

Abstract
A study of the mechanism of the oxidative deamination of the N-nitroso-N-acetyl sialyl glycosides leading with overall retention of configuration to the corresponding 2-keto-3-deoxy-d-glycero-d-galacto-nonulopyranosidonic acid (KDN) glycosides is described, making use of a series of differentially O-protected N-nitroso-N-acetyl sialyl glycosides and of isotopic labelling studies. No evidence is found for stereodirecting participation by ester groups at the 4- and 7-positions. Comparisons are drawn with oxidative deamination reactions of 4-amino-4-deoxy and 2-amino-2-deoxy hexopyranosides and a common mechanism is formulated involving the intermediacy of 1-oxabicyclo[3.1.0]hexyl oxonium ions following participation by the pyranoside ring oxygen. A minor reaction pathway has been uncovered by labelling studies in the β-thiosialosides that results in the exchange of the 4-O-acetyl group by the glacial acetic acid that serves as external nucleophile in the general oxidative deamination process. A mechanism is proposed for this exchange involving participation by the thioglycoside at the level of an intermediate diazoalkane.
Introduction
The sialic acids, a large group of nine carbon sugar acids based on the 2-keto-3-deoxy-d-glycero-d-galacto-nonulopyranosidonic acid framework, are present at the non-reducing end of a myriad of mammalian cell surface glycans.1-3 N-Acetyl neuraminic acid is the most common member of the family, but variation at the 5-position is frequently found with examples including N-glycoyl neuraminic, and 2-keto-3-deoxy-d-glycero-d-galacto-nonulopyranosidonic acid (KDN). The removal of terminal sialic acid glycosides from cell surface glycans by sialidase enzymes is a key step in the replication cycle of influenza viruses, giving rise to the importance of the sialidase inhibitors as antiviral drugs. Increased levels of sialidation in cell surface glycans have led to the development of cancer vaccine candidates based on tumor-associated carbohydrate antigens (TACAs).1-4 Modification of the amido group at the 5-position of N-amidoneuraminic acid derivatives has proved to be beneficial in the development of sialidase inhibitors and of carbohydrate antigens with greater antigenicity.5-9 Such modifications have typically been achieved by N-deacetylation, its glycosides and derivatives, followed by reinstallation of different acyl groups,6-7,10-12 or by use of the biosynthetic machinery when various N-acyl mannosamines are converted enzymatically to the corresponding N-acyl neuraminic acids13 and eventually their glycosides,14-15 or by a combination of the biosynthetic and chemical methods.16 Consequently, modifications are typically limited to amide derivatives or other simple substitutions that are compatible with the biosynthetic machinery such as the conversion of 2-deoxy glucose to 5-desacetamido neuraminic acid.17
The advent of improved chemical methods for the efficient and stereoselective synthesis of the α-sialyl glycosides based on the use of 4-O,5-N-oxazolidinone-protected sialyl donors and their N-acetyl variants18-22 gave rise to the possibility of the effective chemical synthesis of C-5 modified sialyl glycosides provided suitable reactions for post-glycosylation removal of the acetamido function could be identified. Toward this end we have described23-24 the application of the oxidative deamination of neruaminic acid25-27 and N-acetylneuraminic acid28-29 to the synthesis of C-5 modified α-sialosides. We describe here our investigations into the mechanism of stereospecific oxidative deamination of N-nitroso N-acetyl neuraminic acid glycosides making use of a series of differentially O-protected derivatives and isotopic labelling studies. Comparisons are made with oxidative deamination reactions in the 4-amino-4-deoxy and 2-amino-2-deoxy hexopyranosides, leading to the formulation of a general mechanism involving participation by the ring oxygen and the intermediacy of 1-oxabicyclo[3.1.0]hexyl oxonium ions, whose fate is determined by the substituent pattern. We also describe a minor reaction pathway found on oxidative deamination of a N-nitroso-N-acetyl axial sialyl thioglycoside that results in exchange of the 4-O-ester with acetic acid from the medium.
Results
Synthesis
Methyl [methyl (5-acetamido-5-deoxy-β-d-glycero-d-galacto-2-nonulopyranosid)onate] 1 was obtained from N-acetylneuraminic acid under standard conditions,30-31 and was converted by standard methods to the known derivatives 2-5.30,32-34

Removal of the isopropylidene group from 4 with trifluoroacetic acid in wet dichloromethane gave the triol 6, which on acetylation afforded the 4-O-silyl-7,8,9-tri-O-acetyl derivative 7. Treatment with tetrabutylammonium fluoride then afforded 8, which was converted to the corresponding 4-O-benzyl ether 9 by reaction with benzyl trichloroacetimidate and TfOH,35 and to the 4-O-t-butyloxycarbonyl derivative 10 by stirring with Boc2O in the presence of trimethylamine and 4-dimethylaminopyridine, aqueous work-up and purification by chromatography over neutral alumina (Scheme 1). It is noteworthy that attempted purification of 10 by chromatography over silica gel resulted in an isolated yield of 28% of the desired product together with 23% of the N-acetyl oxazolidinone 11. This unanticipated cyclization provides a possible alternative entry into the N-acetyl oxazolidinones, which have found widespread use in the stereocontreolled synthesis of sialic acid glycosides.20-22 Isopropylidene cleavage from 5 gave 12 from which 13 was obtained by acetylation. Subsequent Wittig olefination gave the 4-deoxy-4-alkylidene derivative 14 as the E-isomer as determined by the NOE correlation of the vinylic hydrogen with the H5. Catalytic hydrogenation over palladium charcoal finally gave 15, whose configuration rests on coupling constant analysis in the H3,H4,H5 spin system in the 1H NMR spectrum (Scheme 1).
Scheme 1.

