

Abstract
Purpose
The specialized corneal epithelium requires differentiated properties, specific for its role at the anterior surface of the eye. Thus, tight maintenance of the differentiated qualities of the corneal epithelial is essential. Pinin (PNN) is an exon junction component (EJC) that has dramatic implications for corneal epithelial cell differentiation and may act as a stabilizer of the corneal epithelial cell phenotype. Our studies revealed that PNN is involved in transcriptional repression complexes and spliceosomal complexes, placing PNN at the fulcrum between chromatin and mRNA splicing. Transcriptome analysis of PNN-knockdown cells revealed clear and reproducible alterations in transcript profiles and splicing patterns of a subset of genes that would significantly impact the epithelial cell phenotype. We further investigated PNN’s role in the regulation of gene expression and alternative splicing (AS) in a corneal epithelial context.
Methods
Human corneal epithelial (HCET) cells that carry the doxycycline-inducible PNN-knockdown shRNA vector were used to perform RNA-seq to determine differential gene expression and differential AS events.
Results
Multiple genes and AS events were identified as differentially expressed between PNN-knockdown and control cells. Genes upregulated by PNN knockdown included a large proportion of genes that are associated with enhanced cell migration and ECM remodeling processes, such as MMPs, ADAMs, HAS2, LAMA3, CXCRs, and UNC5C. Genes downregulated in response to PNN depletion included IGFBP5, FGD3, FGFR2, PAX6, RARG, and SOX10. AS events in PNN-knockdown cells compared to control cells were also more likely to be detected, and upregulated. In particular, 60% of exon-skipping events, detected in only one condition, were detected in PNN-knockdown cells and of the shared exon-skipping events, 92% of those differentially expressed were more frequent in the PNN knockdown.
Conclusions
These data suggest that lowering of PNN levels in epithelial cells results in dramatic transformation in the number and composition of splicing variants and that PNN plays a crucial role in the selection of which RNA isoforms differentiating cells produce. Many of the genes affected by PNN knockdown are known to affect the epithelial phenotype. This window into the complexity of RNA splicing in the corneal epithelium implies that PNN exerts broad influence over the regulation and maintenance of the epithelial cell phenotype.
Introduction
The responsibilities of the corneal epithelium require it to maintain differentiated properties, specific for its role at the anterior surface of the eye. Thus, tight regulation and maintenance of the differentiated qualities are essential. Many pathologies of the anterior surface can result in a transformation of the epithelium to a keratinized epithelium, which is a vastly inadequate epithelium for the corneal surface and the avascular cornea. Our studies have recently focused on pinin (PNN), a component of the exon junction complex (EJC), which has dramatic implications for corneal epithelial cell differentiation. We examine in detail the changes in gene expression and splicing events in human corneal epithelial (HCET) cells [1] that carry the doxycycline-inducible PNN-knockdown shRNA vector, to address how PNN may coordinate gene expression and RNA processing in corneal epithelial cells.
Our earlier studies focused on how PNN impacted epithelial biology. Overexpressing PNN in cultured epithelial cells affects epithelial homeostasis and, in turn, drives the epithelial cells to a hyperstable adhesive state and inhibits the transition from quiescence to migratory [2]. However, shRNAi-mediated knockdown of PNN expression leads to a loss of epithelial cell–cell adhesion, changes in cell shape, and movement of cells out of the epithelium [3]. Mice with specific Cre-mediated deletions of PNN (Gene ID:5411; OMIM 603154) revealed perturbations in epithelial differentiation and epithelial phenotype. For example, knocking PNN out in the developing lung interfered with branching morphogenesis and alveolar differentiation, while knocking PNN out in the intestine blocked villi formation [4]. Finally, conditional inactivation of PNN in the anterior eye (lens-cre) resulted in severe disruption in corneal epithelial differentiation [5,6]. Taken together, these data contributed significant support to the hypothesis that PNN may act as a stabilizer of the corneal epithelial cell phenotype.
Based on PNN’s association with several splicing factors, we have determined that PNN is peripherally associated with the EJC [7-10]. Purification of the PNN complex identified several proteins, including components of splicing and transcription (SRSF1, 3 and 4, Dead-box helicases, FUS bp-1, MAGOH, and SAP18). We now know that transcription and splicing are not distinct molecular processes but are tied together and affected by the chromatin structure [11-16]. Interestingly, recent studies identified PNN in complex with two Pro-Trp-Trp-Pro (PWWP) - domain-containing chromatin readers, BS69 [17] and PSIP1 [18], which bind to H3K36me3 [19]. These data are crucial in that they place PNN at the fulcrum between chromatin and mRNA splicing. Perturbations in RNA splicing, through manipulations of splicing-related proteins, such as PNN, may exert broad influence on the regulation of gene expression by impacting RNA diversity through RNA processing and RNA turnover.
Alternative splicing (AS) affects almost all multiexon genes; thus, AS, is one of the main drivers of protein diversity [20]. The estimated 20,000 genes encoded by the human genome are expanded tenfold by AS. It has also been proposed that 50% of disease-causing mutations result in disruption of normal splicing patterns [21]. We are also just now gaining an appreciation of the diversity and relevance of splicing to corneal epithelial biology. We hypothesized that the corneal epithelial identity is linked to AS and vice versa. Coordinated mRNA isoform switching has been observed as cells progress to the differentiated cell populations, resulting in isoform specialization. Interestingly, many genes that encode critical regulators of eye development, for example, OCT4/OCT4a (Gene ID: 5460; OMIM 164177), FOXP1 (Gene ID: 27086; OMIM 605515), FGF4/FGF4si (Gene ID: 2249; OMIM 164980), and PAX6/PAX6(5a) (Gene ID: 5080; OMIM 607108), exhibit isoform-switching phenomena [22-24].
It has been well documented that the coordination and execution of proper RNA processing are sensitive to the levels of expression of core and peripheral splicing factors, such as the EJC components [12,25,26]. Hypomorphic PNN 3f/3f animals displayed broad epithelial defects due to the knockdown instead of the knockout of PNN within the developing mouse embryo, including anterior segment dysgenesis with frequent persistence of the lens stalk (similar to the Peters anomaly [6]. Here we report the evaluation of PNN-knockdown cells revealed clear alterations in transcript profiles, RNA variant switching and splicing patterns of specific subsets of genes, including matrix metalloproteinases, ECM components, cell adhesion-related molecules, and regulators of differentiation. Gene Ontology (GO) analyses of PNN-regulated genes and splicing events indicate that the induced downregulation of PNN leads to loss of differentiation and enhanced cell migration.
Methods
Cell lines
These studies employed an SV40-immortalized human corneal epithelial cell line (HCET), with properties similar to normal corneal epithelial cells, such as cornea-specific keratins and abundant aldehyde dehydrogenase activity [1]. Cell lines were authenticated using short tandem repeat (STR) analysis as described in 2012 in ANSI Standard (ASN-0002) Authentication of Human Cell Lines: Standardization of STR Profiling by the ATCC Standards Development Organization (SDO) and by Capes-Davis et al. [27]. Seventeen STR loci plus the gender-determining locus, amelogenin, were amplified using the commercially available PowerPlex® 18D Kit from Promega, Madison, WI. The cell line sample was processed using the ABI Prism® 3500×l Genetic Analyzer. Data were analyzed using GeneMapper® ID-X v1.2 software (Applied Biosystems, Inc., Grand Island, NY). Appropriate positive and negative controls were run and confirmed for each sample submitted. The HCET cell line used in this study (ATCC FTA bar code STRA0571) was found to be human and a match to two other HCET cell lines, RCB1384 and RCB2280, in the DSMZ STR database (Appendix 1).
For doxycycline-inducible PNN-knockdown HCET cells, human TRIPZ shRNAmir clones (V2THS_170187) were purchased from Open Biosystems (Lafayette, CO). HCET cells transfected with either shRNAmir clone were then selected with 10 μg/ml of puromycin (#61-385-RA, Cellgro, Manassas, VA). To induce shRNA expression, the cells were treated with doxycycline (Doxy) at a concentration of 1 μg/ml with a daily change of fresh doxycycline medium. Using western blots, we detected the typical knockdown at 48 h post-Doxy, D3447, Sigma Aldrich, St. Louis, MO in the range of 46–75% reduction.
RNA-seq library preparation
RNA-seq libraries were prepared beginning with total RNA (RIN 8.60–10) extracted from control and PNN knockdown HCET cells, using RNeasy Plus Mini Kit (Qiagen, Valencia, CA). Before the RNA-seq library was constructed, rRNA was removed from the total RNA samples using an Illumina Ribo-Zero kit. Following rRNA depletion, RNA-seq library construction used ScriptSeq Complete v2 (Illumina, San Diego, CA). The ScriptSeq protocol used sequence-tagged random hexamer priming to produce RNA-seq libraries that retained the identity of the original transcribed strand.
DNA sequence determination
Each library was barcoded and then quantified. The libraries were then pooled into a single pool, and the pool was run on four independent lanes [28] of the Illumina NextSeq 500. Quality control was performed using a combination of FASTQC and Lab Scripts that count the number of duplicate reads, the number of reads with an adaptor, and the number of reads with homopolymer sequences. The results indicated the data were of good quality and all of the libraries were comparable. The native Illumina bcl DNA sequence output format was converted to the fastq format and demultiplexed by index reads using bcl2fastq version 2.15 (Illumina). The average number of reads per sample was about 40 million (average 39,336,132.75 reads/sample), and the distribution of read numbers is given in Appendix 2. All of the reads have been deposited in the SRA (accession # SRP063860).
