

Abstract
Amyloid β (Aβ) damages neurons and triggers microglial inflammatory activation in the Alzheimer disease (AD) brain. BACE1 is the primary enzyme in Aβ generation. Neuroinflammation potentially up-regulates BACE1 expression and increases Aβ production. In Alzheimer amyloid precursor protein-transgenic mice and SH-SY5Y cell models, we specifically knocked out or knocked down gene expression of mapk14, which encodes p38α MAPK, a kinase sensitive to inflammatory and oxidative stimuli. Using immunological and biochemical methods, we observed that reduction of p38α MAPK expression facilitated the lysosomal degradation of BACE1, decreased BACE1 protein and activity, and subsequently attenuated Aβ generation in the AD mouse brain. Inhibition of p38α MAPK also enhanced autophagy. Blocking autophagy by treating cells with 3-methyladenine or overexpressing dominant-negative ATG5 abolished the deficiency of the p38α MAPK-induced BACE1 protein reduction in cultured cells. Thus, our study demonstrates that p38α MAPK plays a critical role in the regulation of BACE1 degradation and Aβ generation in AD pathogenesis.
Keywords: Alzheimer disease, amyloid-β (Aβ), autophagy, β-secretase 1 (BACE1), lysosome, p38 MAPK
Introduction
Alzheimer disease (AD)2 is pathologically characterized by the extracellular deposits of amyloid β peptide (Aβ). Aβ injures neurons in the neocortex and limbic system directly (1) and indirectly by triggering microglial release of various neurotoxic inflammatory mediators, including cytokines (tumor necrosis factor-α and interleukin-1β (IL-1β)) and reactive oxygen species (2). Aβ is generated after serial digestion of Alzheimer amyloid precursor protein (APP) by the membrane-anchored β-site APP-cleaving enzyme (BACE1, β-secretase) and γ-secretase (3). It has been observed that knock-out of BACE1 or administration of the BACE1 inhibitor dramatically decreases Aβ levels in the brain and attenuates behavioral and electrophysiological deficits in APP-transgenic mice (4–6). Thus, extensive investigations have focused on the direct inhibition of BACE1 to reduce Aβ load in the AD brain; however, these studies have unfortunately not yet led to any efficacious therapy for AD patients due to the various physiological roles of BACE1 (7). Using alternative methods to inhibit BACE1 might be a preferable investigative approach.
Inflammatory activation might lead to up-regulation of neuronal BACE1 expression in the AD brain, as NF-κB signaling enhances (8), and PPARγ activation suppresses (9), the activity of bace1 gene promoter. Accumulating evidence has shown that posttranslational modification of BACE1 is extremely important for the activity, intracellular trafficking, and lysosomal degradation of BACE1. For example, phosphorylation of BACE1 at Thr-252 by p25/Cdk5 increases the secretase activity (10), and phosphorylation at Ser-498 facilitates retrograde transport of BACE1 from endosomes to the trans-Golgi network (11). Ubiquitination at Lys-501 targets BACE1 to late endosomes/lysosomes for degradation (12). Finally, bisecting N-acetylglucosamine modification blocks delivery of BACE1 to lysosomes (13).
p38 mitogen-activated protein kinases (p38 MAPKs) are a class of mitogen-activated protein kinases that are responsive to stress stimuli, such as inflammatory cytokines and reactive oxygen species. Phosphorylation of p38 MAPK has been observed in the postmortem brain in the early stages of AD (Braak stages IV–V) (14, 15). Aβ and glutamate have each been shown to activate p38 MAPK in cultured neurons by increasing reactive oxygen species (16, 17), and the Aβ-triggered microglial release of inflammatory mediators, especially IL-1β, is hypothesized to activate neuronal p38 MAPK (18–20). p38α MAPK mediates Aβ-initiated microglial inflammatory activation (21, 22), phosphorylates Tau protein in neurons (20, 23, 24), and mediates Aβ-induced synaptic impairment in the cultured hippocampal slice (25). However, no experiments have yet elucidated whether p38 MAPK phosphorylates BACE1 and regulates Aβ generation.
Previous studies demonstrating the effects of p38 MAPK in hippocampal slices or APP-transgenic mice used p38 MAPK inhibitors, which cannot distinguish neuronal p38 MAPK effects and microglial p38 MAPK effects (22, 25). In attempting to dissect the role of p38 MAPK in AD, it is essential to distinguish between p38α and p38β MAPK enzymes, as they have different functions (26). To investigate the pathogenic function of neuronal p38α MAPK, we used Cre-Lox or knockdown techniques to ablate p38α MAPK specifically in neurons and examined the effect on BACE1 degradation in this study.
Experimental Procedures
Animal Models and Cross-breeding
APP/PS1 double transgenic mice over-expressing human mutated APP (KM670/671NL) and PS1 (L166P) under Thy-1 promoters (27) were kindly provided by M. Jucker, Hertie Institute for Clinical Brain Research, Tübingen, Germany; p38fl/fl mice with loxP site-flanked mapk14 gene were imported from BioResource Center, RIKEN Tsukuba Institute, Japan (28); and Nex-Cre mice expressing Cre recombinase from the endogenous locus of the nex gene that encodes a neuronal basic helix-loop-helix (bHLH) protein were kindly provided by K. Nave, Max-Planck-Institute for Medicine, Göttingen, Germany (29). All three mouse strains were on a C57BL6 genetic background. APP-transgenic mouse models with deletion of p38α MAPK specifically in neurons of the neocortex and hippocampus had been established by mating APP/PS1, p38fl/fl, and Nex-Cre mice. Animal experiments were performed in accordance with all relevant national rules and were authorized by the local research ethical committee.
Tissue Collection for Histological and Biochemical Analysis
Animals were euthanized at 4 months of age by isofluorane inhalation. Mice were perfused with ice-cold PBS, and the brain was removed and divided. The left hemisphere was for immunohistochemistry. A 0.5-μm thick piece of tissue was sagittally cut from the right hemisphere. The cortex and hippocampus were carefully separated and homogenized in TRIzol (Life Technologies) for RNA isolation. The remainder of the right hemisphere was snap frozen in liquid nitrogen and stored at −80 °C until biochemical analysis.
Immunohistological Analysis
For Aβ analysis, the left hemibrains derived from 4-month-old APP-transgenic mice with and without deficiency of p38α MAPK were immediately fixed in 4% paraformaldehyde (Sigma) in PBS for 48 h. The tissue was embedded in paraffin. Serial 50-μm thick sagittal sections were cut and mounted on glass slides. Immunofluorescent staining with the primary antibody, mouse monoclonal anti-human Aβ antibody (clone 6F/3D; Dako Deutschland GmbH, Hamburg, Germany), was performed on these sections. The staining was visualized by incubating sections with Cy3-conjugated goat anti-mouse IgG (Jackson ImmunoResearch Europe Ltd., Suffolk, United Kingdom). All images were acquired by Zeiss AxioImager.Z2 microscope equipped with a Stereo Investigator system (MBF Bioscience, Williston). In the whole hippocampus and cortex, volumes of Aβ and brain tissues were estimated with the Cavalieri probe as we described in the previous study (30) with a 15-μm grid size, which provided coefficient of error estimates of <0.05. The Aβ load was demonstrated as the ratio of Aβ volume to relevant brain tissue volume. To demonstrate Nex-Cre-mediated deletion of p38 MAPK in neurons, the isolated brain was directly embedded in Tissue-Tek® O.C.T. Compound (Sakura Finetek Europe B.V., AJ Alphen aan den Rijn, the Netherlands) and frozen in 2-methylbutane on liquid nitrogen. The brain was cut on a freezing-sliding microtome in 5-μm coronal sections. For immunofluorescent staining, the rabbit polyclonal antibody against p38 MAPK (catalog number 9212, Cell Signaling Technology, Danvers) and Alexa 488-conjugated goat anti-rabbit IgG (Life Technologies) were used. Neurons were identified by staining the tissue with mouse monoclonal antibody against NeuN (clone A60; Merck Chemicals GmbH, Darmstadt, Germany) and Cy3-conjugated goat anti-mouse IgG (Jackson ImmunoResearch). The experimenter was blinded to the genotypes of mice.
Construction of Knockdown and Transgenic Vectors
Two pcDNA6.2-GW/EmGFP-miR vectors (Life Technologies) (kd509 and kd709) were engineered to contain different select hairpins targeting human p38α MAPK-encoding gene, mapk14, using the protocol we established in the previous study (31). Sequences of the DNA oligomers are listed in Table 1. After transfection, the vector will transcribe artificial miRNA, which have 100% homology to the gene sequence of interest, resulting in target RNA cleavage. pcDNA6.2-GW/EmGFP-miR-neg control plasmid (Life Technologies) (kd-ct) containing scrambled sequence was used as a control knockdown vector. pEGFP-hAPP695 transgenic vector was constructed by replacing the eGFP-encoded sequence in pEGFP-N1 vector (Takara Bio Europe/Clontech, Saint-Germain-en-Laye, France) with the PCR products between HindIII and NotI. The PCR was performed using genomic DNA derived from TgCRND8 APP-transgenic mice (32) as the templates and the following primers (Life Technologies) containing HindIII or NotI restriction sites (underlined): 5′-GGGAAGCTTCCCACCATGCTGCCCGGTTTGG-3′ and 5′-ATAAGAATGCGGCCGCTACTAGTTCTGCATCTGC-3′.
TABLE 1.