Synthesis of NeuAc Derivatives Selectively Functionalized at the 4-Position.
Benzylation of 4 with sodium hydride and benzyl bromide in DMF gave the 7-O-benzyl derivative 16, from which the isopropylidene and silyl groups were removed with acetic acid in methanol yielding 17 and, after acetylation 18 (Scheme 2). Treatment of 1 with di-(tert-butyl)-dichlorosilane and imidazole in dichloromethane afforded the 8,9-O-silylene derivative 19, which underwent selective installation of the tert-butyldimethylsilyl group on O4 to give 20. The 7-O-Boc group was then installed with Boc2O to give 21, which on treatment first with TBAF and then acetic anhydride in pyridine afforded 22 (Scheme 2).
Scheme 2.

Synthesis of NeuAc Derivatives Selectively Protected at the 7-Position.
Benzylation of 3 with sodium hydride and benzyl bromide in DMF gave the 4,7-di-O-benzyl ether 23, which on removal of the isopropylidene group with toluenesulfonic acid in methanol and subsequent acetylation gave 24 and 25, respectively (Scheme 3).
Scheme 3.

Synthesis of a NeuAc Derivative Selectively Protected at the 4- and 7-Positions.
Influence of Protecting Groups on Stereoselectivity
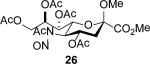
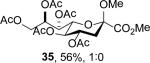
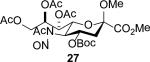
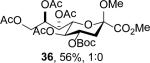
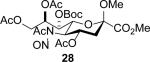
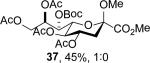
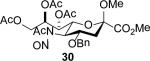
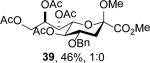
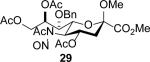
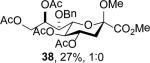
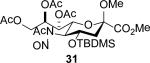
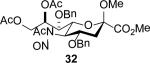
The series of sialyl glycosides 2, 7, 9, 10, 15, 18, 22, 23, and 25 were converted to a series of N-nitroso derivatives 26-34 by treatment with NO+BF4− in a mixture of pyridine and dichloromethane at −10 °C in essentially quantitative yield and taken forward to the next step immediately.23-24 Exposure to trifluoroethanol and sodium isopropoxide in dichloromethane and isopropanol at −10 °C followed by addition of acetic acid under the standard conditions gave the corresponding KDN derivatives and their epimers at the 5-position 35-45 as reported in Table 1.
Table 1.
Influence of Protecting Groups on Stereoselectivity of KDN Formation.a


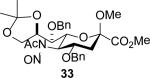


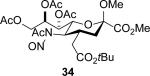
With the exception of entry 9 all reactions were conducted under a standard set of conditions involving treatment of the nitrosoamide in CH2Cl2 at −10 °C with trifluoroethanol and then sodium isopropoxide in isopropanol followed by addition of glacial acetic acid and warming to 0 °C.
The nitrosamide was stored neat at 4 °C in the refrigerator for 12 days. No external reagents were added.
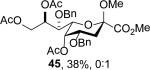
To facilitate isolation from a complex mixture the isopropylidene group was cleaved the initial product 44 and the residual diol acetylated to give 45.
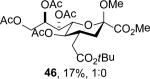
Isolated yield of pure 46 is only 17% owing to difficulties in isolation.
To facilitate interpretation Table 1 is organized in order of decreasing electron-withdrawing effect of their collective protecting group arrays. As observed previously23-24,29 isolated yields of the oxidative deamination products are typically in the range of 50% (Table 1). Inspection of each reaction mixture by mass spectrometry revealed a number of side products typically arising from i) formal elimination of the nitrosoacetamide function to the give an alkene [M - AcNHNO]+;28 ii) substitution of the nitrosoacetamide by trifluoroethoxide [M – AcNNO + CF3CH2O]+; and iii) substitution of the nitrosoacetamide by isopropoxide [M – AcNNO + iPrO]+. As the reaction mixtures were complex and purification of the byproducts tedious, isolation and characterization of such products was not undertaken. Exceptionally, however, for the tert-butyl ester 34, for which the isolated yield of the oxidative amination product 46 was only 17% (Table 1, entry 10), a sample of the alkene 47 was isolated in 6% yield and characterized. In general the KDN derivatives with the equatorial acetoxy group at C5 are distinguished from their 5-epi-KDN isomers with the axial group on the basis of the chemical shift and multiplicity of H5, the multiplicity of H4 and the chemical shift of the axial H3 in the 1H NMR spectra. On this basis, the absence of the 5-epimers in Table 1, entries 1-6 and 10 was readily apparent on inspection of the crude reaction mixtures by 1NMR spectroscopy.

