

Abstract
Epilepsy or seizure disorder is among the least understood chronic medical conditions affecting over 65 million people worldwide. Here, we show that disruption of the polycystic kidney disease 2-like 1 (Pkd2l1 or Pkdl), encoding polycystin-L (PCL), a non-selective cation channel, increases neuronal excitability and the susceptibility to pentylenetetrazol-induced seizure in mice. PCL interacts with β2-adrenergic receptor (β2AR) and co-localizes with β2AR on the primary cilia of neurons in the brain. Pkdl deficiency leads to the loss of β2AR on neuronal cilia, which is accompanied with a remarkable reduction in cAMP levels in the central nervous system (CNS). The reduction of cAMP levels is associated with a reduction in the activation of cAMP response element-binding protein, but not the activation of Ca2+/calmodulin-dependent protein kinase II, Akt or mitogen-activated protein kinases. Our data, thus, indicate for the first time that a ciliary protein complex is required for the control of neuronal excitability in the CNS.
Introduction
Epilepsy is a common and diverse set of chronic neurological disorders characterized by recurrent seizures (1), affecting ∼65 million people worldwide (2). Epilepsy is characterized by an abnormal, excessive and hypersynchronous neuronal activity in the brain (3). Impairment of calcium signaling or other transmembrane balance of ion currents is a frequent cause of neuronal hyperexcitability in the central nervous system (CNS), and is responsible for a variety of epileptic syndromes in humans (4–8). In addition, the second-messenger cyclic adenosine monophosphate (cAMP) signaling pathway plays a crucial role in maintaining the excitability of CNS (9). Pentylenetetrazol (PTZ) is a chemoconvulsant commonly used in mice for discovering novel anti-epileptic compounds. Animals pre-treated with cAMP-lowering agents (reserpine, propranolol or aminophylline) exhibit an enhanced sensitivity to PTZ-induced seizures (10). Conversely, pre-treatment of forskolin, an adenylyl cyclase (AC) activator, protects mice from PTZ-induced seizures by elevating the cAMP levels in the brains (11).
Polycystin-L (PCL), encoded by polycystic kidney disease 2-like 1 (Pkd2l1 or Pkdl), is a Ca2+-activated non-selective cation channel that is permeable to Ca2+, K+ and Na+ (12), and is expressed in the brain (13–16). In the kidney, PCL is detected on the primary cilium of epithelial cells (17), similar to other members of the polycystin family (18,19).
The primary cilium is a polarized organelle projecting from a majority of vertebrate cells (20). It is a microtubule-based structure covered by a specified ciliary membrane with protein content distinct from that of the plasma membrane. The current consensus of the function of the primary cilium is that it acts as a signaling center regulating many crucial signaling pathways and through many sensory functions including mechanosensation, photosensation, osmosensation and hormone sensation (18,21–25). The neuronal primary cilium has been recently identified and appears to be required for normal energy homeostasis, while its defects lead to obesity (26) and abnormal migration of neurons during CNS development (27).
In this study, we found that PCL localizes to neuronal cilia in the brain. To investigate the function of PCL, we generated a Pkdl-deficient mouse model, which manifests an increased susceptibility to PTZ-induced seizures. We show that PCL interacts and co-localizes with β2-adrenergic receptor (β2AR), a G-protein-coupled receptor (GPCR) functioning in cAMP production, on the primary cilia of neuronal cells in the brain. Pkdl deficiency leads to the loss of β2AR on neuronal primary cilia, and a remarkable reduction of cAMP levels in the brain. Taken together, these data suggest, for the first time, that a novel ciliary protein complex regulates neuronal excitability.
Results
PCL localizes to the primary cilium of neurons in the brain
We and others have previously reported high levels of Pkdl mRNA expression in brain tissues (13–15). By immunofluorescence analysis, we observed somatic cytoplasmic labeling of PCL in neurons of the cerebral cortex (Layers II–VI), hippocampus and the thalamus of adult wild-type (WT) mouse brain (Fig. 1A and Supplementary Material, Fig. S1A), this labeling was significantly reduced in Pkdl knockout (KO) mouse brain tissues (Supplementary Material, Fig. S1B). Interestingly, we found PCL on the primary cilia of the primary cultured mouse hippocampal neurons (Fig. 1B), by co-staining with AC3, a primary cilium maker for neuronal cells (28).
Figure 1.

PCL localizes to the primary cilia of neuronal cells in the brain. (A) Confocal imaging indicates PCL expression in the neuronal cells in the cerebral cortex, hippocampus and thalamus. (B) PCL localizes to the primary cilia of hippocampal neurons in a primary culture. (C) PCL localizes to the AC8+ neuronal primary cilia in the mouse cortex and hippocampus. Insets show the cilia indicated by arrows. PCL was stained by antibody 83430. Neurons were detected by neuronal marker NeuN. AC3 was used to mark neuronal primary cilia. AC8 is a new neuronal cilium marker identified in this study.
AC8 has a high-expression level in the CNS (29), and has been reported to localize to the primary cilia of cholangiocytes (30). By co-staining with AC3, we observed intense AC8 signals on most AC3-positive cilia in the mouse brain, such as the hippocampus and the cortex. However, PCL is not required for AC8 ciliary localization in the mouse brain (Supplementary Material, Fig. S2). Since the AC8 antibody recognizes the neuronal primary cilium with much lower cytoplasmic signals compared with AC3, we used AC8 as an additional neuronal ciliary marker in this study. By co-staining PCL with AC8, we further confirmed PCL ciliary localization in brain tissues (Fig. 1C).
Pkdl−/− mice are sensitive to PTZ-induced seizure
To investigate the function of PCL in vivo, we generated a mouse line deficient for PCL by deletion of Exons 3 and 4 (Fig. 2A and B, ‘Materials and Methods’ section). The Pkdl-deficient mice generated by homologous recombination in embryonic stem cells were viable. Reverse transcription polymerase chain reaction (RT-PCR) analysis showed that there was no normal Pkdl mRNA present in the brains of adult Pkdl−/− mice, although a mutant Pkdl mRNA was present (Fig. 2C). Direct DNA sequencing of the PCR products revealed that the mutant transcript in Pkdl−/− mice contains a spliced variant from Exon 2 to Exon 5 and a premature stop codon in Exon 5. No PCL immunoreactivity was detected using antibodies against PCL in testis extracts from Pkdl−/− mice (Fig. 2D), indicating that homozygous mutants are deficient of PCL.