Read alignments for viewing in UCSC Genome Browser
Paired-end reads acquired as described were processed using Trimmomatic version 0.32 [29] to remove oligonucleotide adaptors used during RNA-seq library construction and to trim low-quality sequences. DNA sequence reads that had a minimum length of 50 nt were retained for downstream processing. After filtering, an average of 35 million paired reads were retained per sample. Paired reads were mapped to the GRCh37 human reference genome (Genome) using STAR aligner version 2.0.4c [30] with default mapping parameters. Mapped paired reads were filtered to remove duplicates using the Genome Analysis Toolkit [31]. SAMtools [32] was used to sort mapped reads by chromosomal coordinate, create, and index human chromosome-specific map files, and convert the file format from SAM to BAM. The bam2wig.py Python script from RSeQC [33] was used to create chromosome-specific wig-format files, which were uploaded for viewing in the University of California, Santa Cruz (UCSC) Genome Browser.
The stranded RNA-seq data were aligned to the GRCh37/hg19 version (release 73) of the complete human genome using the BWA-MEM algorithm [34]. Genomic coverage was summarized using AceView annotations [35]. There were 678,664 exons in this annotation, and the genome positions of approximately 68% of these exons overlapped with at least one other exon (e.g., alternative donor/acceptor sites). Overlapping exons in the same direction were combined, and the minimum start and maximum end positions were used to estimate expression in 322,096 unique exonic regions. For ease, these exonic regions are referred to as exons throughout the text. Exons were determined to be “expressed” if they had coverage in at least three samples per group.
Annotating alternative splicing events
We created a complete, annotated catalog of splicing events from genomic feature databases (GFF files) using gene, transcript, and exon-level references for RNA-seq alignments and coverage calculations. We used the latest (2010) release of AceView gene models for the hg19 human genome build [35,36]. A list of possible exon–exon junctions was generated from all logical combinations of exon pairs within a gene in a 5′-to-3′ manner.
For AS event expression analysis, RNA-seq data were aligned to the sequences of all exon junctions and retained intron events using Bowtie [37]. AS event coverage was summarized using the annotations defined above. AS events were deemed “expressed” if they had more than ten aligned reads in at least three samples per group.
We fit the model Yij=μ + tj + ϵij, where Yij is the log2-transformed average depth per nucleotide (APN) for exon or AS event i, sample j, tj is the treatment status of sample j, and ϵij is the random error. A false discovery rate (FDR) was used to correct results for multiple testing [38], and a level of 0.05 was considered to be significant.
Gene ontology analysis
Gene Ontology was analyzed and demonstrated using GOrilla a tool for identifying and visualizing enriched GO terms in ranked lists of genes. GOrilla searches for enriched GO terms that appear densely at the top of a ranked list of genes. The system utilizes 13,033 genes that are associated with a GO term GO database and other sources. Graphical representation of the GO results was accomplished with the Graphviz layout programs, which take descriptions of graphs in a simple text language and create diagrams in useful formats.
Droplet digital PCR
Total RNA was isolated from cultured HCET cells, and semiquantitative reverse transcription (RT)-PCR was performed as described previously [39,40], with RNeasy Plus Mini Kit (#74134, Qiagen, Valencia, CA) and treated with RNase-free DNase I (#79254, Qiagen). One microgram of total RNA was reverse transcribed with the SuperScript III First-Strand Synthesis kit (#18080051, Invitrogen, Carlsbad, CA) using oligo-dT primers. The droplet digital PCR (ddPCR) assays were performed according to the Bio-Rad User Guide. Briefly, each of the 20 μl reactions contained 10 μl ddPCR Supermix (Bio-Rad, Hercules, CA), 250 nM gene-specific primers, and 0.5 μl of the cDNA sample (about 5 ng). Primer sequences for PNN, MMP1 (Gene ID:4312; OMIM 120353), MMP9 (Gene ID:4318; OMIM 120361), MMP13 (Gene ID:4322; OMIM 600108), VEGF (Gene ID:7422; OMIM 192240), and GAPDH are listed in Appendix 3. Each reaction was mixed with 20 μl of Droplet Generation Oil (Bio-Rad), partitioned into about 20,000 droplets in a QX200 Droplet Generator (Bio-Rad), transferred to 96-well plates (Eppendorf, Hauppauge, NY), and sealed. PCR was performed in a PTC-100 Thermal Cycler (Bio-Rad) with the following cycling conditions: 1 × (95 °C for 5 min); 40 cycles: (95 °C for 30 s, 60 °C for 60 s); 1 × (4 °C for 5 min; 90 °C for 5 min); 4 °C (hold). The fluorescence intensity of individual droplets was measured with the QX200 Droplet Reader (Bio-Rad). The data analysis was performed with QuantaSoft droplet reader software (Bio-Rad). Positive and negative droplet populations were detected automatically. The target mRNA concentrations were calculated using the Poisson statistics and background-corrected based on the no template control data. The absolute transcript levels were initially computed in [copies/μl PCR] and in [copies/ng cDNA]. In the latter case, the correction for the input cDNA amount was applied to each sample. The data are presented as expression levels normalized to GAPDH expression, p value, and log2 fold change, for MMP1, MMP9, MMP13, VEGF-A, and PNN.
Immunofluorescence
Doxycycline-inducible PNN-knockdown HCET cells were plated on coverslips in six-well dishes at 2×105 cells per well in a Dulbecco's Modification Eagle's Medium/Ham's F-12 50/50 mix, (#10-090-CV, Cellgro, Manassas, VA); 5% fetal bovine serum [FBS, #35-011-CV, Cellgro], 5 μg/ml insulin, 0.1 μg/ml cholera toxin (C8052, Sigma Aldrich), 10 ng/ml human epidermal growth factor [hEGF, PHG0311, Gibco, Frederick, MD], 0.5% dimethyl sulfoxide [DMSO, D2650, Sigma Aldrich]). After 48 h treatment in media with and without doxycycline (1 μg/ml), cells were fixed in freshly prepared 2% paraformaldehyde (D2650, Sigma Aldrich) in PBS (1X; 137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.4) for 1 h, quenched in 50 mM ammonium chloride (A-661, Fisher Scientific), 0.1%Tween-20 (BP-337, Fisher Scientific) in PBS for 10 min, rinsed and incubated for 2 h at room temperature in 1:200 rabbit anti-MMP9 antibody (#13667, Cell Signaling, Danvers, MA). After rinsing with PBS, the cells were incubated in 1:200 fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit antibody (#F6005, Sigma-Aldrich, St. Louis, MO) for 1 h. The coverslips were mounted in VectaShield with 4',6-diamidino-2-phenylindole (DAPI; #H1200, Vector Labs, Burlingame, CA) and examined using a Leica inverted fluorescent microscope (Model DM-IRBE, Leica Microsystems, Buffalo Grove, IL) and OpenLab (Improvision, Coventry, England) software.
Results
RNA-seq and GO analysis
To evaluate the effect of PNN on mRNA expression and RNA processing on a global scale, we performed mRNA-seq experiments in HCET cells in which the levels of PNN were reduced with specific siRNA. RNA-seq-based transcriptional profiling was employed to compare Wt-HCET cells with doxycycline-inducible PNN-knockdown HCET cells (50% knockdown). Data were deposited in GEO under accession number GSE73060. The RNA-seq profiles of 36,275 transcripts were obtained, out of which 90 are significant at P-adj (p<0.01). Of these, 61 genes exhibited >1.0 log2 fold change, and 28 genes showed <–1.0 log2 fold change. The P-adj is an adjusted p value, taking into account the FDR, which is the proportion of the total rejections that are false positives [41]. The FDR has been widely used for multiple test corrections in various applications, such as when measuring thousands of variables (e.g., gene expression levels) from a small sample set [42]. Table 1 and Table 2 depict the top 20 significant genes with the largest fold changes (supplementary data provided in Appendix 4 and Appendix 5). GO analyses were performed with Gorilla, a tool for identifying and visualizing enriched GO terms in ranked lists of genes [43,44]. Table 3 and Table 4 show the GO analyses for upregulated and downregulated genes, respectively. The upregulated genes included a large proportion of genes that are associated with enhanced cell migration and ECM remodeling processes, including MMPs, ADAMs, HAS2, LAMA3, and UNC5C (Table 3, Figure 1 and supplementary data Appendix 6). In addition, the upregulated genes included members of the CXCR chemokine receptor binding family, metalloendopeptidase activities, and extracellular matrix organization and structure. GO analysis revealed that a significant subset of genes, which impact cellular differentiation, were included in those that exhibited downregulation, such as IGFBP5, FGD3, FGFR2, PAX6, RARG, and SOX10 (Table 4). Taken together, these downregulated genes may contribute to the loss in corneal epithelial differentiation seen in downregulation and knockouts of PNN [3-6,39].
Table 1. Genes upregulated in response to PNN reduction.
| Gene symbol | P-adj | Log2FoldChange | Gene name |
|---|---|---|---|
| ALPK2 |
1.12E-41 |
3.85 |
alpha-kinase 2 |
| UBC |
7.48E-33 |
2.73 |
ubiquitin C |
| MMP13 |
1.69E-28 |
2.63 |
matrix metallopeptidase 13 (collagenase 3) |
| TMPRSS11E |
3.82E-28 |
4.26 |
transmembrane protease, serine 11E |
| MMP1 |
7.32E-20 |
2.22 |
matrix metallopeptidase 1 (interstitial collagenase) |
| BNIP3P4 |
5.40E-14 |
3.54 |
BCL2/adenovirus E1B 19 kDa interacting protein 3 pseudogene 4 |
| EGLN1 |
2.93E-13 |
2.83 |
egl-9 family hypoxia-inducible factor 1 |
| SLC7A11 |
1.09E-10 |
1.57 |
solute carrier family 7 (anionic amino acid transporter light chain, xc- system), member 11 |
| CEACAM6 |
2.38E-10 |
2.12 |
carcinoembryonic antigen-related cell adhesion molecule 6 (non-specific cross reacting antigen) |
| CNGB1 |
5.10E-09 |
1.47 |
cyclic nucleotide gated channel beta 1 |
| NEFL |
1.38E-08 |
3.12 |
neurofilament, light polypeptide |
| MYH15 |
2.83E-08 |
1.76 |
myosin, heavy chain 15 |
| PSAT1 |
2.96E-08 |
1.61 |
phosphoserine aminotransferase 1 |
| KRT6A |
7.54E-08 |
2.98 |
keratin 6A |
| ANKRD22 |
7.54E-08 |
1.70 |
ankyrin repeat domain 22 |
| MMP10 |
1.42E-06 |
2.17 |
matrix metallopeptidase 10 (stromelysin 2) |
| B3GNT2 |
7.58E-06 |
1.19 |
UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 2 |
| ADORA2B |
1.82E-05 |
1.22 |
adenosine A2b receptor |
| LOC102723439 |
1.86E-05 |
1.62 |
uncharacterized LOC102723439 |
| MYO1E | 3.19E-05 | 1.20 | myosin IE |
Table 2. Genes downregulated in response to PNN reduction.
