

Abstract
Purpose
Bcr-Abl, the causative agent of chronic myelogenous leukemia (CML), localizes in the cytoplasm where its oncogenic signaling leads to proliferation of cells. If forced into the nucleus Bcr-Abl causes apoptosis. To achieve nuclear translocation, binding domains for capture of Bcr-Abl were generated and attached to proteins with signals destined for the nucleus. These resulting proteins would be capable of binding and translocating endogenous Bcr-Abl to the nucleus.
Methods
Bcr-Abl was targeted at 3 distinct domains for capture: by construction of high affinity intracellular antibody domains (iDabs) to regions of Bcr-Abl known to promote cytoplasmic retention, via its coiled-coil domain (CC), and through a naturally occurring protein protein interaction domain (RIN1). These binding domains were then tested for their ability to escort Bcr-Abl into the nucleus using a “protein switch” or attachment of 4 nuclear localization signals (NLSs).
Results
Although RIN1, ABI7-iDab, and CCmut3 constructs all produced similar colocalization with Bcr-Abl, only 4NLS-CCmut3 produced efficient nuclear translocation of Bcr-Abl.
Conclusions
We demonstrate that a small binding domain can be used to control the subcellular localization of Bcr-Abl, which may have implications for CML therapy. Our ultimate future goal is to change the location of critical proteins to alter their function.
Keywords: Bcr-Abl, chronic myelogenous leukemia, intracellular domain antibody, subcellular targeting, oncoprotein
INTRODUCTION
It is well established that the fusion between Bcr and Abl transforms the regulated tyrosine kinase activity from c-Abl into constitutive tyrosine kinase activity in Bcr-Abl. In addition to the misregulated kinase activity this fusion also results in a spatial misregulation at the subcellular level (1,2). In healthy cells, c-Abl can shuttle between the cytoplasm and nucleus and plays distinct roles in each subcellular compartment (3–6). This spatial control is important for the role of c-Abl in cell differentiation, division, adhesion, and response to stress signals. In contrast, Bcr-Abl is found to localize exclusively in the cytoplasm where it can be positioned in proximity to the signaling proteins controlled by its activated kinase domain. The combination treatment of Gleevec® and the nuclear export inhibitor Leptomycin B (LMB) has demonstrated the ability to relocate Bcr-Abl to the nucleus, where Bcr-Abl induced apoptosis (7). We have also demonstrated that an exogenous Bcr-Abl construct can be directed to the nucleus through incorporation of nuclear localization signals (NLSs), and established that the induction of apoptosis was dominant over the endogenous Bcr-Abl oncogenic signaling as cytoplasmic depletion of the endogenous Bcr-Abl was not observed (8). Thus, relocalizing Bcr-Abl to the nucleus is a potent method of inducing apoptosis and may be an interesting alternative intervention strategy. However, LMB cannot be used clinically due to neuronal toxicity (9), and treatment with an exogenous Bcr-Abl would also be problematic. In order to harness the apoptotic potential of nuclear Bcr-Abl, an alternative method of repositioning the protein is needed.
In this work, binding domains for capture of Bcr-Abl were identified; two approaches of using these binding domains for escorting Bcr-Abl to the nucleus were compared: a ligand inducible protein switch [37,41] and four SV40 NLSs. The protein switch localizes to the cytoplasm in the absence of the ligand dexamethasone (dex), and translocates to the nucleus upon binding dex. Alternatively, the SV40 NLS is a strong signal; the attachment of four SV40 NLSs to Bcr-Abl sends it to the nucleus [8].
As the ultimate goal is to translocate endogenous Bcr-Abl to the nucleus, a Bcr-Abl binding domain is critical for both of these approaches. The ideal binding motif will exhibit high affinity and specificity, will be stable inside of cells, and will be small in size. These are all attributes of intracellular domain antibodies (iDabs) (10–19). The iDab is a further simplification of an antibody and consists of only one variable domain (from either the heavy or the light chain). The iDab eliminates the necessity for a linker, is half the size of an scFv, and functions in the absence of any disulfide bonds (17). A simple method for isolation of an iDab that will bind to an intracellular target has been termed the third-generation intracellular antibody capture (IAC3) (20), and is based on yeast two-hybrid screening of iDab libraries. The initial libraries consist of randomized amino acids in the CDR3 region, and the initial hits are then randomized in CDR2 and then CDR1 in subsequent rounds of screening for affinity maturation. This method has been used to generate iDabs that bind LMO2, RAS, RAF, and p53 with high affinity (20,21). In line with the goal of escorting Bcr-Abl to the nucleus, we aimed to isolate an iDab that can compete with interactions that anchor Bcr-Abl in the cytoplasm and thus render it more available for transport into the nucleus. One Bcr-Abl interaction of particular interest is actin as it has been demonstrated to play a leading role in the cytoplasmic localization of Bcr-Abl (22–24). As the actin binding domain (ABD) is found at the C-terminus and is contributed by the Abl portion, we also wanted to target a Bcr domain. The Dbl homology and Pleckstrin homology domains (DHPH) are routinely found together and function to bind inositolphospholipids at the inner surface of the membrane (25–30). The interaction with phospholipids provides another plausible contribution to cytoplasmic retention. We thus chose the ABD and DHPH domains as the Bcr-Abl subdomains for targeting by iDabs.
An alternative binding approach is a rationally designed coiled-coil domain based on the coiled-coil (or oligomerization) domain of Bcr-Abl (31–33). Recently, we have designed mutations in this coiled-coil domain (C38A, S41R, L45D, E48R, and Q60E) to improve the interaction with Bcr-Abl (31). Data (manuscript in press, Molecular Pharmaceutics) in our laboratory indicate a set of mutations, containing one additional mutation (K39E) than the previously published CCmut2, results in superior interaction with Bcr-Abl, and has been termed CCmut3.
In addition to an iDab and CCmut3, we also compared the functionality of the Abl binding domain from the Ras and Rab interactor 1 (RIN1) as a binding motif for nuclear escort. This RIN1 domain has been demonstrated to be an efficient binding partner for Bcr-Abl, interacting with the SH3/2 domains and contributing to maintaining Bcr-Abl, in a constitutively active state (34–36). The regions of Bcr-Abl targeted by these four binding domains are illustrated in Fig. 1. As each binding domain targets Bcr-Abl at distinct regions, a combination of binding domains may be employed to achieve a multivalency-type effect. In this report, the IAC3 technology was employed for identifying iDabs targeting Bcr-Abl, and the best binding iDab, CCmut3, and RIN1 were assayed for their ability to escort Bcr-Abl to the nucleus after incorporation into the ligand-inducible protein switch or four NLSs.
