

Abstract
Background
The plant bug Lygus hesperus Knight is a polyphagous pest of many economically important crops. Despite its pest status, little is known about the molecular mechanisms responsible for much of the biology of this species. Earlier Lygus transcriptome assemblies were limited by low read depth, or because they focused on specific conditions. To generate a more comprehensive transcriptome, we supplemented previous datasets with new reads corresponding to specific tissues (heads, antennae, and male reproductive tissues). This transcriptome augments current Lygus molecular resources and provides the foundational knowledge critical for future comparative studies.
Findings
An expanded, Trinity-based de novo transcriptome assembly for L. hesperus was generated using previously published whole body Illumina data, supplemented with 293 million bp of new raw sequencing data corresponding to five tissue-specific cDNA libraries and 11 Illumina sequencing runs. The updated transcriptome consists of 22,022 transcripts (average length of 2075 nt), 62 % of which contain complete open reading frames. Significant coverage of the BUSCO (benchmarking universal single-copy orthologs) dataset and robust metrics indicate that the transcriptome is a quality assembly with a high degree of completeness. Initial assessment of the new assembly’s utility revealed that the length and abundance of transcripts predicted to regulate insect physiology and chemosensation have improved, compared with previous L. hesperus assemblies.
Conclusions
This transcriptome represents a significant expansion of Lygus transcriptome data, and improves foundational knowledge about the molecular mechanisms underlying L. hesperus biology. The dataset is publically available in NCBI and GigaDB as a resource for researchers.
Keywords: Transcriptome, Lygus hesperus, Plant bug, Miridae, RNA-Seq, Trinity
Data description
Background
The western tarnished plant bug Lygus hesperus Knight is a polyphagous pest with an extensive host plant range including many economically important food, fiber, and seed crops [1]. While control measures have traditionally relied on broad-spectrum insecticides, negative ecological ramifications and evolving insecticide resistance have reduced the continued viability of this approach. As a consequence, there is growing interest in biorational-based strategies; however, the development of such approaches requires a comprehensive understanding of a species’ underlying biology. Towards this end, we previously reported on the sequencing and assembly of two L. hesperus transcriptomes: a general Roche 454-based assembly [2], and a second Illumina-based assembly incorporating sequence information from adults under thermal stress [3]. Those databases were developed using sequence data derived from whole bodies. Although this approach yields substantial data, whole body analysis tends to mask underrepresented genes that are expressed primarily in specific tissues or under specific conditions. To generate a more comprehensive transcriptome, here we supplement our previous thermal dataset with reads from specific tissues: heads, antennae, and male reproductive tissues. Incorporation of these new datasets expands the current L. hesperus database, provides greater depth of coverage, and enables new research for the better understanding of Lygus biology.
Samples
All samples and tissues were derived from an L. hesperus laboratory colony maintained at the United States Department of Agriculture-Agricultural Research Service (USDA-ARS) Arid Land Agricultural Research Center (ALARC) in Maricopa, Arizona, USA. The colony was reared at 27–29 °C under 20 % humidity with an L14:D10 photoperiod, and fed an artificial diet [4]. Nymphs and adults used for RNA preparation were from eggs deposited in agar oviposition packets and maintained as described previously [5]. Our initial Illumina-based transcriptome [3] was generated using 10-day old adults exposed for 4 h to one of three temperatures (4 °C, 25 °C, or 39 °C). To provide deeper coverage of transcripts encoding proteins functioning in olfaction, central nervous system-mediated behaviors, and male reproduction, sex-specific antennae, heads, and male accessory glands were dissected and stored at −20 °C in RNALater (Ambion/Life Technologies, Carlsbad, CA). The antennae samples represent ~500 unmated 7–9-day old adult males, and ~600 unmated 7–9-day old adult females. Heads (8–12 per stage/age per replicate) without antennae were collected across three biological replicates from 3rd instar nymphs, 4th instar nymphs, late 5th instar nymphs, and unmated adults of both genders at 1, 3, 7, 10, and 15 days post-eclosion. Accessory glands (30 per replicate) were dissected in phosphate-buffered saline from 7 to 8-day-old adult males 24 h post-mating and from similarly aged unmated cohorts. Total RNA extraction and library generation (TruSeq RNA Sample Preparation Kit v2; Illumina Inc., San Diego, USA) were performed as described previously [3] at the University of Arizona Genomics Center. All samples were sequenced using an Illumina HiSeq2000 or HiSeq2500 in Rapid Run mode (paired-end 100-bp reads).