| Gene symbol | P-adj | Log2FoldChange | Gene name |
|---|---|---|---|
| ABCA4 |
2.33E-15 |
−2.14 |
ATP-binding cassette, sub-family A (ABC1), member 4 |
| ABCA4 |
3.78E-14 |
−2.46 |
ATP-binding cassette, sub-family A (ABC1), member 4 |
| FGD3 |
1.50E-09 |
−2.02 |
FYVE, RhoGEF and PH domain containing 3 |
| INSL4 |
2.80E-06 |
−1.82 |
insulin-like 4 (placenta) |
| IGFL1 |
4.07E-06 |
−1.68 |
IGF-like family member 1 |
| IGFBP5 |
3.46E-05 |
−2.05 |
insulin-like growth factor binding protein 5 |
| VGLL1 |
3.46E-05 |
−1.85 |
vestigial-like family member 1 |
|
PNN |
3.46E-05 |
-1.14 |
pinin |
| UBD |
3.99E-05 |
−1.77 |
ubiquitin D |
| CFI |
9.25E-05 |
−1.61 |
complement factor I |
| PCSK9 |
9.25E-05 |
−1.27 |
proprotein convertase subtilisin/kexin type 9 |
| SLC1A3 |
0.00024818 |
−1.04 |
solute carrier family 1 (glial high affinity glutamate transporter), member 3 |
| DHRS3 |
0.00057796 |
−1.28 |
dehydrogenase/reductase (SDR family) member 3 |
| VGLL1 |
0.00132719 |
−1.65 |
vestigial-like family member 1 |
| CDH5 |
0.00132719 |
−1.49 |
cadherin 5, type 2 (vascular endothelium) |
| RPS24 |
0.00133766 |
−0.99 |
ribosomal protein S24 |
| LINC01559 |
0.00180566 |
−1.05 |
long intergenic non-protein coding RNA 1559 |
| RPS24 |
0.00238888 |
−0.94 |
ribosomal protein S24 |
| CLEC7A |
0.00263968 |
−1.49 |
C-type lectin domain family 7, member A |
| RARRES3 | 0.00294625 | −1.07 | retinoic acid receptor responder (tazarotene induced) 3 |
Table 3. Gene Ontology of genes upregulated in response to PNN reduction.
| GO term | Description | P-value | FDR q-value | Enrichment | Genes |
|---|---|---|---|---|---|
|
Biologic Process | |||||
| GO:0030334 |
regulation of cell migration |
3.28E-08 |
3.79E-04 |
3.73 |
MMP10, EGFR, CXCL3, MMP9, STC1, CXCL1, UNC5C, HAS2, CXCL2, TRIB1, STRAP, LAMA3 |
| GO:0006520 |
cellular amino acid metabolic process |
9.41E-08 |
5.44E-04 |
4.8 |
EGLN1, PSAT1, PLA2G4E, GARS, MTHFD2L, GLS |
| GO:0030198 |
extracellular matrix organization |
1.03E-07 |
3.98E-04 |
5.09 |
MMP13, MMP1, MMP10, MYO1E, ANXA2, MMP9, SERPINb5, CD44, LAMC!, HAS2, LAMC2, LAM3 |
| GO:0043062 |
extracellular structure organization |
1.03E-07 |
2.98E-04 |
5.09 |
MMP1, TIMP1, ACTN1. LAMC2, MMP13, TNC, SERPINB5, CD47, CD44, MMP9, ITGA2, SDC4 |
|
Molecular Function | |||||
| GO:0044822 |
poly(A) RNA binding |
2.03E-09 |
6.63E-06 |
1.63 |
UBC, ANXA2, NQo1, NAA38, RPS16, RRP12, STRAP |
| GO:0045236 |
CXCR chemokine receptor binding |
2.18E-08 |
2.37E-05 |
64.04 |
CXCL3, CXCL1, CXCL2, YARS |
| GO:0004222 | metalloendopeptidase activity | 1.27E-06 | 1.03E-03 | 25.32 | MMP10, ADAMS6, ADAM19, MMP13, MMP1, MMP9 |
Table 4. Gene Ontology of genes downregulated in response to PNN reduction.
| GO term | Description | P-value | FDR q-value | Enrichment | Genes |
|---|---|---|---|---|---|
|
Biologic Process | |||||
| GO:0044283 |
small molecule biosynthetic process |
3.65E-06 |
4.26E-03 |
2.41 |
SLC1A3, MNGCR, EBP, DHCR7,CYP51A1, FADS2, PADI2, HMGSC1 |
| GO:1901624 |
negative regulation of lymphocyte chemotaxis |
9.55E-06 |
9.28E-03 |
57.75 |
KLRK1, CCL2, PADI2 |
| GO:0002376 |
immune system process |
3.52E-05 |
2.28E-02 |
1.37 |
UBD, CFI, RPS24, CLEC7A, IFITM2, ADCY5, IL7R, KITLG |
| GO:2000402 |
negative regulation of lymphocyte migration |
5.61E-05 |
3.27E-02 |
43.31 |
CCL2, PADI2, KLRK1 |
| GO:0045595 |
regulation of cell differentiation |
5.94E-05 |
3.30E-02 |
1.41 |
IGFBP5, IL7R, KITLG |
| GO:0050793 |
regulation of developmental process |
9.10E-05 |
4.82E-02 |
1.33 |
FGD3, IGFB5, COLQ, HMGCR, |
| GO:0032808 | lacrimal gland development | 9.85E-05 | 5.00E-02 | 16.49 | RARG, FGFR2, PAX6, SOX10 |
Figure 1.
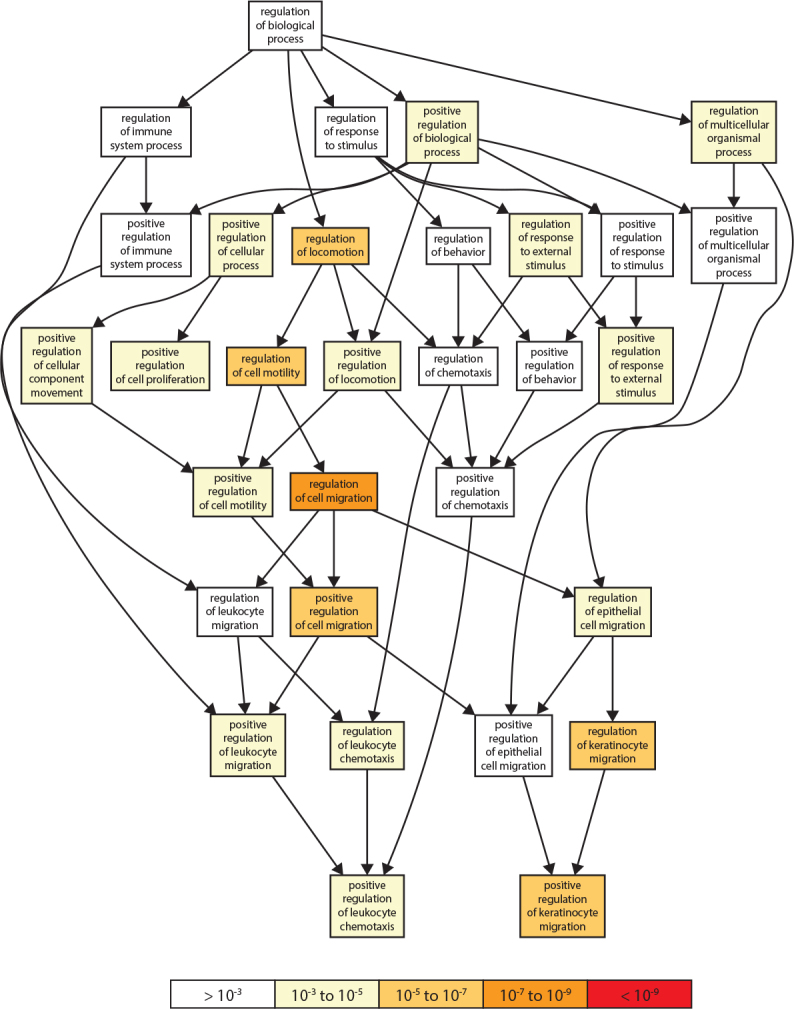
Gene Ontology of genes upregulated in response to PNN knockdown and graphical output of GOrilla for the ranked list of genes upregulated after PNN knockdown. This Gene Ontology (GO) graph generated by GOrilla for identifying and visualizing enriched GO terms in ranked lists of upregulated GOrilla searches for enriched GO terms that appear densely at the top of a ranked list of genes. The system used 13,033 genes that are associated with a GO term GO database and other sources. This figure represents a small portion of the output depicting the pathways associated with enhanced cell migration. The genes identified in this cell behavior are presented in Table 3.