Sequences of DNA oligomers inserted into pcDNA6.2-GW/EmGFP-miR to construct knockdown vectors
| Oligomers | Sequences (from 5′ to 3′) | |
|---|---|---|
| kd509 | Top | TGCTGATGAATGATGGACTGAAATGGGTTTTGGCCACTGACTGACCCATTTCACCATCATTCAT |
| Bottom | CCTGATGAATGATGGTGAAATGGGTCAGTCAGTGGCCAAAACCCATTTCAGTCCATCATTCATC | |
| kd709 | Top | TGCTGATAAGGAACTGAACATGGTCAGTTTTGGCCACTGACTGACTGACCATGCAGTTCCTTAT |
| Bottom | CCTGATAAGGAACTGCATGGTCAGTCAGTCAGTGGCCAAAACTGACCATGTTCAGTTCCTTATC | |
Cell Culture and Establishment of Cell Lines
SH-SY5Y neuroblastoma cells were obtained from LGC Standards GmbH (Wesel, Germany) and maintained in DMEM supplemented with 10% fetal calf serum (FCS; PAN Biotech, Aidenbach, Germany). Different cell lines were established: 1) SH-SY5Y cell lines with knockdown of p38α MAPK expression were established by transfecting cells with kd-ct, kd509, and kd709 vectors; 2) SH-SY5Y cell lines overexpressing wild-type and dominant-negative (DN) human ATG5 were created by transfecting cells with expression gift vectors from N. Mizushima (Addgene plasmids numbers 22948 and 22949) (33). To evaluate the different autophagic activity, these two different cells were treated with 0.2 μg/ml of rapamycin (Sigma) for 24 h; 3) SH-SY5Y cells overexpressing ATG5 (wild-type and DN) were further transfected with kd-ct and kd709 vectors; 4) to investigate the effects of p38α MAPK on Aβ secretion, we transfected SH-SY5Y cells first with pEGFP-hAPP685 and thereafter with p38α MAPK knock-down vectors (kd-ct, kd509, and kd709); 5) to investigate the effects of p38 MAPK on autophagy, the LC3-GFP-RFP-transgenic autophagy reporter cell line was established by transfecting SH-SY5Y cells with a gift vector from T. Yoshimori (Addgene plasmid number 21074) (34). All transfected cells were selected with blasticidin (Life Technologies) and/or G418 (Sigma) until the stable transfection was established. All genetic modifications were confirmed by Western blot detection of their encoding proteins using specific antibodies.
Western Blot Analysis of Aβ Pathology
For detection of Aβ, C99, and APP in the mouse brain, frozen brain tissues derived from p38α MAPK-deficient and wild-type mice were homogenized in 5 ml/g of tissue in radioimmunoprecipitation assay buffer (RIPA; 50 mm Tris, 150 mm NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% Nonidet P-40, and 5 mm EDTA (pH 8.0)) supplemented with protease inhibitor mixture (Roche Applied Science, Mannheim, Germany) on ice and centrifuged at 16,100 × g for 30 min at 4 °C to collect the supernatants. The protein concentration in the supernatant was measured using a Bio-Rad Protein Assay (Bio-Rad). To evaluate the effects of p38α MAPK on Aβ secretion, the FCS-contained culture medium of APP695-transgenic SH-SY5Y cells was replaced with serum-free medium for 16 h. The proteins in the brain homogenate or in the cell culture medium were separated by 10–20% pre-casted Tris-Tricine gels (Anamed Elektrophorese GmbH, Groß-Bieberau/Rodau, Germany). For Western blot, anti-human amyloid β mouse monoclonal antibody (clone W0–2; Merck Chemicals GmbH) and anti-β-actin rabbit monoclonal antibody (clone 13E5; Cell Signaling Technology) were used. Western blots were visualized via the Plus-ECL method (PerkinElmer Life Technologies). Densitometric analysis of band densities was performed with Image-Pro PLUS software version 6.0.0.260 (Media Cybernetics, Inc., Rockville, MD). For each sample, the level of protein in the brain was calculated as a ratio of target protein/β-actin from that sample. In cultured cells, the Aβ level was adjusted by the secreted APP protein level.
Western Blot Analysis of p38α MAPK, BACE1, and Autophagy
The frozen brain or cell pellets were homogenized in RIPA buffer. For the detection of phosphorylated p38α MAPK, the extra phosphatase inhibitors (5 mm NaF, 1 mm Na3VO4, 1 mm EGTA, 50 nm okadaic acid, 5 mm sodium pyrophosphate) and 1 mm DTT were added to the RIPA lysis buffer. The proteins were separated with 10 or 12% SDS-PAGE. Rabbit polyclonal antibodies against phosphorylated (Thr-180/Tyr-182) and total p38α MAPK (catalog numbers 9211 and 9212, respectively; Cell Signaling Technology), rabbit monoclonal antibodies against BACE1, LC3B, beclin1, ATG5, and lysosomal membrane-associated protein 1 (LAMP-1) (clone D10E5, D11, D40C5, D5F5U, and C54H11, respectively; Cell Signaling Technology), and rabbit polyclonal antibody against Gm2 activator protein (Gm2A) (Thermo Scientific, Darmstadt, Germany) were used as the primary antibodies in the Western blot. To confirm the results from Western blot with D10E5 antibody, mouse monoclonal antibody against human/mouse BACE-1 ectodomain (clone number 137612; R&D Systems Inc., Minneapolis, MN) was used. For all Western blot analyses described above, α-tubulin or β-actin were detected as a loading control with the DM1A antibody (Abcam, Cambridge, United Kingdom) and the 13E5 antibody (Cell Signaling Technology).
Measurement of the Half-life of BACE1
To measure the lifetime of BACE1, SH-SY5Y cells were treated with 100 μg/ml of cycloheximide (Sigma) to inhibit protein translation. The cell lysate was harvested at 0, 4, 8, and 12 h after drug treatment. The remaining BACE1 in the cell lysate was analyzed by SDS-PAGE and Western blotting using BACE1 antibody (clone D10E5) and α-tubulin antibody (clone DM1A). Kinetics of the disappearance of BACE1 in p38α MAPK knocked down and control cells were compared.
Preparation and Characterization of Lysosome
The procedure was processed as described (35, 36) with minor modifications. Cultured SH-SY5Y cells were harvested by trypsinization and washed with PBS. All subsequent manipulations were carried out at 4 °C using pre-cooled reagents. Washed cells were homogenized in the HB buffer (0.25 m sucrose, 10 mm Hepes, 1 mm EDTA, (pH 7.4)) supplemented with protease inhibitor mixture (Roche Applied Science) at 1 × 108/ml. Homogenates were spun at 800 × g for 10 min to pellet nuclei and unbroken cells, which were then re-homogenized in a half-volume of HB. The two supernatants were pooled, incubated for 10 min at 37 °C in the presence of 2 mm CaCl2, and then centrifuged at 3,000 × g for 10 min to remove large heavy mitochondria. The resultant supernatant was centrifuged for 10 min at 18,000 × g, obtaining a pellet that was re-suspended in 0.5 ml of HB buffer and layered on 4 ml of iso-osmotic Percoll (GE Healthcare, München, Germany) at a concentration of 30% (pH 7.4). Under Percoll, 0.5 ml of 2.5 m sucrose was laid. Centrifugation was performed at 4 °C for 40 min at 44,000 × g. The subsequent gradients were carefully collected with 0.9 ml/fraction into tubes from the top. Protein concentrations were determined and Western blot analysis was performed using antibodies against the lysosomal marker, LAMP-1 (clone C54H11), and non-lysosomal markers, calnexin (rabbit polyclonal to Calnexin; catalog number ab22595, Abcam) and β-actin (clone 13E5).
Preparation of Membrane Components
To measure β- and γ-secretase activity, membrane components were purified according to the published protocol (37). Briefly, brain tissues or SH-SY5Y cell pellets were transferred into sucrose buffer (10 mm Tris-HCl, pH 7.4, including 1 mm EDTA, and 200 mm sucrose) and homogenized on ice. The homogenate was centrifuged at 1,000 × g for 10 min at 4 °C to delete nuclei. The resulting postnuclear supernatant was transferred to a new tube and centrifuged again at 10,000 × g at 4 °C for 10 min. Finally, the resulting supernatant was centrifuged at 187,000 × g in an Optima MAX Ultracentrifuge (Beckman Coulter GmbH, Krefeld, Germany) for 75 min at 4 °C. The resulting supernatant was discarded, and pellets were re-suspended using cannulas (10 strokes per cannula) of decreasing diameter (0.6, 0.4, and 0.33 mm) in sucrose buffer.
β- and γ-Secretase Activity Assays
β- and γ-secretase activities were measured by incubating the crude membrane fraction with secretase-specific FRET substrates according to our established methods (37). For measurement of β-secretase activity, the crude membrane fraction was resuspended in 500 μl of β-secretase assay buffer (0.1 m sodium acetate, pH 4.5). The final concentrations for the β-secretase assay were: 0.1 mg/ml of membrane protein (125 μg of protein/well in 96-well plates), 10% dimethyl sulfoxide, and 8 μm β-secretase substrate IV (Calbiochem, Darmstadt, Germany). For measurement of γ-secretase activity, the crude membrane fraction was resuspended in 500 μl of γ-secretase assay buffer (50 mm Tris-HCl, pH 6.8, 2 mm EDTA). Final concentrations for the γ-secretase assay were: 1 mg/ml of membrane protein (1250 μg of protein/well in 96-well plates) and 8 μm γ-secretase substrate (Calbiochem). For both secretase assays, kinetics were performed at 37 °C and fluorescence intensity in each well was measured for 73 cycles with intervals of 5 min with Synergy Mx Monochromator-based Multi-mode Microplate Reader (BioTek, Winooski, VT). Fluorescence intensity of the first cycle was considered as background and subtracted for each well.
