalised RDs, specifically leber congenital amaurosis (LCA) and choroideremia. Molecular pathogenesis, application of new genomic technologies for molecular diagnoses and provision of gene-based therapeutic strategies will also be discussed.
Figure 1.
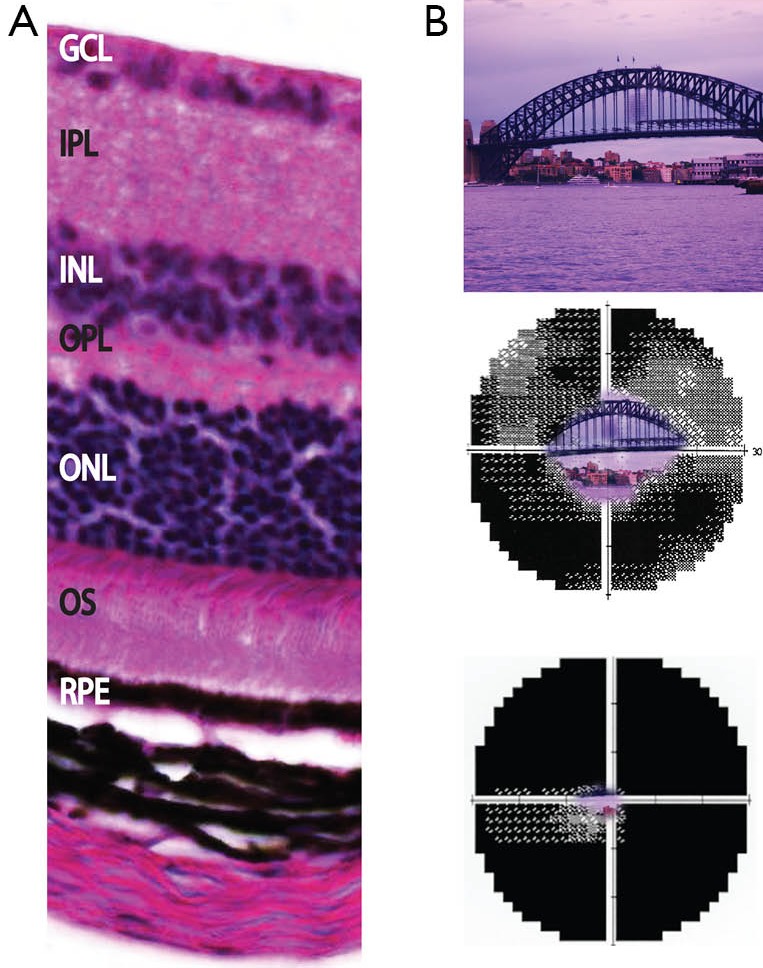
Retinal layers and clinical impact of retinal dystrophy. (A) Expansion of retinal region with histology section showing normal retinal layers of the mouse, which has close homology with histology of the human eye. The ONL contains the nuclei of the photoreceptors, and the OS layer contains the outer segments of the photoreceptors; (B) progressive visual field loss as experienced by retinitis pigmentosa patients. Upper image shows normal full visual field, middle image shows constricted field and lower image shows almost no central vision remaining. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; OS, outer segments of photoreceptors; RPE, retinal pigment epithelium.
Rod and rod-cone dystrophies
The rod and rod-cone dystrophies particularly affect the rod photoreceptors or the rod photoreceptors are the first affected. This group of disorders can be further delineated into progressive degenerative forms including retinitis pigmentosa (RP) and stationary forms called congenital stationary night blindness (CSNB). Syndromic forms of the disease also exist and have clinical presentations which extend to more than the affected retina. All Mendelian inheritance patterns, including autosomal dominant, autosomal recessive and X-linked have been observed.
Retinitis pigmentosa (RP)
The most common clinical manifestation of RD is RP. RP is a progressive non-syndromic rod-cone disease and has high levels of clinical and genetic heterogeneity. Variation exists at multiple levels with locus and allelic heterogeneity, incomplete penetrance and variable expression and penetrance all observed (9). The onset of the disease varies with early onset or juvenile RP sufferers affected from as early as the first years of life whereas adult or late onset RP symptoms develop significantly later. Clinical presentations manifest with progressive deterioration of the ability to see in dim light causing night blindness, followed by loss of peripheral vision that slowly encroaches toward the centre of the visual field resulting in tunnel vision (Figure 1). Later stages of the disease can result in complete blindness where the cone photoreceptors are also implicated. Affected photoreceptors undergo apoptosis, which is evident with the thinning of the outer nuclear layer and pigmented deposits or lesions present in the diseased retina (Figure 2). The loss in visual acuity has been shown to be proportionate to the level of deterioration of the fundus. Other clinical manifestations associated with RP include posterior subcapsular cataracts, dust like particles in the vitreous, white dots deep within the retina and Hyaline bodies affecting the optic nerve. The affected region of the retina may be restricted to a specific site, adding further complexity to disease identification (10).
Figure 2.

Retinitis pigmentosa: fundal images. (A,B) Wide field fundus photography illustrating retinal features of retinitis pigmentosa, including pigmentary changes (arrows), waxy pallor of the optic disc (asterisk) and retinal arteriolar attenuation (arrowheads) in (A), compared with the normal retinal image in (B); (C,D) OCT imaging showing thinning of the rod photoreceptor outer segments in retinitis pigmentosa in (C), compared with normal in (D) (arrows). There is relative preservation of the cone photoreceptor outer segments present in the foveal region in retinitis pigmentosa (arrowhead). OCT, optical coherence tomography.
Over 60 disease genes are reported to associate with RP (Table 1) (5,122). Known functions of the encoded proteins can be grouped into five broad categories including: phototransduction; retinal metabolism; RNA splicing; tissue development and maintenance; and cellular structure. Modes of inheritance vary with 15-20% autosomal dominant, 5-20% autosomal recessive, 5-15% X-linked, and simplex or unknown inheritance observed in 40-50% of cases (9). Digenic inheritance has also been observed where heterozygous mutations in two genes ROM1 and PRPH2 have been shown to cause the RP phenotype (77). Due to such complex clinical presentations and genetic factors, even with the latest genetic diagnostic techniques, including NGS strategies, molecular diagnosis is only achieved in approximately 50% of tested RP patients (123). There is also further genetic heterogeneity with some genes implicated in other forms of RD.
Table 1. Disease genes: human retinitis pigmentosa.
| Gene~ | Inheritance* | Potential function | Estimated frequency | Studies |
|---|---|---|---|---|
| RHO | Dominant & recessive | Phototransduction | 20-30% & <1% | (11,12) |
| GUCA1B | Dominant | Phototransduction | Rare (4-5% in Japan) | (13) |
| RDH12 | Dominant | Phototransduction | Unknown | (14,15) |
| PDE6A | Recessive | Phototransduction | 2-5% | (16,17) |
| PDE6B | Recessive | Phototransduction | 2-8% | (18,19) |
| SAG | Recessive | Phototransduction | 2-3% in Japan | (20) |
| CNGA1 | Recessive | Phototransduction | 1-2% | (21,22) |
| CNGB1 | Recessive | Phototransduction | <1% | (23,24) |
| PDE6G | Recessive | Phototransduction | <1% | (25) |
| RPE65 | Dominant & recessive | Retinal metabolism | Rare & 2-5% | (26,27) |
| LRAT | Recessive | Retinal metabolism | <1% | (28,29) |
| RBP3 | Recessive | Retinal metabolism | <1% | (30,31) |
| RGR | Recessive | Retinal metabolism | <1% | (32) |
| RLBP1 | Recessive | Retinal metabolism | <1% | (33,34) |
| ABCA4 | Recessive | Retinal metabolism | 2-5% | (3,35) |
| MVK | Recessive | Retinal metabolism/unknown | Unknown | (36) |
| IDH3B | Recessive | Citric acid cycle | <1% | (37) |
| PRPF31 | Dominant | Splicing | 5-10% | (38-40) |
| PRPF8 | Dominant | Splicing | 2-3% | (39,41) |
| PRPF3 | Dominant | Splicing | 1% | (39,42) |
| PRPF4 | Dominant | Splicing | Unknown | (43,44) |
| PRPF6 | Dominant | Splicing | Rare | (45) |
| RP9/PAP1 | Dominant | Splicing | Rare | (46,47) |
| SNRNP200 | Dominant | Splicing | 1-2% | (48-50) |
| DHX38 | Recessive | Splicing | Unknown | (51) |
| NR2E3 | Dominant & recessive | Transcription factor | 1-2% & Rare | (52,53) |
| CRX | Dominant | Transcription factor | 1% | (54,55) |
| ZNF513 | Recessive | Transcription factor | <1% | (56,57) |
| RP1 | Dominant & recessive | Tissue development & maintenance | 3-4% & <1% | (58,59) |
| NRL | Dominant & recessive | Tissue development & maintenance | Rare & <1% | (60,61) |
| SEMA4A | Dominant | Tissue development & maintenance | Rare (3-4% in Pakistan) | (62) |
| FAM161A | Recessive | Tissue development & maintenance | <1% | (63,64) |
| TULP1 | Recessive | Tissue development & maintenance | <1% | (65,66) |
| CRB1 | Recessive | Tissue development & maintenance | 6-7% in Spain | (67,68) |
| RP2 | X-linked | Tissue development & maintenance | 10-20% (X-linked) | (69,70) |
| ARL2BP | Recessive | Photoreceptor maintenance & function | Unknown | (71) |
| IMPDH1 | Dominant | Regulates cell growth | 2-3% | (2,72) |
| USH2A | Recessive | Cellular structure | 10-15% | (73,74) |
| FSCN2 | Dominant | Cellular structure | Rare (3% in Japan) | (75,76) |
| ROM1 | Dominant | Cellular structure | Rare | (77,78) |
| IMPG2 | Recessive | Cellular structure | <1% | (79,80) |
| MAK | Recessive | Cellular structure | <1% | (81,82) |
| PROM1 | Recessive | Cellular structure | <1% | (83,84) |
| PRPH2 | Dominant | Photoreceptor OS structure | 5-10% | (85-87) |
| CLRN1 | Recessive | Photoreceptor structure | <1% | (88) |
| DHDDS | Recessive | Photoreceptor structure | <1% | (89,90) |
| MERTK | Recessive | Transmembrane protein | <1% | (91,92) |
| TTC8 | Recessive | Transmembrane protein | <1% | (93) |
| ARL6 | Recessive | Transmembrane protein | <1% | (94,95) |
| BEST1 | Dominant & recessive | Anion channel | Rare & <1% | (96,97) |
| RPGR | X-linked | Intraflagellar transport | 70-90% (X-linked) | (4,98,99) |
| KLHL7 | Dominant | Ubiquitin—proteasome protein degradation | 1-2% | (100,101) |
| TOPORS | Dominant | Ubiquitin-protein ligase | 1% | (102,103) |
| EYS | Recessive | Cell signalling | Common in China (10-30% in Spain) | (104,105) |
| CERKL | Recessive | Cell signalling | 3-4% in Spain | (106,107) |
| KIZ | Recessive | Cell division | <1% in North African Sephardic Jews | (108) |
| NEK2 | Recessive | Cell division | Unknown | (109) |
| CA4 | Dominant | Unknown | Rare | (110,111) |
| C2orf71 | Recessive | Unknown | <1% | (112,113) |
| C8orf37 | Recessive | Unknown | <1% | (114,115) |
| PRCD | Recessive | Unknown | <1% | (116,117) |
| SPATA7 | Recessive | Unknown | <1% | (118,119) |
| EMC1 | Recessive | Unknown | Unknown | (120) |
| GPR125 | Recessive | Unknown | Unknown | (120) |
| KIAA1549 | Recessive | Unknown | Unknown | (120) |
| SLC7A14 | Recessive | Unknown | ~2% | (121) |
~, adapted from sources http://www.ncbi.nlm.nih.gov/books/NBK1417/ & https://sph.uth.edu/Retnet/; *, dominant, autosomal dominant; recessive, autosomal recessive.
Mutations in the gene RHO, encoding rhodopsin which is critical in phototransduction, are a leading cause of RP. Rhodopsin is a 7-transmembrane spanning protein making up approximately 80% of the protein found in the disc membrane of rod outer segments. RHO contains five exons that encode 348 amino acids. The primary function of the protein is to initiate the phototransduction cascade by facilitating the conformational change of 11-cis retinal into 11-trans retinal. Mutations in this gene are seen in ~20-30% of autosomal dominant RP while autosomal recessive inheritance is also observed with certain mutations, but much more rarely. Mutations that lead to recessive forms of inheritance are suggested to still confer a phenotype in the heterozygous state, however it is milder or with onset later in life (124). The severity of the phenotype appears to depend on the location of the mutation in the protein. Patients with mutations leading to abnormal amino acids in the parts of rhodopsin located in the intradiscal space show a less severe phenotype with better visual acuity and improved dark light adaptation, compared with those where mutations affect amino acids in the cytoplasmic space. Patients with mutations occurring within the transmembrane regions of rhodopsin showed an intermediate outcome (125). Intrafamilial variation is also noted amongst patients indicating the likely presence of genetic modifiers and/or environmental factors contributing to the phenotypic effects that are seen (124).