Data filtering
Approximately 438 million reads were obtained, resulting in over 257 GB of 2 x 100 bp paired-end data. Raw read quality was assessed and filtered with a custom pipeline using FastQC (V 0.10.1) and Trimmomatic (V 0.32), using the parameters ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:10 TRAILING:20 SLIDINGWINDOW:4:25 MINLEN:36 to remove adapter sequences and filter by quality score. Short read archive (SRA) accessions for all data are found in Table 1.
Table 1.
Accession numbers for L. hesperus sequence reads and assembled transcripts
| Sample | Short Read Archive | BioSample | BioProject |
|---|---|---|---|
| 10-day-old adultsa | |||
| 4 °C | SRX483635, SRX483674, SRX483877 | SAMN02679940-42 | PRJNA238835 |
| 25 °C | SRX483950, SRX484037, SRX484042 | SAMN02679943-45 | " |
| 39 °C | SRX484076, SRX484077, SRX484079 | SAMN02679946-48 | " |
| Antennae | |||
| Male | SRX317887, SRX317888 | SAMN02222162-63 | PRJNA210219 |
| Female | SRX317885, SRX317886 | SAMN02222160-61 | " |
| Accessory Gland | |||
| Mated | SRX318362, SRX318363 | SAMN02222164-65 | PRJNA210220 |
| Unmated | SRX318364, SRX318365 | SAMN02222166-67 | " |
| Head | SRX1072689, SRX1155625, SRX1155629 | SAMN03792993-95 | PRJNA284294 |
aData from Hull et al. 2014 [3]
Transcriptome assembly
Data used for assembly corresponded to the ~145 million bp of sequence reads generated previously [3], and 293 million bp of new data from 11 Illumina runs covering five tissue-specific libraries. Prior to assembly, the four datasets (thermal-based, head, antennae, and accessory gland) were concatenated, and read abundance was normalized to 50X coverage using the in silico normalization tool in Trinity to improve assembly time and minimize memory requirements. Filtering and normalization reduced the dataset to 15 Gb, comprising approximately 32 million normalized read pairs, which were then assembled using default parameters in Trinity (r2014_07-17). Transcript expression levels were estimated with RSEM [6] and open reading frames (ORFs) were predicted using Transdecoder [7]. Hmmer3 was used to identify additional ORFs matching Pfam-A domains. Following transcriptome assembly, reads were filtered, sorted, and prepared for NCBI transcriptome shotgun assembly (TSA) submission as previously described [8].
Annotation
Functional annotation was performed at the peptide level using a custom pipeline [8] that defines protein products and assigns transcript names. Predicted proteins/peptides were analyzed using InterProScan5, which searched all available databases including Gene Ontology (GO) [9]. BLASTp analysis of the resulting proteins was performed with the UniProt Swiss Prot database (downloaded 11 February 2015). Annie [10], a program that cross-references SwissProt BLAST and InterProScan5 results to extract qualified gene names and products, was used to generate the transcript annotation file. The resulting .gff3 and .tbl files were further annotated with functional descriptors in Transvestigator [8].