Fig. 1.

Bcr-Abl domains and the targeting regions of the binding domains. Numbering indicates amino acidresidue location of each domain in Bcr-Abl (asterisk indicates approximate location). CCmut3 binds the coiled-coil domain, DBI binds the DHPH domains, RINI binds the SH3/SH2 domains, and ABI binds the ABD. CC = coiled-coil domain, S/T kinase = serine/threonine kinase domain, DH #x003; Dbl homology domain, PH = pleckstrin homology domain, SH3 = Src homology 3 domain, SH2 = Src homology 2 domain, Y kinase = tyrosine kinase domain, DBD = DNA binding domain, ABD = actin binding domain, CCmut3 = coiled-coil mutation set 3, RINI = Abl binding domain from RINI, DBI = DHPH binding iDab, ABI = ABD binding iDab, Red P = phosphorylation site.
MATERIALS AND METHODS
Cell Lines and Transformation/Transfection
Transformation into yeast cells (L40 or AH109) was performed by a lithium acetate/carrier DNA/PEG precipitation method as described in the IAC3 protocol (20).
Mammalian two-hybrid assays were performed in murine adenocarcinoma 1471.1 cells, and fluorescence microscopy was performed in simian kidney Cos-7. 1471.1 cells were grown in DMEM (GIBCO, Invitrogen, Carlsbad, CA,USA), and Cos-7 cells were grown in RPMI (GIBCO, Invitrogen) as monolayers. Both DMEM and RPMI were supplemented with 10% FBS (Invitrogen), 1% pen/strep/L-glu (Invitrogen), and 0.1% gentamicin (Invitrogen). 1471.1 cells were transfected using Lipofectamine 2000 (Invitrogen) following the recommended protocol from the manufacturer. Cos-7 cells were transfected with Fugene HD (Promega, Madison, WI, USA) following the recommended protocol from the manufacturer.
Construction of Plasmids and Libraries
The genes encoding the Dbl Homology/Pleckstrin Homology (DHPH) and actin binding (ABD) domains were amplified through PCR using the primers (DHPH) 5′-ACA CACACGAATTCGGCTTGGAGATGAGAAAATGGG T C C T G - 3 ′ a n d 5 ′ - C A A C C C C A G A A T TCCTTCTTCTGCTGCTCCCGGATG-3′ and (ABD) 5 ′ - TATGTCT AGAATTCGCAGGGGACCAGCCGTCTTC-3′ and 5′-CACTCCACGAATTCCTCAG CCACTGTCATGGGTATG-3′ for insertion into the yeast two-hybrid vectors pBTM116 (used in first round screening) and pBD-Gal4-Cam (used in second round screening)at the EcoRI site to create fusions with the LexA and Gal4 DNA binding domains respectively. The first round iDab libraries screened with randomized CDR3 regions were #4320 (9 amino acids randomized) and #4325 (14 amino acids randomized). The construction of these libraries, and the creation of sublibraries for the second round of screening, is described in detail in the IAC3 protocol (20). For the DHPH sublibraries, the iDabs extracted from screening library #4320 and #4325 were kept separate and used to generate two sublibraries. The ligations into pVP16 were transformed into MegaX DH10B™ T1RElectrocomp™ cells (Invitrogen) via electroporation. The transformed cells were plated onto five 25cm×25 cm LB plates containing ampicillin, grown overnight, and then harvested by scraping. The plasmids were isolated with a Qiagen EndoFree Plasmid Maxi kit (Qiagen, Hilden, Germany) and used as the sublibrary for second round screening. The ABD and DHPH sequences were digested out from the pBTM116 plasmids, and ligated into themammalian two-hybrid plasmid pM1 for fusion to the Gal4 DNA binding domain. The plasmids encoding the iDabs isolated after second round screening were digested with SfiI and NotI and ligated into the mammalian two-hybrid plasmid pEFVP16 as fusions to the VP16 activation domain. The ABI7 iDab was amplified through PCR from pEFVP16 ABI7 with the primers 5’ TGCTATCGTCGACAT GGCCGAGGTGCAGCTGTTG-3’ and 5’- TTTACCTGTCGA CCTAGCTCGAGACGGTGACCAGGGTTC-3’ and inserted into pEGFP-C1 (Clontech, Mountain View, CA, USA), pPS (protein switch fused to EGFP) (37), p4NLS (containing 4 SV40t antigen nuclear localization signals fused to EGFP, created through removal of Bcr-Abl from p4NLS Bcr-Abl (8) and re-ligation of the plasmid), and pmCherry-C1 at the SalI site. RIN1 was amplified through PCR from a plasmid containing the human RIN1 gene (NM_004292, OriGene, Rockville, MD, USA) with the primers 5’- TGCTATCGTCGACATGGAAAG CCCTGGAGAGTCAGGC-3’ and 5’- TTTACCTGT CGACCTAGTACCCCACTGAGCTCTCCCTCC-3’ and inserted into pEGFP-C1, pPS, p4NLS, and pmCherry-C1 at the SalI site. pEGFP-CCmut3 was created through site directed mutagenesis of CCmut2 (31) using the primers 5’- CCGCATTCGGCGCCTGGAGCAGCGGGTGAAC 3’ and 5’-GTTCACCCGCTGCTCCAGGCGCCGAATGCGG-3’. CCmut3 was then amplified through PCR and inserted into pEGFP-C1, pPS, p4NLS, and pmCherry-C1 at the XhoI sites. The gene encoding Bcr-Abl was digested from pEGFP Bcr-Abl (8) with EcoRI and inserted into the EcoRI site of pmCherry-C1 (Clontech).
X-gal Assay
The X-gal assay was carried out as described previously in the IAC3 protocol (20). Briefly, yeast cells streaked onto an agarose plate were transferred to a nylon membrane and frozen in liquid nitrogen. After thawing, the membrane was placed on top of a filter paper soaked in a 0.334 mg/mL X-gal (5-bromo-4chloro-indolyl-galactopyranoside) solution and incubated at 30°C for three hours. The appearance of a blue color was then documented.