Other RP disease genes implicated in the phototransduction process have an expected frequency of less than 2-5% amongst affected individuals. Autosomal dominant inheritance is observed due to mutations in RDH12, encoding retinol dehydrogenase-12, which is responsible for metabolising all-trans and -cis retinols (14). GUCA1B encodes guanylate cyclase-activating protein 2, which is responsible for activating photoreceptor guanylate cyclases for the conversion of cGMP to cGTP involved with the hyperpolarisation response to light (13). Mutations in this gene lead to autosomal dominant RP. Autosomal recessive inheritance is seen associated with mutations in PDE6A and PDE6B, which encode phosphodiesterase 6A & 6B responsible for the α and β subunits respectively of a key enzyme that maintains cytoplasmic cGMP concentration crucial for rod cell phototransduction (16,18). Mutations in CNGA1 and CNGB1 also follow an autosomal recessive inheritance pattern and these genes encode the α and β subunits of cyclic nucleotide gated ion channels responsible for opening of calcium channels after binding of the cGMP/cGTP ligand (21,23). The SAG gene encodes the arrestin protein responsible for the inactivation of the phototransduction cascade, specifically acting on the activated rhodopsin molecule. While mutations in SAG are implicated in a form of CSNB, discussed below, a homozygous 1-bp deletion (c.1147delA) has been observed in three unrelated individuals with RP (20).
Mutations in RP disease-causing genes that encode proteins associated with retinal metabolism generally follow an autosomal recessive inheritance pattern. These include ABCA4, which encodes the ATP-binding cassette subfamily A member-4, a transmembrane protein that facilitates the removal of all-trans retinaldehyde from the photoreceptor (3); the LRAT gene which encodes lecithin retinol acyltransferase, an enzyme located in the RPE that initiates the reactions where all-cis retinal is derived from all-trans retinol (vitamin A) (28); the RBP3 gene which encodes retinal binding protein 3 that is secreted from rod photoreceptors and responsible for the transportation of retinoids from the photoreceptor to the RPE and for the binding of fatty acids in the interphotoreceptor matrix (30); the RGR gene which encodes the G protein-coupled receptor retinal that is located in the RPE and preferentially binds all-trans retinal facilitating its conversion into 11-cis retinal (32); and the RLBP1 gene which encodes retinaldehyde-binding protein 1 found in the RPE (33). RPE65 that encodes retinal specific protein 65 kD is responsible for the conversion of all-trans retinyl ester to 11-cis retinol in the RPE. Mutations in this gene are most usually associated with a severe autosomal recessive form of RD called LCA (26). Interestingly, mutations in RPE65 have also been observed segregating in an autosomal dominant pattern in RP, highlighting further the variation of outcomes from mutations within the same gene (27).
Genes encoding splicing factors have also been implicated in the expression of an RP phenotype and all follow an autosomal dominant inheritance pattern. These genes include PRPF31, PRPF8, PRPF3, PAP1, SNRNP200 and PRPF6 (38,41,42,45,46,48). It is interesting that despite deficiencies in splicing having an effect on processes of the entire cell, mutations in the above-mentioned genes only confer an RP phenotype, and there are various reasons considered for this. These include that the splicing factor affected acts specifically on genes which are expressed in the retina, such as RHO (126), or that the splicing of genes outside the retina may be affected, but the phenotype is only evident in the retina due to its specific rapid turnover requirements (127). Some genes may be important in spliceosome assembly and maturation, and studies are ongoing to understand the mechanisms causing the splicing abnormalities (128,129).
Retinal tissue development, differentiation and maintenance is critical for proper photoreceptor function, and mutations in genes encoding factors critical in these processes can cause RP including: RP1, NR2E3, CRX, NRL, SEMA4A, FAM161A, TULP1, RP2, CRB1 and IMPDH1 (2,52,58,60,62,63,65,67,69,130). A subgroup of these genes, namely CRX, NRL and NR2E3 are known to interact during retinal neurogenesis. CRX is a transcription factor that regulates retinal cellular differentiation and also plays a role in the maintenance of neural bipolar cells in the adult retina (54). Interestingly a p.Arg41Gln missense mutation has been shown to cause RP in one study while being associated with cone-rod dystrophy (CORD) in another (55,131). NRL encodes a neural retina leucine zipper transcription factor and NR2E3 encodes a ligand-activated transcription factor. These are exclusively expressed in rod photoreceptors and are required for their differentiation during retinal development (52,60).
The functions of the photoreceptors can only take place through specialised cellular structures and development and maintenance of these are critical for normal retinal function. Mutations in genes encoding proteins in this group that have been associated with autosomal recessive RP include: PROM1, MAK, IMPG2, DHDDS, CLRN1, and USH2A (73,79,81,83,88,89) while genes associated with autosomal dominant RP include: FSCN2, ROM1 and PRPH2 (75,77,85,86). PROM1 encodes Prominin-1, which is a highly conserved protein across the animal kingdom that plays a crucial role in disc membrane morphogenesis in rod photoreceptors (83). MAK encodes the male germ cell-associated kinase involved in regulation of cilium length that connects the outer segments to the cell bodies within the photoreceptor (81). The interphotoreceptor matrix proteoglycan is encoded by the gene IMPG2, and is a part of the extracellular matrix which connects the photoreceptor outer segments to the RPE (79). DHDDS encodes a dehydrodolichyl diphosphate synthase, which localises to the inner segments of the photoreceptor and plays an important role in the glycosylation of rhodopsin (89). CLRN1 and USH2A encode clarin-1 and usherin, respectively, and are both implicated in autosomal recessive forms of syndromic and non-syndromic RP. The clarin-1 protein contains five transmembrane domains that secure the protein into the plasma membrane and is thought to include roles in both hair cell and photoreceptor synapses (88). Usherin is a plasma membrane bound protein with a large extracellular domain, which performs structural and signalling functions through interactions with the cilium of photoreceptors and hair cells (73). Usherin knockout mouse models reveal progressive degeneration of the retina indicating usherin is involved in photoreceptor maintenance (132). Mutations in CLRN1 and USH2A are predominantly associated with Usher syndrome, a disorder which presents with both retinal degeneration and sensorineural hearing loss (132,133).
BEST1 mutations are found in a group of RDs termed ‘Bestrophinopathies’ inherited in both autosomal dominant and recessive patterns (96,134). The BEST1 protein is predominately expressed in the basolateral membrane of the RPE and has been proposed to be an ion channel regulator interacting with calcium activated chloride channels (CaCC). The majority of missense mutations are found in the conserved N-terminal of the protein which contains the membrane spanning and calcium interaction domains (135).
Congenital stationary night blindness (CSNB)
CSNB is a non-progressive form of night blindness, also known as nyctalopia, where patients find it difficult to see in relatively low light intensities. Presentation onset is from an early age and can also include decreased visual acuity, myopia, nystagmus and strabismus (136). Photoreceptor function as measured by electroretinogram (ERG) may indicate absence of rod pathway function, or there may be incomplete rod and cone dysfunction. Varying abnormalities may be observed on fundus examination. Complete CSNB results from defects in bipolar cell signalling pathways, resulting in only one intact alternate pathway (137). There are currently 11 genes where mutations have been identified in patients with CSNB (Table 2). Known functions of these genes include roles in calcium channel function with respect to CACNA1F, CACNA2D4 and TRPM1 (137,141,148), calcium-binding in CABP4 (142), glutamate receptor functions in GRM6 (143) and involvement in the phototransduction cascade in GNAT1, GRK1, PDE6B, SAG and RHO (138-140,146,147).
Table 2. Disease genes: human congenital stationary night blindness.
| Genes~ | Inheritance* | Phenotype | Studies |
|---|---|---|---|
| GNAT1, PDE6B, RHO | Dominant | Night blindness | (138-140) |
| CABP4, CACNA2D4 | Recessive | Incomplete night blindness | (141,142) |
| GRM6, TRPM1 | Recessive | Night blindness | (137,143-145) |
| SAG, GRK1 | Recessive | Oguchi disease | (146,147) |
| CACNA1F | X-Linked | Incomplete night blindness (CSNB2) | (148,149) |
| NYX | X-Linked | Night blindness (CSNB1) | (150) |
~, adapted from source https://sph.uth.edu/Retnet/; *, dominant, autosomal dominant; recessive, autosomal recessive.
The incomplete form of X-linked CSNB (CSNB2) is caused by disruption of the CACNA1F gene, which encodes the α1F subunit of calcium channels located at the synaptic connection between the rod photoreceptor and the bipolar cell (148,149). The calcium channel regulates the release of glutamate into the synaptic cleft depending on the membrane potential of the bipolar cell or photoreceptor. It has been shown that loss of function mutations of the gene disrupt the calcium ion flow at this synapse resulting in a non-functional channel and therefore loss of function of the photoreceptor (151). The CSNB2 phenotype that is observed includes a diminished scotopic b-wave on ERG, indicating a diminution in the signalling between rod photoreceptors and bipolar cells (149). There have been more than 50 reported mutations in this gene that result in various forms of protein truncation and calcium channel loss of function (136,152).
The complete form of X-linked CSNB (CSNB1) is caused by mutations in the gene NYX, which is predominantly expressed in the retina. Although the exact mechanism is yet to be known, the encoded protein nyctalopin is a small leucine-rich proteoglycan that when mutated is believed to disrupt bipolar amacrine and ganglion cell signalling (150). Truncated proteins are typically non-functional and would be expected to cause the phenotype observed. The CSNB1 phenotype is distinguished from CSNB2 with ERG as both rod and cone photoreceptor function is affected (150). Deletions within the gene have also been observed in mice which have resulted in the no b-wave (“nob”) phenotype (153).
Autosomal recessive forms of CSNB can be broken up into 3 clinical sub-types including incomplete forms, complete forms and Oguchi disease. Oguchi disease is clinically distinct from the other autosomal recessive CSNBs due to characteristic ERG abnormalities including an absent rod response with normal cone responses. Fundal changes known as the Mizuo-Nakamura phenomenon are also observed whereby during light exposure and adaption the retina has a metallic, golden brown appearance. This appearance disappears however after the retina is returned to a complete dark adaptation (154). There are now two forms of Oguchi disease known, type 1 and type 2, which are caused by mutations in SAG and the G-protein dependent receptor kinase 1 gene GRK1 respectively. Investigations into a cohort of Japanese patients indicated that arrestin and rhodopsin kinase genes are possible candidates for the phenotype due to their association with the inactivation of rhodopsin in the recovery phase of phototransduction (146). A SAG c.1147delA mutation was observed more frequently in affected unrelated Japanese individuals, which reflects a founder effect (147). Mutations in SAG have also been found in patients with an RP phenotype, suggesting variable expression associated with mutations within the gene (20). GRK1 mutations are mainly reported to cause an Oguchi phenotype, although there have also been reports of an RP phenotype (155).
Autosomal dominant forms of CSNB are caused by mutations in GNAT1, PDE6B, and RHO, all of which are involved in aspects of the phototransduction cascade (138,139,146). GNAT1 encodes Guanine nucleotide-binding protein α, α-transducing activity polypeptide 1 which assembles the α subunit in the rod transducing protein that stimulates the coupling of rhodopsin and GMP during the photoreceptor visual response. PDE6B codes for the β subunit of a membrane bound enzyme, phosphodiesterase 6B, that is responsible for the hyperpolarization of the rod photoreceptor. Mutations in RHO are more usually seen in patients with autosomal dominant RP but have also been identified in CSNB depending on the amino acids that are affected (156). One study identified the RHO c.884C>T:p.Ala295Val missense mutation in a family with autosomal dominant CSNB, where patients had a diminished sensitivity and response to light at lower intensities without any additional retinal degeneration (157).
Cone and cone-rod dystrophies
Cone and cone-rod dystrophies present as more severe compared with the rod or rod-cone dystrophies, as it is high acuity vision and the perception of colour that is lost. Nystagmus and photophobia also occur, and in the cone-rod dystrophies, complete blindness occurs in the later stages because the rod photoreceptors also undergo degeneration (158). As in the rod dominated dystrophies, progressive and stationary forms of cone and cone-rod dystrophies may occur.