The upregulation of MMPs and VEGF mRNA, observed via RNA-seq subsequent to PNN knockdown, was confirmed with quantitative PCR, using droplet digital technology [45]. The downregulation of PNN (log2 fold change, −1.93) was noted at 48 h post-doxycycline induction of PNN-shRNA, as was significant upregulation of MMP1 (log2 fold change, 1.69), MMP9 (log2 fold change, 0.78), MMP13 (log2 fold change, 2.0) and VEGF-A (log2 fold change, 0.49) at 48 h post-Doxy (Table 5). Immunofluorescence microscopy confirmed the upregulation of MMP9 protein in HCETs 48 h after Doxy treatment (Figure 2). Interestingly, when downregulation of PNN was less (log2 fold change, −0.82), there was no observed significant upregulation of MMP1 (log2 fold change, −0.02) or MMP9 (log2 fold change, 0.23), but significant changes were still observed for MMP13 (log2 fold change, 1.73) and VEGF-A (log2 fold change, 0.43) at 48 h post-Doxy (supplementary data in Appendix 7). Although outside the scope of this manuscript, we suggest that these data may reflect a dosage effect of PNN knockdown. Future experiments will explore the dosage response to PNN knockdown, as well as the differential sensitivity seen across the MMPs.
Table 5. Verification of expression changes by droplet digital pcr quantitation.
| Variables | PNN | MMP1 | MMP9 | MMP13 | VEGFa |
|---|---|---|---|---|---|
| Control* |
0.061 |
0.0098 |
0.0025 |
0.0068 |
0.0111 |
| PNN knock down* |
0.016 |
0.0316 |
0.0043 |
0.0272 |
0.0156 |
| P value |
0.0001 |
0.0014 |
0.0093 |
0.0008 |
0.0012 |
| log2 Fold change | -1.93 | 1.69 | 0.78 | 2 | 0.49 |
*Data presented as relative to GAPDH
Figure 2.

MMP9 protein upregulated after PNN knockdown in HCET cells. Immunofluorescence micrographs depicted the upregulation of the MMP9 protein in human corneal epithelial (HCET) cells after pinin (PNN) knockdown.
Annotating alternative splicing events
The evaluation of the RNA-seq analyses of the PNN-knockdown cells revealed the remarkable dynamics of RNA processing of the expressed genes coding and non-coding RNAs. The results are broadly consistent with previous observations from hGlue3_0 transcriptome array analyses cGH arrays [39,40]. One of the main advantages of RNA-seq is the ability to identify and quantify AS events like exon–exon junctions and retained introns. We created a complete, annotated catalog of all possible splicing events using genomic feature databases (GFF files) used when building gene, transcript, and exon-level references used for RNA-seq alignments and coverage calculations. Annotations for each event were classified based on four main alternative splicing event types: alternative donor, alternative acceptor, skipped exon, and retained intron.
For this study, we used the latest (2010) release of AceView gene models for the hg19 human genome build, due to the noted higher accuracy of AceView gene models as well as the higher abundance of gene models per gene compared with Ensembl and RefSeq [35,36]. Our annotation method described here is not specific to the AceView annotations. The scripts to build the AS catalog are available at GitHub and can be used for any existing database of genomic feature annotations.
A list of possible exon–exon junctions were generated from all logical combinations of exon pairs within gene in a 5′-to-3′ manner. Junctions were not generated if they were not biologically plausible (e.g., an exon donor site to an earlier exon acceptor site or a junction between two overlapping exons), nor were junctions created between genes. To facilitate the classification of AS events, overlapping exons were grouped together, and the longest exon in each group was arbitrarily assigned as a reference exon and used to define alternative donor and acceptor sites within each exon group. There were several occurrences where two non-overlapping exons overlapped with another exon. In this case, the longest exon per group was used as the reference, and the junction between the two internal exons was created. We classified junctions as exon skipping if there was at least one exonic region between the exons two exons forming a junction. For each exon-skipping junction, we also generated a list of possible skipped exons. Skipped exons were included on this list if they were unambiguously skipped (i.e., exons overlapping the donor or acceptor exon were not included). We also annotated junctions that were part of at least one known transcript. Intron retention (IR) events were generated by using the donor site of an exon and extending the sequence into the neighboring intron. For groups of overlapping exons, we generated IR events only from the 3′-most exon to ensure that only a uniquely intronic sequence was used for the intronic segment of the event. In total, there were 6,390,703 possible AS junctions and 232,249 IR events annotated in this database: 5,837,064 junctions were annotated as an exon-skipping junction, 1,240,520 junctions were classified as having an alternative donor, 1,257,096 junctions were classified as having an alternative acceptor, and 903,513 junctions were classified as having an alternative donor and an alternative acceptor. In addition, 292,753 junctions were annotated to at least one known transcript. The database is available on Github.
Figure 3 summarizes the impact of PNN knockdown on alternative splicing in HCET cells. We plotted the mean expression (as log2 APN) of AS events detected in PNN knockdown and control cells (58,817 of 80,813 total detected splicing events) and noted that the expression of AS events was generally higher in the PNN-knockdown cells compared to controls (Figure 3). In total, we observed that 5,063 AS events were differentially expressed, with 4,683 AS events (92.5%) significantly higher after PNN knockdown and the remaining 380 significant AS events higher (7.5%) in the control cells, indicating that reduction of PNN expression results in upregulation of AS events (p<0.05; Figure 3). For alternative splicing events detected in both conditions and differentially expressed, all types of AS events were more likely to be upregulated in PNN knockdown HCET cells: 92.5% of all AS events, 91.8% of exon-skipping events (635 AS events were unregulated of the 692 that were significantly different), 94.1% of alternative donor sites (531 AS events were unregulated of the 564 that were significantly different), 93.0% of alternative receptor sites (490 AS events were unregulated of the 527 that were significantly different), 93.7% of AS events involving alternative donors and acceptors (554 AS events were unregulated of the 591 that were significantly different), and 62.9% of intron retention events (95 IR events were unregulated of the 151 that were significantly different). The detailed list of differentially expressed events is presented in the supplementary data (Table 6 and supplementary data in Appendix 8). The knockdown of PNN resulted in a dramatic increase in the number of splicing events, suggesting that altering levels of PNN has a broad influence on splicing. This widespread impact has been observed after the knockdown of other members of the exon junction complex [25]. Interestingly, genes that were upregulated by PNN knockdown displayed a much greater increase in altered splicing events than genes not changed or downregulated. These data speak to the coupling of splicing to transcription.
Figure 3.
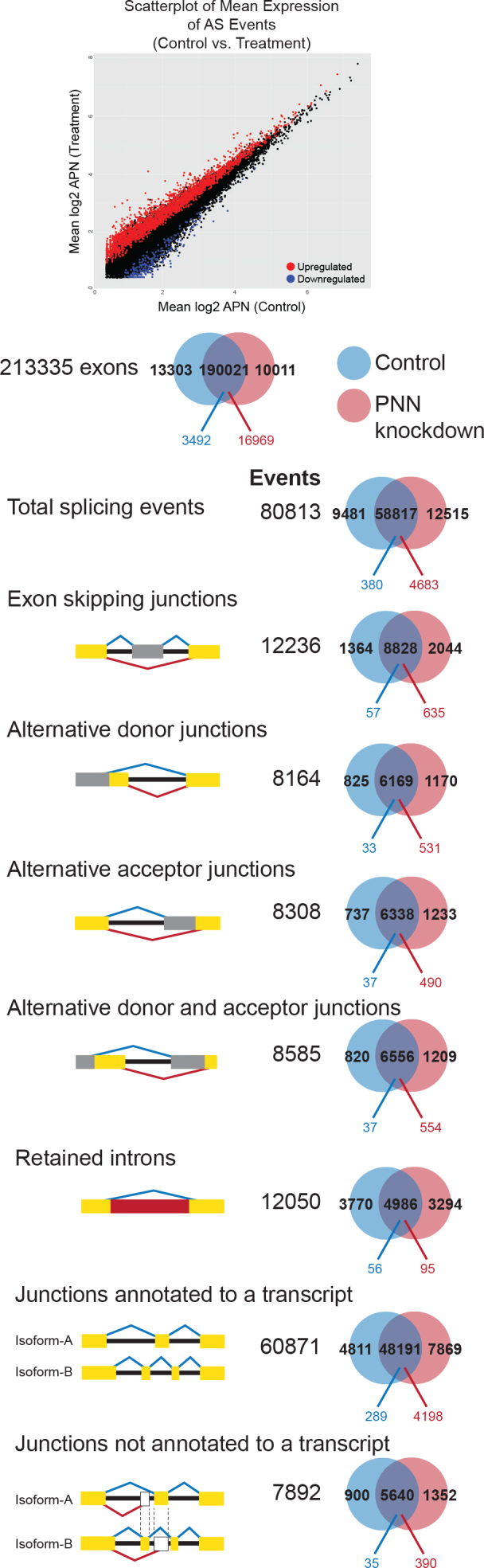
Widespread changes in splicing events after PNN knockdown. A scatterplot of splicing events detected in pinin (PNN) knockdown and control cells (the overlapping area in the Venn diagrams), plotting the mean expression (as log2 APN) of the control cells versus the PNN-knockdown cells. Differentially expressed events are red (upregulated) and blue (downregulated), and non-differentially expressed events are in black. More than 70% of significantly different events are upregulated in PNN (p<0.0001). All types of events are biased toward upregulation in PNN (92.5% of all events, 91.8% of exon-skipping events (635 of 692 differentially expressed [DE] events), 94.1% of alternative donor sites (531 of 564 DE events), 93.0% of alternative receptor sites (490 of 527 DE events), 93.7% of events involving alternative donors and acceptors (554 of 591 DE events), and 62.9% of intron retention events (95 of 151 DE events).