Mammalian Two-Hybrid Assay
Twenty-four hrs after seeding 5×104 1471.1 cells into a white 96-well plate (Cellstar, Greiner Bio-One, Monroe, NC, USA), the media was replaced with DMEM with 10% FBS and without antibiotics, and pM1-ABD or pM1-DHPH was cotransfected with pEFVP16-iDab, pG5-Fluc (Promega), and pRL-CMV (Promega) in a 10:10:10:1 ratio using Lipofectamine 2000. Twenty-four hrs after transfection the firefly and renilla luminescence were measured on PlateLumino (Stratec Biomedical Systems) luminometer using the Dual-Glo Luciferase Assay (Promega) reagents following the manufacturer’s recommendations. The mean from duplicate samples were taken from 3 separate experiments. pAD-SV40 and pBD-p53 (Stratagene) plasmids were used for the positive control, and pM1 was used as the negative control. The luminescence fold induction was calculated by dividing the Firefly: Renilla luminescence ratio by the same ratio for the negative control.
Confocal Microscopy and Colocalization
Twenty-four hrs after transfection into Cos-7 cells seeded in 4-well live cell chambers (Nalge NUNC International, Naper-ville, IL, USA), fluorescence images were captured with a FV1000-XY (Olympus) confocal microscope using a 60X PlanApo oil immersion objective (NA 1.45) and Olympus FluoView software. mCherry was excited at 543 nm (HeNe laser), and a 555–655 nm emission filter was used to select the emitted light. EGFP was excited at 488 nm (Argon laser), and a 500–530 nm emission filter was used to select the emitted light. Images were collected in sequential line mode. The exposure settings and gain of laser were kept constant below the detected pixel saturation, and no bleed-through was observed between channels. Pixel resolution was kept at 1024× 1024 with maximum of 2.5X digital zoom. For each field of view, 6 images were taken in the z-plane. Image analysis was performed using ImageJ software (freeware, NIH) after converting the images to 8 bit format. The background fluorescence was quantified for each image by selecting a region of interest (ROI) that did not contain cells. This background fluorescence was subtracted from each of the images through use of a plugin set to subtract the mean background fluores cence plus three times the standard deviation in the background. Using ROI manager, the parallel images were duplicated and then analyzed for colocalization with the JACoP plugin(http://rsb.info.nih.gov/ij) (38). The quantitative colocalization coefficient was generated with Costes’ automatic threshold (39). All experiments were repeated at least three times. The images shown are representative images that are false colored cyan (binding domain) or magenta (Bcr-Abl) for visualization.
Fluorescence Microscopy and Nuclear Translocation
The day before transfection 2–3×105 Cos-7 cells were seeded in each well of a 2-well live cell chamber (Lab tek II chamber slide system, Nalge NUNC, Rochester, NY, USA). Twenty-four hours following cotransfection of pmCherry-Bcr-Abl and either pPS, pPS-CCmut3, pPS-ABI7or pPS-RIN1 (all fused to EGFP) in duplicate, the media was replaced with phenol red free RPMI, and 10 µL EtOH (control) was added to one replicate and 10 µL of 20 µM dex was added to the other. After 2–4 hr incubation at 37°C, 0.5 µL of 10 mg/mL H33342 (nuclear stain) was added, and the cells were incubated at 37°C for 15 min. Cells were then analyzed with an inverted fluorescence microscope (Olympus IX701F, Scientific Instrument Co., Sunnyvale, CA) with high-quality narrow band GFP filter (excitation HQ480/20 nm, emission HQ510/20 nm, beam splitter Q4951p, Chroma Technology Corp., Brattleboro, VT), high-quality TRITC filter (excitation HQ545/30 nm, emission HQ620/60 nm, beam splitter Q570lp, Chroma Technology Corp.). Cells were photo-graphed with an F-view Monochrome CCD camera using a 60x objective. During the microscopy the cells were maintained at 37°C with an air stream incubator (Nevtek ASI 400, Burnsville, VA) with temperature control. Twenty-four hrs after cotransfection of pmCherry-Bcr-Abl and either p4NLS, p4NLS-ABI7, p4NLS-CCmut3, or p4NLS-RIN1(all fused to EGFP) the cells were imaged using the same microscopy settings and conditions. Following the quantification of the amount of Bcr-Abl in the nucleus as previously described (37,40–42), the amount of Bcr-Abl that translocated to the nucleus was calculated using the following equation:
Statistical Analysis
For the colocalization coefficients and quantification of nuclear Bcr-Abl, statistical significance was determined by one-way analysis of variance (ANOVA) with Tukey’s post test. Each experiment was repeated at least three times (n≥3) and the mean ±S.E.M. was reported for the colocalization coef ficients or mean ±S.D. was graphed for the percentage of nuclear Bcr-Abl.
RESULTS
Third-Generation Intracellular Antibody Capture (IAC3)
Two libraries, one consisting of nine and the other consisting of 14 amino acids randomized in CDR3, were screened with both the ABD and DHPH baits (Fig. 2). For the ABD, 29 colonies were confirmed to be true interactions through the X-gal assay. As the lacZ gene is an alternative genetic reporter controlled by the GAL4/UAS, the X-gal assay provides a second means for determining true interactions and eliminating false positives. Of these 29, 20 colonies were used for creation of the sublibrary. For the DHPH, a much larger number of colonies (159) were validated to exhibit interactions via the X-gal assay. However, the pool was limited to 25 for creation of the sublibrary. For the DHPH bait, two sublibra ries were created, one from the preys extracted from the VP16*-(VH9rdm) screen, and one from the preys extracted from the VP16*-(VH14rdm) screen. The sizes of the sublibraries were estimated to be approximately 7×106. The second round of screening produced more than 800 colonies for both the ABD and DHPH targets. 50 of the largest colonies were chosen for validation in the X-gal assay.
Fig. 2.

Overview of IAC3 screens against ABD and DHPH baits. Two libraries were screened in the first round. 20 and 15 constructs were isolated using the ABD and DHPH as baits, respectively, from a library that contained nine amino acids randomized in CDR3. Zero and 10 constructs were isolated using the ABD and DHPH as baits, respectively, from a library that contained 14 amino acids randomized in CD3. After generation of sublibraries by randomizing the bases encoding CDR2, the top 12 binders were isolated for each bait. Mammalian two hybrid assays then identified the top three ABD binders and the top DHPH binder.
From the 50 assayed for each of the baits, 12 were selected. The sequences of these 24 are tabulated in Tables I and II. The amino acid (or type of amino acids) in at least 6 of the constructs was termed the consensus amino acid at that posi tion and are underlined and summarized in the bottom row of each table ("Consensus"). The sequence of CDR1 was not randomized for screening and is the same for all constructs: GFTFSTFS. The amino acids at positions 1, 2, and 8 of CDR2 were not randomized and are #1-I,#2-S,#8-I. The amino acids at positions 1, 2, 12, 13, and 14 of CDR3 were not randomized and are #1-R, #2-G, #12-F, #13-D, and #14-Y. Although no strong consensus is observed, none is trulyexpected given that each individual iDab may bind at different regions of the target domain.