Progressive cone and cone-rod dystrophies
The onset of progressive cone (COD) and CORD is usually during early childhood or adolescence. Affected individuals usually present with only cone photoreceptor involvement (COD) or cone followed by rod degeneration (CORD). Differences between the two dystrophies are apparent with additional rod involvement leading to an increase in severity with most sufferers reaching legal blindness by the age of 40 (158). On fundus examination, appearance of the macula varies with some cases presenting with an atrophic appearance (Figure 3), or retinal pigment deposits. Currently there are over 30 genes described with reported disease-causing mutations (Table 3), with various roles in similar functional groupings as those described for the rod dystrophies, but with specificity of function to the cone photoreceptors. As shown in previous review of the literature, molecular causes are able to be identified in approximately 20% and 74% of autosomal dominant and X-linked COD/CORD respectively, while 23-25% of autosomal recessive pedigrees can be resolved (200). This indicates that many underlying disease genes remain to be identified especially in the autosomal dominant and recessive forms of these conditions.
Figure 3.

Cone dystrophy: fundal images. (A,B) Wide field fundus photography illustrating retinal features of cone dystrophy, showing macular atrophy in (A) (arrow), compared with the normal macular appearance present in (B); (C,D) OCT in cone dystrophy illustrates the loss of the foveal photoreceptor outer segments in (C), compared with normal in (D) (arrows). OCT, optical coherence tomography.
Table 3. Disease genes: human cone and cone-rod dystrophy.
| Gene~ | Inheritance* | Potential function | Studies |
|---|---|---|---|
| GUCA1A | Dominant | Phototransduction | (159,160) |
| GUCY2D | Dominant | Phototransduction | (161,162) |
| PDE6C | Recessive | Phototransduction | (163) |
| PDE6H | Recessive | Phototransduction | (164,165) |
| CNGB3 | Recessive | Phototransduction | (166,167) |
| PRPH2 | Dominant | Phototransduction | (168,169) |
| ABCA4 | Recessive | Retinal Metabolism | (170,171) |
| RDH5 | Recessive | Retinal metabolism | (172,173) |
| CRX | Dominant | Transcription factor | (130,131) |
| RAX2 | Recessive | Transcription | (174,175) |
| RPGRIP1 | Recessive | Interacts with RPGR | (176) |
| ADAM9 | Recessive | Cell/matrix interaction | (177) |
| AIPL1 | Dominant | Transport, protein trafficking | (178,179) |
| TTLL5 | Recessive | Cilia function | (180) |
| CACNA1F | X-Linked | Calcium channel | (181) |
| CACNA2D4 | Recessive | Ion channel | (142) |
| HRG4 | Dominant | Neurotransmitter release | (182) |
| KCNV2 | Recessive | Ion channel subunit | (183,184) |
| RIMS1 | Dominant | Neurotransmitter release | (185,186) |
| PITPNM3 | Dominant | Transport | (187) |
| SEMA4A | Dominant & recessive | Axon guidance | (62,188) |
| CERKL | Recessive | Cell signalling | (189) |
| RPGR | Dominant & recessive | Intraflagellar transport | (190,191) |
| PROM1 | Dominant | Cellular structure | (192,193) |
| CDHR1 | Recessive | Cellular structure | (194,195) |
| C21orf2 | Recessive | Unknown | (120) |
| C8orf37 | Recessive | Unknown | (114,115) |
| CNNM4 | Recessive | Unknown | (196,197) |
| RAB28 | Recessive | Unknown | (198,199) |
~, adapted from source https://sph.uth.edu/Retnet/; *, dominant, autosomal dominant; recessive, autosomal recessive.
Subgroups of genes where mutations lead to COD and CORD are also associated with other forms of RD, presumably based on their relatively ubiquitous functions across the photoreceptors and/or retina. As an example, ABCA4 mutations are further implicated in RP, and a form of juvenile macular degeneration also known as Stargardts disease (3,201). ABCA4 is the most frequently identified disease gene in COD and CORD of autosomal inheritance with studies reporting its frequency in 9% and 26% of cases respectively (158). Furthermore, truncating mutations involving ABCA4 are more evident in the CORD cohort compared to the COD cohort, reportedly seen in 76% and 63% respectively (158,200).
Achromatopsia
The stationary forms of cone dystrophy can exist in two forms whereby complete or incomplete achromatopsia results in the loss of all colour perception, or the perception of only a specific colour respectively. Tritanopia, or defective blue vision is an autosomal dominant phenotype that is caused by mutations in the gene OPN1SW, which encodes the short wave sensitive opsin that detects blue light (202). Genes currently known to be associated with autosomal recessive complete achromatopsia include CNGA3, CNGB3, GNAT2, PDE6C and PDE6H. CNGB3 alone is responsible for up to 50% of complete achromatopsia in affected individuals (203). CNGA3 and CNGB3 encode the α and β subunits of cGMP-gated channels located in the cone photoreceptor, which are involved in key steps of phototransduction (204,205). GNAT2 encodes the cone-specific α subunit of transducin, which is the cone visual pigment that induces one of the first steps of the phototransduction cascade (206). PDE6C and PDE6H encode the cone specific α and gamma subunits of a cGMP phosphodiesterase respectively, which is an enzyme responsible for the conversion of cGMP to 5’-GMP during light exposure (163,164).
The genetic overlap across the non-syndromic RDs including RP, COD/CORD and the stationary forms is quite complex and is illustrated in Figure 4. Genetic overlap is expected to a certain degree due to fundamental similarities in photoreceptor structures and cellular processes despite independence of their scotopic and photopic roles.
Figure 4.
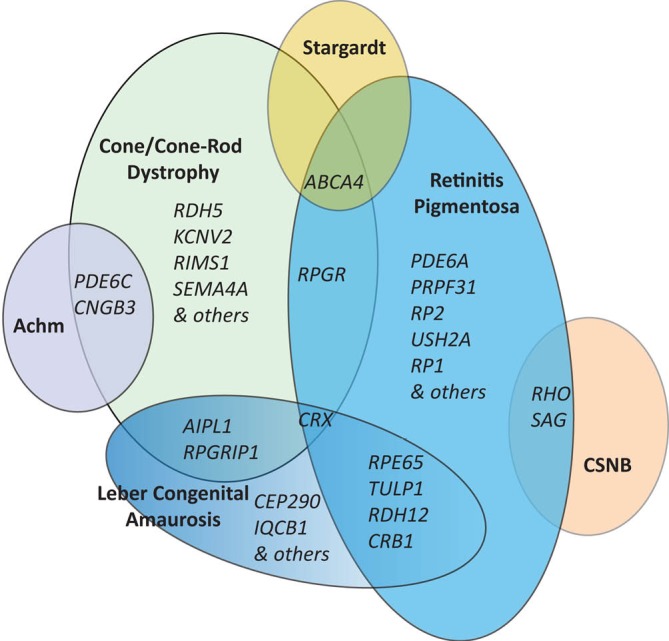
Genetic heterogeneity in retinal dystrophies. Diagrammatic representation of overlap between genetic causes of various forms of retinal dystrophy with some example genes shown. Achm, achromatopsia; CSNB, congenital stationary night blindness.
Generalised non-syndromic RDs
RDs involving the simultaneous degradation of both rod and cone photoreceptor functions are termed generalized RDs. The majority of cases present with progressive, often severe, deterioration of vision. Both syndromic and non-syndromic forms exist.
Leber congenital amaurosis (LCA)
The most common non-syndromic generalised RD is LCA. The onset of LCA is early with affected individuals developing symptoms within the first year of life. Clinical features include poor vision, nystagmus, and no measurable light response on ophthalmic examination with ERG (207). A characteristic finding is Franschetti’s oculo-digital sign where patients repeatedly rub and poke their eyes. The physical appearance of the retina varies in early stages, however retinal pigmentary changes can be observed with progression of disease (207). Interestingly, there have been suggestions of genotype-phenotype patterns of retinal appearance including a translucent RPE appearance with white dots with RPE65 gene mutations and progressive macular atrophy prominent in NMNAT1, but which is also seen in patients with AIPL1 and CRB1 mutations (208). To date there are over 20 genes associated with LCA with nearly all following an autosomal recessive inheritance pattern (Table 4). Despite this, the underlying genetic causes of LCA are not fully known (231), with genetic overlap with other forms of RD present (Figure 4).
Table 4. Disease genes: Leber congenital amaurosis.
| Gene~ | Inheritance* | Potential function | Estimated frequency | Studies |
|---|---|---|---|---|
| GUCY2D | Recessive | Phototransduction | 6-21% | (209,210) |
| RDH12 | Recessive | Phototransduction | ~4% | (14,15) |
| LRAT | Recessive | Retinal metabolism | <1% | (28,211) |
| RPE65 | Recessive | Visual cycle | 3-16% | (209,212) |
| RD3 | Recessive | Splicing | Rare | (213,214) |
| CRX | Dominant & Recessive | Transcription factor | ~3% | (215,216) |
| OTX2 | Dominant | Transcription factor | Rare | (217) |
| CRB1 | Recessive | Tissue development and maintenance | 10% | (209,218) |
| TULP1 | Recessive | Tissue development & maintenance | 1-2% | (209,219) |
| IMPDH1 | Dominant | Regulates cell growth | Rare | (72,220) |
| GDF6 | Recessive | Growth factor | Unknown | (221) |
| CABP4 | Recessive | Cell signalling | Unknown | (222) |
| AIPL1 | Recessive | Transport, protein trafficking | 4-8% | (178,209) |
| CEP290 | Recessive | Centrosomal & ciliary protein | <30% | (223) |
| IQCB1 | Recessive | Interacts with RPGR & connecting cilia | Unknown | (224) |
| LCA5 | Recessive | Centrosome protein with ciliary function | 1-7% | (225,226) |
| NMNAT1 | Recessive | Photoreceptor maintenance | 5% | (208,227) |
| RPGRIP1 | Recessive | Interacts with RPGR | ~5% | (209,228) |
| KCNJ13 | Recessive | Potassium channel | Unknown | (229) |
| DTHD1 | Recessive | Unknown | Unknown | (120) |
| SPATA7 | Recessive | Unknown | 2% | (119,230) |
~, adapted from sources http://www.ncbi.nlm.nih.gov/books/NBK1298/ & https://sph.uth.edu/Retnet/); *, dominant, autosomal dominant; recessive, autosomal recessive.
Choroideremia
The only X-linked form of non-syndromic generalised RD is choroideremia, which is caused by mutations in the gene CHM (232). Patients present during the second decade of life with night blindness and progressive degeneration of photoreceptors, the RPE and the choroid. Characteristically, affected males have an appearance of chorioretinal scalloped atrophy in the midperipheral fundus. CHM encodes REP-1, a subunit of the intracellular trafficking protein rab protein 1 which is responsible for the intracellular transport of proteins and organelles (233). Multiple non-synonymous mutations along with insertions and deletions have been reported to be associated with the disease (234). Heterogeneity is evident among choroideremia patients with some atypical presentations being first identified as RP on clinical examination. Studies using genomic tools to provide molecular diagnosis are proving useful in refining clinical diagnosis (235).
This review of clinical features and underlying genetic causes in the non-syndromic degenerative and stationary RDs including RP, COD/CORD, LCA, CSNB and achromatopsia indicates that while there are specific groupings, there is also a degree of clinical overlap and genetic causes in these conditions (Figure 4). Knowledge of these overlapping clinical features and genetic causes is important in design of molecular diagnostic approaches, molecular genetic result interpretation and work towards therapies in these conditions.
Molecular diagnosis and NGS
Providing a molecular diagnosis in RD is challenging due to the large numbers of genes responsible, variable expression, incomplete penetrance, oligogenic inheritance and frequent clinical and genetic overlap, as discussed above. The value of traditional technologies such as Sanger sequencing for detecting mutations in diseases with high genetic heterogeneity is limited, due to the large amounts of time and labour required to individually sequence many genes and the consequent high costs. Other technologies, such as APEX genotyping microarray chips, can examine multiple variants in multiple genes simultaneously and have provided some advantages in detecting genetic aberrations (236). This technology has been applied to several forms of RD including non-syndromic CORD and syndromic forms of RD such as Usher syndrome (237,238). However, the APEX technology has limited resolving power as the genotyping array only detects a fixed number of mutations from a fixed number of genes. In RD, many of the causative mutations are novel, so the value of a chip with a limited number of known mutations is limited. The contribution of copy number variation (CNV) in RD is yet to be fully explored, with only limited studies so far reported using low resolution comparative genomic hybridisation (CGH) arrays or limited targeted approaches such as MLPA in specific genes (6,239). These technologies are limited in RD diagnosis due to the low effective mean resolution or relatively small number of targets that can be realistically examined.