BACE Phosphorylation Assay
Human BACE1 sequence with UniProt accession number P56817 was used as the query sequence to predict potential p38 MAPK-mediated phosphorylation sites with the NetPhosK 1.0 Server. Thereafter, p38α MAPK knocked down (kd509 or kd709) and wild-type control (kd-ct) cells were lysed in RIPA buffer supplemented with sufficient proteinase and phosphatase inhibitors (50 mm Tris, pH 8.0, 2 mm EDTA, 2 mm EGTA, 50 nm okadaic acid, 5 mm sodium pyrophosphate, 2 mm sodium vanadate, 1 mm DTT, 50 mm NaF, 1% Triton X-100 and protease inhibitor mixture; Roche Applied Science). All operations were on ice and the cell lysate was frozen in liquid nitrogen and stored at −80 °C immediately if not followed by further experiments. The endogenous BACE1 in 200 μl of cell lysate (adjusted to 1 mg/ml) was pulled down with 10 μg of BACE1 antibody (clone D10E5, Cell Signaling Technology) and 50 μl of Dynabeads-conjugated protein G (Life Technologies) for 2 h at 4 °C. Samples were detected with Western blot serially with anti-phosphoserine rabbit polyclonal antibody (catalog number SAB5200086; Sigma) and anti-BACE1 antibody (clone D10E5). The amount of phosphorylated BACE1 was shown as the ratio of total BACE1 protein.
Reverse Transcription and Quantitative PCR for Analysis of Gene Transcripts
Total RNA was isolated from the brain homogenate in TRIzol or from cultured SH-SY5Y cells with RNEasy Plus Mini Kit (Qiagen, Hilden, Germany). First-strand cDNA was synthesized by priming total RNA with hexamer random primers (Life Technologies) and using Superscript III reverse transcriptase (Life Technologies). For quantification of gene transcripts, real-time PCR was performed with SYBR Green (Roche Applied Science) with the 7500 Fast Real-time PCR System (Life Technologies). The primer sequences used for detecting transcripts of mouse and human bace1, ps1, nicastrin, and gapdh are listed in Table 2. The amount of double-stranded PCR product synthesized in each cycle was measured by detecting SYBR Green, which binds to double-stranded DNA. Threshold cycle (Ct) values for each test gene from the replicate PCRs were normalized to the Ct values for gapdh control from the same cDNA preparations. The ratio of each gene transcript was calculated as 2(ΔCt), where ΔCt is the difference Ct (gapdh) − Ct (test gene).
TABLE 2.
Sequences of oligonucleotides used for the real-time quantitative PCR
| Gene | Sense | Anti-sense |
|---|---|---|
| Mouse bace1 | 5′-TTGTAGGGCTAGGGATGGTC-3′ | 5′-CCTAACCCTGCTGGATGAAT-3′ |
| Mouse ps1 | 5′-CAATGGTGTGGTTGGTGAAT-3′ | 5′-GTTCCCAGAACCACTGTCCT-3′ |
| Mouse nicastrin | 5′-ACCAGGTGGAGGATCTTCTG-3′ | 5′-AGGACAACTTCAGGGACACC-3′ |
| Mouse gapdh | 5′-ACAACTTTGGCATTGTGGAA-3′ | 5′-GATGCAGGGATGATGTTCTG-3′ |
| Human bace1 | 5′-TTGAAGCTGCAGTCAAATCC-3′ | 5′-CCAGAAACCATCAGGGAACT-3′ |
| Human ps1 | 5′-CCGAAAGGTCCACTTCGTAT-3′ | 5′-CCACACCATTGTTGAGGAGT-3′ |
| Human nicastrin | 5′-GGGTTCCTGATTAAAGCCAA-3′ | 5′-CGTCACCCAAGTAGGACCTT-3′ |
| Human gapdh | 5′-GAAGGACTCATGACCACAGT-3′ | 5′-GTCATCATATTTGGCAGGTT-3′ |
Confocal Analysis of Autophagic Activity and Colocalization between BACE1 and Autophagic Vacuoles
To investigate the effect of p38α MAPK on autophagy, LC3-GFP-RFP-transgenic SH-SY5Y cells cultured on coverslips in a 24-well plate (BD Bioscience, Heidelberg, Germany) at a density of 1 × 105 cells per well were treated with p38 MAPK inhibitor, SB203580, at 0, 10, and 30 μm for 2 h. The cells were then directly fixed with 4% paraformaldehyde and examined under a Zeiss LSM 510 Meta Confocal Microscope. More than 40 areas under a ×40 objective were randomly chosen and >300 cells were counted. The density of puncta with pure red or green (with week red) fluorescence in each cell was calculated as the total number of puncta divided by the total number of cells. The experiments were repeated independently 3 times. To investigate the relationship between BACE1 and autophagic vacuoles, SH-SY5Y cells were cultured in a 8-well chamber slide (BD Biosciences) at a density of 0.5 × 105 cells per well. The cells were treated with or without 200 nm bafilomycin (Sigma) for 6 h. After fixation in 4% paraformaldehyde at room temperature or in methanol at −20 °C, cells were incubated with 0.3% Triton to increase the permeability and blocked with 5% BSA. The cells were first stained with mouse monoclonal antibody (clone number 137612; R&D Systems) or rabbit polyclonal antibody (catalog number ab10716, Abcam) against BACE1 and Alexa 488-conjugated second antibodies (Life Technologies). After sufficient washing, cells were further incubated with rabbit monoclonal antibody against LC3A/B (Clone D3U4C, Cell Signaling Technology) or mouse monoclonal antibody against SQSTM1/p62 (catalog number ab56416, Abcam) and then Cy3-conjugated second antibodies. Whether BACE1 co-localized with autophagic vacuoles was analyzed under confocal microscopy.
Statistics
Data were presented as mean ± S.E. For multiple comparisons, we used one-way or two-way ANOVA followed by Bonferroni, Tukey, or Dunnett T3 post hoc tests (dependent on the result of Levene's test to determine the equality of variances). Two independent samples of t test were used to compare means for two groups of cases. All statistical analyses were performed with SPSS version 19.0 for Windows (IBM, New York, NY). Statistical significance was set at p < 0.05.
Results
Deletion of p38α MAPK in Neurons Reduces Aβ Load in the Alzheimer Mouse Model
To confirm that p38 MAPK is activated in the AD mouse brain, we compared the phosphorylation levels of p38α MAPK in 4-month-old APP/PS1-transgenic mice and their wild-type littermate controls and observed that phosphorylation levels were significantly higher in the APP/PS1-transgenic mice (Fig. 1, A and B; t test, p < 0.05). We then deleted p38α MAPK in neurons of the neocortex and hippocampus of AD mice (mating APP/PS1 mice, with p38fl/fl, and Nex-Cre mice; see “Experimental Procedures”). Western blot analysis showed that the protein levels of p38α MAPK in the homogenate of whole cortex and hippocampus were significantly decreased in 1-month-old APPtgp38fl/wtNex-Cre+/− (p38α heterozygote) mice compared with APPtgp38fl/wtNex-Cre−/− (p38α wild-type) littermates (Fig. 1, C and D; t test, p < 0.05). In a further experiment, we co-stained p38 MAPK and NeuN, a neuronal marker, in the brain of 4-month-old p38fl/flNex-Cre+/− (p38α homozygote knock-out) and p38fl/wt or p38fl/flNex-Cre−/− (p38α wild-type) littermate mice. In the cortex of p38α MAPK wild-type mice, p38 MAPK protein was stained and shown in puncta around the NeuN antibody-stained nucleus. By contrast, no p38 MAPK was observed in neurons of p38α homozygote knock-out mice (Fig. 1E). In the hippocampal neurons, the fluorescent staining of p38 MAPK was weak in both p38α MAPK-deficient and wild-type mice, which made the comparison of these two groups of mice difficult (data not shown).
FIGURE 1.
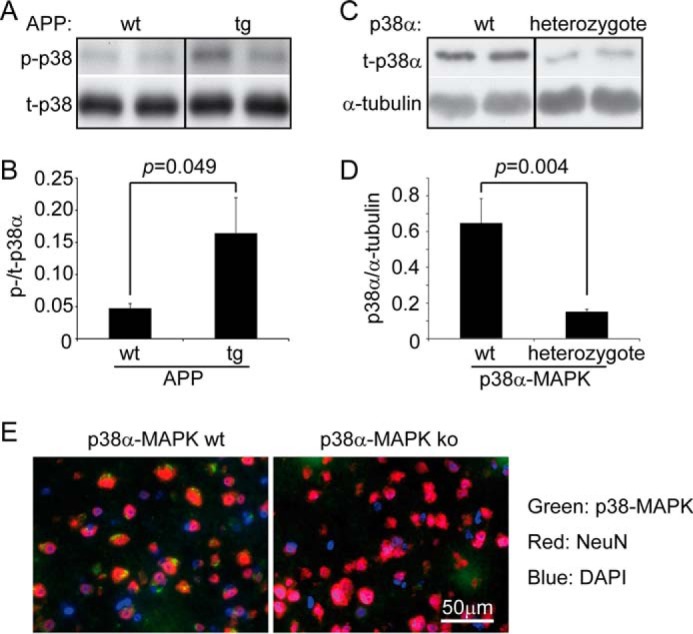
The cerebral p38α MAPK is efficiently ablated by cross-breeding p38fl/fl and Nex-Cre mice. The brain homogenates derived from 4-month-old APP-transgenic (tg) and wild-type (wt) littermates were analyzed for the phosphorylation of p38α MAPK with Western blot (A and B, t test, n ≥ 4 per group). Similarly, the cerebral p38α MAPK protein in 1-month-old APPtgp38fl/wtNex-Cre+/− (p38α MAPK heterozygote) and APPtgp38fl/wtNex-Cre−/− (p38α MAPK wild-type, wt) littermates was detected with Western blot (C and D, t test, n = 7 per group). A or C, grouping images from different parts of the same gel. E, 4-month-old p38fl/flNex-Cre+/− (p38α MAPK ko) and p38fl/wt or p38fl/flNex-Cre−/− (p38α MAPK wt) littermate mice were co-stained with fluorophore-conjugated antibodies against p38 MAPK and NeuN in the brain cortex. p38 MAPK protein was shown in green puncta around the NeuN staining in red, whereas, no p38 MAPK was stained in NeuN staining-positive neurons in p38α MAPK ko mice. Nuclei were stained with DAPI in blue.