Table I.
Amino Acid Sequences of CDR2 and CDR3 Regions of ABD Binding iDabs (ABIs)
| ABI | CDR2 |
CDR3 | ||||
|---|---|---|---|---|---|---|
| Position | 3 | 4 | 5 | 6 | 7 | 3-11 |
| A1 | P | S | G | T | L | PLWSFVRMS |
| A2 | Q | S | G | R | L | PLWSFVRMS |
| A4 | K | D | G | K | A | PLWSFVRMS |
| A5 | P | S | G | Y | S | PLWSFVRMS |
| A7 | K | C | G | H | V | PLWSFVRMS |
| A8 | D | T | G | R | A | PLWSFVRMS |
| A14 | R | T | S | K | T | RF |
| A26 | A | T | G | G | A | PLWSFVRMS |
| A37 | A | K | G | N | N | PLWSFVRMS |
| A45 | G | K | G | D | S | PLWSFVRMS |
| A46 | Q | T | G | S | T | PLWSFVRMS |
| A50 | A | N | S | R | T | PLWSFVRMS |
| Consensus | x | S/T | G | + | φ | PLWSFVRMS |
Table II.
Amino Acid Sequences of CDR2 and CDR3 Regions of DHPH Binding iDabs (DBIs)
| DBI | CDR2 |
CDR3 | ||||
|---|---|---|---|---|---|---|
| Position | 3 | 4 | 5 | 6 | 7 | 3–11 |
| D2 | E | C | L | D | L | RF |
| D5 | D | T | A | N | E | TFFRPPVRA |
| D8 | P | C | C | R | E | TFFRPPVRA |
| D9 | E | Y | G | S | D | TFFRPPVRA |
| D12 | G | E | S | K | D | TFFRPPVRA |
| D13 | S | M | G | E | D | TFFRPPVRA |
| D15 | K | G | W | C | L | GG |
| D29 | D | T | S | H | E | TFFRPPVRA |
| D32 | S | A | S | E | Q | TFFRPPVRA |
| D33 | D | D | S | G | V | TFFRPPVRA |
| D34 | P | D | S | K | E | TFFRPPVRA |
| D37 | G | C | G | R | D | TFFRPPVRA |
| Consensus | x | x | S/G | x | - | TFFRPPVRA |
Screening via Mammalian Two-Hybrid Assay
The sequences of these 24 iDabs were then subcloned into pEFVP16 to generate fusion constructs with theVP16 activation domain in a mammalian expression plasmid for further validation in a mammalian two hybrid assay. As this assay provides a luminescent readout, the binding affinities can be compared relative to each other for determination of the highest affinity binder. As seen in Fig. 3, the ABD screens produced better binders overall, with clone A7 exhibiting the greatest binding. Although none of the DHPH binders dem onstrated highly efficient binding, the top candidate, D5, was further analyzed through colocalization as any ability to compete with Bcr-Abl binding to phospholipids may prove beneficial, and the lack of knowledge regarding the relationshipbetween the mammalian two-hybrid result and the ability to translocate Bcr-Abl justifies further exploration.
Fig. 3.
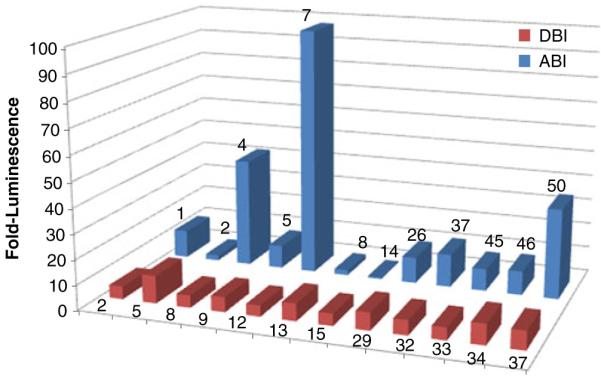
Mammalian two-hybrid assays. Blue columns = ABD binding iDabs (ABI), red columns = DHPH binding iDabs (DBI). Based on these mam malian two hybrid results, the top ABI (ABI7) and the top DBI (DBI5) were selected for further analysis by colocalization with full length Bcr-Abl.
Colocalization (Binding) of Bcr-Abl with ABI7, DBI5, CC mut3 and RINI
The IAC3 screening and mammalian two-hybrid assays were performed using the isolated ABD or DHPH domains. To confirm these interactions carried over to the full length Bcr-Abl protein, and to compare their ability to bind Bcr-Abl with CCmut3 and RIN1, colocalization studies were performed with each binding domainand mCherry Bcr-Abl in Cos-7 cells. A rigorous method for analysis of colocalization, the use of Costes’automatic threshold to determine Pearson’s correlation coeffi cient of colocalization (39), was utilized. In this method, fluores cence signals originating from the same location (indicating the interaction between the proteins fused to them) will result in a value greater than 0.5. Non-interacting proteins will generate values close to zero or negative. As seen in Fig. 4, ABI7, CCmut3, and RIN1 all demonstrated colocalization with Bcr-Abl and confirmed the interactions of these binding motifs with the full protein under biological conditionsinside of cells. In agreement with the mammalian two hybrid result, DBI5 did not result in efficient colocalization (below 0.5 colocalization coefficient with Costes’ automatic threshold) with Bcr-Abl. As no iDab was isolated that efficiently interacted with the DHPH region of Bcr-Abl, as determined by the mammalian two hybrid and colocalization with Bcr-Abl, and due to the other three binding domains functioning superiorly, the DBIswere not analyzed further.
Fig. 4.

Colocalization between Bcr-Abl and the binding domains. Binding domains are in the left columns ( false colored cyan), and Bcr-Abl is in the middle column (false colored magenta). Colormaps are illustrated in the right column with the key at the top of the column (highest colocalization = red). The colocalization coefficients are indicated to the right of the colormaps. The colocalization coefficient for the negative control (EGFP) was found to be -0.11± =.=46. The mean colocalization coefficients were determined after analyzing at least three cells, and the experiment was repeated three times. The values reported arethe means±S.E.M. Statistical significance was determined by one way ANOVA with Tukey’s post test.***p<0.001.