Figure 2.

Pkdl−/− mice are sensitive to PTZ. (A) Targeting strategy for Pkdl and restriction map of the Pkdl gene. The solid boxes represent the exons of the Pkdl gene and the interconnecting lines indicate introns. The probe used for Southern blotting is shown as a bar. (B) Southern blot analysis of mouse tail DNA. The 3.2 and 4.8 kb bands represent the germ-line and targeted allele, respectively. (C) RT-PCR analysis of total RNA extracted from mouse brains using primers mPcLf605 5′ACACAGCCGAGAACAGGGAGCTT3′ and mPcLr611 5′ GCATACGTGTCTGGCTGTTGCAG3′. No normal Pkdl mRNA was detected in the brains of adult Pkdl−/− mice by RT-PCR analysis, but a truncated Pkdl mRNA was present. M, 100 bp DNA marker. (D) Immunoblotting analysis of the PCL protein. Anti-PCL antibody is specific for the N-terminal portion of the PCL protein. Testis extracts from Pkdl+/+, Pkdl+/− or Pkdl−/− mice were immunoprecipitated with anti-PCL and immunoblotted with the same antibody. PCL protein was not detected in Pkdl−/− mice. (E) The severity of seizure increases in KO mice when compared with WT mice (PTZ = 40 μg/g). 0: no response; 1: isolated twitches; 2: tonic-clonic convulsions; 3: tonic extensions and/or death. (F) Latency in KO mice is reduced compared with WT littermates (PTZ = 50 μg/g).
The distinct neuronal expression pattern of PCL led us to examine the role of PCL in the control of neuronal excitability. γ-Aminobutyric acid (GABA) is the main inhibitory neurotransmitter in the mammalian brain. The binding of GABA to its receptor results in the opening of conductances that lead to hyperpolarization and/or membrane shunt, thus reducing neuronal excitability. PTZ is a chemoconvulsant that acts as a GABA-A receptor antagonist and is widely used to study seizures in rodents to discover novel anti-epileptic compounds. Under normal breeding conditions, Pkdl−/− mice are of normal size without discernable appearance and behavior abnormalities. When challenged with a single injection (40 μg/g) of PTZ (31), Pkdl−/− mice showed a right shift in the severity of seizures. We found that ∼79% Pkdl−/− mice (n = 19) showed stage-2 tonic-clonic convulsions (∼47%) or stage-3 tonic-clonic seizures (∼32%) frequently associated with death, while only ∼5% WT littermates (n = 21) showed similar phenotype with a large majority (∼81%) that had no response (Fig. 2E).
A similar result was observed at a higher dosage of PTZ (50 μg/g) (Supplementary Material, Fig. S3), which also revealed reduced latency of Pkdl−/− mice (n = 26) to the first myoclonic jerk, the first observable behavioral response, to 408 s, compared with WT littermates (n = 37) at 594 s (Fig. 2F).
Pkdl−/− mice lose control of excitability
To further characterize the neuronal electric activity in Pkdl−/− mice, we recorded local field potentials (LFPs) simultaneously from neocortex (Cx), hippocampus (Hip) and thalamus (Thal), and compared them with their WT littermates (Fig. 3A). LFPs represent extracellular currents resulting from the combined synaptic activity in thousands of neurons and reflect the state of excitability of local networks. The most noticeable difference was the presence of high amplitude, sharp potentials lasting 0.1–0.4 s in the spontaneous LFPs recorded from all three structures in the Pkdl−/− mice (Fig. 3A, KO, examples indicated with asterisks, n = 10). The sharp potentials were present in all Pkdl−/− mice, but never in the WT littermates (Fig. 3A, WT, n = 6). The background activity under ketamine–xylazine anesthesia was characterized by the presence of the extensively described slow oscillation (32), consisting of negative LFP waves during neuronal depolarization and firing as well as positive LFP waves during neuronal hyperpolarization and the absence of spike firing. These sharp potentials appeared almost exclusively during the negative phases of the slow oscillation, which suggests a poor control of excitability; since the negative LFP phases of the slow oscillation reflect strong synaptic activation (33). The sharp potentials were unevenly distributed and tended to be more prominent in the cortex and the hippocampus than in the thalamus.
Figure 3.

LFP recordings suggest a loss of control of excitability in Pkdl−/− mice. (A) Under ketamine–xylazine anesthesia, EEG recording of spontaneous activity from neocortex (Cx-1, -2 and -3), hippocampus (Hip) and thalamus (Thal) showed the slow oscillation (WT = 0.6 Hz; KO = 0.4 Hz) synchronized among the three structures as shown in the average (AVG, n = 20 cycles) centered on negative peaks of Cx-1. KO showed high-amplitude spikes. (B) Responses to thalamic stimulation of ventrobasal (VB) nucleus of the thalamus. (C) Seizure threshold is lower in KO mice. PTZ (40 μg/g) triggered mild spike–wave seizures in WT mice (n = 6), while KO mice (n = 10) showed severe tonic-clonic seizures.
To compare the slow oscillation between the two genotypes, an averaged cycle of the slow oscillation was constructed for all recording leads using as reference the negative peak of spontaneous cycles (n = 10) from Cx-1. In the WT mice, the average showed a smooth negative wave lasting up to 0.6 s and synchronized across all recording leads. In contrast, in the Pkdl−/− mice, the average revealed the presence of a sharp potential lasting up to 0.2 s (Fig. 3A, AVG).
In order to test the hypothesis that the spontaneous sharp potentials betray enhanced excitability in baseline conditions, we compared responses in the cortex to electrical stimulation of the thalamus between the two types of mice. We used paired-pulse stimuli (Fig. 3B, arrowheads) with 0.1 s period, which is a protocol that generates a well-known form of facilitation termed augmenting that probes the excitability of thalamocortical and corticocortical circuits (34). Two types of differences were observed (Fig. 3B): (i) the amplitude of the averaged responses to both stimuli was higher in the Pkdl−/− mice (first response: 0.21 ± 0.05 mV, mean ± SEM.; second response: 0.48 ± 0.11 mV; n = 10; KO) compared with WT (first: 0.06 ± 0.01 mV; second: 0.12 ± 0.03 mV; n = 6; WT) (first shock: P = 0.0024, Bonferonni corrected: P = 0.0072; second shock: P = 0.015, Bonferonni corrected: P = 0.046, Mann–Whitney U-test with a Bonferonni correction for multiple comparisons) and, (ii) the ratio between the second and first response amplitude (measured from the baseline, dotted line), to the positive peak (double-arrow lines), was higher for the Pkdl−/− mice (2.46 ± 0.44, n = 10) than that for the WT (1.97 ± 0.32, n = 6; ratio from second to first, P = 0.0022, Bonferonni corrected, P = 0.0066). Both differences indicate enhanced excitability in Pkdl−/− mice in baseline conditions.
