

Abstract
Lyngbyastatin 7 (1) is a marine cyanobacteria-derived lariat-type cyclic depsipeptide, of which the macrocyclic core possesses modified amino acids, including a featured 3-amino-6-hydroxy-2-piperidone (Ahp) moiety and a (Z)-2-amino-2-butenoic acid (Abu) moiety. The first total synthesis of 1 was successfully established via 31 steps, and the conditions of several crucial steps were optimized to ensure smooth operations. The previously reported structural assignments and elastase inhibitory activity of the isolated natural product were confirmed. According to the extensive in vitro biological evaluation, compound 1 displayed low nanomolar IC50 in blocking elastase activity, strong ability in protecting bronchial epithelial cells against elastase-induced antiproliferation and abrogating the elastase-triggered induction of pro-inflammatory cytokine expression. Its overall performance was superior over sivelestat, the only approved small molecule drug targeting elastase, which indicated its potential in developing as a pharmacotherapeutic against elastase-mediated pathologies. The success in total synthesis, designed with a novel convergent strategy, not only overcame the supply issue for thorough preclinical studies, but also paved the way for convenient synthesis of analogues with improved potency and drug-like properties.

INTRODUCTION
Human neutrophil elastase (HNE, EC 3.4.21.37) is a serine protease stored at high concentration in azurophilic granules of polymorphonuclear neutrophils. Under normal physiologic conditions, HNE is tightly controlled by its endogenous inhibitors, such as α1-proteinase inhibitor (α1-PI), α2-macroglobulin (α2-MG), secretory leukocyte proteinase inhibitor (SLPI) and elafin. However, vast amounts of reactive oxygen species and proteases released by leukocytes that are recruited to the inflammation sites will inactivate the endogenous inhibitors and result in a protease—antiprotease imbalance. Due to its broad substrate specificity, HNE possesses strong ability to degrade a variety of structural proteins, such as elastin, collagen, fibronectin and other components of the extracellular matrix (ECM), and thus its out-of-balance activity may result in serious tissue damage. The excess of extracellular HNE could also act as a key driver of inflammation, function as a path-clearer for the migration of neutrophil into the sites of inflammation, induce the expression of diverse cytokines and chemokines, cleave surface receptors, and therefore trigger exuberant downstream responses. Various studies have indicated this imbalance is involved in pathogenesis of various pulmonary diseases, including chronic obstructive pulmonary disease (COPD, the fourth leading cause of death), cystic fibrosis (CF), pulmonary hypertension (PH), acute lung injury (ALI), acute respiratory distress syndrome (ARDS), and others.1–3
While current therapies for these life-threatening diseases can only alleviate symptoms, but not halt disease progression, HNE inhibitors appear to be a promising target for developing new therapeutic strategies against these global public health problems. Among all the emerging therapeutic approaches, the most straightforward method is the administration of naturally occurring elastase inhibitors, such as human donor/recombinant α1-PI (52 kD) or SLPI (11.7 kD). However, their large size circumvents the efficacy due to the difficulty for these macromolecular inhibitors to get access to the HNE compartmentalized between neutrophil and ECM. Moreover, due to the challenge of purification, the risk of immunogenic response and the high cost of production of these large molecules, synthetic low molecular weight inhibitors received increasing attention over the last decades.1,4,5 Despite intense efforts in both the pharmaceutical industry and academia, sivelestat sodium hydrate (ONO-5046, Figure 1) is so far the only approved small molecule drug targeting elastase (launched in Japan and Republic of Korea for the treatment of ALI associated with systemic inflammatory response syndrome).6,7 However, its clinical efficacy and safety have not been convincingly demonstrated through multi-country, multi-center clinical trials, which also led to the discontinuation of its development in the United States and Europe.8–10
Figure 1.

Chemical structure of lyngbyastatin 7 (1), somamide A, symplostatin 5 and sivelestat.
Hence, there is still an urgent need to develop powerful elastase inhibitors to control this notorious destructive enzyme and restore the disturbed balance under disease condition. Nature has proven itself to be a reliable source for powerful pharmacotherapeutics: cyanobacteria turn out to be an especially prolific producer of protease inhibitors with striking potency.11,12 Among all the isolated serine protease inhibitors derived from cyanobacteria, one family of compounds stands out due to their ubiquitous existence, structural diversity and outstanding bioactivity. Their attractive molecular architectures are characterized as a 19-membered cyclic depsipeptide core incorporated a featured 3-amino-6-hydroxy-2-piperidone (Ahp) moiety, of which the hydroxy group plays a pivotal role in pre-locking the core ring in its bioactive conformation. The core is decorated with a highly diverse pendant side chain on the Thr amine. Although they generally displayed decent serine protease (mainly trypsin, chymotrypsin and elastase) inhibitory activity, their selectivity profiles were either poor to some extent or had not been extensively investigated until recently.12–14
Among over 100 members of the Ahp-containing cyclic depsipeptides, lyngbyastatin 7 (1), a marine cyanobacteria derived secondary metabolite isolated from a collection of Lyngbya sp. from Summerland Key in the Florida Keys, distinguished itself by showing impressive low nanomolar IC50 against elastase and excellent selectivity over other families of proteases from a panel of 68 proteases.13,14 In addition, according to the X-ray cocrystal analysis regarding of compound 1-procine pancreatic elastase complex (solved at a resolution of 1.55 Å, the best report for elastase-cyclic depsipeptide type inhibitor), except the conserved Ahp residue, the ethylidene of the featured (Z)-2-amino-2-butenoic acid (Abu) moiety and the hydroxy of the N-Me-Tyr unit could provide additional interaction with elastase.14 In terms of the pendant side chain, the polar functionality of Gln and the linear terminal hexanoic acid (Ha) appeared to be more favored in the binding site.12,14
Although at the enzyme level 1 showed great potential in developing as a pharmacotherapeutic against elastase-mediated pathologies (since it displayed superior performance over both sivelestat and symplostatin 5, the best studied Ahp-containing elastase inhibitor14 shown in Figure 1), the minute quantities isolated from the natural source hindered further exploration for its performance at the cellular and organismal level, and therefore justifies the development of a total synthesis methodology for this promising compound.