Incorporation of 13C-Labelled Acetic Acid
The oxidative deamination of 26 was also conducted with acetic acid that was 99% enriched at the carbonyl carbon, resulting in the isolation of 35 carrying the labelled acetate group at the 5-position, with no significant loss of label as determined by mass spectrometry (Scheme 4). The identical experiment was conducted on the nitrosoamide 49, derived from the corresponding α-adamantanyl thioglycoside 48,21,24 with comparable clean and complete incorporation of the labelled acetate at the 5-position in the product 50 (Scheme 4). However, with the isomeric nitrosoamide 51,24 derived from the β-adamantanyl thioglycoside 52,21 mass spectrometric analysis of the crude reaction mixture and of the isolated KDN derivative revealed incorporation of a second isotopically labelled acetoxy group to the extent of approximately 30% (Scheme 4). Careful NMR analysis, particularly correlation of the carbonyl C of the minor enriched acetate with H4 in a HMBC experiment, located this second labelled acetate group at the 4-position of 53 (Scheme 4). The location of the labelled acetate group at the 5-position of the monolabeled products 13C1-35 and 50 was similarly confirmed by HMBC experiments.
Scheme 4.

Incorporation of 13C-Labelled Acetate.
Proton-Deuterium Exchange at the 5-Position
The nitrosylated amide 26 was subjected to the standard Zbiral conditions23-24,29 with the difference that all protic solvents and reagents employed were deuteriated at the exchangeable position. Thus, a solution of 26 and CF3CH2OD in dichloromethane was treated at −10 °C with a solution of sodium isopropoxide in i-PrOD followed by addition of CH3CO2D. Standard work up and chromatographic purification enabled the isolation of the monodeuterio KDN derivative 35-5d in which deuterium was fully and selectively incorporated at the 5-position (Scheme 5).
Scheme 5.

Deuterium Incorporation at the 5-Position of KDN.
Discussion
Mechanism of Oxidative Deamination
An mechanistic overview of the oxidative deamination containing the various hypothetical intermediates is presented in Scheme 6 using the per-O-acetyl N-nitrosoacetamide 26 as example. Trifluoroethoxide generated in situ from trifluoroethanol with sodium isopropoxide, or added as a prepared salt, serves as a mild nucleophile to bring about the selective deacetylation of the nitrosoamide 26 to give the hydroxydiazine 55.36 This step may take place either directly on 26, which is strongly activated because of the presence of the electron-withdrawing nitroso group, or via the intermediacy of the acetoxydiazene 54 with which it is considered to be in equilibrium.36 Other nucleophiles that have been used for this purpose in related systems include amine N-oxides,37 imidazole, and hydroxide.38 On acidification, either by the addition of suitably acidic nucleophile or by the addition of a Brønsted acid in the presence of a nucleophile,24 protonation and elimination of water occurs to give the diazonium ion 56 and its conjugate base the diazoalkane 57.36 These level 1 nitrogenous intermediates the undergo loss of molecular nitrogen to give one or other of the level 2 intermediates, the carbenium ion 58 and the carbene 59. These level two intermediates either proceed directly to the elimination byproducts28 64 and 65, and the substitution product 35, or are stabilized in the form of one or more of the level 3 intermediates 60-63, which ultimately afford the isolated reaction products. Our goal in initiating this project was to determine the reasons underlying the stereoretentive nature of the oxidative deamination reaction; accordingly the focus of this discussion is on the level 2 and level 3 intermediates.
Scheme 6.

