

Abstract
Airway hyperresponsiveness (AHR) affects 55%–77% of children with sickle cell disease (SCD) and occurs even in the absence of asthma. While asthma increases SCD morbidity and mortality, the mechanisms underlying the high AHR prevalence in a hemoglobinopathy remain unknown. We hypothesized that placenta growth factor (PlGF), an erythroblast-secreted factor that is elevated in SCD, mediates AHR. In allergen-exposed mice, loss of Plgf dampened AHR, reduced inflammation and eosinophilia, and decreased expression of the Th2 cytokine IL-13 and the leukotriene-synthesizing enzymes 5-lipoxygenase and leukotriene-C4-synthase. Plgf–/– mice treated with leukotrienes phenocopied the WT response to allergen exposure; conversely, anti-PlGF Ab administration in WT animals blunted the AHR. Notably, Th2-mediated STAT6 activation further increased PlGF expression from lung epithelium, eosinophils, and macrophages, creating a PlGF/leukotriene/Th2-response positive feedback loop. Similarly, we found that the Th2 response in asthma patients is associated with increased expression of PlGF and its downstream genes in respiratory epithelial cells. In an SCD mouse model, we observed increased AHR and higher leukotriene levels that were abrogated by anti-PlGF Ab or the 5-lipoxygenase inhibitor zileuton. Overall, our findings indicate that PlGF exacerbates AHR and uniquely links the leukotriene and Th2 pathways in asthma. These data also suggest that zileuton and anti-PlGF Ab could be promising therapies to reduce pulmonary morbidity in SCD.
Keywords: Genetics, Hematology, Immunology, Inflammation, Pulmonology
Introduction
Sickle cell disease (SCD) is one of the most prevalent genetic disorders worldwide. It is caused by a mutation in the β-globin gene that results in sickle-shaped rbc during deoxygenation (1, 2). Adhesion of rbc and leukocytes to the endothelium, rbc sickling, and adhesion of sickle rbc to wbc result in vascular occlusions and multiple end-organ complications. However, the oxygen tension in lungs is too high for sickle hemoglobin polymerization. Indeed, the lung is the only organ where sickled rbc returning from the venous vasculature can unsickle. Despite this, acute and chronic lung diseases are major complications of SCD, and independent risk factors for morbidity and mortality in these patients (3, 4) and mechanisms behind them are largely unknown. Airway hyperresponsiveness (AHR), a dynamic narrowing of the lower airways, is highly prevalent in patients with SCD, affecting 55%–77% of children (5–7) and 33% of adults with SCD (8, 9). In contrast, the reported prevalence of AHR is 6%–20% in the African-American and black Caribbean pediatric population (6, 10).
Clinically, AHR is a key feature of asthma and is assessed using a provocative challenge (e.g., methacholine challenge) to induce a measurable bronchoconstriction. In SCD, the incidence of AHR is much higher than that of clinically symptomatic asthma (5, 6, 10–12) and is present without clinical features of asthma (wheezing, exhaled NO, and positive skin test to aeroallergens). But unlike asthma, which is a known risk factor for morbidity in SCD (13–16), the mechanism behind high AHR and its clinical significance is unknown. In SCD, biomarkers of hemolysis rather than those of asthma show a strong correlation with AHR (7). This led us to hypothesize that the etiology of the high AHR prevalence in SCD may be related to erythroid cells.
Following allergen stimulation, AHR is driven by a potent Th2 immune response that primes B cells to release allergen-specific IgE and drives lung inflammation. Th2 cytokines, such as IL-13 and IL-4 (17), stimulate airway smooth muscle contraction, recruit inflammatory cells, and increase mucus production; congestion and constriction of the airway lumen contributes to its remodeling (18, 19). In SCD, AHR has been presumed to occur secondarily to the leukocytosis and inflammation present at steady state. Notably, SCD patients also have elevated levels of LTE4 and LTB4 at steady state (20, 21). LTB4, a potent neutrophil inflammatory molecule (22), and cysteinyl LTs (CysLT) have been implicated in different aspects of allergen-induced asthma (18, 23). CysLT receptor inhibitors of 5-lipoxygenase (5-LO, encoded by Alox5) inhibitors are currently licensed for use for the treatment of asthma in humans (24, 25).
Our group previously reported that placenta growth factor (PlGF), an erythroid cell–secreted growth factor that belongs to the vascular endothelial growth factor family, is increased in plasma of patients with SCD and contributes to the baseline inflammation and features of pulmonary hypertension (26, 27). Herein, we hypothesized that PLGF mediates the high prevalence of AHR in SCD. We utilized genetic and pharmacological ablation of Plgf in WT mice and Berkeley sickle mice (hereafter termed HbS mice) (28). We have previously reported that crosses between Plgf KO and HbS mice were born extremely rarely, and the few born were runted and infertile (29). Hence, we studied AHR in Plgf-deficient and HbS mice separately. Herein, we show that Plgf is a novel mediator of AHR.
Results
PlGF deficiency reduces allergen-induced AHR.
To test the functional relevance of PlGF signaling in AHR development, we utilized BALB/c mice genetically lacking Plgf (Plgf–/–) and WT BALB/c controls (Plgf+/+). We first established that Plgf deficiency had no effect on lung mechanics at baseline (Supplemental Figure 1). To generate a model of allergic AHR, Plgf–/– and Plgf+/+ mice were sensitized by i.p. injections with house dust mite (HDM) allergen extract on days 0 and 7, followed by 2 rounds of airway exposure to HDM via intratracheal (i.t.) delivery on days 14 and 19 (Figure 1A). Airway sensitivity was measured on day 21 after the administration of a cholinergic agent using 2 well-established methodologies: the airway pressure time inflation and the forced oscillatory technique, which measure the AHR via airway pressure time index (APTI) (30) and airway resistance (R) (31), respectively.
Figure 1. PlGF deficiency blunts AHR.

(A) Experimental schema of induction of AHR in BALB/c mice. Mice were sensitized to HDM or saline i.p. and then exposed to HDM i.t. at the indicated time points; airway responsiveness was measured 48 hours after the last i.t. HDM exposure. (B) Airway responsiveness in Plgf+/+ or Plgf–/– mice was measured by airway pressure inflation after a challenge with acetylcholine. *P < 0.05 by 1-way ANOVA. (C) Airway responsiveness was also measured in response to increasing concentrations of methacholine using a forced oscillatory technique. HDM-treated Plgf+/+ mice either injected with vehicle (Saline) or αPlGF. *P < 0.05 Plgf+/+ HDM (vehicle) versus Plgf–/– HDM; #P < 0.05 Plgf+/+ HDM (vehicle) versus Plgf+/+ HDM (αPlGF) by 2-way ANOVA. (D and E) Plasma IgE levels in Plgf+/+ and Plgf–/– measured in BALF after the last i.p. sensitization with HDM (D) or 48 hours after the last HDM airway exposure (E). *P < 0.05, **P < 0.01 by 1-way ANOVA. All data are presented as mean ± SEM; each symbol represents and individual mouse; n = 5 animals in the saline-treated groups and 6–10 animals in the HDM-treated groups. Results are from 1 experiment (B) and representative of 2 independent experiments (C–E).
Increased AHR (APTI ≥ 300 cm H2O × s) was detected in Plgf+/+ mice exposed to HDM, as compared with their saline-exposed counterparts (Figure 1B). However, Plgf–/– mice exposed to HDM showed a 2-fold reduction in AHR compared with HDM-exposed Plgf+/+ mice (683 ± 94 cm H2O × s vs. 1,283 ± 229 cm H2O × s, respectively, Figure 1B). We confirmed the decrease in AHR in Plgf–/– mice using the forced oscillatory technique and observed the same results (Figure 1C). PlGF did not directly induce AHR in Plgf+/+ mice, when delivered intranasally into the airways (Supplemental Figure 2). Taken together, our data show that PlGF indirectly exacerbates allergen-mediated AHR.
To determine if PlGF exacerbates allergen sensitization, we measured circulating allergen-specific IgE in the plasma of Plgf+/+ and Plgf–/– mice directly after i.p. allergen sensitization (day 14) or after i.t. allergen exposure (day 21). HDM-specific IgE was significantly lower in Plgf–/– mice than in Plgf+/+ mice within 2 weeks of allergen sensitization, and this effect was more pronounced after airway exposure to HDM (Figure 1, D and E). These data indicate that, in addition to augmenting AHR, PlGF also increased allergen-mediated B cell responses.
We next tested if, beside its impact on sensitization, PlGF would directly contribute to the effector phase by pharmacological blockade of PlGF during allergen challenge. Plgf+/+ mice were injected with a PlGF-neutralizing Ab (αPlGF) during i.t. exposure to HDM (Figure 1A). Plgf+/+ mice injected with αPlGF had significantly diminished R as compared with control Plgf+/+ animals (7.1 ± 0.82 cm H2O × s/ml vs. 12.4 ± 2.9 cm H2O × s/ml) (Figure 1C). Thus, pharmacological neutralization of PlGF in Plgf+/+ mice during allergen challenge reduced AHR to the same degree as seen in mice with genetic deficiency of PlGF (Figure 1C). These findings suggest that PlGF affects AHR, independent of its role in promoting allergic sensitization.
Plgf deficiency results in reduction in airway eosinophilia and mucus production.