The ABD was selected as a target for the potential advantage of competing with actin binding that may preclude nuclear translocation. Interestingly, upon cotransfection of ABI7 with Bcr-Abl, a marked redistributionof the subcellular localization pattern was observed. Alone, Bcr-Abl forms a distinct localization pattern similar to the pattern resulting from actin staining, indicating its interaction with actin (Fig. 5a). However, when ABI7 is coexpressed along with Bcr-Abl this localization pattern shifts to one of punctate dots throughout the cytoplasm (Fig. 5b). This observation may be explained by ABI7 preventing Bcr-Abl from binding actin in the same fashion as normally occurs, and may provide evidence for the hypothesized benefit of rendering Bcr-Abl moreavailable for nuclear translocation. Recently it has been demonstrated that the ABD plays a joint role, along with the tyrosine kinase domain, in regulating the NLSs found in Bcr-Abl (22). A further potential implication of targeting the ABD may be a shift to the NLS-active conformation resulting in nuclear accumulation. Although ABI7 did cause an altered localization pattern of Bcr-Abl in the cyto plasm, no substantial increase in nuclear localization was observed. Interestingly, when CCmut3 was co-expressed with Bcr-Abl a dramatic change in the localization pattern occurred, and Bcr-Abl was found to be diffuse throughout the entire cytoplasm and no longer localized to any particular region (Fig. 5c). This prominent shift may be reflective of the change from tetrameric Bcr-Abl to monomeric Bcr-Abl, and the cor related Bcr-Abl conformations.
Fig. 5.

Redistribution of Bcr-Abl subcellular localization. (a) EGFP Bcr-Abl expressed alone in Cos-7 cells. Bcr-Abl localizes at actin and forms a pattern characteristic of actin filaments. (b) EGFP Bcr-Abl and mCherry ABI7. ABI7 affects Bcr-Abls interaction with actin, and results in a distinct localization pattern. (c) EGFP Bcr-Abl and mCherry CCmut3. CCmut3 causes Bcr-Abl to become diffuse throughout the cytoplasm.
Testing Binding Motifs Fused to Protein Switch
In attempting to use the binding motifs for translocation of Bcr-Abl into the nucleus, they were each subcloned into plasmids as fusions with the localization controllable protein switch (37). One potential advantage of using the protein switch for translocating a protein is the initial localization in the same subcellular compartment as the target protein. This allows equilibration time for the interaction between the target protein and the protein switch before any attempt of translocation. After cotransfecting (in duplicate) mCherry Bcr-Abl with a protein switch construct, ethanol (carrier) or dex was added and fluorescence microscopy used to image the cells 2–4 hrs post ligand induction. The per centage of Bcr-Abl inside the nucleus was determined in the presence of ethanol or dex and used to determine a percent nuclear increase (PNI) upon ligand induction.Attempts to move Bcr-Abl with one binding domain (protein switch fused to either CCmut3, ABI7, or RIN1) did notproduce any nuclear Bcr-Abl (data not shown). However, when two binding domains were used (two separate proteinswitch constructs with different binding domains cotransfected) it was possible to find cells with Bcr-Abl inside the nucleus. Some examples are illustrated in Fig. 6. Nevertheless, the majority of the cells did not have noticeable Bcr-Abl inside the nucleus. While identifying cells with nuclear Bcr-Abl may demonstrate that Bcr-Ablcan be moved with this system, the fact that this occurred on a limited basis indicates the inefficiency of thesystem as applied to Bcr-Abl. As both protein switch constructs are fused to EGFP it is im possible to distinguish cells transfected with only one of the protein switches from those transfected with both protein switch constructs, a fact that may explain why Bcr-Abl could be found in the nucleus of some limited cells. A further note is the level of protein switch that translocated into the nucleus. After incorporation of the binding domain into the protein switch there was a decrease in the nuclear translocation, which may be reflective of the interaction with Bcr-Abl as well as some interference with the protein switch by the binding domain fusion.
Fig. 6.

Nuclear translocation using the protein switch. Left column = EGFP fluorescence from protein switch; Middle column = Nuclei (H33342); Right column = mCherry from Bcr-Abl. Outlines of thenuclei are included for reference in all images. (a) Combination of PS-ABI7 and PS-CCmut3. (b)Combination of PS ABI7 and PS-RINI. (c) Combination of PS CCmut3 and PS RINI.
Testing Binding Motifs Fused to 4NLS
To increase the force driving nuclear localization and simplify the approach to move Bcr-Abl, the binding motifs were subcl oned into a plasmid containing 4 strong NLSs (from SV40 large T antigen). These constructs, in contrast to protein switch constructs that were only marginally nuclear even in the presence of dex, were exclusively localized to the nucleus (Fig. 7a-d, green fluorescence). Nevertheless, 4NLS ABI7 and 4NLS RIN1 were unable to cause a nuclear accumulation of Bcr-Abl (Fig. 7b and c). CCmut3, on the other hand, caused the majority of Bcr-Abl to translocate to the nucleus (Figs. 7d, and 8). The percentage of Bcr-Abl inside the nucleus upon cotransfection with the 4NLS constructs was quantified and is indicated in Fig. 8e. While one possible explanation for the inability of 4NLS ABI7 and 4NLS RIN1 to translocate Bcr-Abl is that their interaction with Bcr-Abl is not strong enough, all of the binding domains generated statistically indistinguish able colocalization coefficients. That only one of these binding domains exhibited the ability to translocate Bcr-Abl supports the concept that the region of Bcr-Abl where the binding domain interacts is also a critical factor.
Fig. 7.

Nuclear translocation using 4NLS binding motif constructs. Bcr-Abl is shown in red (mCherry), and the 4NLS construct is shown in green (EGFP). Images were taken at 60X magnification, and the scale bar is indi cated in the lower right corner. (a) mCherry Bcr-Abl and 4NLS (no binding domain). Bcr-Abl localizes in the cytoplasm with actin. 4NLS is exclusively nuclear. (b) mCherry Bcr-Abl and 4NLS-RIN1. Bcr-Abl remains cytoplasmic and 4NLS-RIN1 localizes to the nucleus. (c) mCherry Bcr-Abl and 4NLS-ABI7. Bcr-Abl does not accumulate in thenucleus as does 4NLS ABI7. (d) mCherry-Bcr-Abl and 4NLS CCmut3. As indicated by the overlapping green and red fluorescence (yellow), Bcr-Abl is translocated into the nucleus along with 4NLS CCmut3.
Fig. 8.