The advent of NGS or massively parallel sequencing (MPS) has seen a rapid increase in the amount of genetic information that can be examined in a single sequencing assay. NGS is a relatively new technology that allows sequencing of targeted exonic regions, whole exomes and whole genomes of patients in a fast and relatively cheaper manner than ever before (240). The ability to simultaneously sequence regions in parallel allows for hundreds or thousands of sequencing fragments or ‘reads’ to cover a single region. This approach achieves accurate large scale sequencing, resulting in possible applications in a diagnostic setting (241). The use of NGS technologies for CNV detection is yet to be fully explored and appears to be a promising method of examining the whole genome at a much higher resolution than previously possible. NGS approaches can be used to detect CNVs via the quantification of the number of reads; however this relies heavily on the depth of coverage and the quality of data (242). Various bioinformatics tools have been designed to aid in the analysis of the large amounts of data generated from NGS (243,244). Some of these including those for: alignment (245), variant annotation (246) and conservation (247) are publicly available or in commercially distributed software packages (245,246). Powerful web tools such as 1,000 genomes (248) allow for minor allele frequencies (MAF) to be obtained while others such as SIFT (249), MutationTaster (250) and PolyPhen2 (251) generate pathogenicity prediction scores, which are essential in prioritising the often extensive list of variants detected.
In the last 5 years, efforts have been focused on applying NGS technology to mutation detection in RD (241,252). Some groups have opted to develop their own purpose-built panels which sequence a predetermined list of known disease genes. This approach provides a significant advance over the previous laborious gene by gene Sanger sequencing approach. It is also an advance on APEX technologies which only examined specific variants in specific genes. It has provided capacity for variant detection in approximately 50% of patients with RP where previously most patients were unable to receive a molecular diagnosis because of the cost and inefficiencies of the previous detection methods (123). By only examining known disease genes in a predetermined list, this approach reduces the number of variants detected, therefore only providing information where a clinical interpretation can be made with relative assurance (252,253). However, novel disease genes in the RDs are being discovered at a rapid rate, so this approach is limited in its flexibility to include more disease genes as they are discovered. In addition, these approaches are limited in their ability to detect variants in regulatory regions that are not included in the targeted strategy or may be less efficient in detection of copy number and structural variants (242).
A more broad-based NGS strategy such as whole exome sequencing (WES) or whole genome sequencing (WGS) can provide an enhanced method of investigation of disease gene regions over a predetermined targeted sequencing and capture approach (254-256). Variants in non-relevant genes can be filtered using bioinformatics strategies to reduce incidental findings. WES and WGS provide the flexibility to examine newly identified disease genes since all genes are captured and the bioinformatic filter can be modified to include examination of novel disease genes. WGS provides additional capacity in identification of copy number and structural variants (257). With multiple commercial companies investing in various types of NGS technology, competition is encouraging regular product improvements, enhancements and lowering of costs, making this a viable diagnostic technology.
Therapies and future directions
Advances in stem cell and genome editing technologies are the catalysing factors in the development of gene-based therapies and treatment options in RD. Various treatment strategies investigating the applications of gene based technologies, cell based therapies and retinal implant or transplantation are actively being sought in the RD’s. The type of RD, the extent of retinal deterioration and the cellular structures or processes affected ultimately determine the appropriateness of which treatment strategy is applied. The rate and amount of disease progression is the limiting factor determining the therapeutic approach which is likely to be successful. Improvements in diagnostic technologies and the understanding of molecular pathogenesis are expected to decrease the time needed to reach a molecular diagnosis, and therefore initiate a treatment before subsequent disease progression. As an example, clinical trials of RPE65 gene replacement therapy delivered via viral vectors have shown promise, with children affected by LCA followed for three years post treatment all showing improvements in rod and cone photoreceptor function (258). In patients with more advanced disease progression, it is likely that a gene therapy approach will not be as effective due to the extent of deterioration. In these cases, a cell replacement or retinal implant approach is likely to be more appropriate, whereby stem cells or a visual prosthesis are delivered to the diseased retina.
Retinal implants or prosthetics are an area actively being explored with the potential of treating the RDs. Also sometimes referred to as ‘bionic eyes’, patients have electronic devices inserted into the retina and utilise the remaining intact neural network to transmit the signals to the visual centres of the brain. The detection of light is performed via a light sensing microchip inserted into the central or peripheral visual field. Implantation of an electrode into the neural layers of the retina enables a connection bridge to the existing neural network (259). Due to the rather intrusive nature of surgery required, applications are often restricted to preserve any remaining limited vision the patient may have (260). This technology has been successfully applied in RP patients with varying stages of disease progression and has resulted in improvements to light detection and some restoration of visual perception (260,261).
More than a decade ago there was success in gene replacement therapies applied to RD using animal models (262). More recently there has been progress in human trials, specifically in the delivery of functional cDNA to the retina via adeno-associated virus (AAV) vectors (263-265). There is also hope in the prospect of use of pluripotent stem cells in replacing disease-affected retinal cells. Stem cells are capable of differentiating into specific retinal cell types and an unlimited number of identical daughter cells can be produced. Work is progressing in delivery of these cells via injection to the accessible eye. Embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs) and retinal progenitor cells (RPCs) are examples of stem cells that have been applied, and have demonstrated regeneration in diseased retinal mouse models (266-268). The use of ESCs is practical and documented in mouse models with success shown by transplantation and restoration of retinal function (269). However the expansion of both ESC and RPC applications using human-derived cells is limited due to ethical considerations. In contrast, iPSCs are derived from child or adult fibroblasts and therefore have the advantage of increased availability, and since they can be derived from an affected patient, there is a reduced risk of host immune system rejection. IPSCs can be differentiated to retinal cells and can deliver healthy retinal cells into the diseased retina in the mouse, facilitating the repair and restoration of function (267). Examples of successful gene therapy, cell replacement and retinal implant strategies are illustrated in Table 5.
Table 5. Examples of therapies applied in the retinal dystrophies.
| Approach | Context | Strategy summary | Studies |
|---|---|---|---|
| Gene therapy | Human clinical trial | RPE65 delivered by Adeno-associated Virus (AAV) | (263) |
| Mouse model | CHM delivered by Adeno-associated Virus (AAV) | (264) | |
| Human clinical trial | CHM delivered by Adeno-associated Virus (AAV) | (270) | |
| In vitro model | CEP290 delivered by Lentivirus vector | (271) | |
| Rat model | MERTK delivered by Adeno-associated Virus (AAV) | (272) | |
| Mouse model | CRB1 and CRB2 delivered by Adeno-associated Virus (AAV) | (273) | |
| Cell replacement | Rat model | Human RPC transplantation restores mature rat retina cells | (274) |
| Mouse model | Adult mouse iPSC differentiate into photoreceptors | (267) | |
| Mouse model | RPCs differentiate and integrate into functional photoreceptors | (269) | |
| Mouse model | RPCs differentiate into photoreceptors | (268) | |
| Retinal implants | Clinical trials | Argus II Retinal Prosthesis System | (260,261) |
Further development of iPSC applications for treatment of RD in human patients, requires use of efficient genome engineering to reverse or alter the mutation. Advances in genome editing tools have allowed in vitro DNA modification to be as precise as to the single base pair level (275). This has important uses in illustrating the mechanisms and pathophysiology of genetic disease with the induction of targeted mutations, while also aiding in the development of treatments and therapies with the modification of pathogenic mutations back to normal. The most promising of these technologies is the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system, whereby precise genomic regions can be targeted through easily synthesised guide RNA (276). The system then edits the genome through inducing double stranded breaks (DSBs) and subsequent homology-directed repair (HDR) at a precise location, resulting in the newly modified target still being in its same position in the genome so still under the influence of its endogenous control elements such as promoters, enhances and repressors. This is particularly important as it prevents incorrect or inappropriate levels of expression of the newly modified gene. Recently, the specificity of the CRISPR/Cas9 system approach has been improved with the modification of the Cas9 nuclease to induce only a single strand break, or nick, as opposed to DSBs where significant misalignment and pairing has been reported. The single strand break approach allows for the endogenous base excision repair pathway to facilitate repair and results in more specific and efficient modification (277).
Application of the CRISPR/Cas9 system in conjunction with stem cell technologies is likely to pave the way for ‘precision medicine’ and catalyse the future understanding and treatment of genetic disease. It is anticipated that future work will be more individualised, whereby a patient has a blood and skin sample collected for the identification of the pathogenic mutation and the generation of a fibroblast cell line. Once the mutation is identified, the effect of therapy and treatment options can be observed in the fibroblast cell line to examine their efficiency. This could be expanded to include the development of iPSCs from the patient fibroblast line, genome editing to correct the DNA mutation, differential to retinal cells and transplant into the affected retina (278). In highly heterogeneous genetic diseases such as RD where the underlying genetic cause is likely to be unique, this approach is one of the most promising avenues of future research and exploration.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Haim M. The epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol Scand Suppl 2002;(233):1-34. [DOI] [PubMed] [Google Scholar]
- 2.Bowne SJ, Sullivan LS, Blanton SH, et al. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet 2002;11:559-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martínez-Mir A, Paloma E, Allikmets R, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet 1998;18:11-2. [DOI] [PubMed] [Google Scholar]
- 4.Meindl A, Dry K, Herrmann K, Manson F, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet 1996;13:35-42. [DOI] [PubMed] [Google Scholar]
- 5.Daiger SP, Rossiter BJ, Greenberg J, et al. RetNet - Retinal Information Network 1998. Data services and software for identifying genes and mutations causing retinal degeneration]. Available online: http://www.sph.uth.tmc.edu/RetNet/ [updated Dec 08, 2014; cited Jan 23, 2015].