The presence of spontaneous sharp potentials in Pkdl−/− mice as well as the increased amplitude and ratio of the augmenting responses to thalamic stimulation strongly suggest increased cortical excitability. Consequently, the Pkdl−/− mice should exhibit a lower threshold for seizure generation. Indeed, a single subcutaneous injection (40 μg/g) of PTZ in WT mice (n = 6) gave rise to the appearance of isolated spikes interspersed with normal electrographic activity or, in the most severe cases (n = 2), to the generation of repetitive large spikes with frequencies of 2–5 Hz (Fig. 3C, WT). This mild seizure-like activity occurred on a background of otherwise normal slow oscillation. Injections in the Pkdl−/− mice (n = 10) readily gave rise to severe seizures similar to those described in the electroencephalogram (EEG) of cats (35) and humans (36). These seizures started with fast runs of 10–15 Hz and ended with repeated spikes at slower frequencies (2–5 Hz). Seizures repeated regularly every 2–8 s and obliterated the slow oscillation becoming the only activity present in the LFPs from the three structures. These results further support the loss of the control of excitability in Pkdl−/− mice.
Reduced cAMP levels in Pkdl−/− mouse brain tissues
The neuronal cAMP homeostasis is crucial in controlling the excitability of CNS (9). Abnormal cAMP production has been reported in polycystic kidney diseases owning to the deficiency of polycystin-1 (PC1) or -2 (PC2) (37). To determine whether PCL is required to maintain the level of cAMP in the CNS, we measured the cAMP levels in mouse forebrain tissues. We found that there was a ∼27% reduction in cAMP levels in adult (4 months old) Pkdl−/− mouse brains when compared with the WT littermates (Fig. 4A), and ∼15% reduction in adolescent (5 weeks old) KO mice (Supplementary Material, Fig. S4). In agreement with this observation, the phosphorylation of cAMP response element-binding protein (CREB) was also significantly reduced in the corresponding brain tissues (Fig. 4B and C).
Figure 4.

cAMP is reduced in Pkdl−/− mouse brain tissues. (A) The cAMP levels in adult Pkdl−/− mouse brains were reduced by ∼27% compared with the WT littermates. (B) Consistently, western blot revealed that pCREBS133 was also significantly reduced in the same brain tissues. (C) The pCREBS133 protein levels were normalized to total CREB protein in each brain tissue. The WT brain tissues were set at 100%, as reference. Error bars represent standard deviation (n = 5). The significance was calculated by Student's t-test (P < 0.01). Three independent experiments were performed. (D) There were no obvious changes in phosphorylation of CaMKII, Akt or MAPK in Pkdl−/− brain tissues compared with WT mice.
Although cAMP is one of the most potent activators of CREB pathway, the phosphorylation of CREB is also subjected to the regulation of Ca2+/calmodulin/CaM kinases II (CaMKII), mitogen-activated protein kinases (MAPKs) and the Akt/protein kinase B pathways in response to various signals. However, we did not observe significant changes in the phosphorylation of CaMKII, MAPK or Akt, which are indicators of the activation of respective signaling pathways, in the Pkdl−/− brain tissues compared with WT mice (Fig. 4D). These results support that the reduced pCREBSer133 levels are because of the reduced neuronal cAMP levels.
PCL interacts with β2AR and is required for the ciliary localization of β2AR in neurons
ARs are a group of GPCRs that are widely distributed throughout the brain and found in regions implicated in regulating neuronal excitability, such as the cortex and the hippocampus. Activated β2AR couples to Gs, which in turn stimulates ACs and increases cAMP production. Moreover, β2AR agonists have anti-convulsion effect to PTZ-induced seizure (38).
To investigate whether PCL can modulate the β2AR expression and localization in the CNS, we immunostained brain sections with β2AR antibody. In the WT brain, β2AR was primarily found in the cytoplasm of neurons. Interestingly, β2AR was also detected on cilium-like structures in most neurons in the cerebral cortex, hippocampus and thalamus. Moreover, cilium-like β2AR signals were remarkably diminished in the Pkdl−/− neuronal cells in the cerebral cortex and hippocampus (Fig. 5A). Co-staining with AC8 verified that these cilium-like structures were neuronal primary cilia (Fig. 5B). The specificity of the ciliary signal of the β2AR antibody was confirmed by antigen blocking experiments (Supplementary Material, Fig. S5). The reduction of ciliary β2AR signals was not likely due to decreased β2AR expression as its protein levels were similar between Pkdl−/− and WT littermate brains (Fig. 4B).
Figure 5.

PCL interacts with β2AR and is required for ciliary localization of β2AR in neurons. (A) β2AR expression on cilium-like structures in hippocampus was remarkably diminished in the KO neurons. (B) β2AR co-localizes with AC8 on the neuronal primary cilia in the dentate gyrus of hippocampus. Cilia from two neighboring cells are shown in insets. (C) PCL and β2AR co-immunoprecipitated each other in HEK293T cells stably expressing PCL-Myc in a tetracycline inducible manner and transiently expressing Flag-β2AR. (D) Reciprocal co-immunoprecipitation of endogenous PCL and β2AR from mouse hippocampal lysates using respective antibodies. (E) PCL co-localizes with β2AR on the neuronal primary cilia in hippocampus. Insets show a cilium indicated by arrows. (B and E) Confocal images.
To investigate how PCL may modulate β2AR ciliary localization, we tested whether these two proteins interact with each other. For this purpose, we established a human embryonic kidney 293T (HEK293T) cell-line stably expressing PCL-Myc in a tetracycline inducible fashion and transiently transfected Flag-β2AR into this cell-line. Immunoprecipitation of PCL-Myc was able to co-immunoprecipitate Flag-β2AR in these cells (Fig. 5C). Flag-β2AR could also co-immunoprecipitate PCL-Myc. To validate this interaction in vivo, we co-immunoprecipitated endogenous PCL and β2AR in mouse hippocampal tissue extracts and observed the same results (Fig. 5D). Consistent with this observed interaction, PCL co-localizes with β2AR on the neuronal primary cilia in the brain (Fig. 5E).