Interestingly, the histological analysis using the stereological Cavalieri method (30) revealed that heterozygous deletion of p38α MAPK in neurons resulted in strongly reduced Aβ levels in both the hippocampus and cortex of APP-transgenic mice, even as early as 4 months of age (APPtgp38fl/wtNex-Cre+/− mice: 0.295 ± 0.036 and 0.565 ± 0.095 (adjusted by the volume of analyzed tissues) in hippocampus and cortex, respectively, and APPtgp38fl/wtNex-Cre−/− littermates: 0.504 ± 0.067 and 0.989 ± 0.188 in hippocampus and cortex, respectively; see Fig. 2, A and B; t test, p < 0.05). Similarly, Western blot analysis with human Aβ-specific antibody showed a 40% reduction with β-actin as an internal control, or a 25% reduction with APP as an internal control in the monomers, dimers, or trimers of Aβ in the homogenate of cortex and hippocampus derived from 4-month-old APPtgp38fl/wtNex-Cre+/− mice compared with APPtgp38fl/wtNex-Cre−/− littermate controls (Fig. 2, C–E; t test, p < 0.05).
FIGURE 2.

Deletion of neuronal p38α MAPK reduces cerebral Aβ load in APP-transgenic mice. Four-month-old APPtgp38fl/wtNex-Cre+/− (p38α heterozygote, ko) and APPtgp38fl/wtNex-Cre−/− (p38α wild-type, wt) littermate mice were analyzed for cerebral Aβ load after immunofluorescent staining with human Aβ-specific antibody (A). The Aβ volume adjusted by relevant brain volume as estimated with Cavalieri method is reduced in p38α MAPK-deficient APP mice (B; t test, n = 10 and 8 for p38 ko and wt groups, respectively). The cerebral Aβ in these APP-transgenic mice was also evaluated by detecting Aβ in the brain homogenate with quantitative Western blot (C–E, t test, n = 7 and 8 for p38 ko and wt groups, respectively). C, the figure is grouping images from different parts of the same gel.
Deletion of p38α MAPK in Neurons Reduces Both Activity and Protein Levels of BACE1 in the Mouse Brain
To investigate the underlying mechanism by which ablation of neuronal p38α MAPK reduces cerebral Aβ levels, we measured β- and γ-secretase activity in 4-month-old littermate mice with the genotypes of p38fl/flNex-Cre+/−, p38fl/wtNex-Cre+/−, and p38fl/wt or p38fl/flNex-Cre−/−, but without the expression of human APP. We observed that neuronal p38α MAPK deletion significantly reduced the activity of β-secretase, but not of γ-secretase, in a copy-dependent manner (Fig. 3, A and B; two-way ANOVA, p < 0.05). However, Bonferroni post hoc tests only showed significant differences between p38fl/flNex-Cre+/− (p38α homozygote knock-out) and p38fl/wt or p38fl/flNex-Cre−/− mice (p38α wild-type), not between p38fl/wtNex-Cre+/− (p38α heterozygote knock-out) and p38α MAPK wild-type mice. Interestingly, the β-secretase activity in the brain tissue of 4-month-old APPtgp38fl/wtNex-Cre+/− mice (p38α heterozygote knock-out) that express human APP was already lower than the β-secretase activity in the brain tissue of APPtgp38fl/wtNex-Cre−/− control mice (p38α wild-type) (Fig. 3C, two-way ANOVA, p < 0.001). Similarly, p38α MAPK deletion in neurons did not affect the γ-secretase activity in APP-transgenic mice (Fig. 3D). Moreover, we observed that brain homogenates derived from 4-month-old APPtgp38fl/wtNex-Cre+/− mice had lower amounts of APP-C99 fragment, a β-secretase product, compared with brain homogenates derived from APPtgp38fl/wtNex-Cre−/− littermates (Fig. 3E, t test, p < 0.05).
FIGURE 3.

Deletion of neuronal p38α MAPK reduces both β-secretase activity and protein amount in the mouse brain. Four-month-old APP-transgenic (APPtg, C–H) or non-transgenic (A and B), p38fl/flNex-Cre+/− (ko), p38fl/wtNex-Cre+/− (heterozygote), and p38fl/wt or p38fl/flNex-Cre−/− (wt) littermate mice were used to prepare membrane components, brain homogenates, and total RNA. β- and γ-secretase activity was measured by incubating membrane components with fluorogenic β- and γ-secretase substrates, respectively (A–D, two-way ANOVA followed by Bonferroni post hoc test between mouse groups with different genotypes; A and B, n ≥ 4 per group; C and D, n ≥ 10 per group). The protein levels of APP, APP-C99, and BACE1 in the APPtg mouse brain homogenate were determined by quantitative Western blots (E–G, n ≥ 4 per group). Moreover, the transcripts of bace1, nicastrin, and ps1 genes in the APPtg mouse brain were measured with real-time PCR, which showed that transcription of bace1 was up-regulated after deletion of p38α MAPK specifically in neurons (H, t test; n ≥ 4 per group). E or G, the figure is grouping images from different parts of the same gel.
We used Western blot analysis to examine β-secretase (BACE1) protein levels in the 4-month-old APP-transgenic mice. As shown in Fig. 3G, p38α MAPK deletion in neurons significantly decreased the amount of BACE1 protein in the brain of APP-transgenic mice compared with wild-type control mice (t test, p < 0.05). Surprisingly, real-time PCR showed that neuronal deletion of p38α MAPK expression up-regulated transcription of the bace1 gene, perhaps indicating compensation following BACE1 protein reduction (Fig. 3H, t test, p < 0.05). Transcription of γ-secretase-encoding genes (like ps1 and nicastrin) was not significantly altered (Fig. 3H). Thus, we hypothesized that deficiency of p38α MAPK in neurons decreases Aβ generation by suppressing β-secretase activity and reducing the levels of BACE1 protein.
In additional experiments, we quantified the protein levels of APP in APPtgp38fl/wtNex-Cre+/− and APPtgp38fl/wtNex-Cre−/− mice with Western blot. We observed no differences (see Fig. 3F, p > 0.05), thereby excluding the possibility that Aβ reduction in the brain observed in our study was due to the decreased protein levels of APP.
Knockdown of p38α MAPK Expression Reduces Both Activity and Protein Amount of β-Secretase in the Neuronal Cells
To confirm the finding that deletion of p38α MAPK in neurons specifically reduces β-secretase activity in APP-transgenic mice, we established both p38α MAPK knockdown and control neuronal cell lines by stably transfecting SH-SY5Y cells with artificial human mapk14 miRNA-transcribing vectors. Vectors kd509 and kd709 were targeted to the mRNA of mapk14 at different sites, and the kd-ct vector transcribed miRNA that was not complementary to any mammalian mRNA (see “Experimental Procedures”). Western blot detection of p38α MAPK protein demonstrated the efficiency of both vectors 509 and 709-induced knockdown of mapk14 gene expression with ∼50% reduction of p38α MAPK protein compared with vector kd-ct-transfected cells (Fig. 4A, one-way ANOVA, p < 0.001). Interestingly, reduction of p38α MAPK resulted in ∼40% reduction of BACE1 levels in the neuronal cell lines (Fig. 4B, one-way ANOVA, p < 0.05) and almost 50% suppression of β-secretase activity (Fig. 4C, two-way ANOVA, followed by Bonferroni post hoc test between cells with different genotypes). We also detected γ-secretase activity in p38α MAPK knocked-down and control cells and observed that deficiency of p38α MAPK did not change γ-secretase activity (Fig. 4D, two-way ANOVA, p > 0.05). With these cell models, we further used real-time PCR to confirm that the knockdown of mapk14-induced reduction of BACE1 proteins was not due to the decreased genetic transcription of BACE1 (Fig. 4E, one-way ANOVA, p > 0.05). The transcription of ps1 was not changed, whereas the transcription of nicastrin was slightly up-regulated (one-way ANOVA for ps1 and nicastrin, p > 0.05 and = 0.024, respectively).
FIGURE 4.

Knockdown of p38α MAPK expression reduces both β-secretase activity and protein amount in SH-SY5Y cells. SH-SY5Y cell lines with knockdown of p38α MAPK expression were established by transfecting artificial miRNA-transcribing vectors targeting the mapk14 gene (kd509 and kd709). The control cell line (kd-ct) was transfected with miRNA-transcribing vectors containing scramble encoding sequence. The protein levels of p38α MAPK and BACE1 were determined by quantitative Western blots (A and B, one-way ANOVA followed by Bonferroni post hoc test; n ≥ 4 per group). For detection of β- and γ-secretase activity, membrane components prepared from different cell lines (kd-ct, kd509, and kd709) were incubated with fluorogenic substrates (C and D, two-way ANOVA followed by Bonferroni post hoc test). The transcripts of bace1, nicastrin, and ps1 genes in the SH-SY5Y cell lines with and without knockdown of the mapk14 gene were also measured with real-time PCR, which showed that transcription of nicastrin was slightly up-regulated after cells were transfected with the vector kd709 (E, one-way ANOVA followed by Tukey post hoc test; n = 4 per group). To investigate the effects of p38α MAPK on Aβ generation, SH-SY5Y cells were transfected first with APP-transgenic vectors and then with p38α MAPK knockdown vectors (kd-ct, kd509, and kd709). The Aβ in the culture medium was detected with quantitative Western blot and adjusted by APP secretion from the same cell line (E, one-way ANOVA followed by Bonferroni post hoc test, n ≥ 6 per group). A, B, or E, grouping images from different parts of the same gel.
In additional experiments, we also knocked down p38α MAPK expression in APP-transgenic SH-SY5Y cells. We confirmed the decreased protein level of p38α MAPK (data not shown) and detected secreted Aβ in the conditional culture medium with quantitative Western blot. As shown in Fig. 4F, significantly less Aβ was released from p38α MAPK-deficient cells than from p38α MAPK-wild-type control cells (one-way ANOVA, p < 0.05).