Given that the delicate macrocyclic core scaffold can be treated as a nature's masterpiece, of which the ideal size and structural complexity could provide spatially distributed interaction with the enzyme while the pre-organized conformation could lower the entropy cost of target binding,15,16 employing it as a pharmacophore was thought to be an advantageous starting point for elastase inhibitor development. Moreover, since the pendant side chain may provide additional assistance during inhibitor-protease recognition and binding process, we envisioned a synthetic approach to further favor this interaction by constructing a library of analogues with diverse side chains, which would also offer an opportunity to modulate the nature of substituents and therefore improve druggability of the lead scaffold.
Two prominent total syntheses of Ahp-containing cyclodepsipeptides, micropeptin T-2017 and somamide A18, have been established. Among these two compounds, somamide A also consists of the featured Abu unit in the macrocyclic ring. Unfortunately, the establishment of this molecule followed a strategy installing the side chain at the very beginning, because the authors discovered the generation of complex mixtures when they attempted to attach the side chain on a small building block containing Abu moiety.18 In order to fuel our aim to build a macrocyclic core-based library and facilitate subsequent in-depth SAR examination, it was necessary to establish a novel scheme constructing the macrocyclic ring first and attaching the side chain at late stage, as indicated in Figure 2.
Figure 2.

Comparison of the reported synthetic strategy of somamide A with our novel designed synthetic strategy for lyngbyastatin 7 (1).
Herein, we report the first total synthesis of 1 in a novel designed strategy and the biological evaluation (the first time reported for synthetic Ahp-containing compounds) of its effects on enzyme, cellular and transcriptional level in comparison with sivelestat and symplostatin 5.
RESULTS AND DISCUSSION
Retrosynthetic Strategy
According to all the aforementioned analyses, the pendant side chain was disconnected from the macrocycle first and the construction of the macrocyclic core was prioritized during the method development. Since one of the most important challenges in macrocyclic depsipeptide synthesis is the macrocyclization, careful decisions in terms of the site and the condition of macrocyclization are required.17–19 Because the nucleophilicity of the amino group is generally higher than that of the hydroxy group, macrolactamization is usually more preferred than macrolactonization. Also, sterically hindered N-methylated amino acids (here, N-Me-Tyr) and β-branched amino acids (here, Val) are not preferable as the cyclization site. Furthermore, embedding the turn-inducing elements, such as N-Me-Tyr, midway along the linear cyclization precursor may assist to pre-arrange the molecule for a more efficient ring closure. Given that the Abu moiety does not possess a stererogenic center, choosing the site between Ahp-precursor N-terminus and Abu C-terminus would effectively reduce the risk of racemization during macrolactamization.
It is also worth pointing out that, since the featured Ahp is generally chemically unstable and sensitive towards both acidic and basic conditions, the construction of this signature moiety was scheduled as the last step. Biosynthetically, this unique Ahp ring can be considered as a moiety derived from glutamate, of which the carboxylic acid was transformed to an aldehyde and then cyclized to form a favored 6-membered hemiaminal ring. This biomimetic strategy to establish the Ahp unit was also employed by Yokokawa et al. in synthesizing micropeptin T-20 and somamide A.17,18
All the above-mentioned considerations led us to dissect the whole structure into four building blocks and the retrosynthetic approach is depicted in Scheme 1.
Scheme 1.

Retrosynthetic Analysis of 1
Total Synthesis of Lyngbyastatin 7 (1)
The synthesis of building block 6 started from converting Boc-(S)-Thr-OH to its corresponding allyl ester 9, as shown in Scheme 2. Esterification of the hydroxy group in 9 with Fmoc-(S)-Val-OH in the presence of N-ethyl-N'-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDCI•HCl) and 4-dimethylaminopyridine (DMAP) quantitatively gave depsipeptide 10. After cleaving the Allyl group of 10 by treating with tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) in the presence of morpholine and removal of the Boc group of 9 under acidic condition, the two resulting deprotected derivatives were connected together in the presence of (benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) and N,N-diisopropylethylamine (DIEA) to afford the tripeptide 11. The stereospecific dehydration was carried out by using Martin's sulfurane18,20 and the configuration of the newly generated double bond in depsipeptide 6 was confirmed by the Nuclear Overhauser Effect (NOE) observed between the signal of Abu-NH at δ 9.27 and Abu-methyl at δ 1.62 after irradiating at either resonance frequencies (without observing the signal of Abu-vinyl H when irradiating at δ 9.27).
Scheme 2.