Next, we assessed the impact of PlGF deficiency on other classic features of asthma, like inflammation, mucus production smooth muscle changes, and Th2 cytokines. In HDM-challenged Plgf–/– mice, there was reduced inflammatory cell infiltration in the peribronchial and perivascular regions and reduced smooth muscle proliferation and goblet cell hyperplasia compared with HDM-challenged Plgf+/+ mice, where these changes were noticeably increased (Figure 2A). Saline-challenged Plgf–/– or Plgf+/+ controls showed no such changes (Supplemental Figure 3). Asthma-related histological changes were graded for smooth muscle hypertrophy/hyperplasia, inflammatory cell infiltration, goblet cell hyperplasia, alveolar septal thickening, vascular thickening, peribronchiolar fibrosis, degree of airway constriction, and perivascular and peribronchial infiltrates, which confirmed a significantly blunted asthma histological score of the HDM-challenged Plgf–/– mice compared with HDM-challenged Plgf+/+ mice (Figure 2B). The decreased mucus formation and a modest decrease in goblet cells in Plgf–/– mice compared with Plgf+/+ mice after HDM exposure was also reflected in lower Muc5AC (Figure 3A) and Clca3 (Figure 3B) immunostaining of lung sections and a trend toward reduction in Muc5AC levels in BALF (P < 0.06, Supplemental Figure 4D). A clear reduction in goblet cell hyperplasia was observed in histology (Figure 2A), but Western blots for Clca3 on whole-lung homogenates from Plgf+/+ and Plgf–/– mice did not demonstrate significant changes in Clca3 levels (Supplemental Figure 4, A–C), likely because combining all the cells in the lungs for protein analysis diluted the goblet cell effect. Overall, we observed a modest reduction in goblet cell hyperplasia and mucus formation in HDM-exposed Plgf-null animals.
Figure 2. PlGF deficiency reduces airway inflammation.

(A) Representative H&E lung sections of Plgf+/+ and Plgf–/– mice after i.p. sensitization and i.t. challenge with HDM depicting goblet cell hyperplasia (arrowheads) and smooth muscle proliferation (red arrows) in the peribronchiolar and perivascular regions, and inflammatory infiltrates in the alveolar sacs (black arrows). (B) Lung histological inflammation score of Plgf–/– and Plgf+/+ mice following HDM exposure; n = 7–10 mice per group. **P < 0.01 by Mann Whitney U test. Data are presented as mean ± SEM. Results are from 2 independent experiments.
Figure 3. PlGF deficiency reduces goblet cell metaplasia and eosinophil recruitment.

(A and B) MUC5AC staining (×20 and ×40 images shown) (A) and CLCA3 (×40) staining (B) of representative lung sections of HDM-exposed Plgf+/+ and Plgf–/– mice. (C) Differential cell counts in the BALF from saline- and HDM-exposed mice. *P < 0.05 by Mann Whitney U test; n = 4–5 animals per group. (D) Fold increase in blood eosinophils counts of Plgf+/+ and Plgf–/– mice following allergen exposure. **P < 0.01 by Mann Whitney U test; n = 3 animals in the saline-treated groups and 6–7 animals in the HDM-treated groups. All data are presented as mean ± SEM. Results are representative of 1 experiment (C and D).
Analysis of the bronchoalveolar lavage fluid (BALF) revealed that the reduction in cellular infiltration into the airways in HDM-exposed Plgf–/– animals was primarily due to reduced eosinophil infiltration in the lungs (Figure 3C). However, levels of the eosinophil chemokines eotaxin-1/2 in BALF were not significantly different from Plgf+/+ animals (P = 0.1, Supplemental Figure 4E). We therefore examined if part of the reason for the reduced eosinophilia in BALF was due to decreased eosinophil production in the Plgf–/– animals. Flow cytometry for blood eosinophils (CD11b+CD11c–SiglecF+ cells; Supplemental Figure 5A), in conjunction with leukocyte counts in Plgf+/+ and Plgf–/– animals, were performed to determine absolute eosinophil numbers in blood. As anticipated, there was no difference in circulating eosinophil levels in saline-challenged controls of either genotype. However, the decrease in BALF eosinophil numbers in HDM-challenged Plgf–/– mice (Figure 3C) was associated with a significant decrease in total circulating eosinophils (Figure 3D), suggesting that impaired recruitment by eotaxin-1/2 may not be the primary reason for reduced BALF eosinophils and that PlGF deficiency affected eosinophil production/recruitment from the BM.
Collectively, these data demonstrate that PlGF enhances allergen-induced eosinophilia in the blood and lungs, modestly affects mucus production, and significantly increases the inflammatory features associated with asthma.
PlGF modestly augments the Th2 immune response.
Analyses of Th2 cytokines in the BALF revealed significantly lower IL-13 levels in Plgf–/– mice (49.6 ± 16.3 pg/ml, n = 22) compared with Plgf+/+ mice exposed to HDM (89.8 ± 16.8 pg/ml, n = 26; P < 0.05) (Figure 4A). Although the average IL-4 levels in Plgf–/– mice were not statistically different from those of Plgf+/+ mice (P = 0.1), the median IL-4 levels in Plgf–/– mice (18.7 pg/ml; n = 22) were nearly half of those seen in Plgf+/+ mice (33.2 pg/ml; n = 26) after HDM exposure (Figure 4B). No significant change was seen in IL-5 levels (Figure 4C) and, as noted earlier, there was a trend toward reduced eotaxin levels in HDM-exposed Plgf–/– mice, albeit not statistically significantly (Supplemental Figure 4E). Additionally, no differences were observed in IFN-γ, IL-1β, MCP-1, IL-10, and IL-17A levels between Plgf–/– and Plgf+/+ mice (Supplemental Figure 6). Further characterization of lung immune cells revealed that, although allergen exposure substantially increased (lung Th2 lymphocytes and DCs, as was expected, no noticeable difference in these cell populations was seen between Plgf+/+ and Plgf–/– mice (Supplemental Figure 7). Taken together, these data suggest that PlGF increases Th2 immune response via IL-13 signaling —but this effect is modest at best — and that PlGF may be utilizing another pathway to exacerbate allergen-mediated pulmonary inflammation and AHR.
Figure 4. PlGF mildly affects IL-13 BALF levels.

(A–C) IL-13, IL-4, and IL-5 levels in the BALF of Plgf+/+ and Plgf–/– mice 48 hours following the last allergen challenge. *P < 0.05 using Mann Whitney U test between Plgf+/+ and Plgf–/– groups of mice. Each symbol represents 1 mouse; mean ± SEM of IL-13, IL-5, and IL-4 levels are indicated by the horizontal line and error bars. Results are from 3 independent experiments.
Allergen exposure induces PlGF in a STAT6-dependent manner.
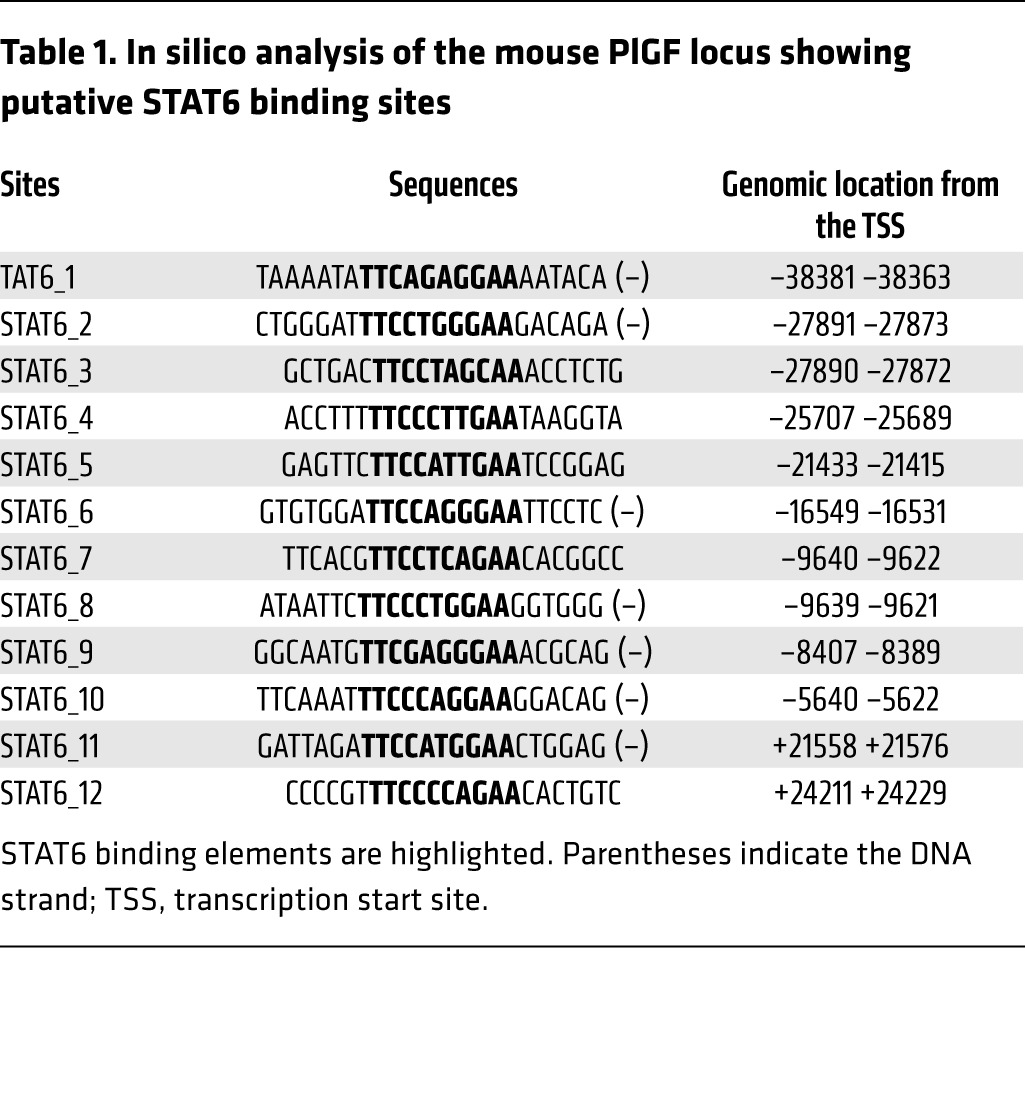
Interestingly, we noted that plasma PlGF levels were upregulated by several-fold in HDM-exposed Plgf+/+ mice compared with saline-exposed Plgf+/+ mice (Figure 5A). It has been reported that PlGF expression is restricted to lung macrophages and epithelial cells during murine (32) and human (33) lung development but is negligible postnatally at steady state (34, 35). Since PlGF modestly increased the Th2 cytokine responses, we tested the involvement of IL-13 and IL-4 signaling in allergen-mediated PlGF upregulation. STAT6 is a known downstream transcriptional effector of canonical IL-13 and IL-4 signaling. An in silico analysis of the PlGF gene promoter showed presence of several STAT6 consensus sequences (Table 1). We therefore hypothesized that Th2 cytokine signaling reactivates PlGF expression postnatally in lungs. We determined Plgf mRNA expression in the lungs of HDM- or saline-exposed Stat6–/– mice. Like the Plgf–/– mice, the Stat6–/– and WT mice were also on a BALB/c background. Plgf mRNA expression was not upregulated in Stat6–/– mice exposed to HDM, in contrast to WT BALB/c mice exposed to HDM (Figure 5B). IHC showed minimal PlGF protein expression in the lungs of Stat6-deficient mice (Figure 5C). In contrast, IHC revealed that lung airway and alveolar epithelial cells produced PlGF in WT mice after allergen exposure (Figure 5C). PlGF was minimally expressed by a few inflammatory cells/alveolar macrophages in Stat6–/– mice, in stark contrast to the high PlGF expression in epithelial cells, inflammatory cells, and alveolar macrophages observed in WT mice (Figure 5C). We sorted alveolar macrophages and eosinophils from lungs of allergen-challenged Plgf+/+ and Plgf–/– mice using flow cytometry (Supplemental Figure 5B) and performed quantitative PCR (qPCR) on RNA from these cell populations. BM (which contains erythroid cells, known to produce PlGF) was used as a control. Interestingly, while increased PlGF expression was seen in isolated alveolar macrophages, lung eosinophils also expressed high levels of PlGF in HDM-exposed Plgf+/+ mice (Supplemental Figure 8A). Plgf–/– mice had no detectable staining and did not show PlGF expression by qPCR on mRNA from any of the cell types examined, as expected (Figure 5C and Supplemental Figure 8A). Hence, we show that PlGF can be expressed by respiratory epithelial cells, alveolar macrophages, and eosinophils following allergen challenge via STAT6 activation, suggesting a positive feedback loop between the Th2 response and PlGF.