Discussion
In this study we report that PCL, a non-selective calcium permeable cation channel, and β2AR are co-localized to the neuronal cilia in the brain for the first time in any species. This is also the first report of the localization of an ion channel to neuronal cilia in mammals and the description of a ciliary protein complex being responsible for increased neuronal excitability in any model system.
We found that PCL is specifically expressed in neurons throughout the cortex, hippocampus and thalamus in the brain. The specific expression of PCL channel in neurons in the brain led us to examine the excitability in the CNS in our newly established Pkdl-deficient mice. As expected, the Pkdl-deficient mice developed increased susceptibility to PTZ-induced seizure, manifested by reduced latency and increased severity of seizures, in comparison with their WT littermates. EEG analyses revealed features of increased cortical excitability and a lower threshold for seizure generation in Pkdl-deficient brains.
The secondary messenger cAMP is known to inhibit neuronal excitability. Pharmaceutically reducing the production of cAMP renders mice more vulnerable to PTZ-induced seizures (10), while increasing cAMP levels protects mice from PTZ-induced seizures (11). Consistent with the presence of increased susceptibility to PTZ-induced seizures, we found a ∼27% reduction in cAMP levels in Pkdl-deficient mouse brains when compared to those of the WT littermates.
Because β2AR couples to Gs whose activity promotes cAMP production, and β2AR agonists have an anti-convulsion effect to PTZ-induced seizure (38), we examined the expression and localization of β2AR in WT and Pkdl-deficient mouse brains. In addition to finding PCL and β2AR co-localization on the neuronal primary cilium in the brain, by co-immunoprecipitation analysis, we discovered that PCL and β2AR interact with each other in vitro and in vivo. Pkdl deficiency does not affect the protein levels of β2AR in the brain, but causes the loss of ciliary β2AR in neurons, suggesting that PCL is required for β2AR ciliary localization.
The loss of β2AR ciliary localization in Pkdl-deficient brains is consistent with a reduction of cAMP levels in Pkdl-deficient mouse brain tissues, compared with those in their WT littermates. Consistent with our data, a recent study of PC2 in kidney epithelial cells suggests that the PC2 channel, which shares high homology with PCL (39), may directly control the cAMP homeostasis in a primary cilium-dependent manner (40). Future analysis is needed to address the precise regulation of β2AR trafficking to the primary cilia and the role of ciliary β2AR in controlling the cAMP signaling cascade. In addition, there are many other GPCRs in the neuronal primary cilium, such as somatostatin receptor 3 (41), serotonin receptor 6 (42), melanin-concentrating hormone receptor 1 (43), dopamine receptor 1 (44) and AC3 (28) and AC8 (this study). More GPCRs and ACs might be detected in the neuronal primary cilia in future. Therefore, the possibility that PCL controls the cAMP signaling cascade through mechanisms in addition to β2AR cannot be excluded.
Reduced cAMP levels are associated with a reduction in the phosphorylation of CREB, a transcription factor. Notably, the activation of CaMKII, Akt and MAPK signaling cascades that also regulate CREB phosphorylation were not affected in our Pkdl-deficient brains, further supporting the reduction of CREB phosphorylation in Pkdl-deficient mouse brains is caused by the decrease in cAMP levels.
Epileptic seizures have been observed in patients with ciliopathies such as Joubert syndrome, Meckel–Gruber syndrome and Bardet–Biedle syndrome. Given that most neurons possess primary cilia (44), this clinical association suggests that primary cilia may control neuronal excitability in the CNS. Here, we provide the first experimental evidence that PCL-β2AR, a ciliary ion channel–receptor complex, regulates cAMP signaling and neuronal excitability (Fig. 6). The human PKDL localizes at Chromosome 10q24 (39), which has been considered inherently unstable, harboring multiple BrdU-inducible fragile sites (45) and containing genes involved in partial epilepsy. Our findings that PCL is widely expressed in neurons of various regions in the brain and its deficiency in mice leads to increased susceptibility to PTZ-induced seizure suggest that PCL is a novel target for epilepsy.
Figure 6.
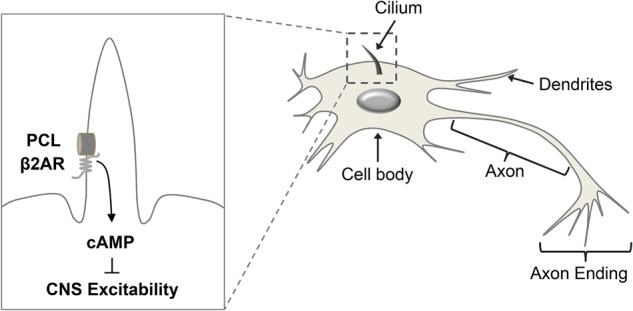
A schematic model of PCL and β2AR localization to the neuronal primary cilia and their interaction. The PCL–β2AR protein complex regulates the cAMP levels and neuronal excitability in the CNS.
The axonal tubulin is stable and enriched in acetylated α-tubulin, a commonly used primary cilium marker in most cells, which prevents the use of acetylated α-tubulin in neuronal cells. Here, we show that AC8 is highly enriched in most AC3-positive neuronal primary cilia, thus identifying AC8 as an additional neuronal primary cilium maker, which will facilitate studies of neuronal primary cilium in vivo.
PCL is expressed in the kidney tubular epithelial cells (13,15,46). However, we did not observe any obvious renal or other phenotypes in our Pkdl-deficient mice. Two recent conjunct studies suggested that PCL might be responsible in modulating hedgehog pathway via regulating the calcium signaling within the primary cilium (46,47). Our RT-PCR results from four pairs of WT and littermate Pkdl-deficient mouse brains did not reveal statistically significant alterations in transcriptional targets of the hedgehog pathway Ptch1 (P = 0.35, t-test) and Gli1 (P = 0.65, t-test), which suggest Pkdl deficiency does not affect the hedgehog pathway in the CNS.
In summary, we have identified a novel ciliary protein complex consisting of a non-selective cation channel and a GPCR (PCL-β2AR), which regulates neuronal excitability. Further studies are required to identify modulators of this PCL complex, which might lead to the identification of novel agents to treat neuronal hyperexcitability disorders in humans.