Knockdown of p38α MAPK Expression Facilitates Lysosomal Degradation of BACE1 in the Neuronal Cells
As reduction of p38α MAPK expression decreased BACE1 proteins in both the mouse brain and cultured cell without down-regulating bace1 gene transcription, we hypothesized that BACE1 degradation was enhanced by p38α MAPK inhibition. As predicted, knockdown of p38α MAPK with either vector (kd509 or kd709) facilitated the degradation of BACE1 and significantly shortened its lifetime in neuronal cells in which protein translation was blocked by cycloheximide treatment (Fig. 5, A and B; two-way ANOVA, p < 0.05).
FIGURE 5.

p38α MAPK deficiency promotes lysosomal degradation of BACE1 in SH-SY5Y cells. p38α MAPK knocked down (kd509 or kd709) and control (kd-ct) cells were treated with cycloheximide. Cell lysates were collected 0, 4, 8, and 12 h thereafter. Quantitative Western blot was used to determine the protein levels of BACE1 and α-tubulin (A and B, two-way ANOVA followed by Tukey post hoc test; n = 3 per group). Lysosomes were isolated from different cell lines using Percoll gradient centrifugation. From the top to bottom, 4 times fractions were collected for Western blot detection of LAMP-1, calnexin, and β-actin (C). In the following experiments, the BACE1 protein was detected and quantified in fraction 4 with LAMP-1 as an internal protein-loading control (D and E, one-way ANOVA followed by Tukey post-hoc test; n = 3 per group). A, C, or D, grouping images from different parts of the same gel.
As BACE1 is degraded by lysosomes (38), we investigated BACE1 protein levels in lysosomes isolated from cultured cells (Fig. 5, C and D). We collected 4 times fractions using Percoll density gradient centrifugation. Western blot analysis showed that LAMP-1, a lysosome-specific protein, was enriched in Fraction 4, whereas non-lysosomal proteins, such as calnexin and β-actin, were nearly absent (Fig. 5C). Thus, we used LAMP-1 as an internal protein control in measuring BACE1 protein in Fraction 4.
Significantly more BACE1 protein were detected in lysosomes derived from p38α MAPK-knocked down cells compared with p38α MAPK-wild-type cells (Fig. 5, D and E, p < 0.05). Interestingly, the lower efficiency of BACE1 degradation in kd509-mediated p38α MAPK knocked down cells in the lifetime assay (Fig. 5B) was correlated with a lower protein level of BACE1 in lysosomes (Fig. 5E) compared with kd709-transfected knockdown cells, which strongly suggested that p38α MAPK knockdown facilitates the lysosomal degradation of BACE1.
Knockdown of p38α MAPK Expression Does Not Change the Phosphorylation of BACE1 in the Neuronal Cells
BACE1 is potentially phosphorylated by p38α MAPK at serine and threonine, which might modulate the intracellular trafficking and degradation of BACE1 (39, 40). Thus, we asked whether the decreased cerebral Aβ load after the inhibition of neuronal p38α MAPK was due to the altered phosphorylation of BACE1. We first utilized NetPhosK 1.0 Server and human BACE1 sequence with UniProt accession number P56817 to predict the phosphorylation sites. We observed that only Ser-83 without any threonine residues in BACE1 was potentially phosphorylated by p38 MAPK. In the following experiments, we only investigated phosphorylation of BACE1 at serine. Phosphorylated serine-specific antibody detected no changes in serine phosphorylation in BACE1 taken from cell lysate derived from p38α MAPK-knocked down and wild-type control cells (Fig. 6, A and B).
FIGURE 6.
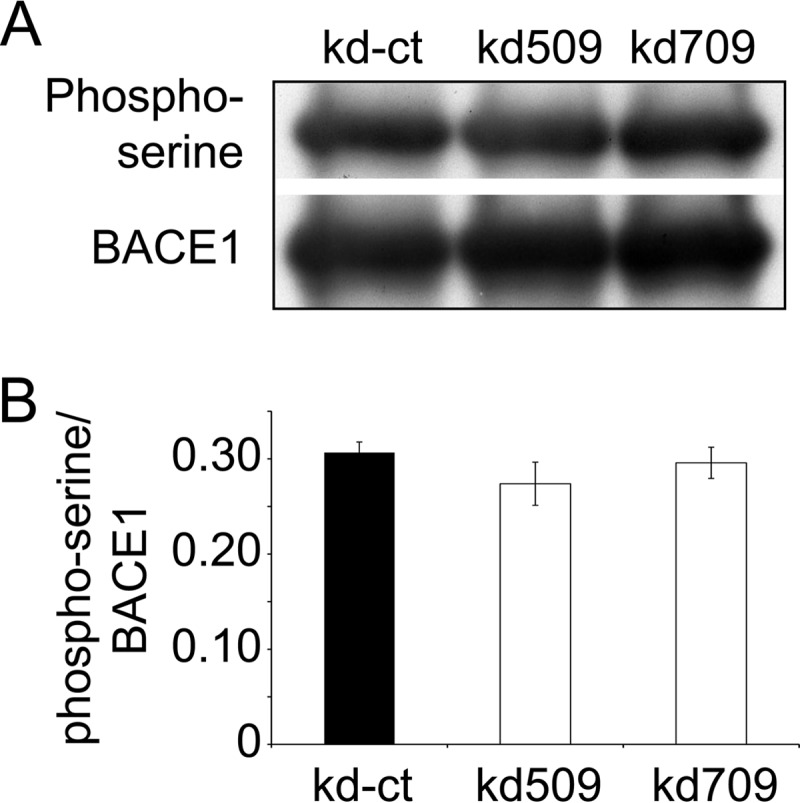
p38α MAPK deficiency does not change the phosphorylation of BACE1 in SH-SY5Y cells. BACE1 proteins were pulled down from the cell lysate derived from p38α MAPK knocked down (kd509 or kd709) or control (kd-ct) cells with anti-BACE1 (C-terminal specific) and protein G; thereafter, they were serially detected with quantitative Western blot using phosphoserine and BACE1-specific antibodies (A and B, one-way ANOVA, p > 0.05, n = 6 per group). A, grouping images from different parts of the same gel.
Deletion of p38α MAPK Enhances Autophagy That Is Involved in the Decrease of BACE1 Protein
Phosphorylation of BACE1 was not altered by p38α MAPK deficiency, suggesting that another mechanism must facilitate the lysosomal degradation of BACE1. p38 MAPK has been observed to block autophagy by phosphorylating ATG5 (41); we asked whether enhanced autophagy mediated p38α MAPK deficiency induced BACE1 degradation.
Autophagic activity was enhanced in APP-transgenic mice with p38α MAPK deletion in neurons, compared with control mice with normal p38α MAPK. Specifically, both the ratio of LC3-II/I protein and the amount of beclin1 protein detected in RIPA buffer-soluble brain homogenate were significantly higher in APPtgp38fl/wtNex-Cre+/− mice than in APPtgp38fl/wtNex-Cre−/− mice (Fig. 7, A–C, t test, p < 0.05). Similarly, we observed that deletion of p38α MAPK in SH-SY5Y cell lines increased both the ratio of LC3-II/I protein and the amount of beclin1 protein (Fig. 7, D–F, one-way ANOVA, p < 0.05). To exclude the possibility that impairment in autophagy or lysosomal digestion caused the increase of LC3, we treated LC3-GFP-RFP-transgenic autophagy reporter SH-SY5Y cells (34) with the p38α MAPK inhibitor SB203580, and observed that inhibition of p38 MAPK significantly increased the numbers of autophagosomes and autolysosmes in a dose-dependent manner (Fig. 7, G–I, one-way ANOVA, p < 0.05). We also used Western blot to compare the protein levels of LAMP-1 and Gm2A, which are degraded by lysosomes (42), in p38α MAPK-deficient and wild-type SH-SY5Y cells. As shown in Fig. 7, J–L, deletion of p38α MAPK does not affect the protein levels of LAMP-1 and Gm2A in SH-SY5Y cells (one-way ANOVA, p > 0.05). We concluded that deficiency of p38α MAPK did enhance neuronal autophagy.
FIGURE 7.

Deletion of p38α MAPK enhances autophagy in both the brain and cell. Autophagy activity was evaluated with quantitative Western blot detection of LC3 and beclin1 in neuronal p38α MAPK-deleted (ko) and wild-type (wt) APP-transgenic mouse brains (A–C), as well as in p38α MAPK knocked down (kd509 or kd709) and control (kd-ct) SH-SY5Y cells (D–F). Deletion of p38α MAPK increased both the ratio of LC3-II/I and beclin1 protein levels (B and C: t test, n = 11 per group; E and F, one-way ANOVA followed by Tukey post-hoc test, n ≥ 5 per group). Additionally, autophagy-reporting cells expressing the fusion protein of LC3-GFP-RFP were treated with the p38 MAPK inhibitor, SB203580, at different concentrations. Autophagosomes are shown as green puncta (overlap with weak red fluorescence, marked with arrowheads) and autolysosomes are shown as red puncta (marked with arrows) (G). The numbers of autophagosomes and autolysosomes are significantly increased by the treatment with SB203580 in a dose-dependent manner (H and I, one-way ANOVA followed by Dunnett T3 post hoc test; n = 4). The lysosomal digestion in p38α MAPK-knocked down (kd509 or kd709) and control (kd-ct) SH-SY5Y cells was evaluated by Western blot detection of LAMP-1 and Gm2A in the cell lysate (J–L, one-way ANOVA, p > 0.05; n ≥ 5 per group). A, D, or J, grouping images from different parts of the same gel.