- 6.Pieras JI, Barragán I, Borrego S, et al. Copy-number variations in EYS: a significant event in the appearance of arRP. Invest Ophthalmol Vis Sci 2011;52:5625-31. [DOI] [PubMed] [Google Scholar]
- 7.Ávila-Fernández A, Cantalapiedra D, Aller E, et al. Mutation analysis of 272 Spanish families affected by autosomal recessive retinitis pigmentosa using a genotyping microarray. Mol Vis 2010;16:2550-8. [PMC free article] [PubMed] [Google Scholar]
- 8.Sung CH, Chuang JZ. The cell biology of vision. J Cell Biol 2010;190:953-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daiger SP, Bowne SJ, Sullivan LS. Perspective on genes and mutations causing retinitis pigmentosa. Arch Ophthalmol 2007;125:151-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet 2006;368:1795-809. [DOI] [PubMed] [Google Scholar]
- 11.Dryja TP, McGee TL, Hahn LB, et al. Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med 1990;323:1302-7. [DOI] [PubMed] [Google Scholar]
- 12.Rosenfeld PJ, Cowley GS, McGee TL, et al. A Null mutation in the rhodopson gene causes rod photoreceptor dysfunction and autosomal recessive retinitis pigmentosa. Nat Genet 1992;1:209-13. [DOI] [PubMed] [Google Scholar]
- 13.Sato M, Nakazawa M, Usui T, et al. Mutations in the gene coding for guanylate cyclase-activating protein 2 (GUCA1B gene) in patients with autosomal dominant retinal dystrophies. Graefes Arch Clin Exp Ophthalmol 2005;243:235-42. [DOI] [PubMed] [Google Scholar]
- 14.Janecke AR, Thompson DA, Utermann G, et al. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet 2004;36:850-4. [DOI] [PubMed] [Google Scholar]
- 15.Perrault I, Hanein S, Gerber S, et al. Retinal Dehydrogenase 12 (RDH12) Mutations in Leber Congenital Amaurosis. Am J Hum Genet 2004;75:639-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang SH, Pittler SJ, Huang X, et al. Autosomal recessive retinitis pigmentosa caused by mutations in the alpha subunit of rod cGMP phosphodiesterase. Nat Genet 1995;11:468-71. [DOI] [PubMed] [Google Scholar]
- 17.Dryja TP, Rucinski DE, Chen S, et al. Frequency of mutations in the gene encoding the alpha subunit of rod cGMP-phosphodiesterase in autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 1999;40:1859-65. [PubMed] [Google Scholar]
- 18.McLaughlin ME, Sandberg MA, Berson EL, et al. Recessive mutations in the gene encoding the beta-subunit of rod photodiesterase in patients with retinitis pigmentosa. Nat Genet 1993;4:130-4. [DOI] [PubMed] [Google Scholar]
- 19.Shen S, Sujirakul T, Tsang SH. Next-generation sequencing revealed a novel mutation in the gene encoding the beta subunit of rod phosphodiesterase. Ophthalmic Genet 2014;35:142-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakazawa M, Wada Y, Tamai M. Arrestin Gene Mutations in Autosomal Recessive Retinitis Pigmentosa. Arch Ophthalmol 1998;116:498-501. [DOI] [PubMed] [Google Scholar]
- 21.Dryja TP, Finn JT, Peng YW, et al. Mutations in the gene encoding the alpha subunit of the rod GMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci U S A 1995;92:10177-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhallan RS, Macke JP, Eddy RL, et al. Human rod photoreceptor cGMP-gated channel: amin acid sequence, gene structure, and functional expression. J Neurosci 1992;12:3248-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bareil C, Hamel CP, Delague V, et al. Segregation of a mutation in CNGB1 encoding the beta-subunit of the rod cGMP-gated channel in a family with autosomal recessive retinitis pigmentosa. Hum Genet 2001;108:328-34. [DOI] [PubMed] [Google Scholar]
- 24.Ardell MD, Bedsole DL, Schoborg RV, et al. Genomic organization of the human rod photoreceptor cGMP-gated cation channel beta-subunit gene. Gene 2000;245:311-8. [DOI] [PubMed] [Google Scholar]
- 25.Dvir L, Srour G, Abu-Ras R, et al. Autosomal-recessive early-onset retinitis pigmentosa caused by a mutation in PDE6G, the gene encoding the gamma subunit of rod cGMP phosphodiesterase. Am J Hum Genet 2010;87:258-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morimura H, Fishman GA, Grover SA, et al. Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci U S A 1998;95:3088-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bowne SJ, Humphries MM, Sullivan LS, et al. A dominant mutation in RPE65 identified by whole-exome sequencing causes retinitis pigmentosa with choroidal involvement. Eur J Hum Genet 2011;19:1074-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson DA, Li Y, McHenry CL, et al. Mutations in the gene encoding lecithin retinol acyltransferase are associated with early-onset severe retinal dystrophy. Nat Genet 2001;28:123-4. [DOI] [PubMed] [Google Scholar]
- 29.Ruiz A, Kuehn MH, Andorf JL, et al. Genomic organization and mutation analysis of the gene encoding lecithin retinol acyltransferase in human retinal pigment epithelium. Invest Ophthalmol Vis Sci 2001;42:31-7. [PubMed] [Google Scholar]
- 30.den Hollander AI, McGee TL, Ziviello C, et al. A homozygous missense mutation in the IRBP gene (RBP3) associated with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci 2009;50:1864-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parker RO, Fan J, Nickerson JM, et al. Normal cone function requires the interphotoreceptor retinoid binding protein. J Neurosci 2009;29:4616-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morimura H, Saindelle-Ribeaudeau F, Berson EL, et al. Mutations in RGR, encoding a light sensitive opsin homologue, in patients with retinitis pigmentosa. Nat Genet 1999;23:393-4. [DOI] [PubMed] [Google Scholar]
- 33.Maw MA, Kennedy B, Knight A, et al. Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet 1997;17:198-200. [DOI] [PubMed] [Google Scholar]
- 34.Eichers ER, Green JS, Stockton DW, et al. Newfoundland rod-cone dystrophy, an early-onset retinal dystrophy, is caused by splice-junction mutations in RLBP1. Am J Hum Genet 2002;70:955-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang N, Tsybovsky Y, Kolesnikov AV, et al. Protein misfolding and the pathogenesis of ABCA4-associated retinal degenerations. Hum Mol Genet 2015. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siemiatkowska AM, van den Born LI, van Hagen PM, et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology 2013;120:2697-705. [DOI] [PubMed] [Google Scholar]
- 37.Hartong DT, Dange M, McGee TL, et al. Insights from retinitis pigmentosa into the roles of isocitrate dehydrogenases in the Krebs cycle. Nat Genet 2008;40:1230-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vithana EN, Abu-Safieh L, Allen MJ, et al. A human homolog of yeast pre-mRNA splicing gene PRP31, underlies autosomal dominant retinitis pigmentosa on chromosome 19q13.4. Mol Cell 2001;8:375-81. [DOI] [PubMed] [Google Scholar]
- 39.Farkas MH, Lew DS, Sousa ME, et al. Mutations in pre-mRNA processing factors 3, 8, and 31 cause dysfunction of the retinal pigment epithelium. Am J Pathol 2014;184:2641-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vithana EN. Expression of PRPF31 mRNA in Patients with Autosomal Dominant Retinitis Pigmentosa: A Molecular Clue for Incomplete Penetrance? Invest Ophthalmol Vis Sci 2003;44:4204-9. [DOI] [PubMed] [Google Scholar]
- 41.McKie AB, McHale JC, Keen TJ, et al. Mutations in the pre-mRNA splicing factor gene PRPC8 in autosomal dominant retinitis pigmentosa (RP13). Hum Mol Genet 2001;10:1555-62. [DOI] [PubMed] [Google Scholar]
- 42.Chakarova CF, Hims MM, Bolz H, et al. Mutations in HPRP3, a third member of pre-mRNA splicing factor genes implicated in autosomal dominant retinitis pigmentosa. Hum Mol Genet 2002;11:87-92. [DOI] [PubMed] [Google Scholar]
- 43.Linder B, Hirmer A, Gal A, et al. Identification of a PRPF4 loss-of-function variant that abrogates U4/U6.U5 tri-snRNP integration and is associated with retinitis pigmentosa. PloS one 2014;9:e111754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen X, Liu Y, Sheng X, et al. PRPF4 mutations cause autosomal dominant retinitis pigmentosa. Hum Mol Genet 2014;23:2926-39. [DOI] [PubMed] [Google Scholar]
- 45.Tanackovic G, Ransijn A, Ayuso C, et al. A missense mutation in PRPF6 causes impairment of pre-mRNA splicing and autosomal-dominant retinitis pigmentosa. Am J Hum Genet 2011;88:643-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keen TJ, Hims MM, McKie AB, et al. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur J Hum Genet 2002;10:245-9. [DOI] [PubMed] [Google Scholar]
- 47.Maita H, Kitaura H, Keen TJ, et al. PAP-1, the mutated gene underlying the RP9 form of dominant retinitis pigmentosa, is a splicing factor. Exp Cell Res 2004;300:283-96. [DOI] [PubMed] [Google Scholar]
- 48.Zhao C, Bellur DL, Lu S, et al. Autosomal-dominant retinitis pigmentosa caused by a mutation in SNRNP200, a gene required for unwinding of U4/U6 snRNAs. Am J Hum Genet 2009;85:617-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Benaglio P, McGee TL, Capelli LP, et al. Next generation sequencing of pooled samples reveals new SNRNP200 mutations associated with retinitis pigmentosa. Hum Mutat 2011;32:E2246-58. [DOI] [PubMed] [Google Scholar]
- 50.Bowne SJ, Sullivan LS, Avery CE, et al. Mutations in the small nuclear riboprotein 200 kDa gene (SNRNP200) cause 1.6% of autosomal dominant retinitis pigmentosa. Mol Vis 2013;19:2407-17. [PMC free article] [PubMed] [Google Scholar]
- 51.Ajmal M, Khan MI, Neveling K, et al. A missense mutation in the splicing factor gene DHX38 is associated with early-onset retinitis pigmentosa with macular coloboma. J Med Genet 2014;51:444-8. [DOI] [PubMed] [Google Scholar]
- 52.Coppieters F, Leroy BP, Beysen D, et al. Recurrent mutation in the first zinc finger of the orphan nuclear receptor NR2E3 causes autosomal dominant retinitis pigmentosa. Am J Hum Genet 2007;81:147-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bernal S, Solans T, Gamundi MJ, et al. Analysis of the involvement of the NR2E3 gene in autosomal recessive retinal dystrophies. Clin Genet 2008;73:360-6. [DOI] [PubMed] [Google Scholar]
- 54.Chen S, Wang Q, Nie Z, et al. Crx, a novel otx-like paired-homeodomain protein, binds to and transactivates photoreceptor cell-specific genes. Neuron 1997;19:1017-30. [DOI] [PubMed] [Google Scholar]
- 55.Sohocki MM, Sullivan LS, Mintz-Hittner HA, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet 1998;63:1307-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Nakaya N, Chavali VR, et al. A mutation in ZNF513, a putative regulator of photoreceptor development, causes autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2010;87:400-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Naz S, Riazuddin SA, Li L, et al. A novel locus for autosomal recessive retinitis pigmentosa in a consanguineous Pakistani family maps to chromosome 2p. Am J Ophthalmol 2010;149:861-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pierce EA, Quinn T, Meehan T, et al. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet 1999;22:248-54. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan LS, Heckenlively JR, Bowne SJ, et al. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet 1999;22:255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bessant DA, Payne AM, Mitton KP, et al. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet 1999;21:355-6. [DOI] [PubMed] [Google Scholar]
- 61.Nishiguchi KM, Friedman JS, Sandberg MA, et al. Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc Natl Acad Sci U S A 2004;101:17819-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abid A, Ismail M, Mehdi SQ, et al. Identification of novel mutations in the SEMA4A gene associated with retinal degenerative diseases. J Med Genet 2006;43:378-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Langmann T, Di Gioia SA, Rau I, et al. Nonsense mutations in FAM161A cause RP28-associated recessive retinitis pigmentosa. Am J Hum Genet. 2010;87:376-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bandah-Rozenfeld D, Mizrahi-Meissonnier L, Farhy C, et al. Homozygosity mapping reveals null mutations in FAM161A as a cause of autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2010;87:382-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Banerjee P, Kleyn PW, Knowles JA, et al. TULP1 mutation in two extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nat Genet 1998;18:177-9. [DOI] [PubMed] [Google Scholar]
- 66.Ajmal M, Kahn MI, Micheal S, et al. Identification of recurrent and novel mutations in TULP1 in Pakistani families with early-onset retinitis pigmentosa. Mol Vis 2012;18:1226-37. [PMC free article] [PubMed] [Google Scholar]
- 67.den Hollander AI, ten Brink JB, de Kok YJ, et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet 1999;23:217-21. [DOI] [PubMed] [Google Scholar]
- 68.Yang L, Wu L, Yin X, et al. Novel mutations in CRB1 in Chinese families presenting with retinal dystrophies. Mol Vis 2014;20:359-67. [PMC free article] [PubMed] [Google Scholar]
- 69.Schwahn U, Lenzner S, Dong J, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet 1998;19:327-32. [DOI] [PubMed] [Google Scholar]
- 70.Pomares E, Riera M, Castro-Navarro J, et al. Identification of an intronic single-point mutation in RP2 as the cause of semidominant X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 2009;50:5107-14. [DOI] [PubMed] [Google Scholar]