Global Mechanism for Oxidative Deamination with Three Levels of Hypothetical Intermediates
The high level of incorporation of 13C enriched acetic acid in to the product 35 (Scheme 4) under the typical reaction conditions necessitates an intermolecular process rather than any intramolecular rearrangement with concomitant loss of nitrogen from 26, 54, or the ion pair 56. If the carbenium ion 58 or one of its stabilized forms 60, 61 and 63 are on the reaction pathway, the complete incorporation of deuterium at the 5-position when the reaction is conducted with deuterated reagents (Scheme 5) requires that the diazonium ion 56 and the diazoalkane 57 be in dynamic equilibrium. Alternatively the incorporation of deuterium (Scheme 5) can be accommodated by the intermediacy of the carbene 59, or its stabilized equivalent, the ylid 62. Other carbohydrate-based diazoalkanes generated from N-nitroacetamides have been previously shown to participate in typical carbene-type intermolecular C-H insertions.39-40 The very high levels of overall retention of configuration typically seen in these reactions, both past23-24,28-29,41 and present (Table 1, entries 1-6), argue against the formation of the substitution product 35 directly from the carbenium ion 58 or the carbene 59, although it remains possible that the elimination products 64 and 65 are formed from 58 or 59 by deprotonation or C-H insertion, respectively. Rather, it is likely that the origin of stereoselectivity lies with the intermediacy of one or other of the level 3 intermediates formed either directly from the level 1 intermediates 56 and 57 in a concerted manner or via the transient level 2 intermediates 58 and 59.
Stereodirecting neighboring group participation by esters through the intermediacy of dioxalenium ions is a well-established phenomenon.42-47 Comparable participation of more remote esters through six-membered dioxenium or larger cyclic ions is hotly debated in carbohydrate chemistry,48-52 but the premise of dioxenium ions as intermediates in simple systems rests securely on a labelling study.53 In the present oxidative deamination reaction participation through the dioxenium ion 61 would appear to be especially favorable in view of the extended zig-zag conformation of the side chain54-60 and the ideal location it imposes on the 7-O-acetyl group. Notwithstanding this precedent, the complete retention of configuration observed when the 4-O or 7-O-acetyl groups are replaced by benzyl ethers or a silyl group (Table 1, entries 4-6) excludes the need for ester participation through either a dioxalenium or dioxenium ion. That the 4-O and 7-O-tert-butoxycarbonyl protected systems 27 and 28 (Table 1, entries 2 and 3) also afford the standard substitution products in the typical yields with no indication of the type of cyclic carbonates50,61 anticipated from Boc participation corroborates this conclusion. Similarly, we could find no indication of lactone formation62 resulting from participation by the tert-butyl ester 34 (Table 1, entry 10) with loss of the tert-butyl group.
The evidence, therefore, points overwhelmingly toward participation by the ring oxygen as in the oxabicyclo[3.1.0]hexyl oxonium ion 63. This bicyclic oxonium ion may be formed directly from the diazonium ion 56, via the intermediacy of the carbenium ion 58, or by protonation of the carbene-derived ylid 62 (Scheme 6). Indeed, an early study on the oxidative deamination of 1, under conditions of simple thermal decomposition, reported the isolation of a ring-contracted furanosyl byproduct such as would arise from competing nucleophilic ring opening at the 6-position of 63, albeit neither characterization data nor a mechanism were advanced.28 Although we have not isolated and characterized such ring contraction products in the present study, it remains possible that they are present among the several byproducts formed in minor amounts. Kinetic evidence for the intermediacy of oxabicyclo[3.1.0]hexyl oxonium ions in the solvolysis of 2-oxa-6-norbornyl brosylates, in comparison to that of 2-norbornyl brosylates, is found in the work of Kirmse and Mrotzeck.63
When both the 4-O- and 7-O-acetyl groups are replaced by less electron-withdrawing benzyl ethers (32, Table 1, entry 7), minor stereochemical leakage is observed in that the still major equatorial acetate 41 was formed in a 4.5:1 ratio with its axial isomer 42. When additionally the 8-O- and 9-O-acetyl groups are replaced by a less electron-withdrawing isopropylidene group (33, Table 1, entry 8) the stereoselectivity of the reaction is fully compromised and the equatorial and axial acetates 43 and 44, respectively, are formed in equimolar quantities. These observations support the existence of an equilibrium between carbenium ion 66 and oxabicyclo[3.1.0]hexyl oxonium ions 67 (Scheme 7) with the latter favored under typical conditions in the presence of multiple electron-withdrawing protecting groups. With only two remote electron-withdrawing esters to destabilize it, as in 32, the carbenium ion is sufficiently populated to cause erosion of stereoselectivity. In the complete absence of electron-withdrawing esters, the carbenium ion is sufficiently stable that it dominates the stereochemical outcome of the reaction. The very high degree of retention of configuration observed with the branched carbon analog 34 (Table 1, entry 1) indicates that the presence of three electron-withdrawing esters in the side chain is sufficient to compensate for the replacement of the C4-O bond by the more electron-donating C-C bond such that the oxonium ion mechanism is still favored for substitution.
Scheme 7.

Equilibrating Carbenium and Bridged Oxonium Ions and the Influence of Protecting Groups.
In an attempt to prepare and store larger quantities of N-nitrosoamides, 33 was accessed on 400 mg scale and stored as a solid in the refrigerator at 4 °C. On examination after 12 days the sample was found to have decomposed to give the typical mixture of substitution and elimination products as determined by mass spectrometry. Inspection of the NMR spectra of the crude mixture of products revealed the substitution product to consist exclusively of the 5-epi-KDN derivative 44, which was isolated as the derivative 45 in 38% yield after removal of the isopropylidene group and acetylation (Table 1, entry 10). Because it takes place in the solid phase, this rearrangement necessarily arises by an intramolecular process and most likely involves passage through an ion pair related to the level 1 intermediate 56 (Scheme 6) with concerted displacement of nitrogen by the incoming acetate. The possibility of an intramolecular SN2 process at the level of the Z-isomer of the acetoxydiazene related to 54, formally a 6-endo-tet process,64 is considered unlikely on the grounds of the endocyclic restriction test.65-66 It is noteworthy that the corresponding peracetyl N-nitrosoamide 26 did not undergo decomposition on standing in the refrigerator at 4 °C for one month, thereby reaffirming the role of protecting groups on the stability of the nitrosoamides.
Comparison with the 4-Deoxy-4-acetamidoglucopyranoside and 2-Deoxy-2-acetamidoglucopyranoside Series
Comparison with older studies on the oxidative deamination of 4- and 2-acetamidopyranosides is instructive. Nitrous acid-promoted deamination of methyl 4-amino-4-deoxy-α-d-glucopyranoside gives methyl α-d-glucopyranoside as the major product together with methyl β-L-altrofuranoside and 4,5-anhydro-d-galactose, all of which are suggestive of the involvement of the ring oxygen and of a bicyclic oxonium ion intermediate (Scheme 8). A further product of this reaction, 3-deoxy-3-C-formyl α-d-xylofuranoside, arises from the migration of C2.67 Although product distributions were substrate and protecting group dependent, comparable products were observed in the nitrous acid-mediated deamination of methyl 4-amino-4-deoxy-2,3-O-isopropylidene-α-d-rhamnopyranoside.68 Methyl 4-amino-4,6-dideoxy-d-mannopyranoside is also reported to give D-rhamnopyranose on nitrous acid deamination and hydrolysis.69 On the other hand, methyl 4-amino-4,6-dideoxy-2,3-O-isopropylidene-α-d-talopyranoside, with an axial amine at the 4-position, does not undergo ring contraction to a significant extent under the same conditions, implying the absence of participation by the ring oxygen, but gives mainly the products of substitution with inversion of configuration.68 The analogous dependence on configuration is observed in the NO-mediated deamination of the 4-amino-4-deoxyglycopyranoside residues of various bacterial oligosaccharides.70
Scheme 8.