We next investigated the effects of inhibition of autophagy on the decrease in BACE1 protein caused by p38α MAPK deficiency. We established cell lines overexpressing wild-type and dominant-negative (DN) human ATG5 by stably transfecting SH-SY5Y cells with the established expression vectors (33). After adding 0.2 μg/ml of rapamycin in the culture medium for 24 h, LC3-II/I levels were significantly lower in ATG5DN-transgenic cells than in wild-type ATG5-overexpressing cells, demonstrating ATG5DN-induced inhibition of autophagy (Fig. 8, A and B, t test, p < 0.05). We then further reduced p38α MAPK expression in both ATG5DN and ATG5 (wild-type)-transgenic cells by transfecting cells with the miRNA-transcribing vector kd709. We also inhibited autophagy with treatment of 1 mm 3-methyladenine (3-MA) in human ATG5-transgenic SH-SY5Y cells with and without knockdown of p38α MAPK. Interestingly, both approaches to autophagic inhibition (overexpression of ATG5DN and 3-MA treatment) increased BACE1 protein levels and abolished the p38α MAPK knockdown-induced decrease of BACE1 protein in SH-SY5Y cells (Fig. 8C, t test, p < 0.05).
FIGURE 8.
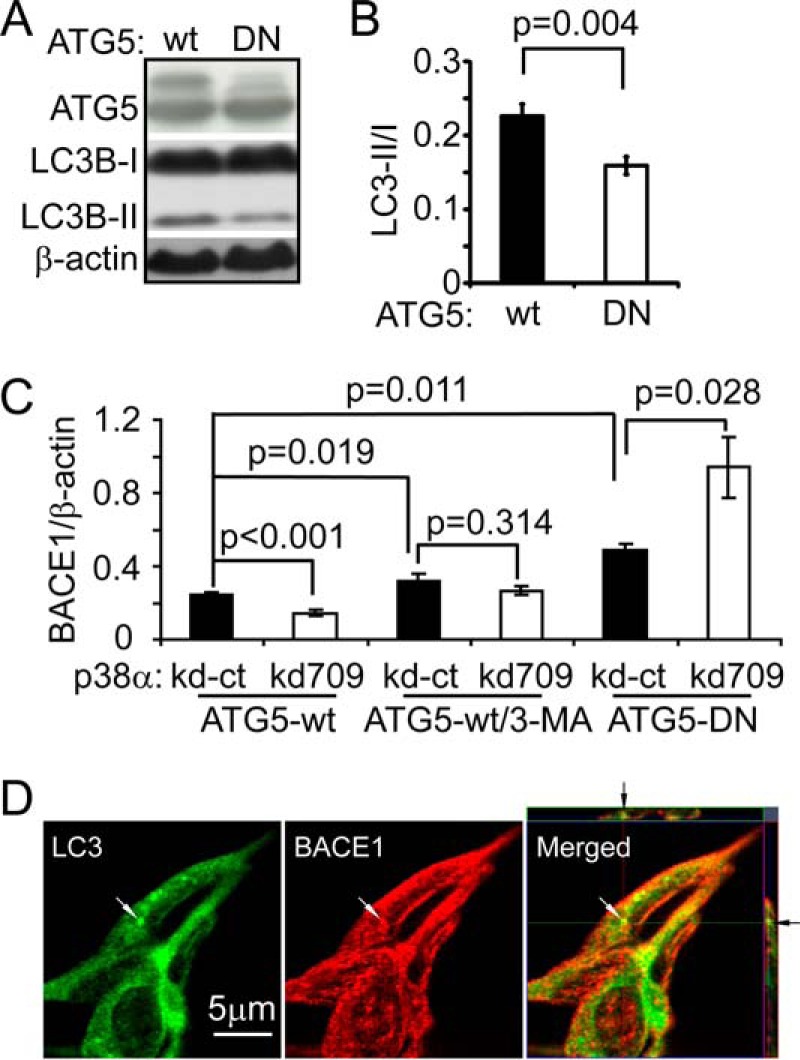
Autophagy inhibition abolishes p38α MAPK deficiency induced BACE1 reduction in SH-SY5Y cells. Dominant-negative (DN) and wild-type (wt) human ATG5-overexpressing cell lines were created. ATG5 and LC3B in the cell lysate were detected with Western blot (A). Autophagy activity was evaluated with the ratio of LC3B-II/I in cells after the treatment of 0.2 μg/ml of rapamycin for 24 h (A and B, t test; n = 3 per group). p38α MAPK expression in ATG5-DN and wt cells was further knocked down (kd709). ATG5-wt cells were also treated with 1 mm 3-MA to inhibit autophagy. The BACE1 protein in knockdown cells was quantified with Western blot and compared with that in relevant p38α MAPK normally expressed control cells (kd-ct). Inhibition of autophagy with both treatment of 3-MA and overexpressing ATG5-DN increases BACE1 protein levels and abolishes the deficiency of p38α MAPK-induced reduction of BACE1 amount in the cells (C, t test; n ≥ 3 per group). The relationship between BACE1 and autophagosome/autolysosome was also analyzed by confocal microscopy. BACE1 was stained with red fluorescence and autophagic vacuoles were visualized by staining LC3A/B with green fluorophore-conjugated antibodies. Co-localization of BACE1 and LC3 was shown by yellow fluorescence (arrow), superimposing fluorescent images of BACE1 and LC3. A, grouping images from different parts of the same gel.
In further experiments, we investigated the co-localization of BACE1 and autophagic vacuoles under confocal microscopy. BACE1 rarely co-localized with autophagosomes or autolysosomes as stained with antibodies against LC3A/B or p62. Treatment of bafilomycin, which was supposed to block the fusion of autophagosomes with lysosomes (43), did not change the results of our observation. Here we showed the co-localization of BACE1 and LC3A/B within one cell that had not been treated with bafilomycin (Fig. 8D).
Discussion
The pathogenic molecule, Aβ, which is produced from the β- and γ-cleavages of APP (7), damages neurons and induces inflammatory activation in the brain (1, 2). Our study demonstrates for the first time that partial deletion (via heterozygous knock-out or knockdown) of the inflammation-sensitive kinase, p38α MAPK, in neurons reduces Aβ generation and decreases cerebral Aβ load by promoting macroautophagy (hereafter referred to as autophagy)-associated BACE1 degradation.
Microglial inflammatory activation is correlated with disease progression in AD patients (44, 45). p38 MAPK, which responds to inflammatory and oxidative stress, is activated in the AD brain (14, 15), and mediates Aβ-induced inflammatory activation in cultured microglia (21). Studies have suggested that inhibition of p38α MAPK retards AD pathogenesis; for example, oral administration of p38α MAPK inhibitor successfully reduced neuroinflammation and synaptic dysfunction in APP-transgenic mice (22, 46). However, the effect of p38α MAPK on Aβ pathology was not examined.
Multiple binding sites for the inflammatory and oxidative stimuli-responsive transcriptional activators, including AP1, AP2, NF-κB, STAT1, and SP1 have been identified within the promoter region of the bace1 gene, suggesting that neuroinflammation may increase BACE1 expression (8, 9, 47, 48). Elevated BACE1 protein expression and activity have been observed in sporadic AD cases (49) and during aging (50). The up-regulated expression of BACE1 tends to further increase Aβ generation, creating a BACE1-based vicious cycle in AD pathogenesis (51). Our study demonstrates that reducing p38α MAPK expression in neurons facilitates BACE1 degradation and decreases Aβ generation. Moreover, this effect is more apparent in APP-transgenic mice than in their non-APP-transgenic littermates.
Our study further demonstrates that reduction of p38α MAPK expression enhances autophagy in neurons. Autophagy is a basic cellular catabolic mechanism, through which the unnecessary or dysfunctional cellular proteins and organelle components are degraded through the lysosomal machinery (52). Growing evidence has shown that autophagy is impaired in AD (53–55). Histological analysis shows accumulation of autophagosomes and autolysosomes in neurons of AD brains (56). These accumulated autophagic vacuoles potentially provide membrane structures to produce Aβ, as BACE1-cleaved APP and γ-secretase components are enriched in autophagic vacuoles (57). Moreover, impaired autophagy was reported to promote the intracellular accumulation of Aβ and subsequent neuronal degeneration (58). Thus, inhibition of p38α MAPK may act to repair dysfunctional autophagy in the AD brain. Furthermore, increased autophagy is helpful in the degradation of APP and its C-terminal fragment C99, as well as phosphorylated Tau proteins (59–61).
How p38α MAPK regulates autophagy is unclear. p38 MAPK inhibits starvation-induced autophagy by interfering with the binding between ATG9 and p38-interacting protein in HEK293 cells (62), and by phosphorylating ATG5 in fibroblasts and macrophages (41); however, p38 MAPK also mediates interferon-γ or oxidative stress-triggered autophagy in macrophages or HeLa cells (63, 64). Thus, the effect of p38α MAPK on autophagy is likely cell type-specific and may even depend on the type of stimuli that activate autophagy. In our neuronal cell models, inhibition of p38α MAPK appears to speed delivery of BACE1 to lysosomes in an ATG5-dependent pattern, but the mechanism by which enhanced autophagy facilitates the lysosomal degradation of BACE1 remains unclear. We have observed that BACE1 rarely co-localizes with autophagic vacuoles. This evidence is not sufficient to support the hypothesis that BACE1 is directly degraded through autophagy. However, this result may be misleading, as it could not be excluded that the BACE1 epitope for the antibodies used in this study was potentially damaged or lost in the autophagic vacuoles, rendering the intravacuolar BACE1 unrecognizable by the antibodies. Consequently, questions concerning the p38α MAPK deficiency promoted autophagic degradation of BACE1 must await clarification of the mechanisms through which BACE1 is delivered to the autophagic pathway.
For transport to lysosomal degradation, BACE1 needs to be carried by its sorting adaptor proteins, especially GGA1 and GGA3 (65, 66). BACE1 contains an acidic di-leucine (DXXLL) motif at the C terminus, which binds the VHS domain in the sorting adaptor proteins. This binding is strongly enhanced by phosphorylation of Ser-498 in the DXX(S)LL motif (40, 67). We have observed that knockdown of p38α MAPK does not affect BACE1 phosphorylation. Thus, we should further investigate whether p38α MAPK deficiency reduces the phosphorylation of GGA1 or GGA3 and subsequently alters GGA1 or GGA3-mediated BACE1 degradation, in the following projects.