Synthesis of Building Block 6
As depicted in Scheme 3, the required fragment 7 was prepared from Boc-(S)-N-Me-Tyr(Bzl)-OH by conversion to its 2,2,2-trichloroethyl protected ester derivative 12, which was subsequently subjected to a Pd-catalyzed hydrogenolytic debenzylation and followed by protection with tert-butyldiphenylsilyl (TBDPS) group to form the intermediate 13. After undergoing acidic cleavage of Boc group by employing 30% trifluoroacetic acid (TFA) in CH2Cl2, the liberated amine was smoothly coupled with Fmoc-(S)-Phe-OH by using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) as coupling reagent to yield the dipeptide 7.
Scheme 3.

Synthesis of Building Block 7
Based on the method established by Yokokawa et al.,18 the generation of building block 8 shown in Scheme 4 started from the commercially available (S)-Glu-OBzl, of which the free amino group was protected by Alloc group and the side chain carboxy group was then reduced to an alcohol by taking it through a two-step reduction to afford 14. The newly established hydroxy group was further protected as a tert-butyldimethylsilyl (TBS) ether to give the fragment 8.
Scheme 4.

Synthesis of Building Block 8
As indicated in Scheme 5, the preparation of the side chain was initiated by esterifying Fmoc-(S)-Gln(Trt)-OH to give compound 15. Following removal of the Fmoc group in the presence of Et2NH in CH2Cl2, coupling with the hexanoyl chloride was carried out to provide the building block 4.
Scheme 5.

Synthesis of Building Block 4
With the required building blocks in hand, fragment condensation (depicted in Scheme 6) was initiated by removal of the Fmoc group in 6 and the Tce group in 7, followed by coupling of the two freshly deprotected fragments in the presence of HATU to give the intermediate 16. After removal of Fmoc from 16 by treating with Et2NH, the liberated amino group was coupled with the deprotected acid derived from 8 by using diethyl phosphorocyanidate (DEPC) as coupling reagent, which ensured a good coupling efficiency and led to the linear macrocyclization precursor 5. In order to set the stage for the macrocyclization, deprotection of the N-terminal Alloc group and the C-terminal Allyl group was carried out simultaneously by treating with Pd(PPh3)4 in the presence of dimethylamine borane complex. Different Allyl group scavengers,21,22 such as morpholine, N-methyl aniline, dimedone and dimethylamine borane complex (Me2NH•BH3), were screened to eliminate the formation of side products (Supporting Information Figure S1), and Me2NH•BH3 turned out to furnish the best result. At the stage of macrolactamization, the resulting deprotected peptide was cyclized under high dilution conditions (in order to avoid intermolecular reactivity) by employing HATU/DIEA/CH2Cl2, chosen after rigorous exploration of various conditions23 (summarized in Table 1) aimed at maximizing the yield of macrocyclic core 3.
Scheme 6.

Total synthesis of 1
Table 1.
Screening of Coupling Reagents for Macrolactamization to Prepare the Macrocyclic Core 3
| entry | coupling reagent | yield (%)a |
|---|---|---|
| 1 | FDPP | 16 |
| 2 | PyBOP | 33 |
| 3 | PyAOP | 35 |
| 4 | HATU/HOAT | 31 |
| 5 | HATU | 49 |
The yield was determined after purification by preparative thin layer chromatography.
Cleavage of the macrocycle's Boc group and attachment of the side chain to form compound 3 turned out to be the most challenging steps in the reaction sequence, which were thought to be a result of the steric effects of the large cyclic structure. Moreover, the existence of an α,β-unsaturated system and an easily accessible primary hydroxy group in the macrocyclic core was also suspected to affect the amide bond formation. Most conditions were ineffective and resulted in no reaction or decomposition of the macrocyclic ring. Monitored by MS, HATU/DIEA/CH2Cl2, HATU/DIEA/DMF, EDCI•HCl/DIEA/DMF, DEPC/triethylamine (TEA)/DMF, BOP/DIEA/DMF, 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT)23,24/TEA/THF afforded no desired product at all. After persistent exploration, we finally identified conditions capable of mediating these two reactions. 50 eq trimethylsilyl trifluoromethanesulfonate (TMSOTf)/62.5 eq 2,6-lutidine/CH2Cl2 was used as the deprotection condition (see Table 2 for condition screening) while DEPBT/DIEA/DMF was chosen as the coupling condition to connect the deprotected side chain derived from building block 4. Since removal of the trityl group under commonly used conditions was found to generate several side products, employing the optimized acid concentration (1:7 TFA:CH2Cl2 accompanied with triisopropylsilane (TIPS)), reaction temperature (0 °C) and time (2 h) afforded a higher yield of compound 2. Finally, the establishment of the Ahp moiety was performed by converting the primary alcohol to an aldehyde using the mild oxidant 1-hydroxy-1,2-benziodoxol-3(1H)-one 1-oxide (IBX) in DMSO, and the resulting aldehyde derivative was immediately exposed to tetra-n-butylammonium fluoride (TBAF)18 solution to afford the hemiaminal Ahp moiety in a regio- and stereo-selective manner and simultaneously cleave the remaining TBDPS group to generate the desired final product 1.
Table 2.