Figure 5. STAT6 mediates IL-13–induced increases in PlGF production.

(A) HDM sensitization further increases PlGF production in Plgf+/+ mice compared with control (saline-treated) mice; n = 3 animals in the saline-treated group and 5–9 animals in the HDM-treated group. **P < 0.01 by Mann-Whitney test. (B) Relative Plgf mRNA expression in the lungs of Stat6+/+ and Stat6–/– mice after HDM or saline exposure. ***P < 0.001 by Mann-Whitney U test; n = 6–8 mice per group. (C) Expression of PlGF protein in airway, alveolar epithelial cells, and alveolar macrophages in WT mice (top left panel), as compared with Plgf–/– mice (top right panel); ×20 images. A magnified view (×40) showing increased PlGF expression in alveolar epithelial cells and macrophages in WT mice following Th2-driven STAT6 activation (bottom left panel), compared with PlGF expression in Stat6–/– (bottom middle panel) and Plgf–/– (bottom right panel) alveolar epithelial cells and macrophages. All data are presented as mean ± SEM. Results are representative of 2 independent experiments.
Table 1. In silico analysis of the mouse PlGF locus showing putative STAT6 binding sites.
PlGF-deficient mice have reduced activation of the leukotriene pathway.
We have recently shown that PlGF induces expression of HIF-1α and Egr-1 transcription factors to mediate an increase in FLAP and Alox5 mRNA (key players in the leukotriene [LT] pathway), respectively, from human lung endothelial cells and monocytes in vitro (36, 37). Therefore, we investigated whether PlGF also exacerbated AHR via activation of the LT pathway in vivo in Plgf–/– and Plgf+/+ mice.
Allergen-exposed PlGF-deficient mice have nearly half the Alox5 mRNA expression compared with their WT counterparts. Control (saline-exposed) animals showed no difference in Alox5 mRNA levels (Figure 6A). Surprisingly, no difference in Flap mRNA levels were observed in the lung tissue between Plgf+/+ and Plgf–/– mice (Supplemental Figure 8B), unlike our in vitro findings in human endothelial cells (37). The transcriptional increase in Alox5 was also confirmed at the protein level, as IHC revealed markedly decreased staining for 5-LO in lung endothelial cells and alveolar macrophages of HDM-exposed Plgf–/– mice versus that of Plgf+/+ mice (Figure 6B). Consequently, urinary CysLTs, the end products of the LT pathway, were significantly reduced in Plgf–/– mice exposed to HDM (Figure 6C). In addition, Plgf+/+ mice exposed to allergen also had significantly higher CysLT receptor-1 (CysLTR1) expression in lungs than their Plgf–/– counterparts (Figure 6D). Moreover, pharmacological inhibition of 5-LO activity with zileuton mirrored the reduction in AHR severity observed in anti–PlGF-treated Plgf+/+ mice (Supplemental Figure 9). Taken together, a loss-of-function analysis suggests that the PlGF mediates its pulmonary effects via the LT pathway.
Figure 6. PlGF activates the LT pathway.

(A) The relative Alox5 mRNA expression in the lungs of Plgf+/+ and Plgf–/– BALB/c mice 48 hours after the last i.t. HDM exposure. *P < 0.05, **P < 0.01 by ANOVA (Bonferroni). (B) IHC showing 5-LO staining in lungs of saline and HDM-exposed Plgf+/+ and Plgf–/– BALB/c mice; magnification ×50. (C) CysLT levels in Plgf+/+ and Plgf–/– BALB/c mice measured in urine collected 24 hours after the last HDM exposure. (D) Cysltr1 mRNA expression in the lungs of HDM-exposed Plgf+/+ and Plgf–/– BALB/c mice. *P < 0.05 by Mann Whitney U test. All data are presented as mean ± SEM. Results are representative of 1 experiment (A, B, and D) and from 2 independent experiments (C).
To confirm the results from loss-of-function studies, we performed a gain-of-function experiment. HDM-exposed Plgf–/– mice were also given a CysLT cocktail i.t. and compared with vehicle-treated HDM-challenged Plgf+/+ controls (Supplemental Figure 10A). Unfortunately, after this dual treatment of HDM and CysLT, which involved repeated i.t. instillations, mice were too bronchoconstricted to get accurate AHR measurements. However, we were able to perform lung histology and blood flow cytometry for eosinophils after the HDM and CysLT exposures. CysLT treatment strikingly rescued the Plgf-null phenotype, as the asthma histology grading (performed by a pathologist blinded to the experimental treatment and mouse genotype) was now similar or even higher than that seen in HDM-challenged WT mice (Supplemental Figure 10, B–D). Furthermore, blood eosinophils were significantly increased in allergen-exposed Plgf–/– mice given i.t. CysLT compared with allergen-exposed Plgf–/– mice treated with vehicle control (Supplemental Figure 10E). CysLT have been reported to induce eosinophil production and blood and airway eosinophilia in mice and humans (38–40), which is abolished by blockade of CysLTR1 (40).
Taken together, our data suggests that, at least in the BALB/c strain, PlGF mediates increased blood and lung eosinophilia via increased eosinophil production and recruitment from BM, and this effect is mediated via both increased CysLT production and upregulation of CysLTR1. Collectively, we show that PlGF contributes to the upregulation of the production of both the ligand (CysLT) and receptor (CysLTR1) expression in the LT pathway to accentuate the allergen-induced AHR in mice.
PlGF and its downstream target gene expression is increased in nasal respiratory epithelial cells of patients with asthma.
Since PlGF expression was highly upregulated by IL-13–mediated STAT6 activation, we wanted to determine the expression of PlGF in a well-characterized cohort of patients with and without asthma (41). RNA was extracted from nasal airway epithelial cells of patients that either had (i) no history of asthma and were confirmed to be nonatopic by skin tests to common allergens (normal controls) or (ii) asthma patients with a clinical history of asthma and a positive skin test to one of the common allergens. The details of the eligibility criteria and methodology of gene expression analysis have been previously described in detail (41). Expression of PLGF was significantly higher in patients with asthma compared with normal controls in nasal airway epithelial cells (Figure 7A). In addition, expression of the downstream target transcription factors that are upregulated by PlGF, Egr-1, and HIF-1α (36, 37) were increased in patients with asthma, as was the expression of Alox5, FLAP, and CYSLTR1 (Figure 7B). Although the expression of Alox5, FLAP, and CYSLTR1 could result from allergen-induced asthma and not be specific to PlGF, these data confirm our prior studies on PlGF and the murine studies performed herein on PlGF-deficient animals. Moreover, we have previously reported that PlGF upregulates endothelin-1 expression in the lung and increases EGR1 transcription (26, 27, 42); this data set also showed significantly increased endothelin-1 and EGR1 expression in respiratory epithelial cells, a further validation that the PlGF signaling pathway is activated in asthma. In experimental models of asthma, the LT pathway and Th2 cytokines are known to potentiate each other (43–46). Our data suggests that PlGF may be the novel link that connects these 2 pathways.
Figure 7. Expression of PLGF mRNA and its downstream target genes is increased in respiratory epithelial cells of patients with asthma.