Representative images of nuclear Bcr-Abl resulting from 4NLS-CCmut3, and quantification of nuclear Bcr-Abl from all 4NLS constructs. (a) To more clearly show the nuclear accumulation of Bcr-Abl upon cotransfection with 4NLS-CCmut3, an image of only the red fluorescence (Bcr-Abl) is shown. (b) The image shown in (a) is shown with the nuclear stain (H33342, blue). (c) Resulting Bcr-Abl localization after transfecting 4NLS ABI7 with 4NLS CCmut3. (d) Image from (c) with nuclear stain. (e) The percentages of Bcr-Abl found in the nucleus after cotransfection with the indicated constructs was quantified and graphed. At least five cells from each experiment were imaged and analyzed. Each construct was cotransfected with pmCherry Bcr-Abl three times. Statistical analysis was determined by one-way ANOVA with Tukey’s post test. *p <0.05, ***p< 0.001.
In spite of the fact that minimal Bcr-Abl translocated with 4NLS ABI7, the combination of 4NLS ABI7 and4NLS CCmut3 did result in enhanced nuclear accumulation of Bcr-Abl and only minimal levels were detected outside of the cytoplasm (Fig. 8c, d, and e). This validates the concept that binding Bcr-Abl through multiple regions is advantageous for nuclear translocation.
DISCUSSION
Both the aberrant tyrosine kinase activity and the cytoplas mic localization are important outcomes of the fusion be tween the Bcr and Abl proteins. Current CML treatments target the tyrosine kinase activity, and an interesting alter native that has not been highly explored is repositioning Bcr-Abl into the nucleus where itis known to be an activator of apoptosis. In this report we have demonstrated the ability to escort Bcr-Abl to the nucleus through Bcr-Abl binding domains.
In order to complement the regions of Bcr-Abl already targeted by CCmut3 and RIN1, we screened for iDabs that would bind at the ABD and DHPH. While the protocol outlined for IAC3 describes the screening of 17 libraries in the first round, followed by screening sublibraries in subse quent second and third rounds of screening, we were able to identify an iDab with efficient binding to Bcr-Abl through screening of only two libraries in the first round and one sublibrary. This demonstrates the versatility and usefulness of the IAC3 technology. As speculated, this iDab (ABI7) caused a redistribution of Bcr-Abl from its regular localization pattern indicating the ability to interfere with actin interactions. However, ABI7 was unable to efficiently translocate Bcr-Abl to the nucleus without CCmut3. Further affinity maturation of ABI7 could be performed through creation of a randomized CDR1 sublibrary and additional screening. Nevertheless, the combination of 4NLS ABI7 and 4NLS CCmut3 produced remarkable accumulation of Bcr-Abl in the nucleus.
Attempts to move Bcr-Abl to the nucleus with the protein switch were unsuccessful with one binding domain, but through two protein switch constructs, each fused to differ ent binding domains it was possible to identify some cells wherein Bcr-Abl had been transported into the nucleus. The numerous interactions and large sizeof Bcr-Abl make it a challenging protein to redirect to alternative subcellular localizations, and other proteins may be more readily trans located. However, the incorporation of the binding domains into the protein switchdecreased the nuclear translocation, a factor contributing to the inefficient translocation of Bcr-Abl. This finding highlights the potential need to optimize the protein switch with the binding domain fused to it. The strength of the NES and NLS used in the protein switch could be re-optimized as well as the linker fusing the protein switch to the binding domain and terminus where the binding domain is fused to the protein switch.
In stark contrast to the protein switch constructs, 4NLS-CCmut3 and 4NLS ABI7+4NLS-Cmut3 produced an impressive re-localization of Bcr-Abl to the nucleus. We have long speculated on the advantage and need for some mean residency time in the location of the target protein to allow the interaction to occur. This meanresidency time is afforded by the controlled translocation in the protein switch. However, that a 4NLS construct (4NLS-CCmut3) was the only construct tested to demonstrate efficient nuclear accumulation of Bcr-Abl stronglysupports the claim that mean residency time is not needed. CML cells where Bcr-Abl is present, and not entirelybeing co translated with 4NLS-CCmut3, may exhibit a different dynamic wherein the mean residency time is more important. Dismissal of the use fulness of the protein switch based on these experiments is not entirely justified, and further exploration into the ability to translocate endogenous Bcr-Abl in CML cells will be needed.
In these experiments four binding domains that bind Bcr-Abl at distinct regions were explored. Interestingly, only one of three binding domains that demonstrated efficient colocaliza tion with Bcr-Abl resulted in efficient nuclear translocation of Bcr-Abl. This suggests the binding affinity is not the only consideration, and may not be the most important consideration, in determining the ability to translocate Bcr-Abl. Speculatively, the resulting Bcr-Abl conformation upon being bound by the binding domain may contribute to the ability of one domain to translocate Bcr-Abl and not another. RIN1 interacts with the SH3/2 domains of Bcr-Abl, and is known to bind in such a way that Bcr-Abl is maintained in an active conformation (34,36,43,44). This active conformation is cor related with protein interactions thought to cause cytoplasmic retention and inhibition of the Bcr-Abl NLSs (22). However, CCmut3 binds the coiled-coil domain and interferes with the oligomeric state of Bcr-Abl (31–33,45). The homo-oligomerization of Bcr-Abl is directly correlated with Bcr-Abl activity, and the formation of hetero-oligomers with CCmut3 decreases its activity (manuscript in press, Molecular Pharma ceutics). This inactive conformation may relieve the cytoplas mic retention as well as activate the Bcr-Abl NLSs. Thus, it is easy to speculate on why CCmut3 was able to translocate Bcr-Abl and RIN1 was not, in spite of the high affinity of RIN1 for Bcr-Abl. As demonstrated with these binding domains, not only binding, but binding Bcr-Abl in a particular fashion and/or conformation, is critical to the ability to translocate Bcr-Abl.
Capture and escort of Bcr-Abl to the nucleus is an interesting concept for turning the disease causing agent against the diseased cells. Here we demonstrate Bcr-Abl can be redirected to the nucleus by NLSs attachedto a Bcr-Abl binding domain. Further, this work validates CCmut3 as an efficient binding partner of Bcr-Abl which can be used for controlling the subcellular localization of Bcr-Abl. Capture and escort of Bcr-Abl would accomplish two goals: nuclear accumulation of endogenous Bcr-Abl, as well as its cytoplasmic depletion. This ability of CCmut3 to translocate Bcr-Abl has great potential for potent induction of apoptosis in CML cells, and mayextend to the ability to induce apoptosis in resistant CML cells and also CML stem cells. This is the subject of future work in our laboratory.