Materials and Methods
Cloning and disruption of mouse Pkdl
Degenerative PCR primer derived from a human PKDL or PKD2L1 cDNA sequence, and AP1, 5′CCATCCTAATACG ACTCACTATAGGGC3′ (Clontech, Palo Alto, CA, USA), was used to carry out rapid amplification of cDNA ends (RACE) on mouse brain marathon-ready™ cDNA. AdvanTaq™ DNA polymerase (Clontech) was used to perform touch-down PCR with cycling parameters as follows: initial denaturation at 94°C for 30 s; 5 cycles of 94°C for 10 s, 72°C for 4 min; 5 cycles of 94°C for 10 s, 70°C for 4 min; 25 cycles of 94°C for 5 s, 68°C for 4 min and final extension at 68°C for 7 min. PCR products were separated on a 1% agarose gel, and all resulting DNA fragments were excised and isolated. The products were purified and sequenced directly. For 5′ RACE amplification to obtain a full-length cDNA sequence, PCR was performed with the primer AP1 and mR902, 5′GTGGATTTCCAGGATTTCCTCCACCA3′. A second amplification was performed using the 2 μl of the diluted first PCR product (1:100) with the nested primers AP2, 5′ACTCACTATAGGGCTCGAGCGGC3′ (Clontech), and mR902. For 3′ RACE amplification, primers AP1 and mF903, 5′CACAAGCTACAGCGGGGGTGGCTACT3′ were used to perform the first PCR on the mouse brain cDNA. AP2 and mF902, 5′TACAGCGGGGGTGGCTACTACTTGGA3′, were used to perform nested PCR for 3′ RACE. 5′ and 3′ RACE products were cloned and both strands sequenced. The sequences were aligned to give an overall consensus sequence. A 10 kb NotI fragment was isolated from a mouse genomic library. The 7 kb NotI–NsiI fragment containing intron 2 of the Pkd2l1 gene was used to insert into a vector containing a neomycin-resistant gene with a PGK promoter, serving as a long arm of homologous recombination. A 0.9 kb short arm was created with the PCR method containing parts of intron 4 and exon 5, and inserted into the upstream of PGK–Neo cassette. Therefore, in this strategy, exons 3 and 4 were deleted. The Pkd2l1 targeting vector was linearized with NotI and used at a concentration of 22 mg/ml for electroporation. REK 3 embryonic stem cells were used at a concentration of 1.2 × 107 cells/ml. Colonies that survived 10 days in G418 (0.3 mg/ml; 66% active; GIBCO) were screened by PCR and Southern blot. Two recombinant cell lines were injected into C57BL/6 blastocysts to generate chimeric mice, and contributed to the germ-line.
All procedures were approved by the Institutional Animal Care and Use Committee at the National Institutes of Health and the Harvard Medical School.
Genotyping of mice
The mice were number-tagged and genotyped by the PCR method. Primers MPcLF904 5′AAGATCAGCTCCCCTTTGGACCT3′ and MPcLR905 5′CCCAGCTCATTCTGGGAATG GT3′ were used to amplify the WT allele; primers Neo1 5′TGCGAGGCCAGAGGCCACTTG TGTAGC3′ and Z6 5′CTGTGC GGATCTGCCAGGATG3′ were used to amplify the mutant allele.
Seizure studies
Age-matched (10–14 weeks) WT and mutant littermates weighing 20–30 g were used. Animals were housed in a room with controlled light–dark cycle (12 h light and 12 h dark) and temperature (23°C). All experiments were performed between 1 pm and 3 pm. Animals were given a single intraperitoneal injection with PTZ (Sigma, St Louis, MO, USA) in phosphate-buffered saline (PBS) (40 μg/g for lower dosage or 50 μg/g for higher dosage) and observed in a transparent cage without prior knowledge of their genotypes. The severity of seizures was rated by using published criteria (31).
LFP measurements of polycystin-L deficient mouse
Experiments were conducted in agreement with ethics guidelines of the NIH and the University of Pennsylvania. Mice (20–30 g) were anesthetized with a ketamine–xylazine mixture (100–20 mg/kg, i.p., respectively). The level of anesthesia was determined by continuously monitoring of the EEG contralateral to the recording site. Supplemental doses of ketamine–xylazine (15–3 mg/kg, i.p., respectively) were given to maintain a synchronized EEG pattern. The bone and dura overlying the somatomotor and parietal cortex was removed. LFP recordings from the cortex, thalamus and hippocampus were made with bipolar tungsten electrodes (tip resistances ∼1 MΩ; tip separation: cortex = 0.7 mm vertically, 0.4 mm in thalamus and 0.5 mm in hippocampus). Polarity is negative down. Signals were digitized at 250 Hz for off-line computer analysis. Filtering of the data was performed digitally. Stimulation of the thalamus was performed using bipolar concentric electrodes (FHC). Analyses were performed offline with the software packages Igor (WaveMetrics), DataWave and Matlab (Mathworks).
Western blotting
Brain tissues were homogenized with either T-PER (Pierce) or radioimmunoprecipitation assay lysis buffer (Millipore) containing protease inhibitors (Roche). Cell lysates were made using M-PER (Pierce) with protease inhibitors. Lysates were separated on either 8, 10 or 12% sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS–PAGE) gels. The proteins were transferred onto nitrocellulose membranes (Amersham). The membranes were then blocked with 5% non-fat dry milk or 5% bovine serum albumin (BSA) in PBST for 60 min at room temperature and incubated overnight at 4°C with primary antibodies. After three washes with PBST, the membranes were incubated with horse reddish peroxidase-conjugated goat anti-mouse or goat anti-rabbit IgG (1:5000, Amersham) for 1 h at room temperature. Finally, the blots were developed by the enhanced chemiluminescence method.
Co-immunoprecipitation
HEK293 cells were homogenized with M-PER (Pierce) containing protease inhibitors (Roche) and 1 mm dithiothreitol (DTT). For the mouse hippocampal tissue extracts, the hippocampus were isolated from WT mice and homogenized with T-PER (Pierce) containing protease inhibitors (Roche) and 1 mm DTT. For co-immunoprecipitation, 2–4 mg of the lysate were incubated with either 2–4 μg antibodies, or an equal amount of normal mouse or rabbit IgG, or anti-Flag M2 Affinity Gel (Sigma-Aldrich) with gentle rocking overnight at 4°C. Protein A Sepharose beads (Invitrogen) were added and incubated for another 2 hours at 4°C. The washed beads were solubilized in an equal volume of 2× SDS sample buffer.
