

Abstract
Cerebral malaria (CM) is the most severe complication of human infection with Plasmodium falciparum. The mechanisms predisposing to CM are still not fully understood. Proinflammatory immune responses are required for the control of blood-stage malaria infection but are also implicated in the pathogenesis of CM. A fine balance between pro- and anti-inflammatory immune responses is required for parasite clearance without the induction of host pathology. The most accepted experimental model to study human CM is Plasmodium berghei ANKA (PbANKA) infection in C57BL/6 mice that leads to the development of a complex neurological syndrome which shares many characteristics with the human disease. We applied this model to study the outcome of PbANKA infection in mice previously infected with Mycobacterium tuberculosis, the causative agent of tuberculosis. Tuberculosis is coendemic with malaria in large regions in the tropics, and mycobacteria have been reported to confer some degree of unspecific protection against rodent Plasmodium parasites in experimental coinfection models. We found that concomitant M. tuberculosis infection did not change the clinical course of PbANKA-induced experimental cerebral malaria (ECM) in C57BL/6 mice. The immunological environments in spleen and brain did not differ between singly infected and coinfected animals; instead, the overall cytokine and T cell responses in coinfected mice were comparable to those in animals solely infected with PbANKA. Our data suggest that M. tuberculosis coinfection is not able to change the outcome of PbANKA-induced disease, most likely because the inflammatory response induced by the parasite rapidly dominates in mice previously infected with M. tuberculosis.
INTRODUCTION
Malaria is the most common and most deadly parasitic infection in the world. The vector-borne disease is caused by apicomplexan parasites of the genus Plasmodium and transmitted by Anopheles mosquitoes. Clinical manifestations in humans range from self-resolving malaria to life-threatening disease. Malaria tropica, the most severe form, is caused by Plasmodium falciparum and accounts for the majority of malaria-related deaths. A fine balance between pro- and anti-inflammatory immune responses is required for parasite clearance without the induction of host pathology associated with life-threatening complications such as respiratory distress, metabolic acidosis, severe malarial anemia, and cerebral malaria (CM). The precise mechanisms and factors predisposing to CM are far from being understood. Human studies are obviously limited by the fact that cerebral pathology can be analyzed only postmortem. By utilizing susceptible mouse strains, it is possible to study the events that lead to cerebral pathology. Infection of C57BL/6 mice with Plasmodium berghei ANKA (PbANKA) causes experimental cerebral malaria (ECM), which shares many characteristics with the human disease (1, 2). While the exact mechanisms that lead to the development of both human and experimental CM are not fully understood, it is thought that the combination of sequestration of parasitized red blood cells (pRBCs) and a strong inflammatory immune response involving cytokines such as gamma interferon (IFN-γ), lymphotoxin alpha (LT-α), tumor necrosis factor alpha (TNF-α), and both CD8+ and CD4+ T cells causes CM (3–10). While CD4+ T cells are required during the early induction phase of ECM, CD8+ T cells mediate late-stage immunopathology and seem to directly contribute to blood-brain barrier damage (4, 11). In fact, antigen-specific CD8+ T cells seem to be of major importance as they are activated during PbANKA infection in the spleen and migrate to the brain just before the onset of neurological symptoms (12). A recent study identified for the first time a conserved and highly immunogenic CD8 epitope which is cross-presented by brain microvessels during PbANKA infection (13). It has been postulated that a certain number of parasites in the brain is required for the full activation of cytotoxic CD8+ T cells (13, 14), suggesting that, indeed, coinciding parasite and CD8+ T cell sequestration causes ECM.
Several groups have shown that the modulation of parasite burden protects against ECM (4, 8, 15, 16). This might at least in part be explained by the reduced availability of parasite antigen in the brain microvasculature (13). We and others have found some degree of nonspecific protection against rodent Plasmodium parasites in the presence of mycobacterial infection (17–21). This protective effect is mainly reflected by reduced parasitemia in coinfected compared to singly infected mice and is believed to be mediated by the mycobacterium-induced proinflammatory immune response (17, 19, 20). Of interest, simultaneous infection of C57BL/6 mice with PbANKA and the closely related PbK173 strain, which does not induce cerebral symptoms, could protect mice from the development of ECM (16). In this coinfection model, early IFN-γ production induced by PbK173 has been associated with protection from ECM (16). Mycobacterium tuberculosis, the causative agent of tuberculosis (Tb), is coendemic with P. falciparum in many regions in the world, and M. tuberculosis is a potent inducer of type I (Th1) immune responses (22), including large amounts of IFN-γ, the hallmark Th1 cytokine which is crucial for protection. Therefore, we were interested to see whether coinfection with M. tuberculosis would actually reduce the risk of development of PbANKA-induced ECM in susceptible C57BL/6 mice. In order to study this, we used a murine coinfection model where mice were infected with M. tuberculosis followed by inoculation with PbANKA. Contrary to our hypothesis, we found no impact of concurrent M. tuberculosis infection on the outcome of PbANKA infection in C57BL/6 mice. All mice developed similar levels of parasitemia and succumbed to ECM. The immunological environments in spleen and brain did not differ between singly infected and coinfected animals; instead, the overall cytokine and T cell responses in coinfected mice were comparable to those in animals solely infected with PbANKA. Our study demonstrates that a preexisting proinflammatory immune environment does not necessarily have a beneficial effect on the outcome of concurrent malaria. Moreover, our data indicate that the elicited immune response to PbANKA which is implicated in disease pathogenesis rapidly dominates in mice previously infected with M. tuberculosis.
MATERIALS AND METHODS
Ethics statement.
Animal experiments were approved by the Ethics Committee for Animal Experiments of the Ministry for Agriculture, Environment, and Rural Areas of the State of Schleswig-Holstein (Kommission für Tierversuche/Ethik-Kommission des Landes Schleswig-Holstein) under licenses 33-3/10 (“Die Auswirkung von Tuberkulose auf die Pathogenese und Immunantwort bei Malaria im Rahmen einer Koinfektion in der Maus”/“The impact of tuberculosis on pathogenesis and immune responses to malaria in an experimental coinfection mouse model”) and 51-5/14 [“Charakterisierung der durch Koinfektion mit Mycobacterium tuberculosis bedingten Immunmodulation während der experimentellen zerebralen Malaria (ECM)”/“Characterization of the immunomodulation induced by coinfection with Mycobacterium tuberculosis in the course of experimental cerebral malaria (ECM)”].
Mice, bacterial infection, and CFU.