Table 6. RNA Splicing events.
| Gene | Event | Description | P-value | Log2 Fold Change |
|---|---|---|---|---|
| RAD54L2andTEX264 |
EJ |
E-E j. between exon(s) 22 and exons(s) 23 of RAD54L2andTEX264 |
0.0001 |
1.28 |
| KIAA0368andOR2K2 |
EJ |
E-E j. between exon(s) 11|13 and exons(s) 14|15 of KIAA0368andOR2K2 |
0.0001 |
1.31 |
| BUB1 |
EJ |
E-E j. between exon(s) 28|29 and exons(s) 30 of BUB1 |
0.0001 |
1.66 |
| ITGA6 |
EJ |
E-E j. between exon(s) 7|8 and exons(s) 9 of ITGA6 |
0.0001 |
1.73 |
| MARK3 |
EJ: AD, AA |
E-E j. between exon(s) 26 and exons(s) 29 of MARK3 |
0.0001 |
1.74 |
| ERO1L |
EJ |
E-E j. between exon(s) 11|12 and exons(s) 13 of ERO1L |
0.0001 |
1.76 |
| NPAS2 |
EJ: AD |
E-E j. between exon(s) 6|8|9 and exons(s) 10 of NPAS2 |
0.0001 |
1.95 |
| MALT1 |
EJ |
E-E j. between exon(s) 9 and exons(s) 10 of MALT1 |
0.0001 |
2.23 |
| NEDD9 |
EJ |
E-E j. between exon(s) 6 and exons(s) 7|8 of NEDD9 |
0.0002 |
0.27 |
| FAM73A |
EJ |
E-E j. between exon(s) 12 and exons(s) 13 of FAM73A |
0.0002 |
1.30 |
| PRKDC |
EJ: AD |
E-E j. between exon(s) 96 and exons(s) 98 of PRKDC |
0.0002 |
1.36 |
| RDX |
EJ |
E-E j. between exon(s) 22 and exons(s) 23 of RDX |
0.0002 |
1.52 |
| IFT140 |
EJ |
E-E j. between exon(s) 31 and exons(s) 32 of IFT140 |
0.0002 |
1.55 |
| RDX |
EJ: ES [1], AD |
E-E j. between exon(s) 18 and exons(s) 21 of RDX, skipping 1 exon |
0.0002 |
1.60 |
| LAMC1 |
EJ |
E-E j. between exon(s) 33|34 and exons(s) 35 of LAMC1 |
0.0002 |
1.60 |
| MYO1E |
EJ |
E-E j. between exon(s) 19 and exons(s) 20 of MYO1E |
0.0002 |
1.64 |
| RPS5 |
EJ: ES [1] |
E-E j. between exon(s) 7|9 and exons(s) 12|13|14 of RPS5, skipping 1 exon |
0.0002 |
1.70 |
| SNRNP200 |
EJ |
E-E j. between exon(s) 52 and exons(s) 53 of SNRNP200 |
0.0002 |
1.80 |
| EPRS |
EJ |
E-E j. between exon(s) 27 and exons(s) 28 of EPRS |
0.0002 |
1.90 |
| KIAA1632 | EJ | E-E j. between exon(s) 13 and exons(s) 14 of KIAA1632 | 0.0002 | 1.94 |
(E-E j.=exon-exon junction, EJ=exon junction, ES=exon-skipping, AD=alternative donor, AA=alternative acceptor, IR=intron retention)
Transcript assembly and quantification with RNA-seq revealed bone fide isoform switching and the generation of unannotated transcripts subsequent to PNN knockdown. Table 7 (more complete list, supplementary data (Appendix 9) reveals the observed splicing events (p<0.05) associated with splicing variants after PNN knockdown. The upregulated variant and log fold change are listed on the left, while the downregulated variant and fold change are listed on the right in the table. Interestingly, in addition to variants in the ADAM family (membrane disintegrin and metalloproteinase), many splicing variations occur in the abundant solute carrier (SLC) group of membrane transport proteins after PNN knockdown. The observed high correlation with SLC group members may reflect the abundance of SLC mRNA and proteins in mammals.
Table 7. Splicing events correlated to change in transcripts known splice isoforms.
| Genes | P-value | Gene description | Log Fold change | Gene description | Log fold change |
|---|---|---|---|---|---|
| NF2 |
0.0129 |
NF2, transcript variant 5 |
3.87 |
NF2, transcript variant 8 |
−0.71 |
| PCNX |
0.0099 |
PCNX, transcript variant X10 |
3.59 |
PCNX, transcript variant X2 |
−1.67 |
| SRPK2 |
0.043 |
SRPK2, transcript variant 2 |
3.32 |
SRPK2, transcript variant X3 |
−2.68 |
| FAM73A |
0.0139 |
FAM73A, transcript variant 1 |
3.09 |
FAM73A, transcript variant X3 |
−2.86 |
| MTHFD2L |
0.0179 |
MTHFD2L, transcript variant X2 |
2.73 |
MTHFD2L, transcript variant X1 |
−0.38 |
| ABHD2 |
0.0242 |
ABHD2, transcript variant X1 |
2.51 |
ABHD2, transcript variant X2 |
−1.34 |
| C1orf27 |
0.0196 |
C1orf27, transcript variant 3 |
2.17 |
C1orf27, transcript variant X4 |
−1.29 |
| NAE1 |
0.0055 |
NAE1, transcript variant X2 |
1.93 |
NAE1, transcript variant 2 |
−0.99 |
| SMEK2 |
0.0288 |
SMEK2, transcript variant X6 |
1.80 |
SMEK2, transcript variant X7 |
−0.09 |
| FBXO38 |
0.0356 |
FBXO38, transcript variant 3 |
1.75 |
FBXO38, transcript variant X3 |
−1.17 |
| FKBP14 |
0.0362 |
FKBP14, transcript variant 2, non-coding RNA |
1.74 |
FKBP14, RefSeqGene (LRG_454) on chromosome 7 |
−0.02 |
| GTF2H1 |
0.0475 |
GTF2H1, transcript variant 2 |
1.64 |
GTF2H1, transcript variant X1 |
−0.50 |
| HUWE1 |
0.0004 |
HUWE1, transcript variant X5 |
1.59 |
HUWE1, transcript variant X2 |
−0.28 |
| SLC38A9 |
0.0457 |
SLC38A9, transcript variant X2 |
1.59 |
SLC38A9, transcript variant X8, misc_RNA |
−0.62 |
| SLC7A11 |
0.0174 |
SLC7A11 |
1.57 |
SLC7A11 transcript variant X1 |
−0.04 |
| SRGAP2 |
0.0091 |
SRGAP2, transcript variant X7 |
1.47 |
SRGAP2, RefSeqGene on chromosome 1 |
−0.22 |
| ADAM19 |
0.0346 |
ADAM19 |
1.47 |
ADAM19, transcript variant X1 |
−0.23 |
| FANCL |
0.0354 |
FANCL, transcript variant X5, misc_RNA |
1.42 |
LRG_501 on chromosome 2 |
−0.35 |
| FOSL1 | 0.0433 | FOSL1, transcript variant 1 | 1.40 | FOSL1, transcript variant 5 | −1.39 |
RNA splicing occurs cotranscriptionally, and we hypothesize that PNN may serve as a functional bridge between transcription and RNA splicing. Therefore, we examined the correlation of change in gene expression and the occurrence of altered RNA splicing. Table 8 and Table 9 and supplementary data (Appendix 10) show the splicing events (p<0.05) that are correlated with upregulated and downregulated genes, respectively. Most interestingly, a large proportion of the upregulated genes, which GO analyses indicate are involved in enhanced cell migration such as MMPs, ADAMs, SERPINs, ECM, and matrix receptors, also demonstrated altered RNA splicing. Although the list of downregulated genes with correlated alteration RNA events is smaller, of significant note is the change in splicing events of two unexpected cadherins in PNN knockdown, endothelial cadherin 5 and minor cadherin 11. Although the roles of these cadherins in the corneal epithelial context is uncertain, these data are similar to that we reported previously for the coregulation of transcription and splicing of E-cadherin by PNN [46] and may indicate a more global role of PNN in RNA processing on cadherin genes.
Table 8. Splicing events correlated to upregulated genes.
| Genes | P-value | Gene description | GENE UP Pval | Log Fold change |
|---|---|---|---|---|
| MMP13 |
0.0003 |
matrix metallopeptidase 13 (collagenase 3; MMP13) |
1.08E-32 |
2.63 |
| MMP1 |
0.0002 |
matrix metallopeptidase 1 (interstitial collagenase; MMP1), transcript variant 1 |
7.83E-24 |
2.22 |
| MYH15 |
0.0214 |
myosin, heavy chain 15 (MYH15) |
1.03E-11 |
1.76 |
| PSAT1 |
0.0009 |
phosphoserine aminotransferase 1 (PSAT1), transcript variant 1 |
1.14E-11 |
1.61 |
| ANKRD22 |
0.0035 |
ankyrin repeat domain 22 (ANKRD22) |
3.11E-11 |
1.70 |
| MYO1E |
0.0001 |
myosin IE (MYO1E) |
1.84E-08 |
1.20 |
| ADAMTS6 |
0.0067 |
ADAM metallopeptidase with thrombospondin type 1 motif, 6 (ADAMTS6) |
3.67E-08 |
1.12 |
| FERMT1 |
0.0089 |
fermitin family member 1 (FERMT1) |
3.93E-08 |
1.39 |
| EGFR |
0.0005 |
epidermal growth factor receptor (EGFR), transcript variant 1 |
5.97E-08 |
1.49 |
| EFNB2 |
0.002 |
ephrin-B2 (EFNB2) |
0.000000136 |
1.06 |
| AGPAT9 |
0.0115 |
1-acylglycerol-3-phosphate O-acyltransferase 9 (AGPAT9), transcript variant 1 |
0.000000144 |
3.05 |
| SLC20A1 |
0.0034 |
solute carrier family 20 (phosphate transporter), member 1 (SLC20A1) |
0.000000388 |
1.44 |
| STC2 |
0.0131 |
stanniocalcin 2 (STC2) |
0.000000727 |
1.00 |
| XPOT |
0.003 |
exportin, tRNA (XPOT) |
0.00000128 |
0.98 |
| GARS |
0.0083 |
glycyl-tRNA synthetase (GARS), transcript variant X1 |
0.00000147 |
0.97 |
| STC1 |
0.0032 |
stanniocalcin 1 (STC1) |
0.00000168 |
1.11 |
| NQO1 |
0.0055 |
NAD(P)H dehydrogenase, quinone 1 (NQO1), transcript variant 1 |
0.00000456 |
1.33 |
| CD55 |
0.0039 |
CD55 molecule, decay accelerating factor for complement (Cromer blood group; CD55), transcript variant 1 |
0.00000608 |
1.08 |
| SERPINB5 |
0.0015 |
serpin peptidase inhibitor, clade B (ovalbumin), member 5 (SERPINB5) |
0.00000683 |
0.90 |
| TMTC2 |
0.0446 |
transmembrane and tetratricopeptide repeat containing 2 (TMTC2), transcript variant X1 |
0.00000708 |
1.68 |
| CD44 | 0.041 | CD44 molecule (Indian blood group; CD44), RefSeqGene on chromosome 11 | 0.000014 | 0.87 |
Table 9. Splicing events correlated to downregulated genes.