Deamination Reactions in the 4-Amino-4-deoxyglucose Series
The ring contraction of 4-O-sulfonyl derivatives of rhamnopyranose in the presence of azide in DMF at reflux also is known to give the 5,6-dideoxy-5-azido-talofuranose skeleton as the major product requiring overall inversion at C4 and retention (double inversion) at C5.71-73 As with the deamination reactions, this ring contraction with inversion of configuration at C4 in suggests participation by the ring oxygen leading again to an intermediate oxabicyclo[3.1.0]hexyl oxonium ion (Scheme 9).
Scheme 9.

Ring Contraction of a Rhamnosyl 4-O-Sulfonate
Nitrosylation of N-acetyl-β-d-glucosamine tetraacetate gives an isolable N-nitroso acetamide that on standing undergoes decomposition to give β-d-glucose pentaacetate, by a process considered to involve rearrangement to the diazoacetate and subsequent SNi, possibly with participation by the ring oxygen.74-75 On standing in chloroform N-acetyl-N-nitroso-α-d-glucosamine tetraacetate, but not its β-anomer, similarly affords β-d-glucose pentacetate leading to the suggestion of neighboring group participation and ultimate migration by the axial anomeric acetate.76 Alternatively, treatment of N-acetyl-N-nitroso-α- or β-d-glucosamine tetraacetate with potassium isopropoxide followed by acetylation gives 3,4,5-tri-O-acetyl-1,2-dideoxy-d-erythro-pent-1-ynitol by a process considered to involve diazoalkane formation, loss of nitrogen to give the carbene, hydrogen migration and C1-C2 bond cleavage (Scheme 10).74-75 In aqueous acetone both anomers of N-acetyl-N-nitroso-d-glucosamine tetraacetate undergo ring contraction with inversion of configuration at C2 to give triacetyl-2,5-anhydro-d-mannose (chitose) implying transannular attack by the ring oxygen and the intermediacy of an oxabicyclo[3.1.0]hexyl oxonium ion (Scheme 10).76 N-Acetylglucosamine and its glycosides undergo the analogous rearrangement on N-nitrosylation;77 the process has been employed in the depolymerization of glycoaminoglycans70 and in the cleavage of ring I from the aminoglycoside antibiotic paromomycin.78
Scheme 10.

Deamination Reactions in the Glucosamine Series
Overall, a consistent pattern emerges in which a tetrahydropyran carrying an equatorial leaving group undergoes transannular participation by the ring oxygen with inversion of configuration to give an 1-oxoabicyclo[3.1.0]hexyl oxonium ion (Scheme 11). In most cases participation by the ring oxygen predominates over participation by ester substituents on the tetrahydropyran framework. The subsequent chemistry of the 1-oxoabicyclo[3.1.0]hexyl oxonium ion is dictated by the reaction conditions and above all by the substituent pattern. Nucleophilic attack at the bridgehead carbon leads to the substitution product with overall inversion of configuration, whereas nucleophilic attack in the smaller bridge affords ring contraction products. The partitioning between these two pathways depends on the overall substituent pattern, the nucleophile, and the reaction conditions. In the sialic acid series, with the larger more heavily substituted side chain in the small bridge, attack at the bridgehead predominates (Table 1 and Scheme 6). Conversely, in the 4-aminoglucosyl series and related compounds (Schemes 8 and 9) with a smaller and less electron-withdrawing side chain in the small bridge, ring contraction is more common. In the 2-aminoglucosyl series (Scheme 10), the small bridge is the anomeric carbon and ring contraction is facilitated by the presence of glycosidic oxygen.
Scheme 11.