For all experiments, female C57BL/6 mice aged between 6 and 8 weeks, obtained from Charles River Laboratories, were used. Mice were maintained under specific barrier conditions in biosafety level 3 (BSL3) facilities.
M. tuberculosis H37Rv was grown in Middlebrook 7H9 broth (BD Biosciences) supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC) enrichment medium (BD Bioscience). Bacterial cultures were harvested and resuspended in phosphate-buffered saline (PBS)–10% glycerol, and aliquots were frozen at −80°C until later use. Viable cell counts in thawed aliquots were determined by plating serial dilutions of cultures onto Middlebrook 7H11 agar plates followed by incubation at 37°C.
For infection of experimental animals, M. tuberculosis stocks were diluted in sterile distilled water at a concentration providing an uptake of 200 viable bacilli per lung. Infection was performed via the respiratory route by using an aerosol chamber (Glas-Col, Terre-Haute, IN, USA). Animals were exposed for 40 min to an aerosol generated by nebulizing the prepared M. tuberculosis suspension (23). The inoculum size was quantified 24 h after infection by determining bacterial loads in the lungs of infected mice. Bacterial loads in lung and spleen were evaluated at different time points after aerosol infection by mechanical disruption of the organs in 0.05% (vol/vol) Tween 20–PBS containing a proteinase inhibitor cocktail (Roche) prepared according to the manufacturer's instructions. Tenfold serial dilutions of organ homogenates in sterile water–1% (vol/vol) Tween 80–1% (wt/vol) albumin (WTA) were plated on Middlebrook 7H11 agar plates and incubated at 37°C. Colonies were enumerated after 3 to 4 weeks.
Parasitic infection and evaluation of disease.
PbANKA was maintained by alternating cyclic passage of the parasites in Anopheles stephensi mosquitoes and BALB/c mice at the mosquito colony of the Bernhard Nocht Institute for Tropical Medicine. Blood was collected from highly parasitemic mice, and aliquots were stored in liquid nitrogen in a solution of 0.9% NaCl, 4.6% sorbitol, and 35% glycerol.
Experimental naive mice or animals preinfected for 15 or 30 days with M. tuberculosis were infected intraperitoneally (i.p.) with 1 × 105 or 1 × 104 PbANKA-infected red blood cells (RBCs) from a homologue donor, which had been infected from frozen stock. Parasitemia was determined on Giemsa-stained blood smears from tail blood. Mice were monitored twice daily after day 5 (d5) postinfection (p.i.) for clinical ECM evaluation according to the severity of the symptoms, including gait, motor performance, limb strength, body position, weight loss, pinna reflex, and grooming. Animals with severe ECM were euthanized to avoid unnecessary suffering, and the time point that followed was denoted the time of death.
Adoptive T cell transfer.
OT-I mice were sacrificed, and spleens were removed. To obtain a single-cell suspension, spleens were passed through a 100-μm-pore-size cell strainer and erythrocytes were lysed (using 155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA, with H2O added to achieve the desired volume). The isolation of CD8+ T cells was performed by magnetic activated cell sorting (MACS) (Miltenyi) using a Pan T cell II isolation kit according to the manufacturer's instruction (negative selection). OT-I T cells (2 × 106) were adoptively transferred intravenously (i.v.) 3 days after PbANKA ovalbumin (PbANKA-Ova) infection.
Cell isolation and purification from brains and spleens.
Mice were sacrificed at different time points p.i. with PbANKA and perfused intracardially with 20 ml PBS to remove circulating leukocytes from the tissue. Brains and spleens were then removed and passed through a 100-μm-pore-size cell strainer to obtain single-cell suspensions. Brains were further passed through a 70-μm-pore-size cell strainer. Remaining erythrocytes in spleen suspensions were lysed (using 155 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA, with H2O added to achieve the desired volume), and cells were resuspended in RPMI 1640 supplemented with 2 mM glutamine–1% (vol/vol) HEPES–50 μM β-mercaptoethanol–10% (vol/vol) heat-inactivated fetal calf serum (complete RPMI 1640 medium).
Flow cytometry.
For flow cytometric analysis of surface markers and intracellular cytokines, single-cell suspensions of brains and spleens were stained with optimal concentrations of the following specific antibodies (Abs): CD45-V450, CD4-V500, CD8a fluorescein isothiocyanate (FITC), CD44-peridinin chlorophyll protein (PerCP)-Cy5.5, CD62L-allophycocyanin (APC), CXC chemokine receptor 3 (CXCR3)-phycoerythrin (PE), CD3e-APC-Cy7, IFN-γ–APC, and interleukin-2 (IL-2)–PE–Cy7 from BD Biosciences; CD8a-Pacific Blue, CD80-AF488, CD11c-PE, CD11b-PerCP-Cy5.5, I-A/I-E–PE-Cy7, CD86–APC, Ly6G-APC-Cy7, CD19-PE, TNF-α–Pacific Blue, and IL-10–PE from BioLegend; CD90.2-eFluor780 from eBioscience; and major histocompatibility complex class I (MHC-I) Ova Pentamer from ProImmune (Oxford, United Kingdom). Data were acquired on a FacsCantoII flow cytometer (BD Biosciences) equipped with a 405-, 488-, and 633-nm-wavelength laser and analyzed with the FCSExpress software (DeNovo Software).
Intracellular cytokine staining.
Single-cell suspensions of spleen (1 × 106) were stimulated for 4.5 h with anti-CD3e/anti-CD28 (BioLegend; 5 μg/ml, respectively) in the presence of GolgiPlug (BD Biosciences) (contains brefeldin A). Nonspecific antibody binding was blocked by incubation with a cocktail containing anti-FcγRIII/II monoclonal Ab (BioLegend) and mouse, hamster, and rat serum. Subsequently, cells were stained with directly labeled anti-CD90.2, anti-CD44, anti-CD4, and anti-CD8a antibodies for 20 min at 4°C. After washing was performed, cells were fixed and permeabilized overnight with Cytofix/Cytoperm (BD Biosciences). Cells were washed with Perm/Wash buffer (BD Biosciences) and stained with directly labeled anti-IFN-γ, anti-IL-10, anti-IL-2, and anti-TNF-α antibodies for 45 min at 4°C.
Multiplex cytokine assay.
The concentrations of IL-10, IFN-γ, and TNF-α in spleen homogenates and serum were determined by cytometric bead array (mouse inflammation kit; BD Biosciences) or Legendplex (mouse inflammation panel; BioLegend) according to the manufacturer's protocol.
RNA isolation, cDNA synthesis, and quantitative real-time PCR.