Note that, the pull-down samples were not heated before loading to the SDS–PAGE gel. Thus, IgG was kept as a dimer and ran at a size of 100 kDa. Because we used anti-flag antibody directly conjugated with beads for immunoprecipitation of Flag-β2AR, the IgG band is not detectable in western analysis.
Immunostaining and imaging
Animals were anesthetized by Isoflurane (Baxter), sacrificed by cardiac puncture and perfused with PBS followed by 4% paraformaldehyde. The brains were cryoprotected in 30% sucrose, frozen, embedded in optimal cutting temperature compound and sectioned in a cryostat at a thickness of 10 μm. For immunofluorescent experiment, sections were permeabilized with 0.3% Triton X-100 in PBS with 10% donkey serum, 5 mg/ml BSA and 0.02% sodium azide. All incubations and washes were carried out in PBS with 2% donkey serum, 1 mg/ml BSA and 0.02% sodium azide. All primary antibody incubations were carried out for 16–24 h at 4°C, and all secondary antibody incubations were carried out for 1 h at room temperature.
Cultured neurons were briefly washed with pre-warmed PBS two times, then fixed with 4% paraformaldehyde with 2% sucrose, permeabilized with 0.3% Triton X-100 in PBS for 10 min, and followed by blocking in 0.3% Triton X-100 in PBS with 10% goat serum, 50 mg/ml BSA and 0.02% sodium azide for 1 h at room temperature. All incubations and washes were carried out in 0.1% Triton X-100 in PBS with 2% goat serum, 10 mg/ml BSA, and 0.02% sodium azide. All primary antibody incubations were carried out for 16–24 h at 4°C, and all secondary antibody incubations were carried out for 1 h at room temperature.
Primary antibodies used for immunofluorescent staining were: rabbit anti-adenylyl cyclase III antibody (sc-588; Santa Cruz Biotechnology), goat anti-adenylyl cyclase VIII antibody (sc-1967; Santa Cruz Biotechnology), rabbit anti-PCL antibody (83430; generated by our group), rabbit anti-β2-AR antibody (sc-570; Santa Cruz Biotechnology) and mouse anti-NeuN antibody (MAB377, clone A60; Millipore). Secondary antibodies included Alexa Fluor 405- and 546-conjugated goat anti-mouse IgG (Invitrogen), Alexa Fluor 488- and 546-conjugated goat anti-rabbit IgG (Invitrogen), Alexa Fluor 546-conjugated donkey anti-goat IgG (Invitrogen) and Alexa Fluor 488-conjugated donkey anti-rabbit IgG (Invitrogen). Nuclei were visualized by 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen).
For double labeling with two rabbit primary antibodies, samples were fixed, permeabilized and incubated with the first primary antibody and an Alexa Fluor-conjugated goat anti-rabbit IgG secondary antibody as described above. The samples were then incubated for 1 h at room temperature in 5% normal rabbit serum (Jackson ImmunoResearch, West Grove, PA, USA) with 10 mg/ml BSA in PBS, to block any IgG-binding sites remaining on the first secondary antibody. PBS washes were then followed by incubation in 100 μg/ml goat anti-rabbit Fab (Jackson ImmunoResearch) for 1 h at room temperature to further block any remaining IgG-binding sites on the first primary or secondary antibodies. PBS washes were then followed by incubation with the second primary antibody for 16–24 h at 4°C. The second primary antibody was then labeled with a different Alexa Fluor-conjugated goat anti-rabbit IgG secondary antibody, as described above, followed by final PBS washes. Before being mounted with ProLong antifade media, the cells were incubated with DAPI for 5 min. A Zeiss Axioskop 2 Plus fluorescence microscope (Carl Zeiss) and the SPOT camera system were used for image analysis.
Neuronal cell culture
Hippocampal neurons were prepared as described previously (48,49). Briefly, hippocampi were dissected from mouse pups on the day of their birth (P0) and placed in a sterile solution of Leibovitz L-15 medium (Invitrogen, La Jolla, CA, USA) containing 0.375 mg/ml BSA. The meninges were removed and the hippocampal tissue was torn into small pieces. The tissue was then transferred into the 1× Trypsin-ethylenediaminetetraacetic acid solution (Gibco) and incubated for 15 min at 37°C with 95% O2/5% CO2 blowing gently over the surface of the solution. After incubation, the tissue was washed three times with pre-warmed seeding medium [Dulbecco's modified Eagle's medium (DMEM) (Gibco) containing 10% fetal bovine serum, 10% horse serum (Gibco), 2 mm l-glutamate and 1% Ampicillin (Gibco)] and triturated with a series of Pasteur pipettes of decreasing diameters. Hippocampal neurons were plated onto poly-l-lysine-coated coverslips (BD Biosciences, Bedford, MA, USA) in 12-well dishes in seeding medium. Cells from each hippocampus were plated onto two coverslips. After the first 24 h, the seeding medium was changed to the culture medium [DMEM medium (Gibco) containing 10% horse serum (Gibco), 2 mm l-glutamate, 1% N2 (Gibco), 2% B-27 (Gibco) and 1% Ampicillin (Gibco)]. After the first 48–72 h, cells were changed to neurobasal medium with 2% B-27, 2 mm l-glutamate, 1% Ampicillin (Gibco) and 1% insulin-transferrin-sodium selenite (Sigma-Aldrich), with the addition of cytosine arabinofuranoside (ARA-C; Sigma-Aldrich) to a final concentration of 10 μm to prevent glial cell replication. Cells were cultured for 9 days with replenishing fresh medium every other day for differentiation.
cAMP measurements
cAMP were measured as described previously with modifications (50). Briefly, whole brains (without olfactory bulb and cerebellum) from C57BL/6 mice were harvested and subsequently homogenized in 0.1 m HCl. Total cyclic AMP was measured in the resulting lysates using an enzymatic immunoassay (Sigma) according to the manufacturer's protocol. The assay is based on competition between unlabeled cAMP and peroxidase-labeled cAMP for a limited number of sites on an immobilized cAMP-specific antibody. The amount of peroxidase-labeled ligand bound by the antibody is inversely proportional to the concentration of unlabeled cAMP. Standard curves were generated for each experiment.