1-Oxabicyclo[3.1.0]hexyl Oxonium Ion Formation and Opening.
Influence of the anomeric Substituent and Configuration
Finally we return to the formation of the doubly labelled KDN derivative 13C2-53 identified as byproduct on use of 13 C-labelled acetic acid as nucleophile in the oxidative deamination of the β-thiosialoside 51, and the absence of comparable products in the reactions of the epimeric substrate 48 or the O-glycoside 26 (Scheme 4). We suggest that incorporation of the label at the 4-position is best explained by the series of equilibria outlined in Scheme 12. Thus the diazoalkane A, whose formation is dictated by the deuterium incorporation experiment (Scheme 6), exists in equilibrium with the zwitterion B arising from participation by the axial thioglycoside. O→N Acetate transfer then occurs via a six-membered cyclic transition state from O4 to give the N-acyl diazene C, which undergoes deacetylation on reaction with the labelled acetic acid present in the reaction mixture to give D and 13C-labelled acetic anhydride. The reverse process returns either A or 13C1-A depending on which of the two carbonyl groups in the mono 13C-labelled acetic anhydride serves as electrophile. With the label introduced at O4, 13C1-A then enters the standard mechanism in parallel with its isotopomer A leading overall to the formation of 13C2-53 and 13C1-53, respectively.
Scheme 12.