Total RNA from brain was extracted using TRIzol reagent (Invitrogen) and a Direct-zol RNA MiniPrep kit (Zymo Research) as recommended by the manufacturer. For quantitative real-time PCR, 400 ng of total RNA was reverse transcribed (RT) using a Maxima First Strand cDNA synthesis kit for RT-quantitative PCR (RT-qPCR) (Life Technologies) according to the manufacturer's instruction at 25°C for 10 min, 55°C for 30 min, and 85°C for 3 min. RT-qPCRs were performed using LightCycler 480 SYBR green I Master (Roche). PCR amplifications were performed in duplicates in a total volume of 10 μl, containing 1 μl of cDNA sample, 0.2 μl of primer pairs (10 μM), 5 μl of SYBR green mix, and 3.8 μl of RNase/DNase-free water. Data analysis was performed using a LightCycler 480 instrument. The PCR cycling protocol entailed 1 cycle at 95°C for 10 min and 45 cycles at 95°C for 10 s, 58 to 63°C for 10 s, and 78°C for 8 s as well as 1 cycle each at 72°C for 1 s, 95°C for 10 s, and 65°C for 10 s. Analysis of the relative changes was performed using LightCycler480 1.5.0 SP4 software (Version 1.5.0.39; Roche). All quantifications were normalized to the level of hypoxanthine guanine phosphoribosyltransferase (HPRT) gene expression (housekeeping gene). The following primers were used: HPRT forward (TCCTCCTCAGACCGCTTTT) and reverse (CATAACCTGGTTCATCATCGC); IFN-γ forward (TCAAGTGGCATAGATGTGGAAGAA) and reverse (TGGCTCTGCAGGATTTTCATG); TNF-α forward (CCACCACGCTCTTCTGTCTAC) and reverse (AGGGTCTGGGCCATAGAACT); IL-12 forward (CATCATCAAACCAGACCCGCCCAA) and reverse (AACTTGAGGGAGAAGTAGGAATGG); IL-10 forward (GGTTGCCAAGCCTTATCGGA) and reverse (ACCTGCTCCACTGCCTTGCT); monocyte chemotactic protein 1 (MCP-1) forward (CCTGCTGTTCACAGTTGCC) and reverse (ATTGGGATCATCTTGCTGGT); keratinocyte-derived chemokine (KC) forward (ACCCAAACCGAAGTCATAGC) and reverse (TCTCCGTTACTTGGGGACAC); and PbANKA 18S rRNA forward (AAGCATTAAATAAAGCGAATACATCCTTAC) and reverse (GGAGATTGGTTTTGACGTTTATGTG).
Statistical analysis.
Statistical analysis was performed using the Mann-Whitney test or Kruskal-Wallis test followed by Dunn's multiple-comparison test as described in the figure legends. Statistical analysis of survival curves was performed using the log rank test. All data were analyzed using GraphPad Prism 5 (GraphPad Software, Inc.).
RESULTS
Coinfected mice succumb to ECM.
We hypothesized that M. tuberculosis coinfection would influence PbANKA-induced parasitemia and disease outcome in ECM-susceptible C57BL/6 mice. Therefore, mice were infected via the aerosol route with M. tuberculosis H37Rv, and 30 days later, when the immune response to M. tuberculosis was fully established and lung CFU controlled at around 2 × 106 (see Fig. SA1A in the supplemental material), mice were infected with 1 × 105 PbANKA pRBCs i.p. Parasitemia was monitored daily on Giemsa-stained thin blood smears starting 4 days after PbANKA infection. We did not detect significant differences in parasitemia or weight loss between singly infected and coinfected animals (Fig. 1A and B). Independently of M. tuberculosis infection, all PbANKA-infected mice developed neurological symptoms such as ataxia, tremor, and loss of motor function between day 6 and 8 indicative of ECM. Consequently, mice succumbed to PbANKA infection after 6 to 8 days, without a significant difference between singly infected and coinfected mice (Fig. 1C).
FIG 1.

Influence of M. tuberculosis coinfection on the development of ECM in PbANKA-infected mice. (A to C) C57BL/6 mice were infected via the aerosol route with M. tuberculosis (M.tb) H37Rv (d1 CFU, 150) and 30 days later with 1 × 105 pRBCs i.p. (groups of 5 to 10 mice; results of one experiment representative of two are shown). (D to F) C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv (d1 CFU, 300) and 30 days later with 1 × 104 pRBCs i.p. (groups of 9 to 10 mice). (G to I) C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv (d1 CFU, 120) and 15 days later with 1 × 105 pRBCs i.p. (groups of 9 to 10 mice; note that survival data represent groups of 4 to 5 mice). Parasitemia was monitored daily on Giemsa-stained thin blood smears starting 4 days after PbANKA infection. Statistical analysis was performed using the Mann-Whitney test for parasitemia and weight loss (data represent means ± standard deviations [SD]) and the log-rank test for survival rates. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
As described here and elsewhere, the onset of neurological symptoms after PbANKA infection is very rapid, usually resulting in death of ECM-susceptible animals after between 6 and 9 days. This time window might be too narrow to allow detection of any potential effects of concurrent M. tuberculosis infection on the development of ECM. In order not to miss any potential M. tuberculosis-related alterations in PbANKA-induced disease outcome, we reduced the number of parasites used to infect mice 10-fold, aiming at a delayed onset of neurological symptoms. As expected, mice displayed signs of ECM from day 7 and died between 7 and 11 days after PbANKA infection except for 1 mouse in the PbANKA group, which did not develop any neurological symptoms. Despite the fact that M. tuberculosis coinfection significantly increased body weight loss, no significant differences in survival compared to mice infected with PbANKA alone were observed (Fig. 1D to F).
In order to appreciate how the different immunological environments that are present at different times of M. tuberculosis infection influence the outcome of PbANKA infection, we next challenged M. tuberculosis-infected mice with PbANKA after 15 days (lung CFU, 2.3 × 104 ± 7,720). During this acute phase of M. tuberculosis infection, adaptive immunity is not fully established and mycobacterial replication not yet controlled. Again, the course of PbANKA infection did not differ in coinfected compared to singly infected mice. All mice showed comparable levels of parasitemia and weight loss over time (Fig. 1G and H), developed neurological symptoms, and died from ECM between days 6 and 7 (Fig. 1I).
Taken together, these data suggest that concurrent M. tuberculosis infection does not influence the development of ECM induced by PbANKA infection in C57BL/6 mice. Mycobacterial loads in lung, spleen, and liver were not changed by concurrent PbANKA infection, most likely because the short time course was not sufficient to induce regrowth of chronic infection-stage M. tuberculosis (data not shown).