| Genes | P-value | Gene description | GENE DOWN Pval | Log Fold change |
|---|---|---|---|---|
| FGD3 |
0.0044 |
FYVE, RhoGEF and PH domain containing 3 (FGD3), transcript variant X2, misc_RNA |
4.16E-13 |
−2.02 |
| CDH5 |
0.0141 |
cadherin 5, type 2 (vascular endothelium; CDH5), mRNA |
0.00000179 |
−1.49 |
| CASP1 |
0.0096 |
caspase 1, apoptosis-related cysteine peptidase (CASP1), RefSeqGene on chromosome 11 |
0.003233781 |
−0.85 |
| C3 |
0.0033 |
complement component 3 (C3), mRNA |
0.004117402 |
−0.57 |
| RDH10 |
0.0204 |
retinol dehydrogenase 10 (all-trans; RDH10), mRNA |
0.009398676 |
−0.70 |
| NEO1 |
0.041 |
neogenin 1 (NEO1), RefSeqGene on chromosome 15 |
0.00995951 |
−0.55 |
| CDH11 |
0.0139 |
cadherin 11, type 2, OB-cadherin (osteoblast; CDH11), RefSeqGene on chromosome 16 |
0.01161935 |
−0.52 |
| PCTP |
0.0151 |
phosphatidylcholine transfer protein (PCTP), transcript variant X4, misc_RNA |
0.014216733 |
−1.20 |
| SP110 |
0.0143 |
SP110 nuclear body protein (SP110), RefSeqGene (LRG_109) on chromosome 2 |
0.015998943 |
−0.50 |
| NIPSNAP1 |
0.0173 |
nipsnap homolog 1 (C. elegans; NIPSNAP1), transcript variant X1, mRNA |
0.016613623 |
−0.47 |
| BCAS1 |
0.0022 |
breast carcinoma amplified sequence 1 (BCAS1), transcript variant X9, mRNA |
0.025350687 |
−4.06 |
| GABRP |
0.0273 |
gamma-aminobutyric acid (GABA) A receptor, pi (GABRP), transcript variant 2, mRNA |
0.031249095 |
−2.79 |
| SYCE1L |
0.0262 |
synaptonemal complex central element protein 1-like (SYCE1L), mRNA |
0.032750969 |
−0.98 |
| PROM1 |
0.0258 |
prominin 1 (PROM1), RefSeqGene on chromosome 4 |
0.038456406 |
−0.54 |
| BRAF |
0.029 |
B-Raf proto-oncogene, serine/threonine kinase (BRAF), RefSeqGene (LRG_299) on chromosome 7 |
0.042463081 |
−0.53 |
| SIAE |
0.0287 |
sialic acid acetylesterase (SIAE), RefSeqGene on chromosome 11 |
0.047026974 |
−0.54 |
| VIPR1 |
0.0068 |
vasoactive intestinal peptide receptor 1 (VIPR1), RefSeqGene on chromosome 3 |
0.047041988 |
−0.49 |
| BAZ2B | 0.0057 | bromodomain adjacent to zinc finger domain, 2B (BAZ2B), transcript variant 3, non-coding RNA | 0.047636931 | −0.73 |
Discussion
The way in which we view the transcriptome has undergone a revolution with the advent of massively parallel DNA sequencing associated with robust computational approaches. Recent RNA-seq efforts have demonstrated that AS generates broad variation in functional mRNAs, in a cell-specific and stage-specific manner [47]. AS includes cassette exon-skipping, alt 5′ and 3′ ends, mutually exclusive exon inclusions, and, more rarely, IRs. We have focused on the AS changes that are central to establishing and maintaining the corneal epithelial phenotype and maintenance of the normal physiology of the cornea and anterior segment [2-4].
We explored the involvement of PNN in gene expression and alternative pre-mRNA splicing in the corneal epithelial context, by using HCET cells that harbored doxycycline-inducible shRNA against PNN. Whole transcriptome RNA-seq analysis of PNN knockdown HCET cells revealed clear and reproducible alterations in the transcript profiles and splicing patterns of specific subsets of genes and RNA splicing complexities, including MMPs, ADAMs, HAS2, LAMA3, and UNC5C. Most interestingly, transcript assembly and quantification by RNA-seq revealed bone fide isoform switching and generation of unannotated transcripts subsequent to PNN knockdown. A large proportion of the upregulated genes, such as ADAMs, SERPINs, ECM, and matrix receptors, also demonstrated altered RNA splicing. Importantly, our work has implicated RNA processing, itself, as a potential regulator of epithelial identity.
Strikingly, PNN knockdown resulted in the upregulation of genes that have been shown to be associated with molecular processes that contribute to tissue plasticity, enhanced cell migration, and ECM remodeling, including MMPs, ADAMs, HAS2, LAMA3, UNC5C, and members of the CXCR chemokine receptor binding family. These data are consistent with our previous work pertaining to PNN’s impact on epithelial phenotype stabilization [3-6,39] and epithelial cell migration [2,48]. We also previously demonstrated that varied epithelial-derived cancers demonstrate absent or greatly reduced expression of PNN, and methylation analyses revealed that aberrant methylation of PNN CpG islands was correlated with decreased/absent PNN expression in a subset of tumor tissues [49]. Interestingly, transfection of such cancer cells with full-length PNN cDNA demonstrated inhibition of anchorage-independent growth and alteration of the expression of a specific subset of genes, p21, CDK4, and CPR2, and cell migration-related genes, such as RhoA, CDK5, TIMPs, and MMPs [48]. Taken together, these observations support our contention that driving PNN expression leads to epithelial stability, while interfering with PNN expression leads to loss of differentiation, loss of cell adhesion, and enhanced cell migration. One intriguing upregulated gene was the serine-threonine kinase receptor-associated protein, STRAP (Gene ID:11171; OMIM: 605986). STRAP has been reported to be associated with the SMN-complex (RNA splicing) and has been reported to influence metaplasia and cancer development [50]. The upregulation of STRAP has been reported to be involved in maintaining the mesenchymal morphology and enhanced cell migration. In contrast, the knockdown of STRAP resulted in the induction of E-cadherin and the adoption of a more-stabile less migratory behavior [50].
Here, we also report that developmental regulatory genes are downregulated in response to PNN depletion, including FGD3 (Gene ID:89846; OMIM:609197), FGFR2 (Gene ID:2263; OMIM:176943), PAX6 (Gene ID:5080; OMIM 607108), RARG (Gene ID:5916; OMIM:180190), IGFBP5 (Gene ID:3488; OMIM:146734), and SOX10 (Gene ID:6663; OMIM:602229). Although the significance of the downregulation of these genes in the corneal epithelial context requires additional study, of specific note, along with the downregulation of PAX6, we previously identified an increase in the splicing isoform of PAX6, PAX6(5a) [39]. PAX6, the paired domain-homeobox transcription factor, is referred to as the master regulator of eye development and is expressed throughout the ocular surface, where this transcription factor plays a major role in early embryonic development and postnatal maturation of the corneal and conjunctival epithelia [51-59]. In the adult, PAX6 continues to be expressed in the corneal epithelium [60] and plays a major role in the maintenance of the ocular surface [61]. The lower expression level of PAX6 may have profound implications on corneal epithelial homeostasis. PAX6 dosage levels have profound impact on eye development in general (including Peters anomaly as seen in PNN hypomorphs); [62]. However, IGFBP5 expression has been linked with enhanced keratinocyte migration [63], the observed downregulation of IGFBP5 with concurrent upregulation of cell migration-associated genes seems to run contrary to these studies on keratinocytes.
Interestingly, cadherins 5 and 11 also demonstrated downregulation accompanied by the change in splicing patterns in PNN knockdown. These data are similar to our previous observations that transcriptional repressor CtBP and PNN can differentially modulate E-cadherin mRNA splicing [46]. Our studies indicated that PNN is associated with RNAPII on the E-cadherin promoter and causes differential modulation of E-cadherin expression and mRNA splicing, thus serving as an interface between transcription, and splicing during RNA elongation [64].
AS enables individual genes to generate multiple protein products (isoforms) that differ in structure and function. These differences occur through the regulated insertion or deletion of functional domains encoded by alternative exons. In addition, AS may regulate the level of protein expression through IR. Post-transcriptional RNA processing can thus modulate essential protein functions and expressions, according to the specific physiologic requirements of the corneal epithelial cell. Our studies provide a window into the molecular processes by which corneal epithelial identity is established and maintained at least in part through AS, and thus identifies potential targets for future therapies that will allow the prevention and reversal of corneal epithelial metaplasia. In addition, our studies, insomuch that they address fundamental processes and mechanisms that control cell- and tissue-specific splicing decisions, have profound relevance for all differentiating systems within all tissues of the eye. Thus, the molecular details pertaining to how the corneal epithelial identity is established and maintained are central to vision research. Normal development of the epithelium relies on a cascade of epithelial cell-type-specific splicing events [65-67], and in the adult, coordinated mRNA splicing variation has been observed as cells progress from the stem cell population to the differentiated cell [22-24].