Quality, completeness and depth of the comprehensive L. hesperus transcriptome
To assess the relative quality and completeness of our assembly, we compared core statistics for published Lygus transcriptomes [2, 3, 11] with those of the L. hesperus transcriptome described in this study (Table 2). The total number of sequence reads used in the current assembly represent 1660 and 300-fold increases over those used in the L. lineolaris transcriptome [11] and the initial Roche 454-based L. hesperus transcriptome [2] respectively. The expansion of read inputs resulted in average transcript lengths increasing from 725 to 2075 bp, and a larger percentage of transcripts with BLAST hits and assigned GO terms. Compared with the previously published Illumina transcriptome, inclusion of nearly three times the number of reads had little effect on average transcript length, and only marginally increased the N50 for the longest transcript per unigene (Table 2). However, low abundance isoforms were specifically removed during data normalization in the expanded assembly, a process that was modified from that used in the construction of the previous Illumina assembly. Consequently, while the expanded assembly represents less overall “gene space” than the previous assembly, it likely provides a more accurate reflection of the transcript landscape. More importantly, the expanded dataset increases overall coverage of transcripts critical to tissue-specific functions.
Table 2.
Transcriptome assembly and annotation statistics compared with previous Lygus transcriptomes
| Transcriptome | ||||
|---|---|---|---|---|
| L. lineolaris a | L. hesperus (454)b | L. hesperus (thermal)c | L. hesperus (current)d | |
| Assembly | ||||
| Total no. read pairs | 262,555 | 1,429,818 | 144,898,116 | 437,850,562 |
| Normalized reads (in silico normalization) | - | - | 16,191,383 | 32,342,216 |
| Total no. transcripts | 6970 | 36,131 | 45,706 | 22,022 |
| Average transcript length | 392 (100–3466) | 725 (2–13,480) | 2237 (300–23,322) | 2073 (297–23,350) |
| Total assembled bases (all transcripts) | - | 32,252,977 | 102,246,199 | 45,687,929 |
| Total assembled bases (longest transcript per unigene) | - | 28.8 Mb | 39.8 Mb | 31.6 Mb |
| N50 (all transcripts) | - | 2430 | 2989 | 2610 |
| N50 (longest transcript per unigene) | - | 1849 | 2638 | 2726 |
| %GC | - | 0.41 | 0.44 | 0.45 |
| Proteins with complete ORF (%) | - | - | - | 13,689 (62.1 %) |
| Annotation | ||||
| No. transcripts with a BLAST hit | 3126 (44.9 %) | 19,393 (54 %) | - | 16,942 (76.9 %) |
| No. transcripts with GO term | 2196 (31.5 %) | 7898 (21 %) | - | 12,114 (54.9 %) |
| PFAM | - | 3705 (22.2 %) | - | 14,575 (66.1 %) |
The respective L. hesperus assemblies were also evaluated using the BUSCO (benchmarking universal single-copy orthologs) arthropod gene set [12], which uses 2675 near-universal single-copy orthologs to assess the relative completeness of genome and transcriptome assemblies. The percentage of conserved genes identified in the new L. hesperus assembly compares favorably with metrics reported for a number of insect transcriptomes and model insect genome assemblies (Table 3). Compared with the previous Illumina assembly, BUSCO genes in the new L. hesperus assembly were less fragmented, indicating the presence of more full-length sequences. The relatively high number of duplicates identified in the L. hesperus assemblies likely reflect isoforms of single unigenes, rather than true gene duplications.
Table 3.