M. tuberculosis coinfection does not alter cytokine responses.
Although we did not find changes in the outcome of PbANKA-induced ECM in mice that had been preinfected with M. tuberculosis, we were interested in analyzing immune responses in co- and singly infected animals. Our aim was to find out to what extent M. tuberculosis infection did modulate cytokine and T cell responses classically associated with the induction of ECM but without having any impact on disease outcome. We performed all analysis in animals which had been infected with M. tuberculosis 30 days before PbANKA infection because we considered this the most relevant model. After 30 days, the adaptive immune response to M. tuberculosis is established and bacterial replication in the lungs well controlled. However, by that time mycobacteria have disseminated to other sites of the body, including the spleen (see Fig. SA1B in the supplemental material), thereby inducing not only a local immune response in the lung but also a systemic one which might interfere with immune responses to the malaria parasite in the spleen.
Compared to naive mice, M. tuberculosis-infected mice showed significant production of IFN-γ in serum and spleen at the time of PbANKA infection (Fig. 2, day 0 [d0]) and significant although low production of TNF-α and IL-10 in the spleen. The IFN-γ and TNF-α proinflammatory cytokines have been closely linked both to malaria immunity and to the manifestations of CM (2, 6, 9, 24–26). In contrast, IL-10 is associated with protection mediated by counteracting proinflammatory cytokine production (27–29). However, the different cytokine milieu at the time of PbANKA infection was lost in the course of the PbANKA infection (Fig. 2, d4 and d6). We determined cytokine concentrations in serum and spleen 4 days after PbANKA infection and in moribund mice sacrificed between 6 and 8 days after PbANKA infection. We did not find significant differences in the levels of any of the mediators in coinfected compared to PbANKA singly infected animals (Fig. 2). Moreover, overall cytokine profiles in coinfected animals were comparable to those in animals solely infected with PbANKA, indicating that PbANKA-induced cytokine responses rapidly dominated in mice previously infected with M. tuberculosis.
FIG 2.

Cytokine responses induced by infection with M. tuberculosis or PbANKA or both. C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv and 30 days later with 1 × 105 pRBCs i.p. Cytokine levels were measured in serum and spleen lysates of naive, M. tuberculosis-infected, and coinfected mice at the indicated times after PbANKA infection as described in Materials and Methods. Data from two independent experiments are shown (n = 5 to 15, mean ± SD, Kruskal-Wallis test with Dunn′s posttest). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The local cerebral environment is crucial in the pathogenesis of ECM. Therefore, we analyzed the expression of certain immune mediators in the brains by qRT-PCR 6 days after PbANKA infection in singly infected and coinfected animals. Again, no differences in the levels of expression of the studied mediators (IFN-γ, TNF-α, IL-12, IL-10, MCP-1, and KC; Fig. 3) were observed. As expected, expression of these mediators in brains of animals infected with M. tuberculosis alone was very low.
FIG 3.

Expression pattern of chemokines and cytokines in the brain. C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv and 30 days later with 1 × 105 pRBCs i.p. Perfused brains were collected on day 6 after PbANKA infection for RNA isolation followed by cDNA synthesis. qRT-PCR was used to analyze cyto- and chemokine expression relative to expression of the HPRT housekeeping gene. Symbols and bars represent individual mice and means, respectively. Data from two independent experiments are shown (n = 8 to 10, mean ± SD, Kruskal-Wallis test with Dunn′s posttest). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In conclusion, chronic M. tuberculosis infection had no impact on the cytokine environment induced by subsequent PbANKA infection.
Coinfection does not affect the activation status of CD11chi DCs in the spleen.
Dendritic cells (DCs) are crucial for priming of parasite-specific T cells during PbANKA infection, and depletion of conventional DCs (cDCs) has been shown to prevent ECM (30). We wondered whether the cDC compartment in the spleen was affected by concurrent M. tuberculosis infection and determined the presence and activation status of CD11chi DCs 4 days after PbANKA infection. We found similar numbers of CD11chi DCs and no differences in the levels of induction of MHC-II molecules or costimulatory receptors in the presence or absence of M. tuberculosis (Fig. 4). These results suggested that cDCs in M. tuberculosis-infected animals were not impaired in T cell priming upon PbANKA infection.
FIG 4.

Coinfection does not affect the activation status of DCs in the spleen. C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv and 30 days later with 1 × 105 pRBCs i.p. Spleens were collected 4 days after PbANKA infection, and single-cell suspensions were analyzed for the presence and activation status of CD11bint CD11chi DCs by flow cytometry. (A) Splenocytes were gated on CD45 cells and further on Ly6G-negative cells and analyzed for the presence of CD11bint CD11chi DCs. (B to E) CD11bint CD11chi DCs were analyzed for their expression of CD80 and CD86 and of MHC-II. Data from two independent experiments are shown as box and whisker plots with medians indicated (A to D) or as means ± SD (E) (n = 6 to 9, Kruskal-Wallis test with Dunn′s posttest). *, P < 0.05. For full gating strategies, see Fig. SA2 in the supplemental material.
T cell responses in spleen of PbANKA-infected mice are not affected by concurrent M. tuberculosis infection.
Next, we investigated singly infected and coinfected animals to dissect T cell responses in the spleen, which is proposed to be the site of initial T cell priming during PbANKA infection (30–32). Changes in T cell activation in the spleen might have an impact on the subsequent recruitment of lymphocytes to the brain. To assess any potential impact of M. tuberculosis coinfection before and at the onset of neurological symptoms, we analyzed CD8+ and CD4+ T cell responses 4 and 6 days after PbANKA infection. We found similar numbers of CD8+ and CD4+ T cells in the absence or presence of M. tuberculosis infection and no differences in the amounts of effector memory CD8+ T cells (CD62L− CD44+; Fig. 5A) at the two time points. In contrast, the numbers of effector memory CD4+ T cells were significantly increased at day 4 of PbANKA infection when M. tuberculosis infection was concurrent. This difference was, however, lost at day 6 of PbANKA infection. CXC chemokine receptor 3 (CXCR3) expression on splenic T cells is responsible for their recruitment to the brain in response to IFN-γ-inducible protein-10 (IP-10) (7, 33). Concurrent M. tuberculosis infection transiently increased the numbers of CXCR3+ cells expressing CD4 but not the numbers of CXCR3+ CD8+ T cells in coinfected compared to PbANKA singly infected mice (Fig. 5A, d4). The difference was no longer apparent as PbANKA infection developed. At day 6, when animals showed neurological symptoms, the numbers of CXCR3-expressing CD8+ and CD4+ T cells were comparable between singly infected and coinfected animals. Intracellular cytokine staining of splenic CD8+ and CD4+ T lymphocytes restimulated with anti-CD3/anti-CD28 revealed no differences in the levels of production of IFN-γ, TNF-α, IL-10, and IL-2 between PbANKA singly infected and coinfected animals except for the production of IFN-γ by CD4+ T cells on day 4 of PbANKA infection (Fig. 5B), which was significantly increased in the presence of M. tuberculosis. This difference was, however, no longer apparent on day 6. Overall cytokine profiles of CD4+ and CD8+ T cells from coinfected animals were rather similar to those from animals solely infected with PbANKA, indicating that PbANKA infection overwrites T cell responses induced by M. tuberculosis infection.