(A) Relative quantification of PLGF mRNA expression in nasal epithelial cells of human subjects with no asthma (n = 9) and those with asthma (n = 18). **P < 0.01 by Mann Whitney U test. All data are presented as mean ± SEM. (B) Heat map of PlGF pathway genes in the microarray profile of some of the subjects shown in A (n = 4 normal, and n = 8 asthmatic). The normalized expression intensity is indicated by the color code: low (blue), medium (yellow to orange), and high (red). *P < 0.05 by unpaired t test. The fold increase is shown as mean ± SEM in parenthesis. Results are from of 2 experiments (A) and 1 experiment (B).
PlGF is critical for allergen-induced AHR in sickle mice.
Of all hematopoietic cells, PlGF is elaborated from erythroid cells (26, 47). Due to the expanded erythropoiesis, hypoxia, and erythropoietin in SCD, blood PlGF levels are increased (26, 48). Similar to patients, mice with SCD have naturally elevated levels of circulating PlGF that secreted from the hyperplastic erythroblasts (27). The prevalence of AHR is also high in patients with SCD (5–9). We therefore sought to determine whether (i) PlGF is the molecule that results in AHR in SCD and (ii) AHR was solely driven by sickle erythropoiesis.
To this end, we utilized the well-established Berkeley mouse model of SCD. Heritable components are known determinants of the susceptibility to allergen-induced AHR in different mouse strains (30, 49, 50), and the HbS mice are relatively outbred; they are originally derived from a mixture of 5 parental strains (28, 51), and in this mixed background, unidentified heritable loci prevent AHR development (Supplemental Figure 12, A and B). Therefore, we utilized a BM transplant approach to generate mice that were completely chimeric for sickle hematopoiesis on a homogenous-mouse genetic background. The predominant strain in HbS mice is C57BL/6, and therefore HbS BM was transplanted into C57BL/6 mice (Figure 8A). This approach allowed us to test whether sickle erythroid cells/rbc (erythropoietic cells) resulted in AHR and also allowed studies on numerous age-matched chimeric HbS mice concurrently, which were not otherwise possible with the fragile and difficult-to-breed HbS mice. Sickle BM cells from HbS mice (that were screened to express the CD45.2+ isoform on their leukocytes) were transplanted into lethally irradiated WT C57BL/6 ByJ (BL/6; CD45.1+) mice (Figure 8A). BM from WT C57BL/6 (BL/6; CD45.2) mice transplanted into congenic C57BL/6 ByJ mice (CD45.1 on the same BL/6 background) served as controls. We confirmed that HbS/WT mice were fully chimeric for donor-derived sickle or normal hematopoiesis that completely replaced the normal recipient hematopoiesis in the blood and the lungs of recipient mice. Sixteen weeks after transplantation, full reconstitution of the recipient hematopoietic system by the donor hematopoietic cells was confirmed by flow cytometry for the exclusive presence of donor leukocytes (CD45.2) in the lung and human sickle hemoglobin in the blood (Supplemental Figure 11, A and B).
Figure 8. PlGF and cytokine levels in chimeric SCD mice.

(A) Experimental schema showing generation of chimeric HbS (Chi-HbS) and BL/6 (Chi-BL/6) mice by BM transplantation. (B) Plasma PlGF levels of HDM- and saline-exposed Chi-HbS and Chi-BL/6 mice. *P < 0.05 by Mann Whitney U test; n = 17–19 animals per group. (C–F) Eotaxin, IL-13, IL-4, and IL-5 levels in the BALF of saline- and HDM-exposed chimeric mice. All data are presented as mean ± SEM; each symbol represents 1 mouse. Results are from 3 independent experiments (B) or representative of 1 experiment (C and D).
Chimeric HbS-BL/6 mice (Chi-HbS mice) developed hematological parameters characteristic of SCD: anemia, reticulocytosis, and leukocytosis (Supplemental Figure 11C). Like the native HbS mice (27), Chi-HbS mice developed elevated PlGF levels compared with chimeric BL/6 mice (Chi-BL/6; Figure 8B), confirming that sickle erythroid cells resulted in elevated PlGF in SCD. We next determined whether the transplanted mice, especially the Chi-HbS mice, were able to mount a robust Th2 response, and indeed, they exhibited classic features of allergen-induced Th2 response, including the induction of Th2 cytokines and the expansion of Th2 lymphocytes in the lung (Figure 8, C–F, and Supplemental Figure 13, A–F).
Next, we exposed Chi-HbS mice to HDM and determined HDM-specific B cell responses and airway reactivity: Both Chi-BL/6 and Chi-HbS mice were able to mount allergen-specific B cell responses. HDM-specific IgE trended higher in HDM-exposed Chi-HbS mice compared with Chi-BL/6 controls, albeit not statistically significantly (P = 0.1; Supplemental Figure 14C).
After HDM airway exposure, Chi-HbS mice developed more severe AHR than their corresponding Chi-BL/6 controls exposed to HDM (Figure 9A). Notably, increased AHR was present in Chi-HbS mice, even at baseline, without exposure to methacholine (Figure 9A). However, this was not due to a higher Th2 response in these Chi-HbS mice: Chi-BL/6 and Chi-HbS mice had very similar Th2 cytokine levels in saline- and allergen-exposed animals. IL-13 levels trended lower in Chi-HbS mice (P = 0.06) but were not significantly different. Notably, the number of lung DCs and Th2 lymphocytes were also similar between Chi-BL/6 and Chi-HbS mice (Supplemental Figure 13, A–F, and Supplemental Figure 14, A and B), excluding the possibility that Chi-HbS mice have an intrinsically skewed Th2 immune response favoring allergic responses.
Figure 9. PlGF mediates AHR seen in SCD mice through a LT-dependent pathway.

(A) Airway responsiveness to methacholine of saline- or HDM-exposed Chi-BL/6 and Chi-HbS mice. *P < 0.05, HDM-exposed Chi-BL/6 versus HDM-treated Chi-HbS group, 2-way ANOVA. (B) Airway responsiveness to methacholine of HDM-exposed Chi-BL/6 and Chi-HbS mice treated with vehicle control, αPlGF, or zileuton. ***P < 0.001, HDM-exposed Chi-HbS (vehicle) versus HDM-exposed Chi-HbS (αPlGF); ###P < 0.001, HDM-exposed Chi-HbS (vehicle) versus HDM-exposed Chi-HbS (zileuton); ##P < 0.01, HDM-treated Chi-HbS (vehicle) versus Chi-BL/6 by 2-way ANOVA. n = 6–8 animals per group. (C) Urine cysteinyl LTs of HDM-exposed Chi-BL/6 and Chi-HbS mice treated with vehicle control, αPlGF, or zileuton. *P < 0.05, ***P < 0.001 by 1-way ANOVA; n = 5–6 animals per group. All data are presented as mean ± SEM. Results are from 2 independent experiments.
Nonetheless, the exaggerated AHR in Chi-HbS mice compared with Chi-BL/6 mice was mediated by PlGF, since treatment of HDM-exposed Chi-HbS mice with αPlGF blocked the airway reactivity to levels seen in vehicle-treated Chi-HbS mice and vehicle-treated Chi-BL/6 mice (Figure 9B). These data highlight the critical role of PlGF in AHR in SCD and suggest a mechanism of action other than the classic allergic Th2 pathway.
PlGF mediates AHR in HbS sickle mice through a LT-dependent pathway.
Chi-HbS mice had higher PlGF levels in circulation, as expected, due to a markedly expanded sickle erythropoiesis, which increases 5-LO and CysLTR1 expression. We found that zileuton treatment reduced AHR in HDM-exposed Chi-HbS mice to levels comparable to αPlGF-treated Chi-HbS mice or vehicle-treated Chi-HbS controls (Figure 9B). This was accompanied with a marked reduction in urinary CysLTs (Figure 9C), more so with zileuton than with αPlGF. It was noteworthy that, similar to BALB/c Plgf+/+ mice, Chi-HbS mice showed increased LT production when compared with Chi-BL/6 mice exposed to allergen (Figure 9C). In both animal models, blocking PlGF function by an Ab or blocking LT production by zileuton resulted in reduced AHR (Figure 9B).
Since zileuton had a greater effect than PlGF Ab, in addition to 5-LO, we also evaluated expression of LTC4 synthase (Ltc4s), a key CysLT synthetic enzyme downstream of 5-LO. There was enhanced expression of Ltc4s in non–allergen-exposed Chi-HbS mice compared with Chi-Bl/6 controls, which explains the AHR in Chi-HbS mice at baseline. The expression of Ltc4s increased further after allergen challenge in Chi-HbS mice (Supplemental Figure 15A), but no such effect was seen in Chi-Bl/6 mice, suggesting that this was a sickle-specific effect. Remarkably, blockade of PlGF with a PlGF-neutralizing mAb significantly reduced Ltc4s expression in HDM-challenged Chi-HbS to the level of AHR seen in allergen/saline-treated Chi-BL/6 mice, suggesting LTC4S is also regulated by PlGF (Supplemental Figure 15A). We also studied Ltc4s gene expression in PlGF-deficient and WT mice. Mean Ltc4s expression trended lower in saline-treated or HDM-challenged Plgf–/– mice compared with WT mice (Supplemental Figure 15B). Our data suggests that there may be PlGF-dependent regulation of LTC4S (Figure 10) and the greater effect of zileuton in lowering CysLT levels in Chi-HbS mice is a PlGF-independent effect. Collectively, our data demonstrate that PlGF enhances allergen-induced AHR in sickle mice through 5-LO– and CysLTR1-mediated LT production.
Figure 10. Schematic showing the mechanism of PlGF-induced AHR and the positive feedback loop between IL-13 and PlGF.