We suggest that during corneal epithelial development and the progression from the adult stem cell populations to the differentiated cells of the mature corneal epithelium, coordination of specific mRNA splicing decisions is crucial. Our studies of epithelial identity and RNA processing exploited the nuclear protein PNN. Although PNN is a peripheral component of the EJC, we postulate that PNN in addition functions through these molecular connections, such as PWWP-containing H3K36me3-chromatin readers, PSIP1 and BS69 [17,18], to coordinate elongating RNAPII–CTD with associated SR proteins (serine-arginine rich proteins) to specific chromatin readers at specific spliced junctions, and thus modulates splice site selection and splicing execution. We suggest that the mechanism by which PNN impacts epithelial cell phenotype is through its activity in RNA processing. Our studies have provided a window into the AS events and splice variant richness within the corneal epithelial cell.
The investigation of PNN-knockdown cells revealed significant mechanistic insight into the regulation of AS and identified numerous aberrant splicing events, along with strong candidate splicing variants and pathways that may have a fundamental impact on the epithelial phenotype [40,68]. However, we still lack an understanding of the global impact of AS on the in vivo corneal epithelial biology. We have hypothesized that there will be a broad and rich variety of AS-derived RNA variants in the corneal epithelial progenitor cells, and that, during the process of differentiation, the diversity of RNA variants will become more restricted. We also hypothesize that perturbations of PNN will drive greater diversity in AS and, in turn, drive the cells to altered differentiation. These data presented are derived from a tightly regulated in vitro setting, using an inducible expression system within the SV40-immortalized HCET. It will be essential to extend these studies to the in vivo setting, such as our PNN knockout mouse models. Most importantly, it will be essential to explore the potential impact of PNN and other RNA processing components in the establishment and maintenance of the human corneal epithelium. Specifically, it will be crucial to better understand RNA processing and its perturbations associated with pathologies of the anterior ocular surface.
Acknowledgments
Supported by NIH Grant R01EY007883, P30EY021721
Appendix 1. STR analysis
To access the data, click or select the words “Appendix 1.”
Appendix 2. Total number of paired-end (PE) reads per sample
To access the data, click or select the words “Appendix 2.”
Appendix 3. Primers used for RT–PCR
To access the data, click or select the words “Appendix 3.”
Appendix 4. Genes upregulated in response to PNN reduction
To access the data, click or select the words “Appendix 4.”
Appendix 5. Genes downregulated in response to PNN reduction
To access the data, click or select the words “Appendix 5.”
Appendix 6. Gene Ontology of genes upregulated in response to PNN knockdown
The full graphical output of GOrilla for the ranked list of genes upregulated after PNN-knockdown. This GO-graph generated by GOrilla was generated for identifying and visualizing enriched GO terms in ranked lists of upregulated genes. To access the data, click or select the words “Appendix 6.”
Appendix 7. Verification of expression changes by droplet digital PCR quantitation
To access the data, click or select the words “Appendix 7.”
Appendix 8. RNA Splicing events
To access the data, click or select the words “Appendix 8.”
Appendix 9. Splicing events correlated to change in transcripts known splice isoforms
To access the data, click or select the words “Appendix 9.”
Appendix 10. Splicing events correlated to upregulated genes
To access the data, click or select the words “Appendix 10.”
References
- 1.Araki-Sasaki K, Ohashi Y, Sasabe T, Hayashi K, Watanabe H, Tano Y, Handa H. An SV40-immortalized human corneal epithelial cell line and its characterization. Invest Ophthalmol Vis Sci. 1995;36:614–21. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=7534282&dopt=Abstract [PubMed] [Google Scholar]
- 2.Shi Y, Tabesh M, Sugrue SP. Role of cell adhesion-associated protein, pinin (DRS/memA), in corneal epithelial migration. Invest Ophthalmol Vis Sci. 2000;41:1337–45. [PubMed] [Google Scholar]
- 3.Joo JH, Alpatov R, Munguba GC, Jackson MR, Hunt ME, Sugrue SP. Reduction of Pnn by RNAi induces loss of cell-cell adhesion between human corneal epithelial cells. Mol Vis. 2005;11:133–42. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15735603&dopt=Abstract [PubMed] [Google Scholar]
- 4.Joo J-H, Taxter TJ, Munguba GC, Kim YH, Dhaduvai K, Dunn NW, Degan WJ, Oh SP, Sugrue SP. Pinin modulates expression of an intestinal homeobox gene, Cdx2, and plays an essential role for small intestinal morphogenesis. Dev Biol. 2010;345:191–203. doi: 10.1016/j.ydbio.2010.07.009. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20637749&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Joo J-H, Kim YH, Dunn NW, Sugrue SP. Disruption of mouse corneal epithelial differentiation by conditional inactivation of pnn. Invest Ophthalmol Vis Sci. 2010;51:1927–34. doi: 10.1167/iovs.09-4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joo JH, Lee YJ, Munguba GC, Park S, Taxter TJ, Elsagga MY, Jackson MR, Oh SP, Sugrue SP. Role of Pinin in neural crest, dorsal dermis, and axial skeleton development and its involvement in the regulation of Tcf/Lef activity in mice. Dev Dyn. 2007;236:2147–58. doi: 10.1002/dvdy.21243. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17654715&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 7.Zimowska G, Shi J, Munguba G, Jackson MR, Alpatov R, Simmons MN, Shi Y, Sugrue SP. Pinin/DRS/memA interacts with SRp75, SRm300 and SRrp130 in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2003;44:4715–23. doi: 10.1167/iovs.03-0240. [DOI] [PubMed] [Google Scholar]
- 8.Sakashita E, Tatsumi S, Werner D, Endo H, Mayeda A. Human RNPS1 and its associated factors: a versatile alternative pre-mRNA splicing regulator in vivo. Mol Cell Biol. 2004;24:1174–87. doi: 10.1128/MCB.24.3.1174-1187.2004. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=14729963&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murachelli AG, Ebert J, Basquin C, Le Hir H, Conti E. The structure of the ASAP core complex reveals the existence of a Pinin-containing PSAP complex. Nat Struct Mol Biol. 2012;19:378–86. doi: 10.1038/nsmb.2242. [DOI] [PubMed] [Google Scholar]
- 10.Costa E, Canudas S, Garcia-Bassets I, Pérez S, Fernández I, Giralt E, Azorín F, Espinás ML. Drosophila dSAP18 is a nuclear protein that associates with chromosomes and the nuclear matrix, and interacts with pinin, a protein factor involved in RNA splicing. Chromosome Res. 2006;14:515–26. doi: 10.1007/s10577-006-1046-5. [DOI] [PubMed] [Google Scholar]
- 11.Alexander R, Beggs JD. Cross-talk in transcription, splicing and chromatin: who makes the first call? Biochem Soc Trans. 2010;38:1251–6. doi: 10.1042/BST0381251. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20863294&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 12.Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–69. doi: 10.1016/j.cell.2013.02.034. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23498935&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown SJ, Stoilov P, Xing Y. Chromatin and epigenetic regulation of pre-mRNA processing. Hum Mol Genet. 2012;21(R1):R90–6. doi: 10.1093/hmg/dds353. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22936691&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Almeida SF, Carmo-Fonseca M. Design principles of interconnections between chromatin and pre-mRNA splicing. Trends Biochem Sci. 2012;37:248–53. doi: 10.1016/j.tibs.2012.02.002. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22398209&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 15.Fox-Walsh K, Fu X-D. Chromatin: the final frontier in splicing regulation? Dev Cell. 2010;18:336–8. doi: 10.1016/j.devcel.2010.03.002. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20230741&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hnilicová J, Staněk D. Where splicing joins chromatin. Nucleus. 2011;2:182–8. doi: 10.4161/nucl.2.3.15876. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21818411&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, Xu W, Mu S, Wen H, Qiu J, Wang Z, Yang P, Wu F, Hui J, Fu X, Shi X, Shi YG, Xing Y, Lan F, Shi Y. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell. 2014;56:298–310. doi: 10.1016/j.molcel.2014.08.022. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25263594&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012;8:e1002717. doi: 10.1371/journal.pgen.1002717. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22615581&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu H, Zeng H, Lam R, Tempel W, Amaya MF, Xu C, Dombrovski L, Qiu W, Wang Y, Min J. Structural and histone binding ability characterizations of human PWWP domains. PLoS ONE. 2011;6:e18919. doi: 10.1371/journal.pone.0018919. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=21720545&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen K, Dai X, Wu J. Alternative splicing: An important mechanism in stem cell biology. World J Stem Cells. 2015;7:1–10. doi: 10.4252/wjsc.v7.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.López-Bigas N, Audit B, Ouzounis C, Parra G, Guigo R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005;579:1900–3. doi: 10.1016/j.febslet.2005.02.047. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15792793&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 22.Chepelev I, Chen X. Alternative splicing switching in stem cell lineages. Front Biol. 2013;8:50–9. doi: 10.1007/s11515-012-1198-y. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23399987&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horiguchi K, Sakamoto K, Koinuma D, Semba K, Inoue A, Inoue S, Fujii H, Yamaguchi A, Miyazawa K, Miyazono K, Saitoh M. TGF-β drives epithelial-mesenchymal transition through δEF1-mediated downregulation of ESRP. Oncogene. 2012;31:3190–201. doi: 10.1038/onc.2011.493. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22037216&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vitting-Seerup K, Porse BT, Sandelin A, Waage J. spliceR: an R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinformatics. 2014;15:81. doi: 10.1186/1471-2105-15-81. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24655717&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Murigneux V, Le Hir H. Transcriptome-wide modulation of splicing by the Exon Junction Complex. Genome Biol. 2014;15:551. doi: 10.1186/s13059-014-0551-7. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25476502&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci USA. 2010;107:12186–91. doi: 10.1073/pnas.1007336107. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20566848&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Capes-Davis A, Reid YA, Kline MC, Storts DR, Strauss E, Dirks WG, Drexler HG, MacLeod RA, Sykes G, Kohara A, Nakamura Y, Elmore E, Nims RW, Alston-Roberts C, Barallon R, Los GV, Nardone RM, Price PJ, Steuer A, Thomson J, Masters JR, Kerrigan L. Match criteria for human cell line authentication: where do we draw the line? Int J Cancer. 2013;132:2510–9. doi: 10.1002/ijc.27931. [DOI] [PubMed] [Google Scholar]