- 71.Davidson AE, Schwarz N, Zelinger L, et al. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2013;93:321-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bowne SJ, Sullivan LS, Mortimer SE, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and leber congenital amaurosis. Invest Ophthalmol Vis Sci 2006;47:34-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rivolta C, Sweklo EA, Berson EL, et al. Missense Mutation in the USH2A Gene: Association with Recessive Retinitis Pigmentosa without Hearing Loss. Am J Hum Genet 2000;66:1975-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Seyedahmadi BJ, Rivolta C, Keene JA, et al. Comprehensive screening of the USH2A gene in Usher syndrome type II and non-syndromic recessive retinitis pigmentosa. Exp Eye Res 2004;79:167-73. [DOI] [PubMed] [Google Scholar]
- 75.Wada Y, Abe T, Takeshita T, et al. Mutation of the human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2001;42:2395-400. [PubMed] [Google Scholar]
- 76.Gamundi MJ, Hernan I, Maseras M, et al. Sequence variations in the retinal fascin FCSN2 gene in a Spanish population with autosomal dominant retinitis pigmentosa or macular degeneration. Mol Vis 2005;11:922-8. [PubMed] [Google Scholar]
- 77.Kajiwara K, Berson EL, Dryja TP. Digenic RP due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994;264:1604-8. [DOI] [PubMed] [Google Scholar]
- 78.Sakuma H, Iana G, Murakami A, et al. A heterozygous putative null mutation in ROM-1 without a mutation in peripherin/RDS in a family with retinitis pigmentosa. Genomics 1995;27:384-6. [DOI] [PubMed] [Google Scholar]
- 79.Bandah-Rozenfeld D, Collin RW, Banin E, et al. Mutations in IMPG2, encoding interphotoreceptor matrix proteoglycan 2, cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2010;87:199-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.van Huet RA, Collin RW, Siemiatkowska AM, et al. IMPG2-associated retinitis pigmentosa displays relatively early macular involvement. Invest Ophthalmol Vis Sci. 2014;55:3939-53. [DOI] [PubMed] [Google Scholar]
- 81.Tucker BA, Scheetz TE, Mullins RF, et al. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. Proc Natl Acad Sci U S A 2011;108:E569-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Stone EM, Luo X, Heon E, et al. Autosomal recessive retinitis pigmentosa caused by mutations in the MAK gene. Invest Ophthalmol Vis Sci 2011;52:9665-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maw MA, Corbeil D, Koch J, et al. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum Mol Genet 2000;9:27-34. [DOI] [PubMed] [Google Scholar]
- 84.Zhang Q, Zulfiqar F, Xiao X, et al. Severe retinitis pigmentosa mapped to 4p15 and associated with a novel mutation in the PROM1 gene. Hum Genet 2007;122:293-9. [DOI] [PubMed] [Google Scholar]
- 85.Farrar GJ, Kenna P, Jordan SA, et al. A three-base-pair deletion in hte peripherin-RDS gene in one form of retinitis pigmentosa. Nature 1991;354:478-80. [DOI] [PubMed] [Google Scholar]
- 86.Kajiwara K, Hahn LB, Mukai S, et al. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature 1991;354:480-3. [DOI] [PubMed] [Google Scholar]
- 87.Manes G, Guillaumie T, Vos WL, et al. High prevalence of PRPH2 in autosomal dominant retinitis pigmentosa in france and characterization of biochemical and clinical features. Am J Ophthalmol 2015;159:302-14. [DOI] [PubMed] [Google Scholar]
- 88.Khan MI, Kersten FF, Azam M, et al. CLRN1 mutations cause nonsyndromic retinitis pigmentosa. Ophthalmology 2011;118:1444-8. [DOI] [PubMed] [Google Scholar]
- 89.Zelinger L, Banin E, Obolensky A, et al. A missense mutation in DHDDS, encoding dehydrodolichyl diphosphate synthase, is associated with autosomal-recessive retinitis pigmentosa in Ashkenazi Jews. Am J Hum Genet 2011;88:207-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Züchner S, Dallman J, Wen R, et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am J Hum Genet 2011;88:201-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mackay DS, Henderson RH, Sergouniotis PI, et al. Novel mutations in MERTK associated with childhood onset rod-cone dystrophy. Mol Vis 2010;16:369-77. [PMC free article] [PubMed] [Google Scholar]
- 92.Gal A, Li Y, Thompson DA, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet 2000;26:270-1. [DOI] [PubMed] [Google Scholar]
- 93.Riazuddin SA, Iqbal M, Wang Y, et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am J Hum Genet. 2010;86:805-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pretorius PR, Aldahmesh MA, Alkuraya FS, et al. Functional analysis of BBS3 A89V that results in non-syndromic retinal degeneration. Hum Mol Genet 2011;20:1625-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aldahmesh MA, Abu Safieh L, Alkuraya H, et al. Molecular characterization of retinitis pigmentosa in Saudi Arabia. Mol Vis 2009;15:2464-9. [PMC free article] [PubMed] [Google Scholar]
- 96.Petrukhin K, Koisti MJ, Bakall B, et al. Identification of the gene responsible for Best macular dystrophy. Nat Genet 1998;19:241-7. [DOI] [PubMed] [Google Scholar]
- 97.Davidson AE, Millar ID, Urquhart JE, et al. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. Am J Hum Genet 2009;85:581-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roepman R, Bauer D, Rosenberg T, et al. Identification of a gene disrupted by a microdeletion in a patient with X-linked retinitis pigmentosa (XLRP). Hum Mol Genet 1996;5:827-33. [DOI] [PubMed] [Google Scholar]
- 99.Roepman R, van Duijnhoven G, Rosenberg T, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology with the guanine-nucleotide-exchange factor RCC1. Hum Mol Genet 1996;5:1035-41. [DOI] [PubMed] [Google Scholar]
- 100.Wen Y, Locke KG, Klein M, et al. Phenotypic characterization of 3 families with autosomal dominant retinitis pigmentosa due to mutations in KLHL7. Arch Ophthalmol 2011;129:1475-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Friedman JS, Ray JW, Waseem N, et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet. 2009;84:792-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chakarova CF, Khanna H, Shah AZ, et al. TOPORS, implicated in retinal degeneration, is a cilia-centrosomal protein. Hum Mol Genet 2011;20:975-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chakarova CF, Papaioannou MG, Khanna H, et al. Mutations in TOPORS cause autosomal dominant retinitis pigmentosa with perivascular retinal pigment epithelium atrophy. Am J Hum Genet 2007;81:1098-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Abd El-Aziz MM, Barragan I, O'Driscoll CA, et al. EYS, encoding an ortholog of Drosophila spacemaker, is mutated in autosomal recessive retinitis pigmentosa. Nat Genet 2008;40:1285-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Collin RW, Littink KW, Klevering BJ, et al. Identification of a 2 Mb human ortholog of Drosophila eyes shut/spacemaker that is mutated in patients with retinitis pigmentosa. Am J Hum Genet 2008;83:594-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tang Z, Wang Z, Wang Z, et al. Novel compound heterozygous mutations in CERKL cause autosomal recessive retinitis pigmentosa in a nonconsanguineous Chinese family. Arch Ophthalmol 2009;127:1077-8. [DOI] [PubMed] [Google Scholar]
- 107.Tuson M, Marfany G, Gonzalez-Duarte R. Mutation of CERKL, a Novel Human Ceramide Kinase Gene, Causes Autosomal Recessive Retinitis Pigmentosa (RP26). Am J Hum Genet 2004;74:128-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.El Shamieh S, Neuille M, Terray A, et al. Whole-exome sequencing identifies KIZ as a ciliary gene associated with autosomal-recessive rod-cone dystrophy. Am J Hum Genet 2014;94:625-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nishiguchi KM, Tearle RG, Liu YP, et al. Whole genome sequencing in patients with retinitis pigmentosa reveals pathogenic DNA structural changes and NEK2 as a new disease gene. Proc Natl Acad Sci U S A 2013;110:16139-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alvarez BV, Vithana EN, Yang Z, et al. Identification and characterization of a novel mutation in the carbonic anhydrase IV gene htat causes retinitis pigmentosa. Invest Ophthalmol Vis Sci 2007;48:3459-68. [DOI] [PubMed] [Google Scholar]
- 111.Rebello G, Ramesar R, Vorster A, et al. Apoptosis-inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci U S A 2004;101:6617-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Collin RW, Safieh C, Littink KW, et al. Mutations in C2ORF71 cause autosomal-recessive retinitis pigmentosa. Am J Hum Genet 2010;86:783-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nishimura DY, Baye LM, Perveen R, et al. Discovery and functional analysis of a retinitis pigmentosa gene, C2ORF71. Am J Hum Genet 2010;86:686-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.van Huet RA, Estrada-Cuzcano A, Banin E, et al. Clinical characteristics of rod and cone photoreceptor dystrophies in patients with mutations in the C8orf37 gene. Invest Ophthalmol Vis Sci 2013;54:4683-90. [DOI] [PubMed] [Google Scholar]
- 115.Estrada-Cuzcano A, Neveling K, Kohl S, et al. Mutations in C8orf37, encoding a ciliary protein, are associated with autosomal-recessive retinal dystrophies with early macular involvement. Am J Hum Genet 2012;90:102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nevet MJ, Shalev SA, Zlotogora J, et al. Identification of a prevalent founder mutation in an Israeli Muslim Arab village confirms the role of PRCD in the aetiology of retinitis pigmentosa in humans. J Med Genet 2010;47:533-7. [DOI] [PubMed] [Google Scholar]
- 117.Zangerl B, Goldstein O, Philp AR, et al. Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics 2006;88:551-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kannabiran C, Palavalli L, Jalali S. Mutation of SPATA7 in a family with autosomal recessive early-onset retinitis pigmentosa. J Mol Genet Med 2012;6:301-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang H, den Hollander AI, Moayedi Y, et al. Mutations in SPATA7 cause Leber congenital amaurosis and juvenile retinitis pigmentosa. Am J Hum Genet 2009;84:380-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Abu-Safieh L, Alrashed M, Anazi S, et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res 2013;23:236-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jin ZB, Huang XF, Lv JN, et al. SLC7A14 linked to autosomal recessive retinitis pigmentosa. Nature communications 2014;5:3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fahim A, Daiger SP, Weleber RG. Retinitis Pigmentosa Overview 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1417/ [updated Mar 21, 2013; cited Jan 23, 2015].
- 123.Neveling K, Collin RW, Gilissen C, et al. Next-generation genetic testing for retinitis pigmentosa. Hum Mutat 2012;33:963-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Neidhardt J, Barthelmes D, Farahmand F, et al. Different amino acid substitutions at the same position in rhodopsin lead to distinct phenotypes. Invest Ophthalmol Vis Sci 2006;47:1630-5. [DOI] [PubMed] [Google Scholar]
- 125.Sandberg MA, Weigel-DiFranco C, Dryja TP, et al. Clinical Expression Correlated With Location of Rhodopsin Mutation in Dominant Retinitis Pigmentosa. Invest Ophthalmol Vis Sci 1995;36:1934-42. [PubMed] [Google Scholar]
- 126.Yuan L, Kawada M, Havlioglu N, et al. Mutations in PRPF31 inhibit pre-mRNA splicing of rhodopsin gene and cause apoptosis of retinal cells. J Neurosci 2005;25:748-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Travis GH. Mechanisms of Cell Death in the Inherited Retinal Degenerations. Am J Hum Genet 1998;62:503-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Boon KL, Grainger RJ, Ehsani P, et al. prp8 mutations that cause human retinitis pigmentosa lead to a U5 snRNP maturation defect in yeast. Nat Struct Mol Biol 2007;14:1077-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ivings L, Towns KV, Matin MA, et al. Evaluation of splicing efficiency in lymphoblastoid cell lines from patients with splicing-factor retinitis pigmentosa. Mol Vis 2008;14:2357-66. [PMC free article] [PubMed] [Google Scholar]
- 130.Freund CL, Gregory-Evans CY, Furukawa T, et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997;91:543-53. [DOI] [PubMed] [Google Scholar]
- 131.Swain PK, Chen S, Wang Q, et al. Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron 1997;19:1329-36. [DOI] [PubMed] [Google Scholar]
- 132.Liu X, Bulgakov OV, Darrow KN, et al. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci U S A 2007;104:4413-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ebermann I, Wilke R, Lauhoff T, et al. Two truncating USH3A mutations, including one novel, in a German family with Usher syndrome. Mol Vis 2007;13:1539-47. [PubMed] [Google Scholar]
- 134.Marquardt A, Stöhr H, Passmore LA, et al. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best's disease). Hum Mol Genet 1998;7:1517-25. [DOI] [PubMed] [Google Scholar]
- 135.Pasquay C, Wang LF, Lorenz B, et al. Bestrophin 1 - Phenotypes and Functional Aspects in Bestrophinopathies. Ophthalmic Genet 2013. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 136.Boycott KM, Maybaum TA, Naylor MJ, et al. A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants. Hum Genet 2001;108:91-7. [DOI] [PubMed] [Google Scholar]
- 137.van Genderen MM, Bijveld MM, Claassen YB, et al. Mutations in TRPM1 are a common cause of complete congenital stationary night blindness. Am J Hum Genet 2009;85:730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Dryja TP, Hahn LB, Reboul T, et al. Missense mutation in the gene encoding the a subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet 1996;13:358-60. [DOI] [PubMed] [Google Scholar]
- 139.Gal A, Orth U, Baehr W, et al. Heterozygous missense mutation in the rod cGMP phosphodiesterase β-subunit gene in autosomal dominant stationary night blindness. Nat Genet 1994;7:64-8. [DOI] [PubMed] [Google Scholar]