FIG 5.
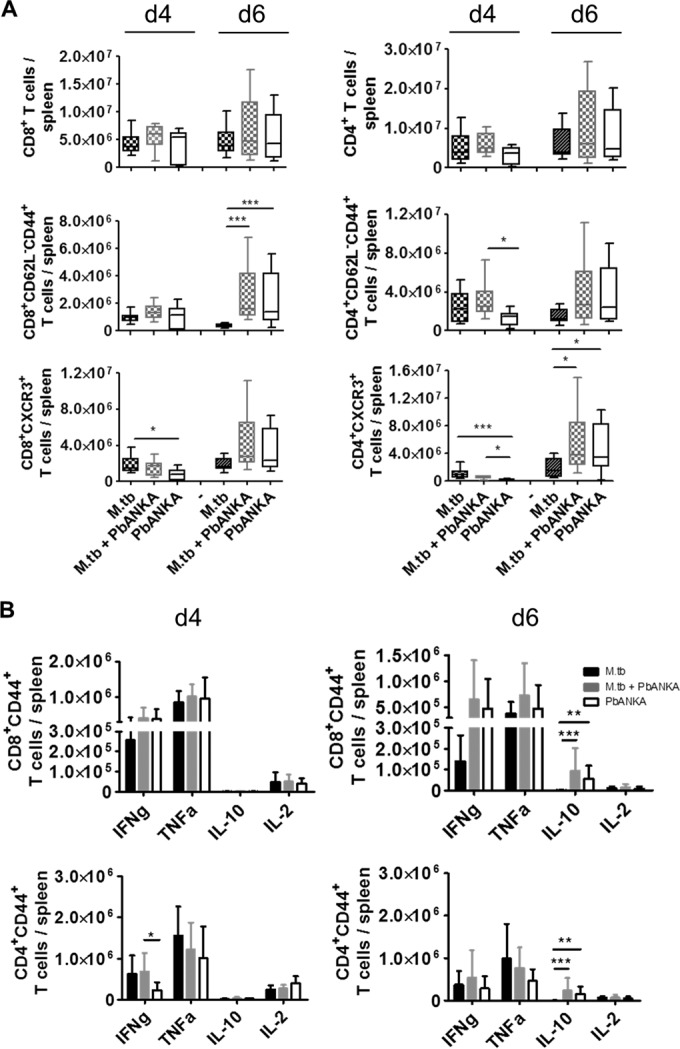
T cell responses in the spleen after infection with PbANKA in the presence or absence of M. tuberculosis. C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv and 30 days later with 1 × 105 pRBCs i.p. Spleens were collected 4 or 6 days after PbANKA infection, and single-cell suspensions were analyzed for the presence and activation status of CD4+ and CD8+ T cells by flow cytometry. (A) Splenocytes were gated on CD90.2 and analyzed for the total numbers of CD4+ and CD8+ T cells, for the numbers of effector memory T cells (CD62L− CD44+), and for the expression of CXCR3. Data are presented as box and whisker plots with medians. (B) Spleen cells were restimulated ex vivo with anti-CD3 and anti-CD28 (5 μg/ml, respectively) and analyzed by flow cytometry for the presence of IFN-γ-, TNF-α-, IL-10-, or IL-2-producing CD44+ CD4+ and CD44+ CD8+ T cells gated for CD90.2 (presented as means ± SD). Data from two (d4) or 3 (d6) independent experiments are shown (n = 8 to 15, Kruskal-Wallis test with Dunn′s posttest). *, P < 0.05; **, P < 0.01; ***, P < 0.001. For full gating strategies, see Fig. SA3 in the supplemental material.
Sequestration of parasites and antigen-specific CD8+ T cells in the brains of PbANKA-infected mice occurs independently of concurrent M. tuberculosis infection.
ECM is associated with leukocyte sequestration in the brain (7, 11, 34). Parasite-specific CD8+ T cells in particular are required for late-stage immunopathology during ECM as they can damage the blood-brain barrier due to their cytotoxic activity (4, 13). Therefore, we analyzed whether M. tuberculosis coinfection altered T cell infiltration of brains from PbANKA-infected mice.
4 days after PbANKA infection, the numbers of brain-sequestered T cells were very low and we could not detect any differences between brains from coinfected mice and brains from those infected with PbANKA alone (Fig. 6A). Moreover, we found no differences in the overall numbers of CD8+ and CD4+ T cells in brains with single infections versus coinfections and in their effector memory phenotypes at the onset of disease symptoms 6 days after PbANKA infection (Fig. 6B). Furthermore, the levels of expression of CXCR3 seen with brain CD8+ and CD4+ T cells were similar in the presence and absence of M. tuberculosis.
FIG 6.

Sequestration of T cells and parasites in the brain of PbANKA-infected mice in the presence and absence of M. tuberculosis. (A and B) C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv and 30 days later with 1 × 105 pRBCs i.p. Perfused brains were collected 4 days (A) or 6 days (B) after PbANKA infection and analyzed for the presence and activation status of CD4+ and CD8+ T cells by flow cytometry. Brain cells were gated on CD45 and analyzed for the total numbers of CD4+ and CD8+ T cells, for the numbers of effector memory T cells (CD62L− CD44+), and for the expression of CXCR3. Data from two independent experiments are presented as box and whisker plots with medians (n = 8 to 10 in panel A and n = 10 in panel B, Kruskal-Wallis test with Dunn's posttest). *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv and 30 days later with 1 × 105 PbANKA or PbANKA-Ova pRBCs i.p. (groups of 4). At 3 days later, 2 × 106 OT-I T cells were adoptively transferred i.v. Perfused brains were collected 6 days after PbANKA-Ova infection and analyzed for the presence of CD45+ CD8+ MHC-I pentamer+ T cells. Data are presented as box and whisker plots with medians. (D) C57BL/6 mice were infected via the aerosol route with M. tuberculosis H37Rv and 30 days later with PbANKA (1 × 105 pRBC i.p.). Perfused brains were collected 6 days after PbANKA infection, and brain parasite load was determined based on parasite-specific 18S rRNA transcription. Data from two independent experiments are shown as box and whisker plots with medians (n = 9 or 10, Kruskal-Wallis test with Dunn′s posttest). *, P < 0.05; **, P < 0.01; ***, P < 0.001. For full gating strategies, see Fig. SA4 in the supplemental material.