PlGF is secreted by erythroid progenitors from the BM in response to hypoxia and erythropoietin (Epo) in SCD. PlGF stimulates the expression of 5-LO, which generates CysLT, and increases CysLTR1 expression. Sickle hematopoietic cells additionally increase LTC4S expression, which further contributes to CysLT generation. LT and IL-13 are key effectors of AHR. PlGF blockade or 5-LO inhibition reduces AHR. IL-13 levels activate its downstream effector molecule, STAT6, which in turn further augments PlGF production from pulmonary airway epithelial cells, eosinophils, and alveolar macrophages. The latter explains the elevated PlGF levels in allergen-challenged WT mice and in patients with asthma.
Discussion
Using genetic and pharmacological approaches, we show a distinct and significant exacerbation of AHR by PlGF. This effect occurs primarily via its upregulation of 5-LO and LT production and, to a lesser extent, upregulation of IL-13–mediated Th2 response, which further upregulates PlGF production from lung epithelial cells, eosinophils, and alveolar macrophages. Indeed, respiratory epithelial cells from patients with asthma showed significantly increased Plgf mRNA and an increase expression of PlGF target genes. Since PlGF is also an erythroid cell–derived factor that is increased in SCD, we transplanted sickle hematopoiesis into WT mice and showed that the high AHR is mediated through sickle erythroid cell–induced high PlGF and is therefore transplantable and completely abrogated by either inhibiting PlGF or the LT pathway.
Our results contrast to a previous report, which suggested that PlGF has no pathogenic effect on AHR in ovalbumin allergen–induced experimental AHR (52). This apparent discrepancy may be due to differences in allergens and mouse strains. The activity of specific pathways required for AHR severity has been shown to be mouse strain dependent (30, 49). Given that HDM inhalation is more relevant to human pathology, our finding of the pathogenic role of PlGF in the context of HDM-induced AHR is clinically relevant to human disease. Our data support and extend the prior work by Pritchard et al., who showed that chimeric sickle mice have baseline lung pathology characterized by increased R (53). In agreement with their study, we showed that sickle mice have an increased R at baseline, in the absence of the bronchoconstrictor challenge with methacholine. Our study further demonstrates the mechanism of increased AHR in SCD is via CysLT induction, as abrogation of the augmented AHR occurred by αPlGF or zileuton. Since Th2 responses are unchanged in chimeric sickle mice compared with controls, some innate effector cells stimulated by sickle hematopoiesis may also contribute to the production of CysLT. While we have shown that PlGF induces 5-LO to generate LTs, pulmonary expression of Ltc4s was substantially increased in Chi-HbS at baseline and further induced after HDM challenge, a phenomenon not seen in Chi-BL/6 mice. Moreover, the ability of PlGF Ab to lower Ltc4s levels in HDM-exposed Chi-HbS mice indicates some form of PlGF-dependent regulation of Ltc4s surge at baseline and during allergen challenge. These data suggest that this phenomenon may be specific to sickle hematopoietic cell–secreted PlGF. While Ltc4s induction is achieved through direct regulation by sickle hematopoiesis–induced PlGF, an innate cell may also contribute to the increased AHR and affect CysLT levels in sickle mice; the particular cell type or pathway affected is an exciting new area for exploration, and future studies will be necessary to dissect these complexities of PlGF-dependent and -independent effects in asthma and in AHR in SCD.
In BALB/c mice, although PlGF enhanced the allergen-induced AHR severity, its expression was not necessary for AHR initiation, since AHR developed in HDM-exposed Plgf–/– mice. However, here AHR was significantly blunted compared with HDM-challenged Plgf+/+ mice. Zileuton had a much greater effect than αPlGF in reducing CysLT production in Chi-HbS mice. This is not surprising, since HDM can mediate LT production via pathways other than PlGF (54). Whether another pathway besides the ones identified herein plays a role in increasing CysLT production in SCD mice remains to be investigated.
Interestingly, PlGF significantly augmented allergen-specific IgE production even during i.p. sensitization. AHR- and allergen-mediated IgE responses have been reported to be significantly blunted in Alox5-deficient mice (55, 56). The same phenotype in PlGF-deficient mice suggests that PlGF is an upstream activator of 5-LO. Besides augmenting the LT pathway, we observed that levels of the Th2 cytokine IL-13 were also significantly more exaggerated in HDM-exposed Plgf+/+ mice compared with Plgf–/– mice. IL-13 has been shown to upregulate CysLT receptor expression (43, 44); conversely, there are also reports of CysLT-mediated increased Th2 response and eosinophilic infiltration in the lung (23, 57). Indeed, CysLTR1 expression was significantly lower in Plgf–/– mice. CysLT are also known potent contractile agonists of the airway smooth muscle cells (58) and vascular mural cells (59). Together, these interconnected LT and Th2 cytokine pathways are accentuated to increase AHR and inflammation in Plgf+/+ mice. IL-13 levels were not significantly different in HbS mice, suggesting that either IL-13 does not play a role in AHR in SCD or that this is a mouse strain–specific effect. Importantly, both models show the primary involvement of the LT pathway for AHR based on the results in Plgf–/– mice, or in Plgf+/+ and HbS mice given αPlGF. Other studies have reported the development of CysLT-induced AHR in the presence of normal IL-13 induction (54, 55).
Our data show that the LT signaling axis is interrupted in the absence of PlGF and is exaggerated in SCD, where PlGF concentrations are increased. Notably, zileuton had the same effect as αPlGF. Moreover, the PlGF-null mouse AHR phenotype was rescued to levels similar to Plgf+/+ mice by i.t. instillation of CysLT. Collectively, our data shows that the LT pathway was the primary downstream effector pathway of PlGF, with secondary activation of an IL-13–mediated T cell (Th2) and B cell (IgE) response. Taken together, this study provides novel mechanistic evidence of activation of both LT and Th2 pathways by PlGF.
In BALB/c mice, PlGF effect on Th2 cytokines was limited to IL-13, but not IL-4, IL-5, or eotaxin, the latter being the same despite high eosinophils. While there may have been a kinetic delay between the peak of eotaxin production in the lung (10 hours) following allergen exposure (60) and our sampling time (48 hours), IL-5– and IL-4–independent allergen-induced lung eosinophilia and airway hyperreactivity have been described in BALB/c mice (61) and asthma patients (62). More importantly, loss- and gain-of-function studies show that PlGF mediates AHR and blood and lung eosinophila primarily via CysLT that are known to increase eosinophil production from BM and induce blood and airway eosinophilia in mice and in humans (38–40), and this effect is abolished by a blockade of CysLTR1 (40). Moreover, the IL-13–restricted effect of PlGF may suggest participation of PlGF in previously described IL-13–mediated IL-4/eotaxin–independent AHR (63), which is directed at augmenting the airway smooth muscle responsiveness (63, 64).
Another finding was that PlGF expression was further upregulated in the lung epithelial cells, eosinophils, and macrophages via a Th2-driven STAT6 induction, creating a positive feedback loop (shown schematically in Figure 10). The human PlGF promoter showed several putative STAT6 binding consensus sequences (Table 1). However, further studies are needed to determine if PlGF is a direct downstream target of STAT6. Regardless, induction of PlGF via a Th2 pathway and vice versa has not been previously reported and has much broader implications than SCD, since the Th2 response is classically upregulated in asthma. Indeed, interrogation of PlGF expression in respiratory epithelial cells in a well-characterized asthma and control cohort we have previously published (41) showed significantly increased PLGF mRNA expression in patients with asthma compared with controls. High PlGF levels in asthma patients were associated with the upregulation of expression of PlGF signaling partners and target genes we have previously identified or those identified herein: EGR1, HIFA, endothelin-1 (ET1), and Alox5 (36, 37, 42). PlGF is expressed largely in the placenta. We have previously shown that, in SCD, excessive expansion of the erythroid precursors, erythropoietin, and hypoxia drive the rise in erythroid cell–secreted PlGF (26, 48). While PlGF expression has been shown to be upregulated in lung cancer and a variety of other cancers (65, 66), its expression in the normal lung has only been documented during mouse and human lung development (33–35). Herein, we show that PlGF expression can be upregulated in the postnatal/adult lung alveolar macrophages/eosinophils and epithelial cells, as well as via allergen-mediated Th2 cytokines/STAT6 in experimentally induced asthma.
Transgenic mice that overexpress PlGF develop emphysema (35), an effect abrogated in elastase-treated Plgf–/– mice (32). PlGF is elevated in patients with chronic obstructive pulmonary disease (COPD), where emphysema is a key component, and its levels were shown to be associated with higher pulmonary morbidity (67). Furthermore, high PlGF levels have been reported in the sputum of COPD patients with lower FEV1 (52, 67). However, its expression in normal adult (postnatal) pulmonary eosinophils, alveolar macrophages, and epithelial cells; its role in exacerbating AHR and asthma; and its mechanism of action were hitherto unexplored. To our knowledge, this is the first report showing a mechanism of increased AHR by PlGF in SCD that also explains its association with acute asthma, and conceivably in obstructive airway physiology in COPD.
We have previously shown that PlGF contributes to the chronic inflammatory state in SCD via activation of monocytes and increased inflammatory cells (48, 68, 69). Indeed, SCD is characterized by increased blood leukocytes at baseline (70, 71), a major source of LTs in vivo. Targeting the LT pathway using αPlGF/zileuton may decrease both PlGF-dependent and PlGF-independent AHR and pulmonary morbidity in patients with SCD. Furthermore, LT plays a role in inflammation (58), a major player in SCD pathophysiology. Hence, LT-targeted therapy such as zileuton may provide benefits beyond the AHR lung disease spectrum in SCD.
Increased hemolysis triggers increased erythropoiesis and may be associated with increased PlGF production, priming the lung to AHR. Importantly, acute chest syndrome (ACS), a life-threatening complication of SCD, has been shown to be associated with asthma in children with SCD (72). However, ACS also occurs in a vast majority of SCD patients who do not have clinically diagnosed asthma. It is conceivable that individuals with high PlGF levels predisposed to ACS by increasing AHR in the absence of asthma, and here, pharmacological blockade of either PlGF or LT may help prevent this life-threatening complication of SCD. Pulmonary complications are important risk factors for the high morbidity in SCD (3, 4, 73); therefore, targeting PlGF or LT can improve the overall SCD outcome. We have previously reported elevated PlGF expression in β-thalassemia (26), another chronic hemolytic anemia disorder. It is possible PlGF may play a similar detrimental role in β-thalassemia that remains to be explored.