ACKNOWLEDGMENTS
This work was funded by NIH R01-CA129528 (AD, JC, CL), MRC and LLR UK (TR), and the University of Leeds(TT). We acknowledge the use of DNA/Peptide Core (NCI Cancer Center Support Grant P30 CA042014, HuntsmanCancer Institute). We would like to thank Dr. Warren Voth, Dr. Jared Rutter, and Danny Li for technical assistance in work involving yeast, and Rian Davis, Mohanad Mossalam, Benjamin Bruno, Geoffrey Miller, David Woessner,Abood Okal, and Karina Matissek for scientific discussions.
ABBREVIATIONS
- Bcr
Abl breakpoint cluster region/Abelson oncogene
- CC
coiled coil
- CML
chronic myelogenous leukemia
- iDab
intracellular domain antibody
- NLS
nuclear localization signal
Footnotes
DISCLOSURES
The authors declare no conflict of interest or disclosures.
Contributor Information
Andrew S. Dixon, Department of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, University of Utah, 421 Wakara Way, Rm. 318, Salt Lake City, Utah 84108, USA
Jonathan E. Constance, Department of Pharmacology and Toxicology, College of Pharmacy University of Utah, Salt Lake City, Utah 84108, USA
Tomoyuki Tanaka, Section of Experimental Therapeutics, Leeds Institute of Molecular Medicine, St. James's University Hospital, Leeds LS9 7TF, UK.
Terence H. Rabbitts, Section of Experimental Therapeutics, Leeds Institute of Molecular Medicine, St. James’s University Hospital, Leeds LS9 7TF, UK
Carol S. Lim, Department of Pharmaceutics and Pharmaceutical Chemistry, College of Pharmacy, University of Utah, 421 Wakara Way, Rm. 318, Salt Lake City, Utah 84108, USA, carol.lim@pharm.utah.edu
REFERENCES
- 1.Wetzler M, Talpaz M, Van Etten RA, Hirsh-Ginsberg C, Beran M, Kurzrock R. Subcellular localization of Bcr, Abl, and Bcr-Abl proteins in normal and leukemic cells and correlation of expression with myeloid differentiation. J Clin Invest. 1993;92(4):1925–39. doi: 10.1172/JCI116786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dhut S, Chaplin T, Young BD. BCR-ABL and BCR proteins: biochemical characterization and localization. Leukemia. 1990;4(11):745–50. [PubMed] [Google Scholar]
- 3.Taagepera S, McDonald D, Loeb JE, Whitaker LL, McElroy AK, Wang JY, et al. Nuclear-cytoplasmic shuttling of C-ABL tyrosine kinase. Proc Natl Acad Sci U S A. 1998;95(13):7457–62. doi: 10.1073/pnas.95.13.7457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vella V, Zhu J, Frasca F, Li CY, Vigneri P, Vigneri R, et al. Exclusion of c-Abl from the nucleus restrains the p73 tumor suppression function. J Biol Chem. 2003;278(27):25151–7. doi: 10.1074/jbc.M301962200. [DOI] [PubMed] [Google Scholar]
- 5.Wang JY. Regulation of cell death by the Abl tyrosine kinase. Oncogene. 2000;19(49):5643–50. doi: 10.1038/sj.onc.1203878. [DOI] [PubMed] [Google Scholar]
- 6.Zhu J, Wang JY. Death by Abl: a matter of location. Curr Top Dev Biol. 2004;59:165–92. doi: 10.1016/S0070-2153(04)59007-5. [DOI] [PubMed] [Google Scholar]
- 7.Vigneri P, Wang JY. Induction of apoptosis in chronic myelogenous leukemia cells through nuclear entrapment of BCR-ABL tyrosine kinase. Nat Med. 2001;7(2):228–34. doi: 10.1038/84683. [DOI] [PubMed] [Google Scholar]
- 8.Dixon AS, Kakar M, Schneider KM, Constance JE, Paullin BC, Lim CS. Controlling subcellular localization to alter function: Sending oncogenic Bcr-Abl to the nucleus causes apoptosis. J Control Release. 2009;140(3):245–9. doi: 10.1016/j.jconrel.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newlands ES, Rustin GJ, Brampton MH. Phase I trial of elactocin. Br J Cancer. 1996;74(4):648–9. doi: 10.1038/bjc.1996.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli. Nature. 1989;341(6242):544–6. doi: 10.1038/341544a0. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka T, Lobato MN, Rabbitts TH. Single domain intracellular antibodies: a minimal fragment for direct in vivo selection of antigen-specific intrabodies. J Mol Biol. 2003;331(5):1109–20. doi: 10.1016/s0022-2836(03)00836-2. [DOI] [PubMed] [Google Scholar]
- 12.Colby DW, Chu Y, Cassady JP, Duennwald M, Zazulak H, Webster JM, et al. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc Natl Acad Sci U S A. 2004;101(51):17616–21. doi: 10.1073/pnas.0408134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paz K, Brennan LA, Iacolina M, Doody J, Hadari YR, Zhu Z. Human single-domain neutralizing intrabodies directed against Etk kinase: a novel approach to impair cellular transformation. Mol Cancer Ther. 2005;4(11):1801–9. doi: 10.1158/1535-7163.MCT-05-0174. [DOI] [PubMed] [Google Scholar]
- 14.Verheesen P, de Kluijver A, van Koningsbruggen S, de Brij M, de Haard HJ, van Ommen GJ, et al. Prevention of oculopharyngeal muscular dystrophy-associated aggregation of nuclear polyA-binding protein with a single-domain intracellular antibody. Hum Mol Genet. 2006;15(1):105–11. doi: 10.1093/hmg/ddi432. [DOI] [PubMed] [Google Scholar]
- 15.Gueorguieva D, Li S, Walsh N, Mukerji A, Tanha J, Pandey S. Identification of single-domain, Bax-specific intrabodies that confer resistance to mammalian cells against oxidative-stress-induced apoptosis. Faseb J. 2006;20(14):2636–8. doi: 10.1096/fj.06-6306fje. [DOI] [PubMed] [Google Scholar]
- 16.Serruys B, Van Houtte F, Verbrugghe P, Leroux-Roels G, Vanlandschoot P. Llama-derived single-domain intrabodies inhibit secretion of hepatitis B virions in mice. Hepatology. 2009;49(1):39–49. doi: 10.1002/hep.22609. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka T, Rabbitts TH. Functional intracellular antibody fragments do not require invariant intra-domain disulfide bonds. J Mol Biol. 2008;376(3):749–57. doi: 10.1016/j.jmb.2007.11.085. [DOI] [PubMed] [Google Scholar]