Exploring the Conditions of Removing the Boc and TBS Group in 3
| MS profileb-relative height | ||||
|---|---|---|---|---|
| entry | reagentsa | peak at 961 Dac | peak at 1061/1083 Dad | other peaks |
| 1 | 13% TFA in 5:95 H2O:THF mixture |
1 | 6.4/8.6 | low |
| 2 | 23% TFA in 5:95 H2O:THF mixture |
1 | 5.8/4.8 | 1157/1179=high |
| 3 | 33% TFA in 5:95 H2O:THF mixture |
1 | 4.2/0.5 | 1057=high 1157/1179=high |
| 4 | 39% TFA in 5:95 H2O:THF mixture |
1 | 2.3/0.3 | 1057=high 1157/1179=high |
| 5 | 44% TFA in 5:95 H2O:THF mixture |
detected | no | complex |
| 6 | 4 M HCl/EtOAc | detected | no | complex |
| 7 | 2-bromo-1,3,2-benzodioxaborole in CH2Cl2 |
detected | no | complex |
| 8 | 12 eq. TMSOTf /15 eq. 2,6-lutidine in CH2Cl2 |
1 | <0.5 | starting material=detected others=low |
| 9 | 20 eq. TMSOTf /25 eq. 2,6-lutidine in CH2Cl2 |
1 | <0.5 | low |
| 10 | 26 eq. TMSOTf /32 eq. 2,6-lutidine in CH2Cl2 |
1 | <0.5 | low |
| 11 | 40 eq. TMSOTf /52 eq. 2,6-lutidine in CH2Cl2 |
1 | <0.3 | low |
| 12 | 50 eq. TMSOTf /62.5 eq. 2,6-lutidine in CH2Cl2 |
1 | <0.1 | low |
Reactions were carried out using different concentrations of TFA in 5:95 H2O:THF mixture in room temperature for entries 1–5. For entry 6, compound 3 was directly treated with 4 M HCl/EtOAc at 0 °C. For entry 7, compound 3 was treated with 2-bromo-1,3,2-benzodioxaborole in CH2Cl2 at 0 °C. For entries 8–12, reactions were performed with different equivalents of TMSOTf and 2,6-lutidine at room temperature.
Reactions were monitored by MS after 2 h of treatment. Data was depicted as the height ratio of peak at 961 Da to peak at 1061/1083 Da, or the detectability of peaks within 500–1500 Da.
Product peak, detected as [M + H]+.
Side product of which the molecular weight was matched with the molecule failed to cleave the Boc group (detected as [M + H]+ and/or [M + Na]+).
Compared with the original isolated lyngbyastatin 7, our synthetic sample showed identical 1H and 13C NMR spectra (Supporting Information Figures S2 and S3), comparable optical rotation value ([α]20D − 10.0 (c 0.04, MeOH), lit [α]20D −7.4 (c 0.27, MeOH)13), as well as the same retention time on the Phenomenex Synergi 4μ Hydro-RP 80 Å column (250 × 4.68 mm, 4 μm) (Supporting Information Figures S4–S7). Therefore, we could confidently conclude that our synthetic product is completely identical in all respects with the naturally occurring lyngbyastatin 7, confirming the originally reported structure.
Biological Evaluation
The in vitro antiproteolytic activity of the synthetic compound 1 against porcine pancreatic elastase (PPE) (Figure 3A) and disease-relevant human neutrophil elastase (HNE) (Figure 3B) was evaluated in direct comparison with symplostatin 5 and sivelestat. BEAS-2B25, a human bronchial epithelial cell line transformed by the adenovirus 12-SV 40 hybrid virus, was chosen as a model system (which maintains the epithelial cell characteristics while extending in vitro lifespan to over 1 year) that was also previously employed in studies of symplostatin 514. At the cellular level, the evaluation regarding the cytoprotective ability of these three compounds towards attenuating the HNE-induced antiproliferation was further performed (Figure 3C).
Figure 3.
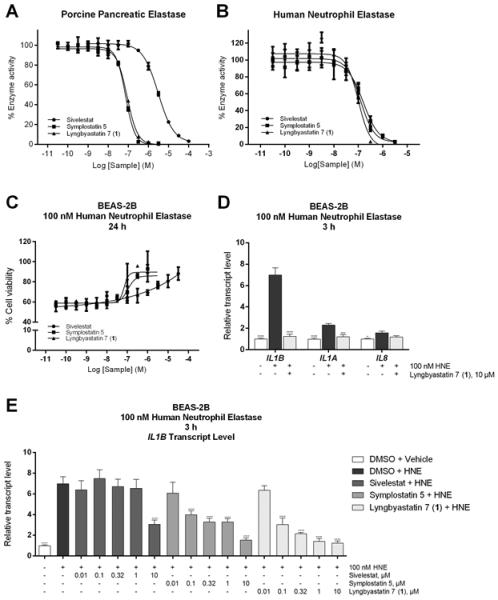
In vitro biological evaluation of compound 1, symplostatin 5 and sivelestat at the enzyme, cellular and transcriptional level. (A) In PPE enzyme assay, PPE was first incubated with compound 1, symplostatin 5 and sivelestat respectively for 15 min in Tris-HCl (pH 8.0) and then N-succinyl-Ala-Ala-Ala-p-nitroanilide was used as substrate to monitor the enzyme activity. The IC50 value for each compound is 88, 73 and 3080 nM respectively. (B) In HNE enzyme assay, HNE was first incubated with compound 1, symplostatin 5 and sivelestat respectively for 15 min in 0.1 M Tris-0.5 M NaCl (pH 7.5) and then N-(OMe-succinyl)-Ala-Ala-Pro-Val-p-nitroanilide was used as substrate to monitor the enzyme activity. The IC50 value for each compound is 100, 161 and 123 nM respectively. (C) In the cell viability assay, BEAS-2B cells were cotreated with compound 1, symplostatin 5, sivelestat or solvent control and 100 nM HNE or vehicle for 24 h. The cell viability was monitored by using MTT reagent. The IC50 value for each compound is 70, 106 and 861 nM respectively. (D) In order to monitor the changes in the transcript levels of IL1B, IL1A and IL8, total RNA was extracted after BEAS-2B cells were cotreated with 10 μM compound 1 or solvent control and 100 nM HNE or vehicle for 3 h. After cDNA synthesis, qPCR was carried out while using GAPDH as endogenous control. (E) In order to monitor the changes in transcript level of IL1B in the presence of different compounds in various concentrations, total RNA was extracted after BEAS-2B cells were cotreated with compound 1, symplostatin 5 and sivelestat or solvent control and 100 nM HNE or vehicle for 3 h. After cDNA synthesis, qPCR was carried out while using GAPDH as endogenous control. Data are presented as mean ± SD, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 compared to HNE-treated cells using ANOVA, Dunnett's t test (n=3).