- 140.Dryja TP, Berson EL, Rao VR, et al. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat Genet 1993;4:280-3. [DOI] [PubMed] [Google Scholar]
- 141.Zeitz C, Kloeckener-Gruissem B, Forster U, et al. Mutations in CABP4, the Gene Encoding the Ca2+-Binding Protein 4, Cause Autosomal Recessive Night Blindness. Am J Hum Genet 2006;79:657-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wycisk KA, Zeitz C, Feil S, et al. Mutation in the Auxiliary Calcium-Channel Subunit CACNA2D4 Causes Autosomal Recessive Cone Dystrophy. Am J Hum Genet 2006;79:973-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dryja TP, McGee TL, Berson EL, et al. Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci U S A 2005;102:4884-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zeitz C, van Genderen M, Neidhardt J, et al. Mutations in GRM6 cause autosomal recessive congenital stationary night blindness with a distinctive scotopic 15-Hz flicker electroretinogram. Invest Ophthalmol Vis Sci 2005;46:4328-35. [DOI] [PubMed] [Google Scholar]
- 145.Audo I, Kohl S, Leroy BP, et al. TRPM1 is mutated in patients with autosomal-recessive complete congenital stationary night blindness. Am J Hum Genet 2009;85:720-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yamamoto S, Sippel KC, Berson EL, et al. Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 1997;15:175-8. [DOI] [PubMed] [Google Scholar]
- 147.Fuchs S, Nakazawa M, Maw M, et al. A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet 1995;10:360-2. [DOI] [PubMed] [Google Scholar]
- 148.Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Loss-of-function mutations in a calcium-channel α1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet 1998;19:264-7. [DOI] [PubMed] [Google Scholar]
- 149.Strom TM, Nyakatura G, Apfelstedt-Sylla A, et al. An L-type calcium-channel gene mutated in incomplete X-linkid congenital stationary night blindness. Nat Genet 1998;19:260-3. [DOI] [PubMed] [Google Scholar]
- 150.Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Mutations in NYX, encoding the leucine-rish proteoglycan nyctalopin, causse X-linked complete congenital stationary night blindness. Nat Genet 2000;26:319-23. [DOI] [PubMed] [Google Scholar]
- 151.Mansergh F, Orton NC, Vessey JP, et al. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum Mol Genet 2005;14:3035-46. [DOI] [PubMed] [Google Scholar]
- 152.Wutz K, Sauer C, Zrenner E, et al. Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina. Eur J Hum Genet 2002;10:449-56. [DOI] [PubMed] [Google Scholar]
- 153.Gregg RG. Identification of the Gene and the Mutation Responsible for the Mouse nob Phenotype. Invest Ophthalmol Vis Sci 2003;44:378-84. [DOI] [PubMed] [Google Scholar]
- 154.Takada M, Otani A, Ogino K, et al. Spectral-domain optical coherence tomography findings in the Mizuo-Nakamura phenomenon of Oguchi disease. Retina 2011;31:626-8. [DOI] [PubMed] [Google Scholar]
- 155.Yamamoto S, Khani SC, Berson EL, et al. Evaluation of the rhodopsin kinase gene in patients with retinitis pigmentosa. Exp Eye Res 1997;65:249-53. [DOI] [PubMed] [Google Scholar]
- 156.Sieving PA, Fowler ML, Bush RA, et al. Constitutive "light" adaption in rods from G90D rhodopsin: a mechanism for human congenital nightblindness without rod cell loss. J Neurosci 2001;21:5449-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zeitz C, Gross AK, Leifert D, et al. Identification and functional characterization of a novel rhodopsin mutation associated with autosomal dominant CSNB. Invest Ophthalmol Vis Sci 2008;49:4105-14. [DOI] [PubMed] [Google Scholar]
- 158.Thiadens AA, Phan TM, Zekveld-Vroon RC, et al. Clinical course, genetic etiology, and visual outcome in cone and cone-rod dystrophy. Ophthalmology 2012;119:819-26. [DOI] [PubMed] [Google Scholar]
- 159.Downes SM, Holder GE, Fitzke FW, et al. Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch Ophthalmol 2001;119:96-105. [PubMed] [Google Scholar]
- 160.Payne AM, Downes SM, Bessant DA, et al. A mutation in guanylate cyclase activator 1A (GUCA1A) in an autosomal dominant cone dystrophy pedigree mapping to a new locus on chromosome 6p21.1. Hum Mol Genet 1998;7:273-7. [DOI] [PubMed] [Google Scholar]
- 161.Kelsell RE, Evans K, Gregory-Evans CY, et al. Localisation of a gene for dominant cone-rod dystrophy (CORD6) to chromosome 17p. Hum Mol Genet 1997;6:597-600. [DOI] [PubMed] [Google Scholar]
- 162.Kitiratschky VB, Wilke R, Renner AB, et al. Mutation analysis identifies GUCY2D as the major gene responsible for autosomal dominant progressive cone degeneration. Invest Ophthalmol Vis Sci 2008;49:5015-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Thiadens AA, den Hollander AI, Roosing S, et al. Homozygosity mapping reveals PDE6C mutations in patients with early-onset cone photoreceptor disorders. Am J Hum Genet 2009;85:240-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kohl S, Coppieters F, Meire F, et al. A nonsense mutation in PDE6H causes autosomal-recessive incomplete achromatopsia. Am J Hum Genet 2012;91:527-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Piri N, Gao YQ, Danciger M, et al. A substitution of G to C in the cone cGMP-phosphodiesterase gamma subunit gene found in a distinctive form of cone dystrophy. Ophthalmology 2005;112:159-66. [DOI] [PubMed] [Google Scholar]
- 166.Nishiguchi KM, Sandberg MA, Gorji N, et al. Cone cGMP-gated channel mutations and clinical findings in patients with achromatopsia, macular degeneration, and other hereditary cone diseases. Hum Mutat 2005;25:248-58. [DOI] [PubMed] [Google Scholar]
- 167.Michaelides M, Aligianis IA, Ainsworth JR, et al. Progressive cone dystrophy associated with mutation in CNGB3. Invest Ophthalmol Vis Sci 2004;45:1975-82. [DOI] [PubMed] [Google Scholar]
- 168.Renner AB, Fiebig BS, Weber BH, et al. Phenotypic variability and long-term follow-up of patients with known and novel PRPH2/RDS gene mutations. Am J Ophthalmol 2009;147:518-30.e1. [DOI] [PubMed]
- 169.Nakazawa M, Kikawa E, Chida Y, et al. Asn244His muttions of peripherin/RDS gene causing autosomal dominant cone-rod degeneration. Hum Mol Genet 1994;3:1195-6. [DOI] [PubMed] [Google Scholar]
- 170.Riveiro-Alvarez R, Lopez-Martinez MA, Zernant J, et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: retrospective analysis in 420 Spanish families. Ophthalmology 2013;120:2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 2000;67:960-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cideciyan AV, Haeseleer F, Fariss RN, et al. Rod and cone visual cycle consequences of a null mutation in the 11-cis-retinol dehydrogenase gene in man. Vis Neurosci 2000;17:667-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nakamura M, Hotta Y, Tanikawa A, et al. A high association with cone dystrophy in fundus albipunctatus caused by mutations of the RDH5 gene. Invest Ophthalmol Vis Sci 2000;41:3925-32. [PubMed] [Google Scholar]
- 174.Yang P, Chiang PW, Weleber RG, et al. Autosomal Dominant Retinal Dystrophy With Electronegative Waveform Associated With a Novel RAX2 Mutation. JAMA Ophthalmol 2015. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 175.Wang QL, Chen S, Esumi N, et al. QRX, a novel homeobox gene, modulates photoreceptor gene expression. Hum Mol Genet 2004;13:1025-40. [DOI] [PubMed] [Google Scholar]
- 176.Hameed A, Abid A, Aziz A, et al. Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J Med Genet 2003;40:616-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Parry DA, Toomes C, Bida L, et al. Loss of the metalloprotease ADAM9 leads to cone-rod dystrophy in humans and retinal degeneration in mice. Am J Hum Genet 2009;84:683-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sohocki MM, Bowne SJ, Sullivan LS, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet 2000;24:79-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sohocki MM, Perrault I, Leroy BP, et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol Genet Metab 2000;70:142-50. [DOI] [PubMed] [Google Scholar]
- 180.Sergouniotis PI, Chakarova C, Murphy C, et al. Biallelic variants in TTLL5, encoding a tubulin glutamylase, cause retinal dystrophy. Am J Hum Genet 2014;94:760-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Jalkanen R, Mantyjarvi M, Tobias R, et al. X linked cone-rod dystrophy, CORDX3, is caused by a mutation in the CACNA1F gene. J Med Genet 2006;43:699-704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kobayashi A, Higashide T, Hamasaki D, et al. HRG4 (UNC119) Mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Invest Ophthalmol Vis Sci 2000;41:3268-77. [PubMed] [Google Scholar]
- 183.Zelinger L, Wissinger B, Eli D, et al. Cone dystrophy with supernormal rod response: novel KCNV2 mutations in an underdiagnosed phenotype. Ophthalmology 2013;120:2338-43. [DOI] [PubMed] [Google Scholar]
- 184.Wu H, Cowing JA, Michaelides M, et al. Mutations in the gene KCNV2 encoding a voltage-gated potassium channel subunit cause "cone dystrophy with supernormal rod electroretinogram" in humans. Am J Hum Genet 2006;79:574-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Sisodiya SM, Thompson PJ, Need A, et al. Genetic enhancement of cognition in a kindred with cone-rod dystrophy due to RIMS1 mutation. J Med Genet 2007;44:373-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Johnson S, Halford S, Morris AG, et al. Genomic organisation and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7). Genomics 2003;81:304-14. [DOI] [PubMed] [Google Scholar]
- 187.Köhn L, Kadzhaev K, Burstedt MS, et al. Mutation in the PYK2-binding domain of PITPNM3 causes autosomal dominant cone dystrophy (CORD5) in two Swedish families. Eur J Hum Genet 2007;15:664-71. [DOI] [PubMed] [Google Scholar]
- 188.Rice DS, Huang W, Jones HA, et al. Severe retinal degeneration associated with disruption of semaphorin 4A. Invest Ophthalmol Vis Sci 2004;45:2767-77. [DOI] [PubMed] [Google Scholar]
- 189.Aleman TS, Soumittra N, Cideciyan AV, et al. CERKL mutations cause an autosomal recessive cone-rod dystrophy with inner retinopathy. Invest Ophthalmol Vis Sci 2009;50:5944-54. [DOI] [PubMed] [Google Scholar]
- 190.Thiadens AA, Soerjoesing GG, Florijn RJ, et al. Clinical course of cone dystrophy caused by mutations in the RPGR gene. Graefes Arch Clin Exp Ophthalmol 2011;249:1527-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): Identification of mutations in RPGR Exon ORF15. Am J Hum Genet 2002;70:1049-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Khan AO, Bolz HJ. Pediatric Cone-rod Dystrophy with High Myopia and Nystagmus Suggests Recessive PROM1 Mutations. Ophthalmic Genet 2014. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 193.Pras E, Abu A, Rotenstreich Y, et al. Cone-rod dystrophy and a frameshift mutation in the PROM1 gene. Mol Vis 2009;15:1709-16. [PMC free article] [PubMed] [Google Scholar]
- 194.Henderson RH, Li Z, Abd El-Aziz MM, et al. Biallelic mutation of protocadherin-21 (PCDH21) causes retinal degeneration in humans. Mol Vis 2010;16:46-52. [PMC free article] [PubMed] [Google Scholar]
- 195.Ostergaard E, Batbayli M, Duno M, et al. Mutations in PCDH21 cause autosomal recessive cone-rod dystrophy. J Med Genet 2010;47:665-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Parry DA, Mighell AJ, El-Sayed W, et al. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am J Hum Genet 2009;84:266-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Polok B, Escher P, Ambresin A, et al. Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am J Hum Genet 2009;84:259-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Riveiro-Álvarez R, Xie YA, López-Martínez MÁ, et al. New mutations in the RAB28 gene in 2 Spanish families with cone-rod dystrophy. JAMA Ophthalmol 2015;133:133-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Roosing S, Rohrschneider K, Beryozkin A, et al. Mutations in RAB28, encoding a farnesylated small GTPase, are associated with autosomal-recessive cone-rod dystrophy. Am J Hum Genet 2013;93:110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Roosing S, Thiadens AA, Hoyng CB, et al. Causes and consequences of inherited cone disorders. Prog Retin Eye Res 2014;42:1-26. [DOI] [PubMed] [Google Scholar]
- 201.Zhou Y, Tao S, Chen H, et al. Exome sequencing analysis identifies compound heterozygous mutation in ABCA4 in a Chinese family with Stargardt disease. PloS one 2014;9:e91962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Weitz CJ, Miyake Y, Shinzato K, et al. Human Tritanopia Associated with Two Amino Acid Substitutions in the Blue-sensitive Opsin. Am J Hum Genet 1992;50:498-507. [PMC free article] [PubMed] [Google Scholar]
- 203.Kohl S, Varsanyi B, Antunes GA, et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J Hum Genet 2005;13:302-8. [DOI] [PubMed] [Google Scholar]
- 204.Kohl S, Baumann B, Broghammer M, et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptorcGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 2000;9:2107-16. [DOI] [PubMed] [Google Scholar]
- 205.Kohl S, Marx T, Giddings I, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet 1998;19:257-9. [DOI] [PubMed] [Google Scholar]
- 206.Kohl S, Baumann B, Rosenberg T, et al. Mutations in the Cone Photoreceptor G-Protein alpha-subunit gene GNAT2 in Patients with Achromatopsia. Am J Hum Genet 2002;71:422-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Weleber RG, Francis PJ, Trzupek KM. Leber Congenital Amaurosis Overview 2004. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1298/ [updated 2013; cited Jan 26, 2015].