BUSCOa analysis of assembly completeness
| Species | Complete (%) | Duplicated (%) | Fragment (%) | Missing (%) |
|---|---|---|---|---|
| L. hesperus Transcriptomes | ||||
| 454-basedb | 56 | 18 | 13 | 29 |
| Illumina-thermalc | 77 | 43 | 11 | 10 |
| Illumina-current | 74 | 33 | 7.3 | 17 |
| Select Insect Transcriptomesd | ||||
| Nilaparvata lugens (GI:604923024) | 64 | - | 19 | 15 |
| Musca domestica (GI:510208131) | 6.4 | - | 6.3 | 87 |
| Spodoptera exigua (GI:556694752) | 73 | - | 11 | 14 |
| Drosophila serrata (GI:570485056) | 8.5 | - | 21 | 70 |
| Select Insect Genomesd | ||||
| Pediculus humanus (PhumU2) | 92 | 3.9 | 6.1 | 1.6 |
| Acyrthosiphon pisum (GCA_000142985.2) | 72 | 6.1 | 15 | 12 |
| Drosophila melanogaster (Dmel_r5.55) | 98 | 6.4 | 0.6 | 0.3 |
Next, we used sequences encoding neuropeptides, G protein-coupled receptors, and chemosensory receptors to more fully evaluate the effect of expanding the current assembly with tissue-specific sequencing data. These gene sets mediate much of insect physiology and behavior, and are frequently characterized by spatially restricted expression. The query sequences used in the tBLASTx analyses are from two insect species (Nilaparvata lugens and Rhodnius prolixus) within the same phylogenetic order (Hemiptera) as L. hesperus. The first analysis, which used the 48 neuropeptide sequences reported in N. lugens [13] as queries, revealed nearly twice as many homologous sequences in the Illumina-based assemblies as in the initial Roche 454 assembly (Fig. 1). Subsequent searches using N. lugens G protein-coupled receptors [13] or Rhodnius prolixus chemosensory receptors [14, 15] as queries identified more transcripts ≥300 nt in length in the new, expanded assembly than in the previous transcriptomes. Based on these comparisons, we conclude that the expanded transcriptome represents a marked improvement over the first 454-based assembly, and provides greater coverage of tissue-specific transcripts, such as chemosensory genes and neuropeptide precursors, relative to the previous Illumina assembly. This expanded assembly extends previous work and provides a more comprehensive resource to facilitate the development of new research avenues into the molecular basis of L. hesperus biology.
Fig. 1.
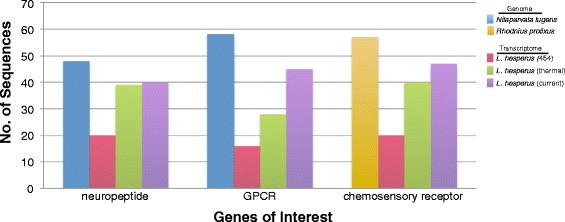
Relative transcript depth of the respective L. hesperus transcriptomes. tBLASTx analyses were performed using queries corresponding to genes of interest identified in genome assemblies of Nilaparvata lugens or Rhodnius prolixus. The L. hesperus transcriptomes analyzed include the initial Roche 454-based assembly [2], an Illumina-based thermal assembly [3], and the current assembly. tBLASTx search criteria for the neuropeptide analysis used an e-value of 10−1, whereas the G protein-coupled receptor (GPCR) and chemosensory receptor analyses used an e-value of 10−5 and transcripts ≥300 nt in length
Availability of supporting data
The filtered and annotated transcriptome was deposited at GenBank as a TSA under the accession GDHC01000000, associated with BioProject PRJNA284294. NCBI accession identifiers for all of the associated SRA, Biosample, and Bioproject data repositories are listed in Table 1. Datasets further supporting the results of this article are available in the GigaScience repository, GigaDB [16].
Acknowledgements
The authors thank both Daniel Langhorst (ALARC) and Lynn Forlow Jech (ALARC) for technical support, as well as Brooks Silversmith (ALARC) and Anna Cervantes (ALARC) for maintaining the L. hesperus colony. The research described in this manuscript was partially supported by funds from Cotton Inc. (CSB, project no. 12–373). Bioinformatic analysis was performed on computing resources at the USDA-ARS Daniel K. Inouye Pacific Basin Agricultural Research Center (Moana cluster; Hilo, HI) and the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575XSEDE under allocation TG-MCB140032 to SMG. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. USDA is an equal opportunity provider and employer.