These data provided no information on the antigen specificity of the recruited T cells. To address the issue of whether M. tuberculosis coinfection alters the sequestration of parasite-specific T cells in the brain, we used a transgenic PbANKA strain expressing MHC-I-restricted epitope SIINFEKL from chicken ovalbumin (Ova) (12), which is recognized by Ova-specific CD8+ (OT-I) T cells. OT-I T cells were adoptively transferred into singly infected or coinfected mice 3 days after PbANKA infection. At 3 days later (d 6 p.i.), brains were analyzed for the presence of OT-I T cells by MHC-I pentamer staining. The number of pentamer-positive (pentamer+) CD8+ T cells was slightly but not significantly enhanced in brains of M. tuberculosis-coinfected mice (Fig. 6C).
ECM is associated with the sequestration not only of leukocytes but also of parasites in the brain (3, 8, 15). We determined parasite numbers in brains of singly infected and coinfected mice by qRT-PCR, which did not reveal any differences in parasite sequestration between coinfected and PbANKA singly infected mice (Fig. 6D).
Taking the data together, parasite and T cell sequestration in the brain was not affected by concurrent M. tuberculosis infection.
DISCUSSION
Mycobacterial protection against infection by different rodent Plasmodium parasites has been described in multiple reports (17–21, 35–37). While the majority of such studies addressed the issue of whether the widely used M. bovis bacillus Calmette-Guérin (BCG) tuberculosis vaccine strain confers nonspecific protection against subsequent Plasmodium infection, a few studies investigated concurrent infection with virulent M. tuberculosis and rodent malaria parasites and noted a beneficial effect of M. tuberculosis coinfection against mouse malaria (17, 20). One of these reports noted that aerosol infection with M. tuberculosis 2 or 8 weeks before challenge with lethal P. yoelii resulted in reduced parasitemia and significantly increased survival of coinfected mice compared to those infected only with P. yoelii XL (20). Our group recently reported that mice presented with significantly reduced parasite numbers in the peripheral blood and reduced liver damage upon PbNK65 infection when the mice had previously been infected with M. tuberculosis (17), also suggesting a beneficial effect of mycobacterial coinfection on the outcome of malaria.
BCG-induced protection against subsequent Plasmodium challenge was demonstrated almost 40 years ago (21, 35). Smrkovski and Strickland reported considerable protection against PbANKA infection in mice that had been previously infected with BCG (35). In their studies, a single dose of BCG administered i.v. 10 days before PbANKA challenge protected 50% of the mice from death. Unexpectedly, and in contrast to the study described above, we did not find an influence of chronic M. tuberculosis infection on PbANKA parasitemia in the present study. Consequently, coinfected mice were not protected but succumbed to ECM like the singly infected controls. However, there are several differences between the two studies. First, while we used pRBC to infect mice with PbANKA, Smrkovski and Strickland used sporozoites. Infection with pRBC, in contrast to sporozoites, gives rise to blood-stage malaria while excluding the liver-stage phase. It is possible that BCG vaccination interfered with subsequent PbANKA infection already at the liver-stage phase, which we would have missed in our study.
Second, Smrkovski and Strickland infected mice with BCG and not with M. tuberculosis, and, more importantly, the route of BCG infection was i.v. and was hence systemically and physiologically not relevant, while we infected mice with M. tuberculosis via the natural aerosol route. Systemic BCG infection rapidly causes high mycobacterial loads and immune activation in spleen, liver, and lung, with the highest bacterial loads found in the spleen (38). BCG titers slowly decline over time, which might explain the temporary effect of BCG-mediated protection against malaria infection, which vanished after 30 days (35). BCG-mediated protection against unrelated pathogens such as Plasmodium parasites is thought to be nonspecific. The data suggest that the presence of BCG bacteria is required for this nonspecific protection, presumably because they stimulate innate immune mechanisms that can counteract parasite infections (36, 39). Aerosol M. tuberculosis infection mainly targets the lung, and mycobacteria disseminate only slowly to other sites such as the lung draining lymph nodes, spleen, and liver. Numbers comparable to those seen after i.v. administration are achieved only after several weeks of low-dose infection. Consequently, we found only moderate levels of IFN-γ and very little TNF-α in serum and spleens of M. tuberculosis-infected mice on the day of PbANKA infection, which might explain why we did not find any protective effect on parasitemia and, eventually, the induction of ECM in coinfected mice. However, Page et al. reported significant protection from lethal P. yoelii infection in mice previously infected with M. tuberculosis by low-dose aerosol challenge (20) that had already occurred after 2 weeks, suggesting that M. tuberculosis-induced immune activation is able to control subsequent malaria infection. Of note, those authors used a different strain of M. tuberculosis (CDC1551) and reported the recovery of high numbers of bacteria from spleens by 2 weeks after M. tuberculosis aerosol infection, a time point at which most of our mice were still negative for mycobacteria in the spleen (data not shown). This might explain why those authors, in contrast to us, found an augmented Th1 immune response to be associated with protection against lethal P. yoelii infection when M. tuberculosis was concurrent. Gene expression analysis revealed that not the quality but the magnitude of the Th1 immune response was crucial for the protective effect. M. tuberculosis coinfection increased the expression of classical Th1 cytokines such as IFN-γ and TNF-α and of chemokines such as CCL5, CXCL9, and CXCL10. As protection against malaria in the coinfected mice occurred before the onset of adaptive immunity to M. tuberculosis, the authors concluded that the effect was most likely not due to cross-reactive memory T cells. They suggested instead that both active and chronic M. tuberculosis infections generate a systemic immunological environment that promotes immune responses to secondary infections by enhancing innate and pathogen-specific responses.