- 28.Auer PL, Doerge RW. Statistical design and analysis of RNA sequencing data. Genetics. 2010;185:405–16. doi: 10.1534/genetics.110.114983. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20439781&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24695404&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23104886&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303. doi: 10.1101/gr.107524.110. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20644199&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19505943&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L, Wang S, Li W. RSeQC: quality control of RNA-seq experiments. Bioinformatics. 2012;28:2184–5. doi: 10.1093/bioinformatics/bts356. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22743226&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 34.Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30:2843–51. doi: 10.1093/bioinformatics/btu356. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24974202&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006;7:S12. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen G, Wang C, Shi L, Qu X, Chen J, Yang J, Shi C, Chen L, Zhou P, Ning B, Tong W, Shi T. Incorporating the human gene annotations in different databases significantly improved transcriptomic and genetic analyses. RNA. 2013;19:479–89. doi: 10.1261/rna.037473.112. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23431329&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22388286&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Benjamini Y, Yekutieli D. Quantitative trait Loci analysis using the false discovery rate. Genetics. 2005;171:783–90. doi: 10.1534/genetics.104.036699. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15956674&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joo JH, Correia GP, Li JL, Lopez MC, Baker HV, Sugrue SP. Transcriptomic analysis of PNN- and ESRP1-regulated alternative pre-mRNA splicing in human corneal epithelial cells. Invest Ophthalmol Vis Sci. 2013;54:697–707. doi: 10.1167/iovs.12-10695. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23299472&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joo J-H, Ryu D, Peng Q, Sugrue SP. Role of Pnn in alternative splicing of a specific subset of lncRNAs of the corneal epithelium. Mol Vis. 2014;20:1629–42. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25489234&dopt=Abstract [PMC free article] [PubMed] [Google Scholar]
- 41.Benjamini Y, Hochberg Y. CONTROLLING THE FALSE DISCOVERY RATE - A PRACTICAL AND POWERFUL APPROACH TO MULTIPLE TESTING. J R Stat Soc, B. 1995;57:289–300. [Google Scholar]
- 42.Noble WS. How does multiple testing correction work? Nat Biotechnol. 2009;27:1135–7. doi: 10.1038/nbt1209-1135. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=20010596&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19192299&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLOS Comput Biol. 2007;3:e39. doi: 10.1371/journal.pcbi.0030039. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=17381235&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDermott GP, Do D, Litterst CM, Maar D, Hindson CM, Steenblock ER, Legler TC, Jouvenot Y, Marrs SH, Bemis A, Shah P, Wong J, Wang S, Sally D, Javier L, Dinio T, Han C, Brackbill TP, Hodges SP, Ling Y, Klitgord N, Carman GJ, Berman JR, Koehler RT, Hiddessen AL, Walse P, Bousse L, Tzonev S, Hefner E, Hindson BJ, Cauly TH, 3rd, Hamby K, Patel VP, Regan JF, Wyatt PW, Karlin-Neumann GA, Stumbo DP, Lowe AJ. Multiplexed target detection using DNA-binding dye chemistry in droplet digital PCR. Anal Chem. 2013;85:11619–27. doi: 10.1021/ac403061n. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=24180464&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 46.Alpatov R, Shi Y, Munguba GC, Moghimi B, Joo JH, Bungert J, Sugrue SP. Corepressor CtBP and nuclear speckle protein Pnn/DRS differentially modulate transcription and splicing of the E-cadherin gene. Mol Cell Biol. 2008;28:1584–95. doi: 10.1128/MCB.00421-07. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18086895&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23222703&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shi Y, Simmons MN, Seki T, Oh SP, Sugrue SP. Change in gene expression subsequent to induction of Pnn/DRS/memA: increase in p21(cip1/waf1). Oncogene. 2001;20:4007–18. doi: 10.1038/sj.onc.1204507. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=11494129&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 49.Shi Y, Shi Y, Ouyang P, Ouyang P, Sugrue SP, Sugrue SP. Characterization of the gene encoding pinin/DRS/memA and evidence for its potential tumor suppressor function. Oncogene. 2000;19:289–97. doi: 10.1038/sj.onc.1203328. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=10645008&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 50.Reiner JE, Datta PK. TGF-beta-dependent and -independent roles of STRAP in cancer. Front Biosci. 2011;16:105–15. doi: 10.2741/3678. Landmark Ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Azuma N, Tadokoro K, Asaka A, Yamada M, Yamaguchi Y, Handa H, Matsushima S, Watanabe T, Kohsaka S, Kida Y, Shiraishi T, Ogura T, Shimamura K, Nakafuku M. The Pax6 isoform bearing an alternative spliced exon promotes the development of the neural retinal structure. Hum Mol Genet. 2005;14:735–45. doi: 10.1093/hmg/ddi069. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15677484&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 52.Chauhan BK, Yang Y, Cveklova K, Cvekl A. Functional properties of natural human PAX6 and PAX6(5a) mutants. Invest Ophthalmol Vis Sci. 2004;45:385–92. doi: 10.1167/iovs.03-0968. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=14744876&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chauhan BK, Yang Y, Cveklova K, Cvekl A. Functional interactions between alternatively spliced forms of Pax6 in crystallin gene regulation and in haploinsufficiency. Nucleic Acids Res. 2004;32:1696–709. doi: 10.1093/nar/gkh334. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15020706&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Collinson JM, Chanas SA, Hill RE, West JD. Corneal development, limbal stem cell function, and corneal epithelial cell migration in the Pax6(+/−) mouse. Invest Ophthalmol Vis Sci. 2004;45:1101–8. doi: 10.1167/iovs.03-1118. [DOI] [PubMed] [Google Scholar]
- 55.Cvekl A, Tamm ER. Anterior eye development and ocular mesenchyme: new insights from mouse models and human diseases. Bioessays. 2004;26:374–86. doi: 10.1002/bies.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dorà N, Ou J, Kucerova R, Parisi I, West JD, Collinson JM. PAX6 dosage effects on corneal development, growth, and wound healing. Dev Dyn. 2008;237:1295–306. doi: 10.1002/dvdy.21528. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18386822&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ito YA, Walter MA. Genomics and anterior segment dysgenesis: a review. Clin Experiment Ophthalmol. 2014;42:13–24. doi: 10.1111/ceo.12152. [DOI] [PubMed] [Google Scholar]
- 58.Li W, Chen Y-T, Hayashida Y, Blanco G, Kheirkah A, He H, Chen S-Y, Liu C-Y, Tseng SCG. Down-regulation of Pax6 is associated with abnormal differentiation of corneal epithelial cells in severe ocular surface diseases. J Pathol. 2008;214:114–22. doi: 10.1002/path.2256. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18027901&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simpson TI, Price DJ. Pax6; a pleiotropic player in development. Bioessays. 2002;24:1041–51. doi: 10.1002/bies.10174. [DOI] [PubMed] [Google Scholar]
- 60.Koroma BM, Yang JM, Sundin OH. The Pax-6 homeobox gene is expressed throughout the corneal and conjunctival epithelia. Invest Ophthalmol Vis Sci. 1997;38:108–20. [PubMed] [Google Scholar]
- 61.Ouyang H, Xue Y, Lin Y, Zhang X, Xi L, Patel S, Cai H, Luo J, Zhang M, Zhang M, Yang Y, Li G, Li H, Jiang W, Yeh E, Lin J, Pei M, Zhu J, Cao G, Zhang L, Yu B, Chen S, Fu X-D, Liu Y, Zhang K. WNT7A and PAX6 define corneal epithelium homeostasis and pathogenesis. Nature. 2014;511:358–61. doi: 10.1038/nature13465. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=25030175&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schedl A, Ross A, Lee M, Engelkamp D, Rashbass P, van Heyningen V, Hastie ND. Influence of PAX6 gene dosage on development: overexpression causes severe eye abnormalities. Cell. 1996;86:71–82. doi: 10.1016/s0092-8674(00)80078-1. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=8689689&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 63.Hyde C, Hollier B, Anderson A, Harkin D, Upton Z. Insulin-like growth factors (IGF) and IGF-binding proteins bound to vitronectin enhance keratinocyte protein synthesis and migration. J Invest Dermatol. 2004;122:1198–206. doi: 10.1111/j.0022-202X.2004.22527.x. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=15140223&dopt=Abstract [DOI] [PubMed] [Google Scholar]
- 64.Alpatov R, Shi Y, Munguba GC, Moghimi B, Joo J-H, Bungert J, Sugrue SP. Corepressor CtBP and nuclear speckle protein Pnn/DRS differentially modulate transcription and splicing of the E-cadherin gene. Mol Cell Biol. 2008;28:1584–95. doi: 10.1128/MCB.00421-07. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=18086895&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dittmar KA, Jiang P, Park JW, Amirikian K, Wan J, Shen S, Xing Y, Carstens RP. Genome-wide determination of a broad ESRP-regulated posttranscriptional network by high-throughput sequencing. Mol Cell Biol. 2012;32:1468–82. doi: 10.1128/MCB.06536-11. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=22354987&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell. 2009;33:591–601. doi: 10.1016/j.molcel.2009.01.025. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19285943&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Warzecha CC, Shen S, Xing Y, Carstens RP. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol. 2009;6:546–62. doi: 10.4161/rna.6.5.9606. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=19829082&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Joo J-H, Correia GP, Li J-L, Lopez M-C, Baker HV, Sugrue SP. Transcriptomic Analysis of PNN- and ESRP1-regulated Alternative pre-mRNA Splicing in Human Corneal Epithelial Cells. Invest Ophthalmol Vis Sci. 2013 doi: 10.1167/iovs.12-10695. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&list_uids=23299472&dopt=Abstract [DOI] [PMC free article] [PubMed] [Google Scholar]