According to the enzyme assays and cell viability assay, all three compounds presented a dose-dependent effect in blocking elastase activity. Compared with sivelestat, compound 1 exhibited 35-fold higher potency in inhibiting PPE (Figure 3A, IC50 values: 88 nM for compound 1 vs. 3080 nM for sivelestat). Although all the compounds displayed similar IC50 against HNE (Figure 3B, IC50 values for compound 1, symplostatin 5 and sivelestat, respectively: 100, 161, 123 nM), the cellular activity for the two natural products (especially for 1) in mitigating the elastase-induced antiproliferative effect was much enhanced compared with sivelestat (Figure 3C, IC50 values for 1, symplostatin 5 and sivelestat, respectively: 70, 106 and 861 nM). While 1 shared similar inhibitory activity with symplostatin 5 in the PPE assay (Figure 3A), 1 appeared to inactivate the human enzyme (HNE) with greater efficiency compared with symplostatin 5 at both the enzyme (Figure 3B) and cellular level (Figure 3C). Morphological change (rounding-up) and cell detachment (floating) could be observed within 2–3 h upon treating with HNE, which indicated the powerful destructive ability of HNE in a different phenotypic readout. All three compounds could prevent these elastase induced-early onset shape changes, again, sivelestat required a higher concentration to completely negate the effect of HNE.
Various studies demonstrated elastase possesses the ability to upregulate the transcript level of several pro-inflammatory cytokines. According to the preceding comprehensive profiling of the transcriptome of BEAS-2B in response to elastase, several interleukin-related genes were identified as majorly upregulated genes after treating with HNE for 3 h.14 Among IL1B, IL1A and IL8, the transcription of IL1B in BEAS-2B cells was significantly induced (7.0-fold) upon challenge with HNE for 3 h, as assessed by reverse transcription and quantitative polymerase chain reaction (RT-qPCR) (Figure 3D). Since IL-1β is considered a key pro-inflammatory cytokine in various pulmonary diseases and also serves as an important biomarker for elastase-mediated effects,26–30 the capability of compound 1 for regulating the transcriptional level of IL1B could benefit the control of inflammation in the progression of related diseases. For the three tested compounds, dose-dependent trends revealed in blocking the proteolytic activity of elastase were observed and, again, compound 1 displayed the best inhibitory activity (maximum efficacy achieved at 1 μM) while sivelestat and symplostatin 5 required a higher concentration to attenuate the elastase-induced transcriptional change (Figure 3E).
CONCLUSION
We have designed a strategy to obtain lyngbyastatin 7 through convergent synthesis that could not only enable easy access to sufficient materials for in-depth preclinical studies, but also facilitate the diversification of the pendant side chain in just five steps. Conditions of several key steps had been optimized to maximize the yield and minimize the generation of side products. In vitro biological evaluations carried out at the enzyme, cellular and transcriptional level emphasized that molecules bearing the signature macrocyclic core, particularly lyngbyastatin 7, would be promising therapeutics to negate the undesired effects induced by the overshooting elastase. Additional research is warranted to provide deeper insights into the SAR and explore the lyngbyastatin 7 scaffold systematically to develop effective cyanobacterial natural product-based therapeutics for elastase-mediated pathologies.