Data analysis and statistics
All values for a statistical significance represent mean ± SEM, and each data set was verified to be normally distributed before analysis. We carried out comparisons between means using Student's t-test. For all comparisons, we carried out power analyses to enable reliable conclusion, and coefficient variances were <10%. All comparisons with negative results had statistical powers of 0.8, and statistical significance implies P < 0.05.
Supplementary Material
Funding
This work was supported by grants from the National Institutes of Health (DK51050, DK40703, DK53357, DK51050, DK099532 and P50DK074030) to J.Z. G.Y. is a recipient of a postdoctoral fellowship from the American Heart Association. C.L. was supported by funding from National Nature Science Foundation of P.R. China (2012AA02A512).
Supplementary Material
Acknowledgements
We thank the members of the Zhou Lab and the Harvard Center for Polycystic Kidney Disease Research for scientific discussions and support.
Conflict of Interest statement. None declared.
References
- 1.Chang B.S., Lowenstein D.H. (2003) Epilepsy. N. Engl. J. Med., 349, 1257–1266. [DOI] [PubMed] [Google Scholar]
- 2.Brodie M.J., Elder A.T., Kwan P. (2009) Epilepsy in later life. Lancet Neurol., 8, 1019–1030. [DOI] [PubMed] [Google Scholar]
- 3.Fisher R.S., van Emde Boas W., Blume W., Elger C., Genton P., Lee P., Engel J. Jr. (2005) Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia, 46, 470–472. [DOI] [PubMed] [Google Scholar]
- 4.Burgess D.L., Noebels J.L. (1999) Voltage-dependent calcium channel mutations in neurological disease. Ann. N. Y. Acad. Sci., 868, 199–212. [DOI] [PubMed] [Google Scholar]
- 5.Steinlein O.K., Noebels J.L. (2000) Ion channels and epilepsy in man and mouse. Curr. Opin. Genet. Dev., 10, 286–291. [DOI] [PubMed] [Google Scholar]
- 6.Kullmann D.M. (2010) Neurological channelopathies. Annu. Rev. Neurosci., 33, 151–172. [DOI] [PubMed] [Google Scholar]
- 7.Lee U.S., Cui J. (2010) BK channel activation: structural and functional insights. Trends Neurosci., 33, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Escayg A., Goldin A.L. (2010) Sodium channel SCN1A and epilepsy: mutations and mechanisms. Epilepsia, 51, 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bloom F.E. (1975) The role of cyclic nucleotides in central synaptic function. Rev. Physiol. Biochem. Pharmacol., 74, 1–103. [DOI] [PubMed] [Google Scholar]
- 10.Gross R.A., Ferrendelli J.A. (1979) Effects of reserpine, propranolol, and aminophylline on seizure activity and CNS cyclic nucleotides. Ann. Neurol., 6, 296–301. [DOI] [PubMed] [Google Scholar]
- 11.Sano M., Seto-Ohshima A., Mizutani A. (1984) Forskolin suppresses seizures induced by pentylenetetrazol in mice. Experientia, 40, 1270–1271. [DOI] [PubMed] [Google Scholar]
- 12.Chen X.Z., Vassilev P.M., Basora N., Peng J.B., Nomura H., Segal Y., Brown E.M., Reeders S.T., Hediger M.A., Zhou J. (1999) Polycystin-L is a calcium-regulated cation channel permeable to calcium ions. Nature, 401, 383–386. [DOI] [PubMed] [Google Scholar]
- 13.Guo L., Chen M., Basora N., Zhou J. (2000) The human polycystic kidney disease 2-like (PKDL) gene: exon/intron structure and evidence for a novel splicing mechanism. Mamm. Genome, 11, 46–50. [DOI] [PubMed] [Google Scholar]
- 14.Wu G., Hayashi T., Park J.H., Dixit M., Reynolds D.M., Li L., Maeda Y., Cai Y., Coca-Prados M., Somlo S. (1998) Identification of PKD2L, a human PKD2-related gene: tissue-specific expression and mapping to chromosome 10q25. Genomics, 54, 564–568. [DOI] [PubMed] [Google Scholar]
- 15.Basora N., Nomura H., Berger U.V., Stayner C., Guo L., Shen X., Zhou J. (2002) Tissue and cellular localization of a novel polycystic kidney disease-like gene product, polycystin-L. J. Am. Soc. Nephrol., 13, 293–301. [DOI] [PubMed] [Google Scholar]
- 16.Li Q., Dai X.Q., Shen P.Y., Wu Y., Long W., Chen C.X., Hussain Z., Wang S., Chen X.Z. (2007) Direct binding of alpha-actinin enhances TRPP3 channel activity. J. Neurochem., 103, 2391–2400. [DOI] [PubMed] [Google Scholar]
- 17.Bui-Xuan E.F., Li Q., Chen X.Z., Boucher C.A., Sandford R., Zhou J., Basora N. (2006) More than colocalizing with polycystin-1, polycystin-L is in the centrosome. Am. J. Physiol. Renal Physiol., 291, F395–F406. [DOI] [PubMed] [Google Scholar]
- 18.Nauli S.M., Alenghat F.J., Luo Y., Williams E., Vassilev P., Li X., Elia A.E., Lu W., Brown E.M., Quinn S.J. et al. (2003) Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet., 33, 129–137. [DOI] [PubMed] [Google Scholar]
- 19.Yoder B.K., Hou X., Guay-Woodford L.M. (2002) The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J. Am. Soc. Nephrol., 13, 2508–2516. [DOI] [PubMed] [Google Scholar]
- 20.Wheatley D.N., Wang A.M., Strugnell G.E. (1996) Expression of primary cilia in mammalian cells. Cell Biol. Int., 20, 73–81. [DOI] [PubMed] [Google Scholar]
- 21.van Reeuwijk J., Arts H.H., Roepman R. (2011) Scrutinizing ciliopathies by unraveling ciliary interaction networks. Hum. Mol. Genet., 20, R149–R157. [DOI] [PubMed] [Google Scholar]
- 22.Gerdes J.M., Davis E.E., Katsanis N. (2009) The vertebrate primary cilium in development, homeostasis, and disease. Cell, 137, 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goetz S.C., Anderson K.V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet., 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee J.E., Gleeson J.G. (2011) Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr. Opin. Neurol., 24, 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lancaster M.A., Gleeson J.G. (2009) The primary cilium as a cellular signaling center: lessons from disease. Curr. Opin. Genet. Dev., 19, 220–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davenport J.R., Watts A.J., Roper V.C., Croyle M.J., van Groen T., Wyss J.M., Nagy T.R., Kesterson R.A., Yoder B.K. (2007) Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr. Biol., 17, 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baudoin J.P., Viou L., Launay P.S., Luccardini C., Espeso Gil S., Kiyasova V., Irinopoulou T., Alvarez C., Rio J.P., Boudier T. et al. (2012) Tangentially migrating neurons assemble a primary cilium that promotes their reorientation to the cortical plate. Neuron, 76, 1108–1122. [DOI] [PubMed] [Google Scholar]