- 208.Perrault I, Hanein S, Zanlonghi X, et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat Genet 2012;44:975-7. [DOI] [PubMed] [Google Scholar]
- 209.Hanein S, Perrault I, Gerber S, et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat 2004;23:306-17. [DOI] [PubMed] [Google Scholar]
- 210.Perrault I, Rozet J, Calvas P, et al. Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet 1996;14:461-4. [DOI] [PubMed] [Google Scholar]
- 211.Dev Borman A, Ocaka LA, Mackay DS, et al. Early onset retinal dystrophy due to mutations in LRAT: molecular analysis and detailed phenotypic study. Invest Ophthalmol Vis Sci 2012;53:3927-38. [DOI] [PubMed] [Google Scholar]
- 212.Marlhens F, Bareil C, Griffoin JM, et al. Mutations in RPE65 cause Leber's congenital amaurosis. Nat Genet 1997;17:139-41. [DOI] [PubMed] [Google Scholar]
- 213.Preising MN, Hausotter-Will N, Solbach MC, et al. Mutations in RD3 are associated with an extremely rare and severe form of early onset retinal dystrophy. Invest Ophthalmol Vis Sci 2012;53:3463-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Friedman JS, Chang B, Kannabiran C, et al. Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am J Hum Genet 2006;79:1059-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Freund CL, Wang Q, Chen S, et al. De nove mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet 1998;18:311-2. [DOI] [PubMed] [Google Scholar]
- 216.Swaroop A, Wang Q, Wu W, et al. Leber congenital amaurosis caused by a homozygous mutation in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of the photoreceptor function. Hum Mol Genet 1999;8:299-305. [DOI] [PubMed] [Google Scholar]
- 217.Henderson RH, Williamson KA, Kennedy JS, et al. A rare de novo nonsense mutation in OTX2 causes early onset retinal dystrophy and pituitary dysfunction. Mol Vis 2009;15:2442-7. [PMC free article] [PubMed] [Google Scholar]
- 218.den Hollander AI, Heckenlively J, Van den Born LI, et al. Leber congenital amaurosis and retinitis pigmentosa with coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet 2001;69:198-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.den Hollander AI, Lopez I, Yzer S, et al. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci 2007;48:5690-8. [DOI] [PubMed] [Google Scholar]
- 220.Chen Y, Zhang Q, Shen T, et al. Comprehensive mutation analysis by whole-exome sequencing in 41 Chinese families with Leber congenital amaurosis. Invest Ophthalmol Vis Sci 2013;54:4351-7. [DOI] [PubMed] [Google Scholar]
- 221.Asai-Coakwell M, March L, Dai XH, et al. Contribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophies. Hum Mol Genet 2013;22:1432-42. [DOI] [PubMed] [Google Scholar]
- 222.Aldahmesh MA, Al-Owain M, Alqahtani F, et al. A null mutation in CABP4 causes Leber's congenital amaurosis like phenotype. Mol Vis 2010;16:207-12. [PMC free article] [PubMed] [Google Scholar]
- 223.den Hollander AI, Koenekoop R, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber Congenital Amaurosis. Am J Hum Genet 2006;79:556-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Estrada-Cuzcano A, Koenekoop R, Coppieters F, et al. IQCB1 mutations in patients with leber congenital amaurosis. Invest Ophthalmol Vis Sci 2011;52:834-9. [DOI] [PubMed] [Google Scholar]
- 225.Corton M, Avila-Fernandez A, Vallespin E, et al. Involvement of LCA5 in Leber congenital amaurosis and retinitis pigmentosa in the Spanish population. Ophthalmology 2014;121:399-407. [DOI] [PubMed] [Google Scholar]
- 226.den Hollander AI, Koenekoop RK, Mohamed MD, et al. Mutations in LCA5, encoding the ciliary protein lebercilin, cause Leber congenital amaurosis. Nat Genet 2007;39:889-95. [DOI] [PubMed] [Google Scholar]
- 227.Falk MJ, Zhang Q, Nakamaru-Ogiso E, et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet 2012;44:1040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dryja TP, Adams SM, Grimsby JL, et al. Null RPGRIP1 alleles in patients with leber congenital amaurosis. Am J Hum Genet 2001;68:1295-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Sergouniotis PI, Davidson AE, Mackay DS, et al. Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis. Am J Hum Genet 2011;89:183-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mackay DS, Ocaka LA, Borman AD, et al. Screening of SPATA7 in patients with Leber congenital amaurosis and severe childhood-onset retinal dystrophy reveals disease-causing mutations. Invest Ophthalmol Vis Sci 2011;52:3032-8. [DOI] [PubMed] [Google Scholar]
- 231.den Hollander AI, Black A, Bennett J, et al. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest 2010;120:3042-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.van Bokhoven H, Schwartz M, Andreasson S, et al. Mutation spectrum in the CHM gene of Danish and Swedish choroidermia patients. Hum Mol Genet 1994;3:1047-51. [DOI] [PubMed] [Google Scholar]
- 233.Seabra MC, Brown MS, Goldstein JL. Retinal Degeneration in Chorioderemia: Deficiency of Rab Geranylgeranyl Transferase. Science 1993:377-81. [DOI] [PubMed] [Google Scholar]
- 234.McTaggart KE, Tran M, Mah DY, et al. Mutational analysis of patients with the diagnosis of choroideremia. Hum Mutat 2002;20:189-96. [DOI] [PubMed] [Google Scholar]
- 235.Li S, Guan L, Fang S, et al. Exome sequencing reveals CHM mutations in six families with atypical choroideremia initally diagnosed as retinitis pigmentosa. Int J Molec Med 2014;34:573-7. [DOI] [PubMed] [Google Scholar]
- 236.Henderson RH, Waseem N, Searle R, et al. An assessment of Apex microarray technology in genotyping patients with Leber Congenital Amaurosis and early-onset severe retinal dystrophy. Invest Ophthalmol Vis Sci 2007;48:5684-9. [DOI] [PubMed] [Google Scholar]
- 237.Yoshida S, Yamaji Y, Kuwahara R, et al. Novel triple missense mutations of GUCY2D gene in Japanese family with cone-rod dystrophy: Possible use of genotyping microarray. Mol Vis 2006;12:1558-64. [PubMed] [Google Scholar]
- 238.Cremers FP, Kimberling WJ, Kulm M, et al. Development of a genotyping microarray for Usher syndrome. J Med Genet 2007;44:153-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Jin ZB, Mandai M, Homma K, et al. Allelic copy number variation in FSCN2 detected using allele-specific genotyping and multiplex real-time PCRs. Invest Ophthalmol Vis Sci 2008;49:3799-805. [DOI] [PubMed] [Google Scholar]
- 240.Lacey S, Chung JY, Lin H. A comparison of whole genome sequencing with exome sequencing for family-based association studies. BMC proceedings 2014;8:S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Audo I, Bujakowska KM, Léveillard T, et al. Development and application of a next-generation-sequencing (NGS) approach to detect known and novel gene defects underlying retinal diseases. Orphanet J Rare Dis 2012;7:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Eisenberger T, Neuhaus C, Khan AO, et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: the example of retinal dystrophies. PloS one 2013;8:e78496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Liu X, Jian X, Boerwinkle E. dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum Mutat 2011;32:894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 2013;34:E2393-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Siepel A, Pollard KS, Haussler D. New Methods for Detecting Lineage-Specific Selection. In: Apostolico A, Guerra C, Istrail S, et al. eds. Research in Computational Molecular Biology. New York: Springer Berlin Heidelberg, 2006:190-205. [Google Scholar]
- 248.1000 Genomes Project Consortium, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56-65. [DOI] [PMC free article] [PubMed]
- 249.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073-81. [DOI] [PubMed] [Google Scholar]
- 250.Schwarz JM, Rodelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods 2010;7:575-6. [DOI] [PubMed] [Google Scholar]
- 251.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Daiger SP, Sullivan LS, Bowne SJ, et al. Targeted high-throughput DNA sequencing for gene discovery in retinitis pigmentosa. Adv Exp Med Biol 2010;664:325-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Jin X, Qu LH, Meng XH, et al. Detecting genetics variations in hereditary retinal dystrophies with next-generation sequencing technology. Mol Vis 2014;20:553-60. [PMC free article] [PubMed] [Google Scholar]
- 254.Guo Y, Prokudin I, Yu C, et al. Advantages of whole exome sequencing over allele-specific and targeted segment sequencing, in detection of novel TULP1 mutation in Leber congenital amaurosis. Ophthalmic Genet 2014. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 255.Prokudin I, Li D, He S, et al. Value of whole exome sequencing for syndromic retinal dystrophy diagnosis in young patients. Clin Experiment Ophthalmol 2015;43:132-8. [DOI] [PubMed] [Google Scholar]
- 256.Martin HC, Kim GE, Pagnamenta AT, et al. Clinical whole-genome sequencing in severe early-onset epilepsy reveals new genes and improves molecular diagnosis. Hum Mol Genet 2014;23:3200-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zhao M, Wang Q, Wang Q, et al. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics 2013;14 Suppl 11:S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 2012;130:9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Zrenner E, Bartz-Schmidt KU, Benav H, et al. Subretinal electronic chips allow blind patients to read letters and combine them to words. Proc Biol Sci 2011;278:1489-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Rizzo S, Belting C, Cinelli L, et al. The Argus II Retinal Prosthesis: 12-month outcomes from a single-study center. Am J Ophthalmol 2014;157:1282-90. [DOI] [PubMed] [Google Scholar]
- 261.Humayun MS, Dorn JD, da Cruz L, et al. Interim results from the international trial of Second Sight's visual prosthesis. Ophthalmology 2012;119:779-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Acland GM, Aguirre GD, Ray JW, et al. Gene therapy restores vision in canine model of childhood blindness. Nat Genet 2001:92-5. [DOI] [PubMed] [Google Scholar]
- 263.Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med 2008;358:2231-9. [DOI] [PubMed] [Google Scholar]
- 264.Black A, Vasireddy V, Chung DC, et al. Adeno-associated virus 8-mediated gene therapy for choroideremia: preclinical studies in in vitro and in vivo models. J Gene Med 2014;16:122-30. [DOI] [PubMed] [Google Scholar]
- 265.Cronin T, Vandenberghe LH, Hantz P, et al. Efficient transduction and optogenetic stimulation of retinal bipolar cells by a synthetic adeno-associated virus capsid and promoter. EMBO Mol Med 2014;6:1175-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Lamba DA, Karl MO, Ware CB, et al. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A 2006;103:12769-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Tucker BA, Park IH, Qi SD, et al. Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. PloS one 2011;6:e18992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Klassen HJ, Ng TF, Kurimoto Y, et al. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-mediated behaviour. Invest Ophthalmol Vis Sci 2004;45:4167-73. [DOI] [PubMed] [Google Scholar]
- 269.MacLaren RE, Pearson RA, MacNeil A, et al. Retinal repair by transplantation of photoreceptor precursors. Nature 2006;444:203-7. [DOI] [PubMed] [Google Scholar]
- 270.MacLaren RE, Groppe M, Barnard AR, et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 2014;383:1129-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Burnight ER, Wiley LA, Drack AV, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene therapy 2014;21:662-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Conlon TJ, Deng WT, Erger K, et al. Preclinical potency and safety studies of an AAV2-mediated gene therapy vector for the treatment of MERTK associated retinitis pigmentosa. Hum Gene Ther Clin Dev 2013;24:23-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Pellissier LP, Quinn PM, Alves CH, et al. Gene therapy into photoreceptors and Muller glial cells restores retinal structure and function in CRB1 retinitis pigmentosa mouse models. Hum Mol Genet 2015. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 274.Luo J, Baranov P, Patel S, et al. Human retinal progenitor cell transplantation preserves vision. J Biol Chem 2014;289:6362-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Esvelt KM, Wang HH. Genome-scale engineering for systems and synthetic biology. Mol Syst Biol 2013;9:641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science 2013;339:823-826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Ran FA, Hsu PD, Lin CY, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013;154:1380-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Tucker BA, Mullins RF, Stone EM. Stem cells for investigation and treatment of inherited retinal disease. Hum Mol Genet 2014;23:R9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]