Incorporation of 13C-Labelled Acetate at O4 in the Axial Thioglycoside 53.
Conclusion
The stereoretentive oxidative deamination of the N-nitroso-N-acetyl sialyl glycosides derives its stereospecificity from participation by the pyranosyl ring oxygen through the intermediacy of a 1-oxabicyclo[3.1.0]hexyl oxonium ion and not via participation by esters at either the 4- or 7-positions. In general the oxidative deamination of the sialic acids and the 4-amino-4-deoxy- and 2-amino-2-deoxy glycopyranosides proceeds through a common pathway with the differences in product distribution, mainly substitution verses ring contraction, determined by the substituents at the level of the 1-oxabicyclo[3.1.0]hexyl oxonium ion intermediate. A minor reaction pathway in the β-thiosialosides has been identified by isotopic labelling studies in which the 4-O-acetate undergoes exchange with external acetic acid in the medium, for which a mechanism is proposed.
Supplementary Material
Acknowledgments
We thank the NIH (GM62160) for support of this work, and acknowledge support from the NSF (MRI-084043) for the purchase of the 600 MHz NMR spectrometer in the Lumigen Instrument Center at Wayne State University.
Footnotes
Supporting Information Available. Full experimental details and copies of 1H and 13C NMR spectra for all new compounds. This information is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Angata T, Varki A. Chem. Rev. 2002;102:439–469. doi: 10.1021/cr000407m. [DOI] [PubMed] [Google Scholar]
- 2.Chen X, Varki AP. ACS Chem. Biol. 2010;5:163–176. doi: 10.1021/cb900266r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varki A, Gagneux P. Ann. N. Y. Acad. Sci. 2012;1253:16–36. doi: 10.1111/j.1749-6632.2012.06517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crocker PR, Kelm S. In: Carbohydrates in Synthesis and Biology. Ernst B, Hart GW, Sinaÿ P, editors. Vol. 4. Wiley-VCH; Weinheim: 2000. pp. 579–596. [Google Scholar]
- 5.Thomson R, von Itzstein M. In: Carbohydraye-Based Drug Discovery. Wong C-H, editor. Vol. 2. Wiley-VCH; Weinheim: 2003. pp. 831–861. [Google Scholar]
- 6.Hsu Y, Ma H-H, Lico LS, Jan J-T, Fukase K, Uchinashi Y, Zulueta MML, Hung S-C. Angew. Chem. Int. Ed. 2014;53:2413–2416. doi: 10.1002/anie.201309646. [DOI] [PubMed] [Google Scholar]
- 7.El-Deeb IM, Guillon P, Winger M, Eveno T, Haselhorst T, Dyason JC, von Itzstein M. J. Med. Chem. 2014;57:7613–7623. doi: 10.1021/jm500759v. [DOI] [PubMed] [Google Scholar]
- 8.Rillahan CD, Schwartz E, McBride R, Fokin VV, Paulson JC. Angew. Chem. Int. Ed. 2012;51:11014–11018. doi: 10.1002/anie.201205831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiefel MJ, von Itzstein M. Chem. Rev. 2002;102:471–490. doi: 10.1021/cr000414a. [DOI] [PubMed] [Google Scholar]
- 10.Smith PW, Starkey ID, Howes PD, Sollis SL, Keeling SP, Cherry PC, von Itzstein M, Wu WY, Jin B. Eur, J. Med. Chem. 1996;31:143–150. [Google Scholar]
- 11.Wang Q, Guo Z. ACS Med. Chem. Lett. 2011;2:373–378. doi: 10.1021/ml100313d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pazynina G, Tyrtysh T, Nasonov V, Belyanchikov I, Paramonov A, Malysheva N, Zinin AI, Kononov LO, Bovin N. Synlett. 2013:226–230. [Google Scholar]
- 13.Horstkorte R, Keppler OT, Reutter W. In: Carbohydrate-Based Drug Discovery. Wong C-H, editor. Vol. 2. Wiley-VCH; Weinheim: 2003. pp. 863–873. [Google Scholar]
- 14.Chokhawala HA, Huang S, Lau K, Yu H, Cheng J, Thon V, Hurtado-Ziola N, Guerrero JA, Varki A, Chen X. ACS Chem. Biol. 2008;3:567–576. doi: 10.1021/cb800127n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem. Biol. 2012;7:1232–1240. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rillahan CD, Macauley MS, Schwartz E, He Y, McBride R, Arlian BM, Rangarajan J, Fokin VV, Paulson JC. Chem. Sci. 2014;5:2398–2406. doi: 10.1039/C4SC00451E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starkey ID, Mahmoudian M, Noble D, Smith PW, Cherry PC, Howes PD, Sollis SL. Tetrahedron Lett. 1995;36:299–302. [Google Scholar]
- 18.Tanaka H, Nishiura Y, Takahashi T. J. Am. Chem. Soc. 2006;128:7124–7125. doi: 10.1021/ja0613613. [DOI] [PubMed] [Google Scholar]
- 19.Farris MD, De Meo C. Tetrahedron Lett. 2007;48:1225–1227. [Google Scholar]
- 20.Crich D, Li W. J. Org. Chem. 2007;72:2387–2391. doi: 10.1021/jo062431r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crich D, Li W. J. Org. Chem. 2007;72:7794–7797. doi: 10.1021/jo7012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu C-H, Chu K-C, Lin Y-S, Han J-L, Peng Y-S, Ren C-T, Wu C-Y, Wong C-H. Chem. Eur. J. 2010;16:1754–1760. doi: 10.1002/chem.200903035. [DOI] [PubMed] [Google Scholar]
- 23.Crich D, Navuluri C. Angew. Chem. Int. Ed. 2010;49:3049–3052. doi: 10.1002/anie.200907178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Navuluri C, Crich D. Angew. Chem. Int. Ed. 2013;52:11549–11552. doi: 10.1002/anie.201303781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isemura M, Schmid K. Biochem. J. 1971;124:591–604. doi: 10.1042/bj1240591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mononen I. Carbohydr. Res. 1981;88:39–50. [Google Scholar]
- 27.Inoue S, Kitajima K. Glycoconj. J. 2006;23:277–290. doi: 10.1007/s10719-006-6484-y. [DOI] [PubMed] [Google Scholar]
- 28.Shirai R, Nakamura M, Hara S, Takayanagi H, Ogura H. Tetrahedron Lett. 1988;29:4449–4452. [Google Scholar]
- 29.Schreiner E, Zbiral E. Liebigs Annalen der Chemie. 1990:581–586. [Google Scholar]