EXPERIMENTAL SECTION
General
All commercially available reagents were used without further purification unless otherwise noted. Solvents were purified according to the guidelines stated in reference.31 CH2Cl2 was distilled from CaH2; tetrahydrofuran (THF) was distilled from sodium chips in the presence of a small amount of benzophenone; MeOH was dried with 3 Å molecular sieves, and N,N-dimethylformamide (DMF) was dried with 4 Å molecular sieves. 4 M HCl solution in EtOAc was prepared by dissolving HCl gas (yielded by dropping aqueous hydrochloric acid (34%) to concentrated sulfuric acid (98%)) to EtOAc. All reactions were performed in heat-gun dried flasks (400 °C under reduced pressure) under an inert atmosphere of anhydrous argon unless otherwise noted. All reactions were monitored by thin layer chromatography (TLC) performed on silica gel 60 Å F254 aluminum sheet with UV light, visualized by phosphomolybdic acid-ethanol solution. Preparative thin layer chromatography was performed on silica gel 60 Å F254 glass plates (layer thick 1000 μm). Flash column chromatography was performed with 170–400 mesh silica gel with the indicated solvent system. HPLC-based compound purification was performed on a liquid chromatography system with a UV/VIS detector (UV detection at 220 and 254 nm). Validation of the purity of compound 1 and comparison of the retention time between natural lyngbyastatin 7 and compound 1 were performed on a liquid chromatography system with a diode array detector (UV detection from 200 to 300 nm). Optical rotation was measured at 589 nm on a polarimeter (Na D line) using a microcell of 1 dm path length. Nuclear magnetic resonance (NMR) spectra were recorded on a 400 MHz or 600 MHz spectrometer as indicated in the Supporting Information. Chemical shifts (δ) for proton nuclear magnetic resonance (1H NMR) spectra are reported in parts per million relative to the solvent residual signal (CDCl3, 7.26 ppm or (CD3)2SO, 2.50 ppm). Chemicals shifts for carbon nuclear magnetic resonance (13C NMR) spectra are reported in parts per million relative to the center line of CDCl3 triplet at 77.16 ppm or that of (CD3)2SO heptet at 39.52 ppm. Multiplicities are given as the following abbreviations: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), ddd (doublet of doublet of doublets), ddt (doublet of doublet of triplets), br s (broad singlet) and br d (broad doublet). Coupling constants (J) are reported in Hertz (Hz). Data are presented as follows: chemical shift (multiplicity, integration, coupling constant). High resolution mass spectra (HRMS) was performed on an ESI-TOF or an MALDI-TOF mass spectrometer. Low resolution mass spectra (LRMS) were obtained using a 3200 QTrap triple quadrupole mass spectrometer with an ESI interface operated in positive/negative mode.
Boc-(S)-Thr-OAllyl (9)
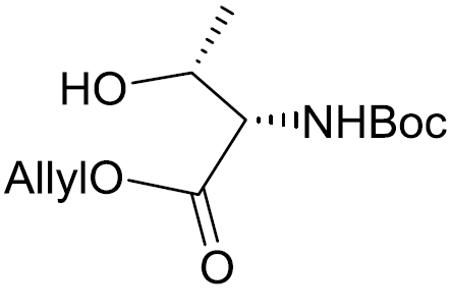
To a stirred solution of Boc-(S)-Thr-OH (10.00 g, 45.61 mmol) and Na2CO3 (9.67 g, 91.23 mmol) in dimethyl sulfoxide (DMSO) (250 mL) was added allylbromide (4.74 mL, 54.74 mmol) at room temperature. After being stirred at the same temperature for 23 h, the reaction mixture was quenched with H2O in an ice bath, and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (35% EtOAc in hexane) to give 9 (10.77 g, 91%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 5.90 (ddt, J = 17.2, 10.5, 5.7 Hz, 1H), 5.35 (br s, 1H), 5.33 (d, J = 17.2 Hz, 1H), 5.24 (d, J = 10.4 Hz, 1H), 4.65 (d, J = 5.7 Hz, 2H), 4.35–4.28 (m, 1H), 4.26 (br d, J = 8.8 Hz, 1H), 2.32 (br s, 1H), 1.44 (s, 9H), 1.24 (d, J = 6.3 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 171.0, 156.2, 131.5, 118.2, 79.7, 67.5, 65.7, 58.9, 28.1, 19.8; HRMS (ESI) m/z calcd for C12H21NO5Na [M + Na]+ 282.1317, found 282.1316.91.
Boc-(S)-Thr[Fmoc-(S)-Val]-OAllyl (10)
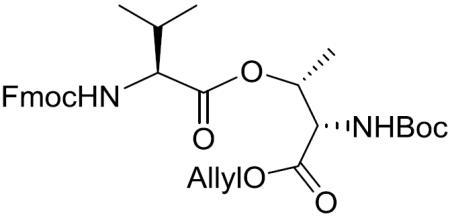
To a stirred solution of 9 (7.06 g, 27.23 mmol) in CH2Cl2 (140 mL) was added Fmoc-(S)-Val-OH (12.01 g, 35.40 mmol), N-ethyl-N'-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDCI•HCl) (7.83 g, 40.85 mmol) and 4-dimethylaminopyridine (DMAP) (665.3 mg, 5.45 mmol) at room temperature. After being stirred at the same temperature for 16 h, the reaction mixture was concentrated in vacuo and diluted with EtOAc. The whole mixture was washed with 0.5 M aqueous KHSO4, saturated aqueous NaHCO3 and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (15% EtOAc in hexane) to give 10 (14.56 g, 92%) as a white amorphous solid: [α]20D +11.2 (c 1.17, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.60 (d, J = 7.3 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 5.85 (ddt, J = 17.5, 11.7, 5.8 Hz, 1H), 5.52–5.44 (m, 1H), 5.41–5.30 (m, 2H), 5.28 (d, J = 17.2 Hz, 1H), 5.20 (d, J = 10.4 Hz, 1H), 4.63–4.57 (m, 2H), 4.51 (br d, J = 9.4 Hz, 1H), 4.46–4.32 (m, 2H), 4.27–4.18 (m, 2H), 2.20–2.08 (m, 1H), 1.45 (s, 9H), 1.33 (d, J = 6.1 Hz, 3H), 0.96 (d, J = 6.4 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.9, 169.6, 156.2, 156.0, 143.8, 141.4, 131.4, 127.8, 127.1, 125.1, 120.1, 119.3, 80.4, 71.9, 67.1, 66.5, 59.0, 57.2, 47.2, 31.2, 28.3, 19.0, 17.5, 16.9; HRMS (ESI) m/z calcd for C32H40N2O8Na [M + Na]+ 603.2682, found 603.2691.