Interestingly, in SCD, the transition from childhood to adulthood is associated with a decline in the incidence of AHR (5, 6, 8, 9) and obstructive lung disease (74, 75) and an increase in restrictive lung disease. While asthma and AHR decline with age due to the increased size of the airways in adults, other factors — including transfusion hemosiderosis, vascular pruning, and pulmonary artery hypertension that develop in adults with SCD — may cause a profibrotic phenotype. More broadly, our studies show a distinct role of PlGF in asthma. Therefore, targeting PlGF may prove beneficial for asthma by blunting the Th2 and LT pathways and for other airway diseases featuring an AHR component, such as COPD and asthma.
In conclusion, we report an erythropoiesis-induced, PlGF-dependent IgE–IL-13–LT axis that contributes to increased AHR in SCD, which in turn further upregulates PlGF production from lungs via Th2 cytokine upregulation. PlGF-exacerbated AHR can be pharmacologically targeted by the licensed 5-LO inhibitor, zileuton, or αPlGF, which is currently in clinical trials for cancer. Thus, this study provides an opportunity for targeting PlGF as an early clinical intervention to effectively reduce the pulmonary morbidity associated with SCD and asthma.
Methods
Supplemental Methods are available online with this article.
Animals.
PlGF KO mice were obtained from Peter Carmeliet’s laboratory (University of Leuven) and back-crossed for 10 generations into a BALB/c background at Cincinnati Children’s Hospital; 10- to 14-week-old male and female Plgf+/+ mice (also referred to as Plgf+/+) with physiological PlGF expression were used for experimentation. Hbatm1Paz Hbbtm1Tow Tg (HBA-HBBs) 41Paz/J or HbS sickle mice were obtained from The Jackson Laboratory and were bred up to the sixth generation under C57BL/6NJ background. HbS sickle mice carry KO for the mouse α-globin and carry a transgene expressing the human βS-globin gene (28). C57BL/6NJ or B6.SJL-Ptprca Pepcb/BoyJ (BL/6; CD45.1 [C57BL/6ByJ]) mice were purchased from The Jackson Laboratory. Stat6 KO mice were provided by Gurjit Khurana Hershey’s laboratory (CCHMC). The Stat6 KO mice were also in a BALB/c background. Chi-HbS were generated by transplanting whole BM from native HbS mice (that expressed CD45.2 on their leukocytes) into lethally irradiated B6.SJL-Ptprca Pepcb/BoyJ (C57BL/6ByJ; CD45.1) recipient mice in a 1:6 donor/recipient ratio. For the generation of control mice, whole BM from C57BL/6 (CD45.2) donor mice was transplanted into lethally irradiated C57BL/6ByJ (CD45.1) mice. Twelve weeks after transplantation, hemoglobin chimerism was assessed by HPLC. Lung mechanics were assessed at 16 weeks after transplantation, at which point, the recipient mice had full donor hematopoiesis. Lungs were dissociated and cells stained for CD45.1 and CD45.2 to determine donor leukocyte chimerism in lungs.
Experimental models of allergen-induced AHR.
To study experimental AHR, we used the HDM extract as the allergen. For the HDM challenge regimen, mice were sensitized on days 0 and 7 with an i.p. dose of 10 μg (100 μl) of HDM (Greer) or with phosphate-buffered saline (saline controls); on days 14 and 19, mice were treated i.t. with 100 μg (40 μl) of HDM or with saline. On day 21 of the challenge, airway reactivity was tested, and thereafter, mice were euthanized.
Measurement of lung mechanics.
AHR was studied in mice 48 hours after the last challenge, using 2 testing methods. For the airway pressure test, mice were anesthetized with sodium pentobarbital (65 mg/ml) and intubated. Respiration rate was maintained at approximately 120 breaths per minute with a constant tidal volume (0.2 ml), while mice were kept paralyzed with decamethonium bromide (25 mg/kg). After a stable baseline was achieved, acetylcholine (50 mg/kg) was injected into the inferior vena cava and dynamic airway pressure (cm H2O × s) was monitored for 5 minutes. In addition, AHR was also measured in parallel experiments using an independent method: the forced oscillation technique, as described previously (76). Mice were anesthetized with a triple sedative solution (ketamine/zylaxine/acepromazine; 4:1:1) and connected to a computer-controlled small animal ventilator (flexiVent, SCIREQ) by intubation. R was assessed following delivery of nebulized saline and increasing doses of the broncho-constrictor agent methacholine (50 mg/ml and 100 mg/ml; acetyl-β-methylcholine chloride, Sigma-Aldrich). For each concentration of methacholine, the maximal R (Rs) values were averaged. Following studies on lung mechanics, blood, bronchoalveolar lavage fluid, and organs were collected for ELISA assays and pathology.
PlGF Ab and zileuton regimens.
PlGF neutralizing Ab (PL5D11D4) was a gift from Bart Jonckx (ThromboGenics NV). The mouse monoclonal anti-PlGF Ab was developed against all mouse PlGF proteins (65). A dose of 50 mg/kg of PlGF-neutralizing Ab or saline was administrated i.p. on days 14, 16, 17, 19, and 20 (relative to the day of the initial administration of HDM) to mice. In one experiment, Plgf+/+ animals challenged with HDM were given either mouse IgG1 (the isotype of αPlGF) or saline i.p. to ensure there was no difference in pulmonary phenotype with either. Doses of 70 mg/kg or 150 mg/kg of zileuton (Zyflo CR) were delivered by gavage into mice 5 hours prior to the first i.t. administration of HDM and an hour after the first HDM challenge. Subsequently, mice received 70 mg/kg or 150 mg/kg (200 μl) of zileuton twice daily until the termination of the experiment on day 21. Zileuton was dissolved in a diluent solution: 10% DMSO in water. An equal volume of the diluent solution was given to control mice. For treatment of Plgf–/– with CysLT, 1 μg of a HPLC-purified CysLT mixture (Cayman Chemical) was instilled daily over 3 days into the trachea of the animals from day 18–20 relative to the day of the initial administration of HDM. The CysLT was prepared by drying the stock of ethanol-diluted CysLT using a nitrogen gas tank, and the reduced powder was resuspended into sterile saline. Control Plgf+/+ and Plgf–/– mice received sterile saline.
Asthma cohort analysis.
Nasal respiratory epithelial cells from asthmatic and nonasthmatic subjects, younger than 18 years of age, were obtained. The selection criteria were described in detail before (41). Briefly, subjects included in the control group had (i) no acute infection or major illnesses and (ii) a negative result to the skin-prick test to a panel of environmental allergens. Subjects in the asthma group included both patients with (i) a physician diagnosis of asthma with no current wheezing episodes and (ii) a positive skin-prick test to environmental allergens.
Nasal epithelial brushings were found to contain 97% respiratory epithelial cells (41). RNA from 4 individuals in the nonasthma group and 8 individuals in the asthma cohort was obtained from these cells and subjected to microarray analysis using the HG-U133A GeneChip (Affymetrix) (41). The expression profile was interrogated for PlGF and its target genes using GeneSpring software. Expression profiles were derived from a microarray database utilized in a previous publication (77).
In silico analysis of STAT6 binding sites.
A search of putative STAT6 binding sites in the murine PlGF locus was performed using MatInspector software (Genomatix); a stringent cut-off allowing a single mismatch was applied. The 2011 mouse genome version GRCm38/mm10 Assembly (www.genome.ucsc.edu; University of California Santa Cruz, Santa Cruz, California, USA) was used for the analysis.
Statistics.
The software GraphPad Prism was used for all statistical analyses. Statistical tests used include 1-way Dunnet’s ANOVA, Sidak 2-way ANOVA, unpaired t test, and Mann-Whitney U test wherever applicable. Data are presented as mean ± SEM and P < 0.05 was considered significant.
Study approval.
Animal protocols were approved by the Institutional Animal Care and Use Committee at CCHMC (IACUC Protocol numbers: 2E07051; 2D07050). For the human studies, patients were recruited after appropriate Institutional Review Board approval at CCHMC (IRB; protocol number 2008-1384). Written informed consent was obtained from all study participants or their guardians.
Author contributions
MSEMM, EBB, PIA, ST, AL, and DP conducted the experiments; MSEMM, PM, EBB, and GKKH designed research studies, MSEMM coordinated all research experiments; MSEMM and EBB plotted and analyzed the data; SKS performed the histopathological staining and interpretation; TR performed the IHC; ML performed the microarray and bioinformatics analysis; BJ provided α-PlGF Ab; PC generated and provided the placenta growth factor–deficient mice; VKK, TDLC, NR, MWK, GKKH, and PM provided intellectual input into the conduct and analysis of the experiments; MSEMM, EBB, GKKH, and PM analyzed and interpreted the data; PM conceived of the project; MSEMM and PM wrote the manuscript; and all authors read and edited the manuscript.
Supplementary Material
Acknowledgments
We would like to thank Katie Burke and Lanxi Song for their technical expertise and help with mouse experiments; Krista Dienger and Alyssa Sproles for their help with APTI experiments; Umasundari Sivaprasad for developing the Muc5AC ELISA in BALF samples; Anil Jegga for his help with the in silico promoter analysis; and Nihal Bakeer for the differential counts. This work was funded by RO1 HL079916 to P. Malik and RO1 HL111372 to P. Malik and V.K. Kalra.