- 28.Berbari N.F., Bishop G.A., Askwith C.C., Lewis J.S., Mykytyn K. (2007) Hippocampal neurons possess primary cilia in culture. J. Neurosci. Res., 85, 1095–1100. [DOI] [PubMed] [Google Scholar]
- 29.Ferguson G.D., Storm D.R. (2004) Why calcium-stimulated adenylyl cyclases? Physiology (Bethesda), 19, 271–276. [DOI] [PubMed] [Google Scholar]
- 30.Masyuk A.I., Gradilone S.A., Banales J.M., Huang B.Q., Masyuk T.V., Lee S.O., Splinter P.L., Stroope A.J., Larusso N.F. (2008) Cholangiocyte primary cilia are chemosensory organelles that detect biliary nucleotides via P2Y12 purinergic receptors. Am. J. Physiol. Gastrointest. Liver Physiol., 295, G725–G734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Signorini S., Liao Y.J., Duncan S.A., Jan L.Y., Stoffel M. (1997) Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc. Natl Acad. Sci. USA, 94, 923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Steriade M., Contreras D., Amzica F. (1994) Synchronized sleep oscillations and their paroxysmal developments. Trends Neurosci., 17, 199–208. [DOI] [PubMed] [Google Scholar]
- 33.Contreras D., Steriade M. (1995) Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J. Neurosci., 15, 604–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steriade M. (1999) Coherent oscillations and short-term plasticity in corticothalamic networks. Trends Neurosci., 22, 337–345. [DOI] [PubMed] [Google Scholar]
- 35.Steriade M., Contreras D. (1998) Spike-wave complexes and fast components of cortically generated seizures. I. Role of neocortex and thalamus. J. Neurophysiol., 80, 1439–1455. [DOI] [PubMed] [Google Scholar]
- 36.Niedermeyer E., Ribeiro M., Hertz S. (1999) Mixed-type encephalopathies: preliminary considerations. Clin. Electroencephalogr., 30, 12–15. [DOI] [PubMed] [Google Scholar]
- 37.Zhou J., Pei Y. (2007) Autosomal dominant polycystic kidney disease. In Mount, D.B. and Pollak, M.R. (eds), The Molecular and Genetic Basis of Kidney Disease. A Companion to Brenner and Rector's The Kidney. Saunders/Elsevier, Philadelphia, pp. 85–171. [Google Scholar]
- 38.Weinshenker D., Szot P., Miller N.S., Palmiter R.D. (2001) Alpha(1) and beta(2) adrenoreceptor agonists inhibit pentylenetetrazole-induced seizures in mice lacking norepinephrine. J. Pharmacol. Exp. Ther., 298, 1042–1048. [PubMed] [Google Scholar]
- 39.Nomura H., Turco A.E., Pei Y., Kalaydjieva L., Schiavello T., Weremowicz S., Ji W., Morton C.C., Meisler M., Reeders S.T. et al. (1998) Identification of PKDL, a novel polycystic kidney disease 2-like gene whose murine homologue is deleted in mice with kidney and retinal defects. J. Biol. Chem., 273, 25967–25973. [DOI] [PubMed] [Google Scholar]
- 40.Choi Y.H., Suzuki A., Hajarnis S., Ma Z., Chapin H.C., Caplan M.J., Pontoglio M., Somlo S., Igarashi P. (2011) Polycystin-2 and phosphodiesterase 4C are components of a ciliary A-kinase anchoring protein complex that is disrupted in cystic kidney diseases. Proc. Natl Acad. Sci. USA, 108, 10679–10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Handel M., Schulz S., Stanarius A., Schreff M., Erdtmann-Vourliotis M., Schmidt H., Wolf G., Hollt V. (1999) Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience, 89, 909–926. [DOI] [PubMed] [Google Scholar]
- 42.Brailov I., Bancila M., Brisorgueil M.J., Miquel M.C., Hamon M., Verge D. (2000) Localization of 5-HT(6) receptors at the plasma membrane of neuronal cilia in the rat brain. Brain Res., 872, 271–275. [DOI] [PubMed] [Google Scholar]
- 43.Berbari N.F., Johnson A.D., Lewis J.S., Askwith C.C., Mykytyn K. (2008) Identification of ciliary localization sequences within the third intracellular loop of G protein-coupled receptors. Mol. Biol. Cell, 19, 1540–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Domire J.S., Green J.A., Lee K.G., Johnson A.D., Askwith C.C., Mykytyn K. (2011) Dopamine receptor 1 localizes to neuronal cilia in a dynamic process that requires the Bardet-Biedl syndrome proteins. Cell Mol. Life Sci., 68, 2951–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Giraud F., Ayme S., Mattei J.F., Mattei M.G. (1976) Constitutional chromosomal breakage. Human Genetics, 34, 125–136. [DOI] [PubMed] [Google Scholar]
- 46.Delling M., DeCaen P.G., Doerner J.F., Febvay S., Clapham D.E. (2013) Primary cilia are specialized calcium signalling organelles. Nature, 504, 311–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeCaen P.G., Delling M., Vien T.N., Clapham D.E. (2013) Direct recording and molecular identification of the calcium channel of primary cilia. Nature, 504, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brewer G.J., Torricelli J.R., Evege E.K., Price P.J. (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res., 35, 567–576. [DOI] [PubMed] [Google Scholar]
- 49.Askwith C.C., Wemmie J.A., Price M.P., Rokhlina T., Welsh M.J. (2004) Acid-sensing ion channel 2 (ASIC2) modulates ASIC1 H+-activated currents in hippocampal neurons. J. Biol. Chem., 279, 18296–18305. [DOI] [PubMed] [Google Scholar]
- 50.Starremans P.G., Li X., Finnerty P.E., Guo L., Takakura A., Neilson E.G., Zhou J. (2008) A mouse model for polycystic kidney disease through a somatic in-frame deletion in the 5′ end of Pkd1. Kidney Int., 73, 1394–1405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.