Boc-(S)-Thr[Fmoc-(S)-Val]-(S)-Thr-OAllyl (11)
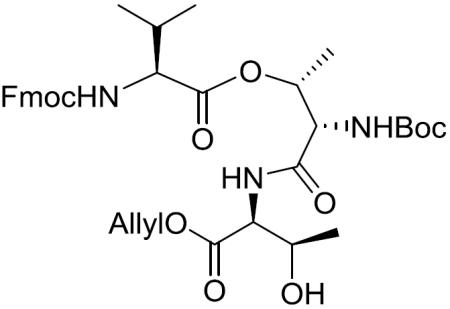
To a stirred solution of 10 (14.56 g, 25.07 mmol) in THF (175 mL) was added morpholine (6.58 mL, 75.20 mmol) and tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) (5.00 g, 5.01 mmol) at room temperature. The reaction was protected with aluminum foil from light. After being stirred at the same temperature for 5 h, the reaction mixture was concentrated in vacuo and diluted with EtOAc. The whole mixture was washed with 0.5 M aqueous KHSO4 and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (4% MeOH in CH2Cl2) to give the deprotected acid as a yellow amorphous solid, which was used without further characterizations.
6 (3.71 g, 14.30 mmol) was treated with 4 M HCl-dioxane (72 mL) dropwise at 0 °C. After being stirred at 0 °C to room temperature for 5 h, the whole mixture was concentrated in vacuo to give the crude deprotected amine as a brown oil, which was used for the next reaction without further purification.
To a stirred solution of the deprotected acid (10.04 g, 18.58 mmol) in CH2Cl2 (160 mL) was added N,N-diisopropylethylamine (DIEA) (9.96 mL, 57.16 mmol), benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) (9.48 g, 21.43 mmol) and the deprotected amine at 0 °C. After being stirred at 0 °C to room temperature for 48 h, the reaction mixture was concentrated in vacuo. The residue was purified by silica gel column chromatography (35% EtOAc in hexane) to give 11 (6.15 g, 63% over 3 steps) as a pale yellow amorphous solid: [α]20D +7.4 (c 1.74, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.4 Hz, 2H), 7.57 (d, J = 7.3 Hz, 2H), 7.43–7.35 (m, 2H), 7.31 (t, J = 7.4 Hz, 2H), 5.88 (ddt, J = 17.1, 11.4, 5.7 Hz, 1H), 5.65–5.45 (m, 2H), 5.45–5.35 (m, 1H), 5.31 (d, J = 17.5 Hz, 1H), 5.21 (d, J = 10.4 Hz, 1H), 4.64 (d, J = 5.6 Hz, 2H), 4.58 (dd, J = 8.7, 2.1 Hz, 1H), 4.50–4.44 (m, 1H), 4.42 (dd, J = 10.5, 7.2 Hz, 1H), 4.36–4.27 (m, 2H), 4.19 (dd, J = 6.9 Hz, 6.9 Hz, 1H), 4.12 (dd, J = 7.5 Hz, 7.5 Hz, 1H), 3.36 (br s, 1H), 2.18–2.02 (m, 1H), 1.45 (s, 9H), 1.30 (d, J = 6.5 Hz, 3H), 1.15 (d, J = 6.4 Hz, 3H), 0.97 (t, J = 6.7 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 171.1, 170.1, 169.2, 157.1, 155.3, 143.6, 141.41, 141.36, 131.7, 127.9, 127.3, 127.2, 125.2, 125.1, 120.12, 120.10, 118.8, 80.5, 71.0, 68.2, 67.6, 66.2, 59.9, 58.3, 56.7, 47.1, 30.6, 28.4, 20.2, 19.1, 18.3, 15.2; HRMS (ESI) m/z calcd for C36H47N3O10Na [M + Na]+ 704.3159, found 704.3167.