Footnotes
Conflict of interest: B. Jonck is an employee and P. Carmeliet is on the board of ThromboGenics NV. ThromboGenics NV provided the anti-PlGF Ab.
Reference information:J Clin Invest. 2016;126(2):571–584. doi:10.1172/JCI77250.
References
- 1.Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956;178(4537):792–794. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 2.Madigan C, Malik P. Pathophysiology and therapy for haemoglobinopathies; Part I: sickle cell disease. Expert Rev Mol Med. 2006;8(9):1–23. doi: 10.1017/S1462399406010659. [DOI] [PubMed] [Google Scholar]
- 3.Platt OS, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639–1644. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 4.Boyd JH, Macklin EA, Strunk RC, DeBaun MR. Asthma is associated with Increased mortality in individuals with sickle cell anemia. Haematologica. 2007;92(8):1115–1118. doi: 10.3324/haematol.11213. [DOI] [PubMed] [Google Scholar]
- 5.Ozbek OY, Malbora B, Sen N, Yazici AC, Ozyurek E, Ozbek N. Airway hyperreactivity detected by methacholine challenge in children with sickle cell disease. Pediatr Pulmonol. 2007;42(12):1187–1192. doi: 10.1002/ppul.20716. [DOI] [PubMed] [Google Scholar]
- 6.Leong MA, Dampier C, Varlotta L, Allen JL. Airway hyperreactivity in children with sickle cell disease. J Pediatr. 1997;131(2):278–283. doi: 10.1016/s0022-3476(97)70166-5. [DOI] [PubMed] [Google Scholar]
- 7.Field JJ, et al. Airway hyperresponsiveness in children with sickle cell anemia. Chest. 2011;139(3):563–568. doi: 10.1378/chest.10-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vendramini EC, Vianna EO, De Lucena Ângulo I, De Castro FB, Martinez JAB, Terra-Filho J. Lung function and airway hyperresponsiveness in adult patients with sickle cell disease. Am J Med Sci. 2006;332(2):68–72. doi: 10.1097/00000441-200608000-00003. [DOI] [PubMed] [Google Scholar]
- 9.Sen N, Kozanoglu I, Karatasli M, Ermis H, Boga C, Eyuboglu F. Pulmonary function and airway hyperresponsiveness in adults with sickle cell disease. Lung. 2009;187(3):195–200. doi: 10.1007/s00408-009-9141-y. [DOI] [PubMed] [Google Scholar]
- 10.Knight-Madden JM, Forrester TS, Lewis NA, Greenough A. Asthma in children with sickle cell disease and its association with acute chest syndrome. Thorax. 2005;60(3):206–210. doi: 10.1136/thx.2004.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koumbourlis AC, Zar HJ, Hurlet-Jensen A, Goldberg MR. Prevalence and reversibility of lower airway obstruction in children with sickle cell disease. J Pediatr. 2001;138(2):188–192. doi: 10.1067/mpd.2001.111824. [DOI] [PubMed] [Google Scholar]
- 12.Sylvester KP, Patey RA, Rafferty GF, Rees D, Thein SL, Greenough A. Airway hyperresponsiveness and acute chest syndrome in children with sickle cell anemia. Pediatr Pulmonol. 2007;42(3):272–276. doi: 10.1002/ppul.20571. [DOI] [PubMed] [Google Scholar]
- 13.Boyd JH, Macklin EA, Strunk RC, DeBaun MR. Asthma is associated with acute chest syndrome and pain in children with sickle cell anemia. Blood. 2006;108(9):2923–2927. doi: 10.1182/blood-2006-01-011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyd JH, DeBaun MR, Morgan WJ, Mao J, Strunk RC. Lower airway obstruction is associated with increased morbidity in children with sickle cell disease. Pediatr Pulmonol. 2009;44(3):290–296. doi: 10.1002/ppul.20998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cohen RT, Madadi A, Blinder MA, DeBaun MR, Strunk RC, Field JJ. Recurrent, severe wheezing is associated with morbidity and mortality in adults with sickle cell disease. Am J Hematol. 2011;86(9):756–761. doi: 10.1002/ajh.22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glassberg JA, Chow A, Wisnivesky J, Hoffman R, DeBaun MR, Richardson LD. Wheezing and asthma are independent risk factors for increased sickle cell disease morbidity. Br J Haematol. 2012;159(4):472–479. doi: 10.1111/bjh.12049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wills-Karp M, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282(5397):2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 18.Vargaftig BB, Singer M. Leukotrienes mediate murine bronchopulmonary hyperreactivity, inflammation, and part of mucosal metaplasia and tissue injury induced by recombinant murine interleukin-13. Am J Respir Cell Mol Biol. 2003;28(4):410–419. doi: 10.1165/rcmb.2002-0032OC. [DOI] [PubMed] [Google Scholar]
- 19.Zhu Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103(6):779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Setty BNY, Stuart MJ. Eicosanoids in sickle cell disease: Potential relevance of neutrophil leukotriene B4 to disease pathophysiology. J Lab Clin Med. 2002;139(2):80–89. doi: 10.1067/mlc.2002.121200. [DOI] [PubMed] [Google Scholar]
- 21.Jennings JE, et al. Elevated urinary leukotriene E4 levels are associated with hospitalization for pain in children with sickle cell disease. Am J Hematol. 2008;83(8):640–643. doi: 10.1002/ajh.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monteiro APT, et al. Leukotriene B4 mediates neutrophil migration induced by heme. J Immunol. 2011;186(11):6562–6567. doi: 10.4049/jimmunol.1002400. [DOI] [PubMed] [Google Scholar]
- 23.Holgate ST, Peters-Golden M, Panettieri RA, Henderson WR., Jr Roles of cysteinyl leukotrienes in airway inflammation, smooth muscle function, and remodeling. J Allergy Clin Immunol. 2003;111(1 suppl):S18–S36. doi: 10.1067/mai.2003.25. [DOI] [PubMed] [Google Scholar]
- 24.Knorr B, et al. Montelukast, a leukotriene receptor antagonist, for the treatment of persistent asthma in children aged 2 to 5 years. Pediatrics. 2001;108(3): doi: 10.1542/peds.108.3.e48. [DOI] [PubMed] [Google Scholar]
- 25.Reiss TF, et al. Montelukast, a once-daily leukotriene receptor antagonist, in the treatment of chronic asthma: A multicenter, randomized, double-blind trial. Arch Intern Med. 1998;158(11):1213–1220. doi: 10.1001/archinte.158.11.1213. [DOI] [PubMed] [Google Scholar]
- 26.Perelman N, et al. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood. 2003;102(4):1506–1514. doi: 10.1182/blood-2002-11-3422. [DOI] [PubMed] [Google Scholar]
- 27.Sundaram N, et al. High levels of placenta growth factor in sickle cell disease promote pulmonary hypertension. Blood. 2010;116(1):109–112. doi: 10.1182/blood-2009-09-244830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pászty C, et al. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278(5339):876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 29.Patel N, Sundaram N, Yang M, Madigan C, Kalra VK, Malik P. Placenta growth factor (PlGF), a novel inducer of plasminogen activator inhibitor-1 (PAI-1) in sickle cell disease (SCD) J Biol Chem. 2010;285(22):16713–16722. doi: 10.1074/jbc.M110.101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lajoie S, et al. Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol. 2010;11(10):928–935. doi: 10.1038/ni.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGovern TK, Robichaud A, Fereydoonzad L, Schuessler TF, Martin JG. Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J Vis Exp. 2013;(75): doi: 10.3791/50172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hou HH, Cheng SL, Liu HT, Yang FZ, Wang HC, Yu CJ. Elastase induced lung epithelial cell apoptosis and emphysema through placenta growth factor. Cell Death Dis. 2013;4: doi: 10.1038/cddis.2013.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janér J, Andersson S, Haglund C, Karikoski R, Lassus P. Placental growth factor and vascular endothelial growth factor receptor-2 in human lung development. Pediatrics. 2008;122(2):340–346. doi: 10.1542/peds.2007-1941. [DOI] [PubMed] [Google Scholar]
- 34.DiPalma T, et al. The placenta growth factor gene of the mouse. Mamm Genome. 1996;7(1):6–12. doi: 10.1007/s003359900003. [DOI] [PubMed] [Google Scholar]
- 35.Tsao PN, et al. Overexpression of placenta growth factor contributes to the pathogenesis of pulmonary emphysema. Am J Respir Crit Care Med. 2004;169(4):505–511. doi: 10.1164/rccm.200306-774OC. [DOI] [PubMed] [Google Scholar]
- 36.Patel N, Gonsalves CS, Yang M, Malik P, Kalra VK. Placenta growth factor induces 5-lipoxygenase–activating protein to increase leukotriene formation in sickle cell disease. Blood. 2009;113(5):1129–1138. doi: 10.1182/blood-2008-07-169821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patel N, Kalra VK. Placenta growth factor-induced early growth response 1 (Egr-1) regulates hypoxia-inducible factor-1alpha (HIF-1α) in endothelial cells. J Biol Chem. 2010;285(27):20570–20579. doi: 10.1074/jbc.M110.119495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laitinen LA, Haahtela T, Vilkka V, Lee TH, Spur BW. Leukotriene E4 and granulocytic infiltration into asthmatic airways. Lancet. 1993;341(8851):989–990. doi: 10.1016/0140-6736(93)91073-U. [DOI] [PubMed] [Google Scholar]