Abbreviations
- ALARC
arid land agricultural research center
- BUSCO
bench-marking universal single-copy ortholog
- GO
gene ontology
- GPCR
G protein-coupled receptor
- ORF
open reading frame
- SRA
short read archive
- TSA
transcriptome shotgun assembly
- USDA-ARS
United States Department of Agriculture-Agricultural Research Service
- XSEDE
extreme science and engineering discovery environment
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JAF, CSB, and JJH conceived and designed the experiments; JAF, CSB and JJH performed the experiments; EET, SMG, BH, and JJH analyzed the data; EET, SMG, JAF, CSB and JJH evaluated the conclusions; and EET and JJH wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Erica E. Tassone, Email: erica.tassone@ars.usda.gov
Scott M. Geib, Email: scott.geib@ars.usda.gov
Brian Hall, Email: bhall7@hawaii.edu.
Jeffrey A. Fabrick, Email: jeff.fabrick@ars.usda.gov
Colin S. Brent, Email: colin.brent@ars.usda.gov
J. Joe Hull, Email: joe.hull@ars.usda.gov.
References
- 1.Scott DR. An annotated listing of host plants of Lygus hesperus Knight. Entomol Soc Am Bull. 1977;23:19–22. [Google Scholar]
- 2.Hull JJ, Geib SM, Fabrick JA, Brent CS. Sequencing and de novo assembly of the western tarnished plant bug (Lygus hesperus) transcriptome. PLoS ONE. 2013;8:e55105. doi: 10.1371/journal.pone.0055105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hull JJ, Chaney K, Geib SM, Fabrick JA, Brent CS, Walsh D, et al. Transcriptome-based identification of ABC transporters in the western tarnished plant bug Lygus hesperus. PLoS ONE. 2014;9:e113046. doi: 10.1371/journal.pone.0113046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Debolt JW. Meridic diet for rearing successive generations of Lygus hesperus. Ann Entomol Soc Am. 1982;75:119–122. doi: 10.1093/aesa/75.2.119. [DOI] [Google Scholar]
- 5.Brent CS, Hull JJ. Characterization of male-derived factors inhibiting female sexual receptivity in Lygus hesperus. J Insect Physiol. 2014;60:104–110. doi: 10.1016/j.jinsphys.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Prot. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sim SB, Calla B, Hall B, DeRego T, Geib SM. Reconstructing a comprehensive transcriptome assembly of a white-pupal translocated strain of the pest fruit fly Bactrocera cucurbitae. GigaScience. 2015;4:14. doi: 10.1186/s13742-015-0053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tate R, Hall B, DeRego, T. Annie the functional annotator – initial release. ZENODO; 2014. Available from: http://doi.org/10.5281/zenodo.10470.
- 11.Magalhaes LC, van Kretschmar JB, Donohue KV, Roe RM. Pyrosequencing of the adult tarnished plant bug, Lygus lineolaris, and characterization of messages important in metabolism and development. Entomol Exp Appl. 2013;146:364–378. doi: 10.1111/eea.12035. [DOI] [Google Scholar]
- 12.Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka Y, Suetsugu Y, Yamamoto K, Noda H, Shinoda T. Transcriptome analysis of neuropeptides and G-protein coupled receptors (GPCRs) for neuropeptides in the brown planthopper Nilaparvata lugens. Peptides. 2014;53:125–133. doi: 10.1016/j.peptides.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 14.Megy K, Emrich SJ, Lawson D, Campbell D, Dialynas E, Hughes DST, et al. VectorBase: improvements to a bioinformatics resource for invertebrate vector genomics. Nucleic Acids Res. 2012;40(Database issue):D729–D734. doi: 10.1093/nar/gkr1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Latorre-Estivalis JM, de Oliveira ES, Beiral Esteves B, Santos Guimarães L, Neves Ramos M, Lorenzo MG. Patterns of expression of odorant receptor genes in a Chagas disease vector. Insect Biochem Mol Biol. Forthcoming 2015. doi:10.1016/j.ibmb.2015.05.002 [DOI] [PubMed]
- 16.Tassone, E, E; Geib, S, M; Hall, B; Fabrick, J, A; Brent, C, S; Hull, J, J (2016): Supporting data for “De novo construction of an expanded transcriptome assembly for the western tarnished plant bug, Lygus hesperus”. GigaScience Database. http://dx.doi.org/10.5524/100172 [DOI] [PMC free article] [PubMed]