In our study, chronic M. tuberculosis infection did not augment proinflammatory immune responses induced by PbANKA infection. We found instead the immunological milieu in coinfected mice to be very similar to that in PbANKA singly infected mice, indicating that responses elicited by PbANKA were predominant. Susceptibility of C57BL/6 mice to ECM is attributed to the strong proinflammatory immune response associated with PbANKA infection. Recently, an Irf8-regulated genomic response has been described which drives the pathological inflammation during ECM in C57BL/6 mice (40). Interestingly, this response substantially overlaps the responses of genes activated following M. tuberculosis infection. Those authors therefore suggested the occurrence of a shared core inflammatory response which is protective against M. tuberculosis infection but deleterious with respect to ECM. These observations might also explain why concurrent M. tuberculosis infection was not able to rescue mice from ECM, because the very same responses that are induced to control M. tuberculosis infection contribute to ECM development.
In conclusion, compared to other studies, mice infected concomitantly with Tb and malaria pathogens here had no significant changes in the course of both infections and in overall morbidity and mortality. Our findings suggest that the concurrent presence of mycobacteria does not necessarily have a beneficial effect on the control of coinfecting malaria parasites, particularly in cases in which the two pathogens are associated with the induction of similar proinflammatory pathways which are, as in the case of PbANKA, implicated in disease pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nadine Harmel and Lars Eggers for excellent technical assistance and Ann-Kristin Mueller, University Hospital Heidelberg, for helpful comments on the manuscript.
B. E. Schneider, J. Blank, and T. Jacobs conceived and designed the experiments. J. Blank and B. E. Schneider performed the experiments. J. Blank, J. Behrends, and B. E. Schneider analyzed the data. B. E. Schneider, T. Jacobs, and J. Blank contributed reagents, materials, or analysis tools. B. E. Schneider and J. Blank wrote the paper.
Funding Statement
This work was supported by a Leibniz Center Infection grant to B. E. Schneider and T. Jacobs.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01290-15.
REFERENCES
- 1.de Souza JB, Hafalla JC, Riley EM, Couper KN. 2010. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology 137:755–772. doi: 10.1017/S0031182009991715. [DOI] [PubMed] [Google Scholar]
- 2.Riley EM, Couper KN, Helmby H, Hafalla JC, de Souza JB, Langhorne J, Jarra WB, Zavala F. 2010. Neuropathogenesis of human and murine malaria. Trends Parasitol 26:277–278. doi: 10.1016/j.pt.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Amante FH, Haque A, Stanley AC, de Labastida Rivera F, Randall LM, Wilson YA, Yeo G, Pieper C, Crabb BS, de Koning-Ward TF, Lundie RJ, Good MF, Pinzon-Charry A, Pearson MS, Duke MG, McManus DP, Loukas A, Hill GR, Engwerda CR. 2010. Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J Immunol 185:3632–3642. doi: 10.4049/jimmunol.1000944. [DOI] [PubMed] [Google Scholar]
- 4.Haque A, Best SE, Unosson K, Amante FH, de Labastida F, Anstey NM, Karupiah G, Smyth MJ, Heath WR, Engwerda CR. 2011. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J Immunol 186:6148–6156. doi: 10.4049/jimmunol.1003955. [DOI] [PubMed] [Google Scholar]
- 5.Villegas-Mendez A, de Souza JB, Murungi L, Hafalla JC, Shaw TN, Greig R, Riley EM, Couper KN. 2011. Heterogeneous and tissue-specific regulation of effector T cell responses by IFN-gamma during Plasmodium berghei ANKA infection. J Immunol 187:2885–2897. doi: 10.4049/jimmunol.1100241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Villegas-Mendez A, Greig R, Shaw TN, de Souza JB, Gwyer Findlay E, Stumhofer JS, Hafalla JC, Blount DG, Hunter CA, Riley EM, Couper KN. 2012. IFN-gamma-producing CD4+ T cells promote experimental cerebral malaria by modulating CD8+ T cell accumulation within the brain. J Immunol 189:968–979. doi: 10.4049/jimmunol.1200688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nie CQ, Bernard NJ, Norman MU, Amante FH, Lundie RJ, Crabb BS, Heath WR, Engwerda CR, Hickey MJ, Schofield L, Hansen DS. 2009. IP-10-mediated T cell homing promotes cerebral inflammation over splenic immunity to malaria infection. PLoS Pathog 5:e1000369. doi: 10.1371/journal.ppat.1000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baptista FG, Pamplona A, Pena AC, Mota MM, Pied S, Vigario AM. 2010. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect Immun 78:4033–4039. doi: 10.1128/IAI.00079-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P. 1987. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 237:1210–1212. doi: 10.1126/science.3306918. [DOI] [PubMed] [Google Scholar]
- 10.Engwerda CR, Mynott TL, Sawhney S, De Souza JB, Bickle QD, Kaye PM. 2002. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J Exp Med 195:1371–1377. doi: 10.1084/jem.20020128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Belnoue E, Kayibanda M, Vigario AM, Deschemin JC, van Rooijen N, Viguier M, Snounou G, Renia L. 2002. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J Immunol 169:6369–6375. doi: 10.4049/jimmunol.169.11.6369. [DOI] [PubMed] [Google Scholar]
- 12.Lundie RJ, de Koning-Ward TF, Davey GM, Nie CQ, Hansen DS, Lau LS, Mintern JD, Belz GT, Schofield L, Carbone FR, Villadangos JA, Crabb BS, Heath WR. 2008. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc Natl Acad Sci U S A 105:14509–14514. doi: 10.1073/pnas.0806727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howland SW, Poh CM, Gun SY, Claser C, Malleret B, Shastri N, Ginhoux F, Grotenbreg GM, Renia L. 2013. Brain microvessel cross-presentation is a hallmark of experimental cerebral malaria. EMBO Mol Med 5:916–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haque A, Best SE, Amante FH, Ammerdorffer A, de Labastida F, Pereira T, Ramm GA, Engwerda CR. 2011. High parasite burdens cause liver damage in mice following Plasmodium berghei ANKA infection independently of CD8(+) T cell-mediated immune pathology. Infect Immun 79:1882–1888. doi: 10.1128/IAI.01210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McQuillan JA, Mitchell AJ, Ho YF, Combes V, Ball HJ, Golenser J, Grau GE, Hunt NH. 2011. Coincident parasite and CD8 T cell sequestration is required for development of experimental cerebral malaria. Int J Parasitol 41:155–163. doi: 10.1016/j.ijpara.