- 39.Gauvreau GM, Parameswaran KN, Watson RM, O’Byrne PM. Inhaled leukotriene E(4), but not leukotriene D(4), increased airway inflammatory cells in subjects with atopic asthma. Am J Respir Crit Care Med. 2001;164(8):1495–1500. doi: 10.1164/ajrccm.164.8.2102033. [DOI] [PubMed] [Google Scholar]
- 40.Nabe T, Yamashita K, Miura M, Kawai T, Kohno S. Cysteinyl leukotriene-dependent interleukin-5 production leading to eosinophilia during late asthmatic response in guinea-pigs. Clin Exp Allergy. 2002;32(4):633–640. doi: 10.1046/j.0954-7894.2002.01329.x. [DOI] [PubMed] [Google Scholar]
- 41.Guajardo JR, et al. Altered gene expression profiles in nasal respiratory epithelium reflect stable versus acute childhood asthma. J Allergy Clin Immunol. 2005;115(2):243–251. doi: 10.1016/j.jaci.2004.10.032. [DOI] [PubMed] [Google Scholar]
- 42.Patel N, Gonsalves CS, Malik P, Kalra VK. Placenta growth factor augments endothelin-1 and endothelin-B receptor expression via hypoxia-inducible factor-1α. Blood. 2008;112(3):856–865. doi: 10.1182/blood-2007-12-130567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thivierge M, Staňková J, Rola-Pleszczynski M. IL-13 and IL-4 up-regulate cysteinyl leukotriene 1 receptor expression in human monocytes and macrophages. J Immunol. 2001;167(5):2855–2860. doi: 10.4049/jimmunol.167.5.2855. [DOI] [PubMed] [Google Scholar]
- 44.Chibana K, Ishii Y, Asakura T, Fukuda T. Up-regulation of cysteinyl leukotriene 1 receptor by IL-13 enables human lung fibroblasts to respond to leukotriene C4 and produce eotaxin. J Immunol. 2003;170(8):4290–4295. doi: 10.4049/jimmunol.170.8.4290. [DOI] [PubMed] [Google Scholar]
- 45.Jackson SE, Holloway JW, Warner JA, Sampson AP. Interleukin-13, but not indomethacin, increases cysteinyl-leukotriene synthesis in human lung macrophages. J Allergy (Cairo) 2012;2012: doi: 10.1155/2012/348741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doherty TA, Khorram N, Lund S, Mehta AK, Croft M, Broide DH. Lung type 2 innate lymphoid cells express cysteinyl leukotriene receptor 1, which regulates TH2 cytokine production. J Allergy Clin Immunol. 2013;132(1):205–213. doi: 10.1016/j.jaci.2013.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tordjman R, et al. Erythroblasts are a source of angiogenic factors. Blood. 2001;97(7):1968–1974. doi: 10.1182/blood.V97.7.1968. [DOI] [PubMed] [Google Scholar]
- 48.Selvaraj SK, Giri RK, Perelman N, Johnson C, Malik P, Kalra VK. Mechanism of monocyte activation and expression of proinflammatory cytochemokines by placenta growth factor. Blood. 2003;102(4):1515–1524. doi: 10.1182/blood-2002-11-3423. [DOI] [PubMed] [Google Scholar]
- 49.Wills-Karp M, Ewart SL. The genetics of allergen-induced airway hyperresponsiveness in mice. Am J Respir Crit Care Med. 1997;156(4):S89–S96. doi: 10.1164/ajrccm.156.4.12-tac-3. [DOI] [PubMed] [Google Scholar]
- 50.Ewart SL, et al. Quantitative trait loci controlling allergen-induced airway hyperresponsiveness in inbred mice. Am J Respir Cell Mol Biol. 2000;23(4):537–545. doi: 10.1165/ajrcmb.23.4.4199. [DOI] [PubMed] [Google Scholar]
- 51.Manci EA, Hillery CA, Bodian CA, Zhang ZG, Lutty GA, Coller BS. Pathology of Berkeley sickle cell mice: similarities and differences with human sickle cell disease. Blood. 2006;107(4):1651–1658. doi: 10.1182/blood-2005-07-2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bobic S, et al. Placental growth factor contributes to bronchial neutrophilic inflammation and edema in allergic asthma. Am J Respir Cell Mol Biol. 2012;46(6):781–789. doi: 10.1165/rcmb.2011-0152OC. [DOI] [PubMed] [Google Scholar]
- 53.Pritchard KA, et al. Effects of experimental asthma on inflammation and lung mechanics in sickle cell mice. Am J Respir Cell Mol Biol. 2012;46(3):389–396. doi: 10.1165/rcmb.2011-0097OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKinley L, Kim J, Bolgos G, Siddiqui J, Remick D. Allergens induce enhanced bronchoconstriction and leukotriene production in C5 deficient mice. Respir Res. 2006;7: doi: 10.1186/1465-9921-7-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Irvin CG, Tu YP, Sheller JR, Funk CD. 5-Lipoxygenase products are necessary for ovalbumin-induced airway responsiveness in mice. Am J Physiol. 1997;272(6 pt 1):L1053–L1058. doi: 10.1152/ajplung.1997.272.6.L1053. [DOI] [PubMed] [Google Scholar]
- 56.Peebles RS, et al. Allergen-induced airway hyperresponsiveness mediated by cyclooxygenase inhibition is not dependent on 5-lipoxygenase or IL-5, but is IL-13 dependent. J Immunol. 2005;175(12):8253–8259. doi: 10.4049/jimmunol.175.12.8253. [DOI] [PubMed] [Google Scholar]
- 57.Shin K, Hwang JJ, Kwon BI, Kheradmand F, Corry DB, Lee SH. Leukotriene enhanced allergic lung inflammation through induction of chemokine production. Clin Exp Med. 2015;15(3):233–244. doi: 10.1007/s10238-014-0292-7. [DOI] [PubMed] [Google Scholar]
- 58.Lewis RA, Austen KF, Soberman RJ. Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. N Engl J Med. 1990;323(10):645–655. doi: 10.1056/NEJM199009063231006. [DOI] [PubMed] [Google Scholar]
- 59.Bellik L, Vinci MC, Filippi S, Ledda F, Parenti A. Intracellular pathways triggered by the selective FLT-1-agonist placental growth factor in vascular smooth muscle cells exposed to hypoxia. Br J Pharmacol. 2005;146(4):568–575. doi: 10.1038/sj.bjp.0706347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pope SM, Zimmermann N, Stringer KF, Karow ML, Rothenberg ME. The eotaxin chemokines and CCR3 are fundamental regulators of allergen-induced pulmonary eosinophilia. J Immunol. 2005;175(8):5341–5350. doi: 10.4049/jimmunol.175.8.5341. [DOI] [PubMed] [Google Scholar]
- 61.Hogan SP, Matthaei KI, Young JM, Koskinen A, Young IG, Foster PS. A novel T cell-regulated mechanism modulating allergen-induced airways hyperreactivity in BALB/c mice independently of IL-4 and IL-5. J Immunol. 1998;161(3):1501–1509. [PubMed] [Google Scholar]
- 62.Flood-Page PT, Menzies-Gow AN, Kay AB, Robinson DS. Eosinophil’s role remains uncertain as anti-interleukin-5 only partially depletes numbers in asthmatic airway. Am J Respir Crit Care Med. 2003;167(2):199–204. doi: 10.1164/rccm.200208-789OC. [DOI] [PubMed] [Google Scholar]
- 63.Eum S-Y, Maghni K, Tolloczko B, Eidelman DH, Martin JG. IL-13 may mediate allergen-induced hyperresponsiveness independently of IL-5 or eotaxin by effects on airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2005;288(3):L576–L584. doi: 10.1152/ajplung.00380.2003. [DOI] [PubMed] [Google Scholar]
- 64.Kuperman DA, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8(8):885–889. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 65.Fischer C, et al. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131(3):463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 66.Li B, et al. Elevated PLGF contributes to small-cell lung cancer brain metastasis. Oncogene. 2013;32(24):2952–2962. doi: 10.1038/onc.2012.313. [DOI] [PubMed] [Google Scholar]
- 67.Cheng S-L, Wang H-C, Yu C-J, Yang P-C. Increased expression of placenta growth factor in COPD. Thorax. 2008;63(6):500–506. doi: 10.1136/thx.2007.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Belcher JD, et al. Transgenic sickle mice have vascular inflammation. Blood. 2003;101(10):3953–3959. doi: 10.1182/blood-2002-10-3313. [DOI] [PubMed] [Google Scholar]
- 69.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11(2):129–151. [PubMed] [Google Scholar]
- 70.Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. J Clin Invest. 2000;106(3):411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kaul DK, Liu X-d, Choong S, Belcher JD, Vercellotti GM, Hebbel RP. Anti-inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2004;287(1):H293–H301. doi: 10.1152/ajpheart.01150.2003. [DOI] [PubMed] [Google Scholar]
- 72.Vichinsky EP, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med. 2000;342(25):1855–1865. doi: 10.1056/NEJM200006223422502. [DOI] [PubMed] [Google Scholar]
- 73.Knight-Madden JM, Barton-Gooden A, Weaver SR, Reid M, Greenough A. Mortality, asthma, smoking and acute chest syndrome in young adults with sickle cell disease. Lung. 2013;191(1):95–100. doi: 10.1007/s00408-012-9435-3. [DOI] [PubMed] [Google Scholar]
- 74.Koumbourlis AC, Hurlet-Jensen A, Bye MR. Lung function in infants with sickle cell disease. Pediatr Pulmonol. 1997;24(4):277–281. doi: 10.1002/(SICI)1099-0496(199710)24:4<277::AID-PPUL6>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 75.Koumbourlis AC, Lee DJ, Lee A. Longitudinal changes in lung function and somatic growth in children with sickle cell disease. Pediatr Pulmonol. 2007;42(6):483–488. doi: 10.1002/ppul.20601. [DOI] [PubMed] [Google Scholar]
- 76.Kramer EL, et al. Early growth response-1 suppresses epidermal growth factor receptor-mediated airway hyperresponsiveness and lung remodeling in mice. Am J Respir Cell Mol Biol. 2009;41(4):415–425. doi: 10.1165/rcmb.2008-0470OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin LJ, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One. 2012;7(4): doi: 10.1371/journal.pone.0033454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.