In summary, we have shown that inhibition of p38α MAPK in neurons enhances autophagy and promotes BACE1 degradation, which thereby reduces neuronal Aβ generation and decreases cerebral Aβ load. p38α MAPK might be a promising target for AD therapy. However, as a signal transduction mediator, p38 MAPK signaling has been implicated in many cellular responses including inflammation, cell cycle, cell death, development, cell differentiation, senescence, and tumor genesis (68). Possible toxic side effects should be carefully evaluated during any translational study of p38 MAPK inhibitors in AD therapy (69).
Author Contributions
Y. L. and K. F. designed research; L. S., W. H., Y. Q., S. L., I. T., X. L., and Y. L. performed research; K. F. contributed analytic tools; L. S. and Y. L. analyzed data; Y. L. wrote the paper.
Acknowledgments
We thank Dr. M. Jucker (Hertie Institute for Clinical Brain Research, Tübingen) for providing APPPS1 transgenic mice; and K. Nave (Max-Planck-Institute for Medicine, Göttingen) for Nex-Cre mice. The floxed-p38α (p38fl/fl) transgenic mice were kindly provided by Dr. K. Otsu (Osaka University) through the RIKEN Bioresearch Center. We appreciate Mirjam Müller for excellent technical assistance.
This work was supported by the Deutsche Forschungsgemeinschaft Grant LI1725/2-1 (to Y. L.), Else Kröner-Fresenius-Stiftung 2012_A247 (to Y. L. and K. F.), and a Prof. Dr. Peter Theiss Wissenschaftspreis 2015 grant (to X. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- AD
- Alzheimer disease
- Aβ
- amyloid β peptide
- APP
- amyloid precursor protein
- BACE1
- β-site APP-cleaving enzyme
- Gm2A
- Gm2 activator protein
- LAMP-1
- lysosomal membrane-associated protein 1
- 3-MA
- 3-methyladenine
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- ANOVA
- analysis of variance
- miRNA
- microRNA.
References
- 1. Mucke L., and Selkoe D. J. (2012) Neurotoxicity of amyloid beta-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med. 2, a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wyss-Coray T., and Rogers J. (2012) Inflammation in Alzheimer disease: a brief review of the basic science and clinical literature. Cold Spring Harb. Perspect. Med. 2, a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Haass C., Kaether C., Thinakaran G., and Sisodia S. (2012) Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2, a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ohno M., Sametsky E. A., Younkin L. H., Oakley H., Younkin S. G., Citron M., Vassar R., and Disterhoft J. F. (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron 41, 27–33 [DOI] [PubMed] [Google Scholar]
- 5. McConlogue L., Buttini M., Anderson J. P., Brigham E. F., Chen K. S., Freedman S. B., Games D., Johnson-Wood K., Lee M., Zeller M., Liu W., Motter R., and Sinha S. (2007) Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J. Biol. Chem. 282, 26326–26334 [DOI] [PubMed] [Google Scholar]
- 6. May P. C., Dean R. A., Lowe S. L., Martenyi F., Sheehan S. M., Boggs L. N., Monk S. A., Mathes B. M., Mergott D. J., Watson B. M., Stout S. L., Timm D. E., Smith Labell E., Gonzales C. R., Nakano M., Jhee S. S., Yen M., Ereshefsky L., Lindstrom T. D., Calligaro D. O., Cocke P. J., Greg Hall D., Friedrich S., Citron M., and Audia J. E. (2011) Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J. Neurosci. 31, 16507–16516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vassar R., Kuhn P. H., Haass C., Kennedy M. E., Rajendran L., Wong P. C., and Lichtenthaler S. F. (2014) Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J. Neurochem. 130, 4–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. He P., Zhong Z., Lindholm K., Berning L., Lee W., Lemere C., Staufenbiel M., Li R., and Shen Y. (2007) Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J. Cell Biol. 178, 829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sastre M., Dewachter I., Rossner S., Bogdanovic N., Rosen E., Borghgraef P., Evert B. O., Dumitrescu-Ozimek L., Thal D. R., Landreth G., Walter J., Klockgether T., van Leuven F., and Heneka M. T. (2006) Nonsteroidal anti-inflammatory drugs repress β-secretase gene promoter activity by the activation of PPARγ. Proc. Natl. Acad. Sci. U.S.A. 103, 443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Song W. J., Son M. Y., Lee H. W., Seo H., Kim J. H., and Chung S. H. (2015) Enhancement of BACE1 activity by p25/Cdk5-mediated phosphorylation in Alzheimer's disease. PLoS ONE 10, e0136950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wahle T., Prager K., Raffler N., Haass C., Famulok M., and Walter J. (2005) GGA proteins regulate retrograde transport of BACE1 from endosomes to the trans-Golgi network. Mol. Cell Neurosci. 29, 453–461 [DOI] [PubMed] [Google Scholar]
- 12. Kang E. L., Cameron A. N., Piazza F., Walker K. R., and Tesco G. (2010) Ubiquitin regulates GGA3-mediated degradation of BACE1. J. Biol. Chem. 285, 24108–24119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kizuka Y., Kitazume S., Fujinawa R., Saito T., Iwata N., Saido T. C., Nakano M., Yamaguchi Y., Hashimoto Y., Staufenbiel M., Hatsuta H., Murayama S., Manya H., Endo T., and Taniguchi N. (2015) An aberrant sugar modification of BACE1 blocks its lysosomal targeting in Alzheimer's disease. EMBO Mol. Med. 7, 175–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hensley K., Floyd R. A., Zheng N. Y., Nael R., Robinson K. A., Nguyen X., Pye Q. N., Stewart C. A., Geddes J., Markesbery W. R., Patel E., Johnson G. V., and Bing G. (1999) p38 kinase is activated in the Alzheimer's disease brain. J. Neurochem. 72, 2053–2058 [DOI] [PubMed] [Google Scholar]
- 15. Sun A., Liu M., Nguyen X. V., and Bing G. (2003) P38 MAP kinase is activated at early stages in Alzheimer's disease brain. Exp. Neurol. 183, 394–405 [DOI] [PubMed] [Google Scholar]
- 16. Chang K. H., de Pablo Y., Lee H. P., Lee H. G., Smith M. A., and Shah K. (2010) Cdk5 is a major regulator of p38 cascade: relevance to neurotoxicity in Alzheimer's disease. J. Neurochem. 113, 1221–1229 [DOI] [PubMed] [Google Scholar]
- 17. Criscuolo C., Fabiani C., Bonadonna C., Origlia N., and Domenici L. (2015) BDNF prevents amyloid-dependent impairment of LTP in the entorhinal cortex by attenuating p38 MAPK phosphorylation. Neurobiol. Aging 36, 1303–1309 [DOI] [PubMed] [Google Scholar]
- 18. Bhaskar K., Konerth M., Kokiko-Cochran O. N., Cardona A., Ransohoff R. M., and Lamb B. T. (2010) Regulation of Tau pathology by the microglial fractalkine receptor. Neuron 68, 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh S., Wu M. D., Shaftel S. S., Kyrkanides S., LaFerla F. M., Olschowka J. A., and O'Banion M. K. (2013) Sustained interleukin-1β overexpression exacerbates Tau pathology despite reduced amyloid burden in an Alzheimer's mouse model. J. Neurosci. 33, 5053–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li Y., Liu L., Barger S. W., and Griffin W. S. (2003) Interleukin-1 mediates pathological effects of microglia on Tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 23, 1605–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bachstetter A. D., Xing B., de Almeida L., Dimayuga E. R., Watterson D. M., and Van Eldik L. J. (2011) Microglial p38α MAPK is a key regulator of proinflammatory cytokine up-regulation induced by Toll-like receptor (TLR) ligands or β-amyloid (Aβ). J. Neuroinflammation 8, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Munoz L., Ralay Ranaivo H., Roy S. M., Hu W., Craft J. M., McNamara L. K., Chico L. W., Van Eldik L. J., and Watterson D. M. (2007) A novel p38α MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer's disease mouse model. J. Neuroinflammation 4, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Reynolds C. H., Nebreda A. R., Gibb G. M., Utton M. A., and Anderton B. H. (1997) Reactivating kinase/p38 phosphorylates Tau protein in vitro. J. Neurochem. 69, 191–198 [DOI] [PubMed] [Google Scholar]
- 24. Zhu X., Rottkamp C. A., Boux H., Takeda A., Perry G., and Smith M. A. (2000) Activation of p38 kinase links Tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 59, 880–888 [DOI] [PubMed] [Google Scholar]
- 25. Li S., Jin M., Koeglsperger T., Shepardson N. E., Shankar G. M., and Selkoe D. J. (2011) Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci. 31, 6627–6638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xing B., Bachstetter A. D., and Van Eldik L. J. (2015) Inhibition of neuronal p38α, but not p38β MAPK, provides neuroprotection against three different neurotoxic insults. J. Mol. Neurosci. 55, 509–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Radde R., Bolmont T., Kaeser S. A., Coomaraswamy J., Lindau D., Stoltze L., Calhoun M. E., Jäggi F., Wolburg H., Gengler S., Haass C., Ghetti B., Czech C., Hölscher C., Mathews P. M., and Jucker M. (2006) Aβ42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishida K., Yamaguchi O., Hirotani S., Hikoso S., Higuchi Y., Watanabe T., Takeda T., Osuka S., Morita T., Kondoh G., Uno Y., Kashiwase K., Taniike M., Nakai A., Matsumura Y., Miyazaki J., Sudo T., Hongo K., Kusakari Y., Kurihara S., Chien K. R., Takeda J., Hori M., and Otsu K. (2004) p38α mitogen-activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol. Cell. Biol. 24, 10611–10620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goebbels S., Bormuth I., Bode U., Hermanson O., Schwab M. H., and Nave K. A. (2006) Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis 44, 611–621 [DOI] [PubMed] [Google Scholar]