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 16.Mitchell AJ, Hansen AM, Hee L, Ball HJ, Potter SM, Walker JC, Hunt NH. 2005. Early cytokine production is associated with protection from murine cerebral malaria. Infect Immun 73:5645–5653. doi: 10.1128/IAI.73.9.5645-5653.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller A-K, Behrends J, Hagens K, Mahlo J, Schaible UE, Schneider BE. 2012. Natural transmission of Plasmodium berghei exacerbates chronic tuberculosis in an experimental co-infection model. PLoS One 7:e48110. doi: 10.1371/journal.pone.0048110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy JR. 1981. Host defenses in murine malaria: nonspecific resistance to Plasmodium berghei generated in response to Mycobacterium bovis infection or Corynebacterium parvum stimulation. Infect Immun 33:199–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsumoto S, Yukitake H, Kanbara H, Yamada H, Kitamura A, Yamada T. 2000. Mycobacterium bovis bacillus Calmette-Guerin induces protective immunity against infection by Plasmodium yoelii at blood-stage depending on shifting immunity toward Th1 type and inducing protective IgG2a after the parasite infection. Vaccine 19:779–787. doi: 10.1016/S0264-410X(00)00257-7. [DOI] [PubMed] [Google Scholar]
- 20.Page KR, Jedlicka AE, Fakheri B, Noland GS, Kesavan AK, Scott AL, Kumar N, Manabe YC. 2005. Mycobacterium-induced potentiation of type 1 immune responses and protection against malaria are host specific. Infect Immun 73:8369–8380. doi: 10.1128/IAI.73.12.8369-8380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark IA, Allison AC, Cox FE. 1976. Protection of mice against Babesia and Plasmodium with BCG. Nature 259:309–311. doi: 10.1038/259309a0. [DOI] [PubMed] [Google Scholar]
- 22.Ernst JD. 2012. The immunological life cycle of tuberculosis. Nat Rev Immunol 12:581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- 23.Mueller A-K, Behrends J, Blank J, Schaible UE, Schneider BE. 2014. An experimental model to study tuberculosis-malaria coinfection upon natural transmission of Mycobacterium tuberculosis and Plasmodium berghei. J Vis Exp 2014:e50829. doi: 10.3791/50829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudin W, Eugster HP, Bordmann G, Bonato J, Muller M, Yamage M, Ryffel B. 1997. Resistance to cerebral malaria in tumor necrosis factor-alpha/beta-deficient mice is associated with a reduction of intercellular adhesion molecule-1 up-regulation and T helper type 1 response. Am J Pathol 150:257–266. [PMC free article] [PubMed] [Google Scholar]
- 25.Rudin W, Favre N, Bordmann G, Ryffel B. 1997. Interferon-gamma is essential for the development of cerebral malaria. Eur J Immunol 27:810–815. doi: 10.1002/eji.1830270403. [DOI] [PubMed] [Google Scholar]
- 26.Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, Billiau A, Vassalli P. 1989. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc Natl Acad Sci U S A 86:5572–5574. doi: 10.1073/pnas.86.14.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niikura M, Inoue S, Kobayashi F. 2011. Role of interleukin-10 in malaria: focusing on coinfection with lethal and nonlethal murine malaria parasites. J Biomed Biotechnol 2011:383962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kossodo S, Monso C, Juillard P, Velu T, Goldman M, Grau GE. 1997. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology 91:536–540. doi: 10.1046/j.1365-2567.1997.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Specht S, Ruiz DF, Dubben B, Deininger S, Hoerauf A. 2010. Filaria-induced IL-10 suppresses murine cerebral malaria. Microbes Infect 12:635–642. doi: 10.1016/j.micinf.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 30.deWalick S, Amante FH, McSweeney KA, Randall LM, Stanley AC, Haque A, Kuns RD, MacDonald KP, Hill GR, Engwerda CR. 2007. Cutting edge: conventional dendritic cells are the critical APC required for the induction of experimental cerebral malaria. J Immunol 178:6033–6037. doi: 10.4049/jimmunol.178.10.6033. [DOI] [PubMed] [Google Scholar]
- 31.Curfs JH, Schetters TP, Hermsen CC, Jerusalem CR, van Zon AA, Eling WM. 1989. Immunological aspects of cerebral lesions in murine malaria. Clin Exp Immunol 75:136–140. [PMC free article] [PubMed] [Google Scholar]
- 32.Hermsen CC, Mommers E, van de Wiel T, Sauerwein RW, Eling WM. 1998. Convulsions due to increased permeability of the blood-brain barrier in experimental cerebral malaria can be prevented by splenectomy or anti-T cell treatment. J Infect Dis 178:1225–1227. doi: 10.1086/515691. [DOI] [PubMed] [Google Scholar]
- 33.Hansen DS, Bernard NJ, Nie CQ, Schofield L. 2007. NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J Immunol 178:5779–5788. doi: 10.4049/jimmunol.178.9.5779. [DOI] [PubMed] [Google Scholar]
- 34.Belnoue E, Potter SM, Rosa DS, Mauduit M, Gruner AC, Kayibanda M, Mitchell AJ, Hunt NH, Renia L. 2008. Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol 30:544–553. doi: 10.1111/j.1365-3024.2008.01053.x. [DOI] [PubMed] [Google Scholar]
- 35.Smrkovski LL, Strickland GT. 1978. Rodent malaria: BCG-induced protection and immunosuppression. J Immunol 121:1257–1261. [PubMed] [Google Scholar]
- 36.Parra M, Liu X, Derrick SC, Yang A, Tian J, Kolibab K, Kumar S, Morris SL. 2013. Molecular analysis of non-specific protection against murine malaria induced by BCG vaccination. PLoS One 8:e66115. doi: 10.1371/journal.pone.0066115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leisewitz AL, Rockett K, Kwiatkowski D. 2008. BCG-malaria co-infection has paradoxical effects on C57BL/6 and A/J mouse strains. Parasite Immunol 30:1–12. [DOI] [PubMed] [Google Scholar]
- 38.Mittrücker HW, Steinhoff U, Kohler A, Krause M, Lazar D, Mex P, Miekley D, Kaufmann SH. 2007. Poor correlation between BCG vaccination-induced T cell responses and protection against tuberculosis. Proc Natl Acad Sci U S A 104:12434–12439. doi: 10.1073/pnas.0703510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iqbal NT, Hussain R. 2014. Non-specific immunity of BCG vaccine: a perspective of BCG immunotherapy. Trials Vaccinol 3:143–149. doi: 10.1016/j.trivac.2014.08.002. [DOI] [Google Scholar]
- 40.Berghout J, Langlais D, Radovanovic I, Tam M, MacMicking JD, Stevenson MM, Gros P. 2013. Irf8-regulated genomic responses drive pathological inflammation during cerebral malaria. PLoS Pathog 9:e1003491. doi: 10.1371/journal.ppat.1003491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.