- 18.Visintin M, Quondam M, Cattaneo A. The intracellular antibody capture technology: towards the high-throughput selection of functional intracellular antibodies for target validation. Methods. 2004;34(2):200–14. doi: 10.1016/j.ymeth.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Visintin M, Settanni G, Maritan A, Graziosi S, Marks JD, Cattaneo A. The intracellular antibody capture technology (IACT): towards a consensus sequence for intracellular antibodies. J Mol Biol. 2002;317(1):73–83. doi: 10.1006/jmbi.2002.5392. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka T, Rabbitts TH. Protocol for the selection of single-domain antibody fragments by third generation intracellular anti-Normal capture. Nat Protoc. 5(1):67–92. doi: 10.1038/nprot.2009.199. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka T, Sewell H, Waters S, Phillips SE, Rabbitts TH. Single domain intracellular antibodies from diverse libraries: emphasizing dual functions of LMO2 protein interactions using a single VH domain. J Biol Chem. Feb 4;286(5):3707–16. doi: 10.1074/jbc.M110.188193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Preyer M, Vigneri P, Wang JY. Interplay between kinase domain autophosphorylation and F-actin binding domain in regulating imatinib sensitivity and nuclear import of BCR-ABL. PLoS One. 6(2):e17020. doi: 10.1371/journal.pone.0017020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hantschel O, Wiesner S, Guttler T, Mackereth CD, Rix LL, Mikes Z, et al. Structural basis for the cytoskeletal association of Bcr-Abl/c-Abl. Mol Cell. 2005;19(4):461–73. doi: 10.1016/j.molcel.2005.06.030. [DOI] [PubMed] [Google Scholar]
- 24.McWhirter JR, Wang JY. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. EMBO J. 1993;12(4):1533–46. doi: 10.1002/j.1460-2075.1993.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kin Y, Li G, Shibuya M, Maru Y. The Dbl homology domain of BCR is not a simple spacer in P210BCR-ABL of the Philadelphia chromosome. J Biol Chem. 2001;276(42):39462–8. doi: 10.1074/jbc.M105484200. [DOI] [PubMed] [Google Scholar]
- 26.McWhirter JR, Wang JY. Effect of Bcr sequences on the cellular function of the Bcr-Abl oncoprotein. Oncogene. 1997;15(14):1625–34. doi: 10.1038/sj.onc.1201342. [DOI] [PubMed] [Google Scholar]
- 27.Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350:1–18. Pt 1. [PMC free article] [PubMed] [Google Scholar]
- 28.Lemmon MA, Ferguson KM, Abrams CS. Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 2002;513(1):71–6. doi: 10.1016/s0014-5793(01)03243-4. [DOI] [PubMed] [Google Scholar]
- 29.Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci U S A. 1995;92(23):10472–6. doi: 10.1073/pnas.92.23.10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007;74:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dixon AS, Pendley SS, Bruno BJ, Woessner DW, Shimpi AA, Cheatham TE, 3rd, et al. Disruption of Bcr-Abl coiled-coil oligomerization by design. J Biol Chem. 2011;286(31):27751. doi: 10.1074/jbc.M111.264903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taylor CM, Keating AE. Orientation and oligomerization specificity of the Bcr coiled-coil oligomerization domain. Biochemistry. 2005;44(49):16246–56. doi: 10.1021/bi051493t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol. 2002;9(2):117–20. doi: 10.1038/nsb747. [DOI] [PubMed] [Google Scholar]
- 34.Afar DE, Han L, McLaughlin J, Wong S, Dhaka A, Parmar K, et al. Regulation of the oncogenic activity of BCR-ABL by a tightly bound substrate protein RIN1. Immunity. 1997;6(6):773–82. doi: 10.1016/s1074-7613(00)80452-5. [DOI] [PubMed] [Google Scholar]
- 35.Lim YM, Wong S, Lau G, Witte ON, Colicelli J. BCR/ABL inhibition by an escort/phosphatase fusion protein. Proc Natl Acad Sci U S A. 2000;97(22):12233–8. doi: 10.1073/pnas.210253497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han L, Wong D, Dhaka A, Afar D, White M, Xie W, et al. Protein binding and signaling properties of RIN1 suggest a unique effector function. Proc Natl Acad Sci U S A. 1997;94(10):4954–9. doi: 10.1073/pnas.94.10.4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kakar M, Davis JR, Kern SE, Lim CS. Optimizing the protein switch: altering nuclear import and export signals, and ligand binding domain. J Control Release. 2007;120(3):220–32. doi: 10.1016/j.jconrel.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006;224:213–32. doi: 10.1111/j.1365-2818.2006.01706.x. Pt 3. [DOI] [PubMed] [Google Scholar]
- 39.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86(6):3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dixon AS, Lim CS. The nuclear translocation assay for intracellular protein-protein interactions and its application to the Bcr coiled-coil domain. Biotechniques. 2010;49(1):519–24. doi: 10.2144/000113452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kanwal C, Mu S, Kern SE, Lim CS. Bidirectional on/off switch for controlled targeting of proteins to subcellular compartments. J Control Release. 2004;98(3):379–93. doi: 10.1016/j.jconrel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 42.Kakar M, Cadwallader AB, Davis JR, Lim CS. Signal sequences for targeting of gene therapy products to subcellular compartments: the role of CRM1 in nucleocytoplasmic shuttling of the protein switch. Pharm Res. 2007;24(11):2146–55. doi: 10.1007/s11095-007-9333-1. [DOI] [PubMed] [Google Scholar]
- 43.Thai M, Ting PY, McLaughlin J, Cheng D, Muschen M, Witte ON, et al. ABL fusion oncogene transformation and inhibitor sensitivity are mediated by the cellular regulator RIN1. Leukemia. 2011;25(2):290–300. doi: 10.1038/leu.2010.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cao X, Tanis KQ, Koleske AJ, Colicelli J. Enhancement of ABL kinase catalytic efficiency by a direct binding regulator is independent of other regulatory mechanisms. J Biol Chem. 2008;283(46):31401–7. doi: 10.1074/jbc.M804002200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McWhirter JR, Galasso DL, Wang JY. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol. 1993;13(12):7587–95. doi: 10.1128/mcb.13.12.7587. [DOI] [PMC free article] [PubMed] [Google Scholar]