- 30. Liu Y., Liu X., Hao W., Decker Y., Schomburg R., Fülöp L., Pasparakis M., Menger M. D., and Fassbender K. (2014) IKKβ deficiency in myeloid cells ameliorates Alzheimer's disease-related symptoms and pathology. J. Neurosci. 34, 12982–12999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu Y., Hao W., Dawson A., Liu S., and Fassbender K. (2009) Expression of amyotrophic lateral sclerosis-linked SOD1 mutant increases the neurotoxic potential of microglia via TLR2. J. Biol. Chem. 284, 3691–3699 [DOI] [PubMed] [Google Scholar]
- 32. Chishti M. A., Yang D. S., Janus C., Phinney A. L., Horne P., Pearson J., Strome R., Zuker N., Loukides J., French J., Turner S., Lozza G., Grilli M., Kunicki S., Morissette C., Paquette J., Gervais F., Bergeron C., Fraser P. E., Carlson G. A., George-Hyslop P. S., and Westaway D. (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J. Biol. Chem. 276, 21562–21570 [DOI] [PubMed] [Google Scholar]
- 33. Mizushima N., Sugita H., Yoshimori T., and Ohsumi Y. (1998) A new protein conjugation system in human: the counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 273, 33889–33892 [DOI] [PubMed] [Google Scholar]
- 34. Kimura S., Noda T., and Yoshimori T. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 [DOI] [PubMed] [Google Scholar]
- 35. Liu R. Q., Zhou Q. H., Ji S. R., Zhou Q., Feng D., Wu Y., and Sui S. F. (2010) Membrane localization of β-amyloid 1–42 in lysosomes: a possible mechanism for lysosome labilization. J. Biol. Chem. 285, 19986–19996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Taguchi T., Akimaru K., Yamasakt M., Ryu S., Miyamoto E., Takano Y., and Sato A. (2000) Isolation of highly purified rat cerebral lysosomes using percoll gradients with a variety of calcium concentrations. Environ. Health Prev. Med. 4, 217–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xie K., Liu Y., Hao W., Walter S., Penke B., Hartmann T., Schachner M., and Fassbender K. (2013) Tenascin-C deficiency ameliorates Alzheimer's disease-related pathology in mice. Neurobiol. Aging 34, 2389–2398 [DOI] [PubMed] [Google Scholar]
- 38. Koh Y. H., von Arnim C. A., Hyman B. T., Tanzi R. E., and Tesco G. (2005) BACE is degraded via the lysosomal pathway. J. Biol. Chem. 280, 32499–32504 [DOI] [PubMed] [Google Scholar]
- 39. von Arnim C. A., Tangredi M. M., Peltan I. D., Lee B. M., Irizarry M. C., Kinoshita A., and Hyman B. T. (2004) Demonstration of BACE (β-secretase) phosphorylation and its interaction with GGA1 in cells by fluorescence-lifetime imaging microscopy. J. Cell Sci. 117, 5437–5445 [DOI] [PubMed] [Google Scholar]
- 40. Walter J., Fluhrer R., Hartung B., Willem M., Kaether C., Capell A., Lammich S., Multhaup G., and Haass C. (2001) Phosphorylation regulates intracellular trafficking of β-secretase. J. Biol. Chem. 276, 14634–14641 [DOI] [PubMed] [Google Scholar]
- 41. Keil E., Höcker R., Schuster M., Essmann F., Ueffing N., Hoffman B., Liebermann D. A., Pfeffer K., Schulze-Osthoff K., and Schmitz I. (2013) Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ. 20, 321–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karaca I., Tamboli I. Y., Glebov K., Richter J., Fell L. H., Grimm M. O., Haupenthal V. J., Hartmann T., Gräler M. H., van Echten-Deckert G., and Walter J. (2014) Deficiency of sphingosine-1-phosphate lyase impairs lysosomal metabolism of the amyloid precursor protein. J. Biol. Chem. 289, 16761–16772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Klionsky D. J., Elazar Z., Seglen P. O., and Rubinsztein D. C. (2008) Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4, 849–850 [DOI] [PubMed] [Google Scholar]
- 44. Cagnin A., Brooks D. J., Kennedy A. M., Gunn R. N., Myers R., Turkheimer F. E., Jones T., and Banati R. B. (2001) In vivo measurement of activated microglia in dementia. Lancet 358, 461–467 [DOI] [PubMed] [Google Scholar]
- 45. Edison P., Archer H. A., Gerhard A., Hinz R., Pavese N., Turkheimer F. E., Hammers A., Tai Y. F., Fox N., Kennedy A., Rossor M., and Brooks D. J. (2008) Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol. Dis. 32, 412–419 [DOI] [PubMed] [Google Scholar]
- 46. Alam J. J. (2015) Selective brain-targeted antagonism of p38 MAPKα reduces hippocampal IL-1β levels and improves Morris water maze performance in aged rats. J. Alzheimers. Dis. 48, 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Christensen M. A., Zhou W., Qing H., Lehman A., Philipsen S., and Song W. (2004) Transcriptional regulation of BACE1, the β-amyloid precursor protein β-secretase, by Sp1. Mol. Cell. Biol. 24, 865–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ge Y. W., Maloney B., Sambamurti K., and Lahiri D. K. (2004) Functional characterization of the 5′ flanking region of the BACE gene: identification of a 91 bp fragment involved in basal level of BACE promoter expression. FASEB J. 18, 1037–1039 [DOI] [PubMed] [Google Scholar]
- 49. Yang L. B., Lindholm K., Yan R., Citron M., Xia W., Yang X. L., Beach T., Sue L., Wong P., Price D., Li R., and Shen Y. (2003) Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4 [DOI] [PubMed] [Google Scholar]
- 50. Fukumoto H., Rosene D. L., Moss M. B., Raju S., Hyman B. T., and Irizarry M. C. (2004) β-Secretase activity increases with aging in human, monkey, and mouse brain. Am. J. Pathol. 164, 719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chami L., and Checler F. (2012) BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer's disease. Mol. Neurodegener. 7, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nixon R. A. (2013) The role of autophagy in neurodegenerative disease. Nat. Med. 19, 983–997 [DOI] [PubMed] [Google Scholar]
- 53. Peric A., and Annaert W. (2015) Early etiology of Alzheimer's disease: tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 129, 363–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nixon R. A., Wegiel J., Kumar A., Yu W. H., Peterhoff C., Cataldo A., and Cuervo A. M. (2005) Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122 [DOI] [PubMed] [Google Scholar]
- 55. Yang D. S., Stavrides P., Mohan P. S., Kaushik S., Kumar A., Ohno M., Schmidt S. D., Wesson D., Bandyopadhyay U., Jiang Y., Pawlik M., Peterhoff C. M., Yang A. J., Wilson D. A., St George-Hyslop P., Westaway D., Mathews P. M., Levy E., Cuervo A. M., and Nixon R. A. (2011) Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain 134, 258–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boland B., Kumar A., Lee S., Platt F. M., Wegiel J., Yu W. H., and Nixon R. A. (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J. Neurosci. 28, 6926–6937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yu W. H., Cuervo A. M., Kumar A., Peterhoff C. M., Schmidt S. D., Lee J. H., Mohan P. S., Mercken M., Farmery M. R., Tjernberg L. O., Jiang Y., Duff K., Uchiyama Y., Näslund J., Mathews P. M., Cataldo A. M., and Nixon R. A. (2005) Macroautophagy: a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J. Cell Biol. 171, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nilsson P., Loganathan K., Sekiguchi M., Matsuba Y., Hui K., Tsubuki S., Tanaka M., Iwata N., Saito T., and Saido T. C. (2013) Aβ secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69 [DOI] [PubMed] [Google Scholar]
- 59. Parr C., Carzaniga R., Gentleman S. M., Van Leuven F., Walter J., and Sastre M. (2012) Glycogen synthase kinase 3 inhibition promotes lysosomal biogenesis and autophagic degradation of the amyloid-β precursor protein. Mol. Cell. Biol. 32, 4410–4418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jo C., Gundemir S., Pritchard S., Jin Y. N., Rahman I., and Johnson G. V. (2014) Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 5, 3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Polito V. A., Li H., Martini-Stoica H., Wang B., Yang L., Xu Y., Swartzlander D. B., Palmieri M., di Ronza A., Lee V. M., Sardiello M., Ballabio A., and Zheng H. (2014) Selective clearance of aberrant Tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol. Med. 6, 1142–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Webber J. L., and Tooze S. A. (2010) Coordinated regulation of autophagy by p38α MAPK through mAtg9 and p38IP. EMBO J. 29, 27–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Matsuzawa T., Kim B. H., Shenoy A. R., Kamitani S., Miyake M., and Macmicking J. D. (2012) IFN-γ elicits macrophage autophagy via the p38 MAPK signaling pathway. J. Immunol. 189, 813–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhong W., Zhu H., Sheng F., Tian Y., Zhou J., Chen Y., Li S., and Lin J. (2014) Activation of the MAPK11/12/13/14 (p38 MAPK) pathway regulates the transcription of autophagy genes in response to oxidative stress induced by a novel copper complex in HeLa cells. Autophagy 10, 1285–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bonifacino J. S., and Traub L. M. (2003) Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 72, 395–447 [DOI] [PubMed] [Google Scholar]
- 66. Tan J., and Evin G. (2012) β-Site APP-cleaving enzyme 1 trafficking and Alzheimer's disease pathogenesis. J. Neurochem. 120, 869–880 [DOI] [PubMed] [Google Scholar]
- 67. Kato Y., Misra S., Puertollano R., Hurley J. H., and Bonifacino J. S. (2002) Phosphoregulation of sorting signal-VHS domain interactions by a direct electrostatic mechanism. Nat. Struct. Biol. 9, 532–536 [DOI] [PubMed] [Google Scholar]
- 68. Zarubin T., and Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 [DOI] [PubMed] [Google Scholar]
- 69. Coulthard L. R., White D. E., Jones D. L., McDermott M. F., and Burchill S. A. (2009) p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 15, 369–379 [DOI] [PMC free article] [PubMed] [Google Scholar]