- 30.Kuhn R, Lutz P, Macdonald DL. Chem. Ber. 1966;99:611–617. doi: 10.1002/cber.19660990235. [DOI] [PubMed] [Google Scholar]
- 31.Chopra P, Thomson RJ, Grice D, von Itzstein M. Tetrahedron Lett. 2012;53:6254–6256. [Google Scholar]
- 32.Baggett N, Marsden BJ. Carbohydr. Res. 1982;110:11–18. doi: 10.1016/0008-6215(82)85022-2. [DOI] [PubMed] [Google Scholar]
- 33.Zbiral E, Phadtare S, Schmid W. Liebigs Ann. Chem. 1987:39–43. [Google Scholar]
- 34.Hartmann M, Zbiral E. Monat. Chem. 1989;120:899–906. [Google Scholar]
- 35.Iversen T, Bundle DR. J. Chem. Soc., Chem. Commun. 1981:1240–1241. [Google Scholar]
- 36.Kirmse W. Angew. Chem. Int. Ed. 1976;15:251–320. [Google Scholar]
- 37.Nikolaides N, Godfrey AG, Ganem B. Tetrahedron Lett. 1990;31:6009–6012. [Google Scholar]
- 38.Challis BC, Jones SP. J. Chem. Soc., Perkin Trans. 1979;2:703–706. [Google Scholar]
- 39.Sarabia F, Martín-Ortiz L, López-Herrera FJ. Org. Biomol. Chem. 2003;1:3716–3725. doi: 10.1039/b307674a. [DOI] [PubMed] [Google Scholar]
- 40.Sarabia-García F, López-Herrer FJ. Tetrahedron. 1996;52:4757–4768. [Google Scholar]
- 41.Zunk M, Williams J, Carter J, Kiefel MJ. Org. Biomol. Chem. 2014;12:2918–2925. doi: 10.1039/c3ob42491j. [DOI] [PubMed] [Google Scholar]
- 42.Frush HL, Isbell HS. J. Research Natl. Bur. Standards. 1941;27:413–428. [Google Scholar]
- 43.Winstein S, Grunwald E, Ingraham LL. J. Am. Chem. Soc. 1948;70:821–828. [Google Scholar]
- 44.Capon B, McManus SP. Neighboring Group Participation. Plenum; New York: 1976. [Google Scholar]
- 45.Paulsen H, Herold C-P. Chem. Ber. 1970;103:2450–2462. [Google Scholar]
- 46.Crich D, Dai Z, Gastaldi S. J. Org. Chem. 1999;64:5224–5229. doi: 10.1021/jo990424f. [DOI] [PubMed] [Google Scholar]
- 47.Nukada T, Berces A, Zgierski MZ, Whitfield DM. J. Am. Chem. Soc. 1998;120:13291–13295. [Google Scholar]
- 48.Komarova BS, Ustyuzhania NE, Tsvetkov YE, Nifantiev NE. In: Modern Synthetic Methods in Carbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates. Werz DB, Vidal S, editors. Wiley; Weinheim: 2014. pp. 125–160. [Google Scholar]
- 49.Kim KS, Suk D-H. Top. Curr. Chem. 2011;301:109–140. doi: 10.1007/128_2010_107. [DOI] [PubMed] [Google Scholar]
- 50.Crich D, Hu T, Cai F. J. Org. Chem. 2008;73:8942–8953. doi: 10.1021/jo801630m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baek JY, Kwon H-W, Myung SJ, Park JJ, Kim MY, Rathwell DCK, Jeon HB, Seeberger PH, Kim KS. Tetrahedron. 2015;71:5315–5320. [Google Scholar]
- 52.Ma Y, Lian G, Li Y, Yu B. Chem. Commun. 2011;47:7515–7517. doi: 10.1039/c1cc11680k. [DOI] [PubMed] [Google Scholar]
- 53.Wilen SH, Delguzzo L, Saferstein R. Tetrahedron. 1987;43:5089–5094. [Google Scholar]
- 54.Grindley TB. In: Glycoscience: Chemistry and Chemical Biology. Fraser-Reid B, Tatsuta K, Thiem J, editors. Vol. 1. Springer; Berlin: 2001. pp. 3–51. [Google Scholar]
- 55.Christian R, Schulz G, Brandstetter HH, Zbiral E. Carbohydr. Res. 1987;162:1–11. doi: 10.1016/0008-6215(87)80195-7. [DOI] [PubMed] [Google Scholar]
- 56.Brown EB, Brey WS, Jr., Weltner W., Jr. Biochim. Biophys. Acta. 1975;399:124–130. doi: 10.1016/0304-4165(75)90218-4. [DOI] [PubMed] [Google Scholar]
- 57.Flippen JL. Acta Cryst. 1973;B29:1881–1886. [Google Scholar]
- 58.Veluraja K, Rao VSR. Biochim. Biophys. Acta. 1980;630:442–446. doi: 10.1016/0304-4165(80)90293-7. [DOI] [PubMed] [Google Scholar]
- 59.Sabesan S, Bock K, Lemieux RU. Can. J. Chem. 1984;62:1034–1045. [Google Scholar]
- 60.Kancharla PK, Crich D. J. Am. Chem. Soc. 2013;135:18999–19007. doi: 10.1021/ja410683y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bartlett PA, Meadows JD, Brown EG, Morimoto A, Jernstedt KK. J. Org. Chem. 1982;47:4013–4018. [Google Scholar]
- 62.Wen P, Crich D. J. Org. Chem. 2015;80:12300–12310. doi: 10.1021/acs.joc.5b02203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kirmse W, Mrotzeck U. Chem. Ber. 1988;121:485–492. [Google Scholar]
- 64.Baldwin JE. J. Chem. Soc., Chem. Commun. 1976:734–736. [Google Scholar]
- 65.Tenud L, Farooq S, Seibl J, Eschenmoser A. Helv. Chim. Acta. 1970;53:2059–2069. [Google Scholar]
- 66.Beak P. Acc. Chem. Res. 1992;25:215–222. [Google Scholar]
- 67.Ng Ying Kin NMK, Williams JM, Horsington A. J. Chem. Soc. C. 1971:1578–1583. [Google Scholar]
- 68.Al-Radhi AK, Brimacombe JS, Tucker LCN. J. Chem. Soc, Perkin Trans. 1972;1:315–320. [Google Scholar]
- 69.Lee C-H, Schaffner CP. Tetrahedron Lett. 1966;7:5837–5840. doi: 10.1016/s0040-4039(00)76093-x. [DOI] [PubMed] [Google Scholar]
- 70.Duan J, Kasper DL. Glycobiology. 2011;21:401–409. doi: 10.1093/glycob/cwq171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stevens CL, Glinski RP, Taylor KG, Blumbergs P, Sirokman F. J. Am. Chem. Soc. 1966;88:2073–2074. [Google Scholar]
- 72.Hanessian S. Chem. Comm. 1966:796–798. [Google Scholar]
- 73.Stevens CL, Glinski RP, Taylor KG, Sirokman F. J. Org. Chem. 1970;35:592–596. [Google Scholar]
- 74.Horton D, Loh W. Carbohydr. Res. 1974;36:121–130. doi: 10.1016/s0008-6215(00)81997-7. [DOI] [PubMed] [Google Scholar]
- 75.Horton D, Loh W. Carbohydr. Res. 1974;38:189–203. doi: 10.1016/s0008-6215(00)81997-7. [DOI] [PubMed] [Google Scholar]
- 76.Llewellyn JW, Williams JM. Carbohydr. Res. 1973;28:339–350. [Google Scholar]
- 77.Horton D, Philips KD. Carbohydr. Res. 1973;30:367–374. [Google Scholar]
- 78.Cassinelli G, Julita P, Arcamone F. J. Antibiotics. 1978;31:382–384. doi: 10.7164/antibiotics.31.382. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.