Boc-(S)-Thr[Fmoc-(S)-Val]-(Z)-Abu-OAllyl (6)
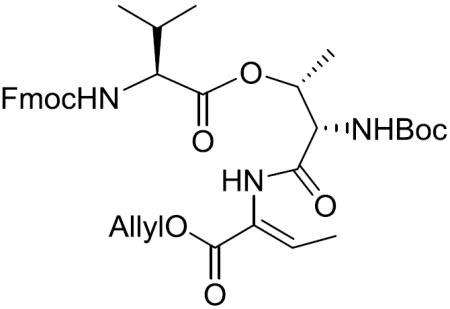
To a stirred solution of 11 (42.7 mg, 0.063 mmol) in CH2Cl2 (1 mL) was added Martin's sulfurane (84.3 mg, 0.125 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 3 h, the reaction mixture was quenched with saturated aqueous NaHCO3 and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (20% EtOAc in hexane) to give 6 (29.1 mg, 70%) as a white amorphous solid: [α]20D +8.3 (c 1.03, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.79 (br s, 1H), 7.75 (d, J = 7.5 Hz, 2H), 7.59 (dd, J = 7.2, 3.3 Hz, 2H), 7.39 (t, J = 7.4 Hz, 2H), 7.30 (t, J = 7.4 Hz, 2H), 6.87 (q, J = 7.1 Hz, 1H), 5.89 (ddt, J = 16.2, 10.9, 5.7 Hz, 1H), 5.63 (br d, J = 8.2 Hz, 1H), 5.49–5.38 (m, 2H), 5.30 (dd, J = 17.2, 1.3 Hz, 1H), 5.20 (d, J = 10.4 Hz, 1H), 4.62 (d, J = 5.7 Hz, 2H), 4.49–4.39 (m, 2H), 4.39–4.29 (m, 1H), 4.25–4.15 (m, 2H), 2.23–2.10 (m, 1H), 1.74 (d, J = 7.1 Hz, 3H), 1.43 (s, 9H), 1.32 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.9, 167.5, 163.9, 156.8, 155.5, 143.9, 141.4, 135.7, 131.9, 127.8, 127.2, 126.1, 125.1, 120.1, 118.6, 80.7, 77.4, 71.0, 67.2, 66.1, 59.7, 57.4, 47.3, 30.7, 28.3, 19.1, 17.9, 15.7, 14.6; HRMS (ESI) m/z calcd for C36H45N3O9Na [M + Na]+ 686.3053, found 686.3064.
Boc-(S)-N-Me-Tyr(Bzl)-OTce (12)
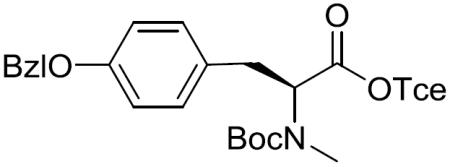
To a stirred solution of Boc-(S)-N-Me-Tyr(Bzl)-OH (4.80 g, 12.45 mmol) in CH2Cl2 (100 mL) was added 2,2,2-trichloroethanol (1.86 mL, 19.46 mmol), DMAP (158.5 mg, 1.30 mmol) and EDCI•HCl (2.98 g, 15.56 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 24 h, the reaction mixture was concentrated in vacuo and diluted with EtOAc. The whole mixture was washed with 5% citric acid solution and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (15% EtOAc in hexane) to give 12 (6.06 g, 94%) as a colorless oil: [α]20D −40.2 (c 1.15, CH2Cl2); 1H NMR (400 MHz, CDCl3, mixture of rotamers) δ 7.45–7.29 (m, 4H), 7.13 (m, 2H), 6.91 (m, 2H), 5.04 (s, 2H), 5.00–4.69 (m, 3H), 3.38–3.24 (m, 1H), 3.09–2.97 (m, 1H), 2.80 (s, 1.5H), 2.75 (s, 1.5H), 1.40 (s, 4.5H), 1.36 (s, 4.5H); 13C NMR (100 MHz, CDCl3) δ 170.1, 169.8, 157.8, 157.7, 155.9, 154.9, 137.2, 137.1, 130.0, 129.4, 128.7, 128.1, 127.6, 115.1, 115.0, 94.8, 94.7, 80.7, 80.3, 74.6, 74.5, 70.1, 61.3, 60.0, 34.6, 34.2, 32.5, 32.3, 28.4; HRMS (ESI) m/z calcd for C24H28Cl3NO5Na [M + Na]+ 538.0931, found 538.0915.
Boc-(S)-N-Me-Tyr(TBDPS)-OTce (13)
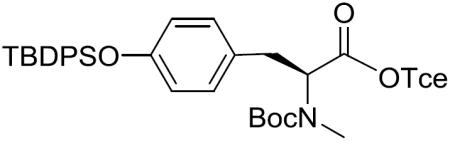
To a stirred solution of 12 (6.06 g) in MeOH (100 mL) was added Pd/C (1.21 g) at room temperature. After being vigorously stirred under an H2 atmosphere (balloon pressure) at the same temperature for 2 h, the reaction mixture was filtered through a Celite pad and the filtrate was concentrated in vacuo. The resultant deprotected product was used for the next reaction without further purification.
To a stirred solution of the deprotected product in CH2Cl2 (70 mL) was added imidazole (389.3 mg, 5.72 mmol), DMAP (69.9 mg, 0.57 mmol) and tert-butyldiphenylsilyl chloride (TBDPSCl) (1.12 mL, 4.29 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 7 h, the reaction mixture was quenched with water and the aqueous layer was extracted with CH2Cl2. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (2% EtOAc in hexane) to give 13 (1.77 g, 23% over 2 steps) as a colorless oil: [α]20D −28.5 (c 1.13, CH2Cl2); 1H NMR (400 MHz, CDCl3, a mixture of rotamers) δ 7.73–7.68 (m, 4H), 7.45–7.33 (m, 6H), 6.96–6.88 (m, 2H), 6.73–6.67 (m, 2H), 4.86–4.60 (m, 3H), 3.29–3.15 (m, 1H), 3.04–2.90 (m, 1H), 2.72 (s, 1.5H), 2.66 (s, 1.5H), 1.38 (s, 4.5H), 1.35 (s, 4.5H), 1.10 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 170.1, 169.8, 155.8, 154.9, 154.6, 154.5, 135.6, 135.4, 134.9, 133.1, 133.0, 130.0, 129.8, 129.6, 129.5, 127.9, 119.94, 119.86, 94.9, 94.7, 80.7, 80.3, 74.6, 74.5, 61.4, 60.2, 34.6, 34.2, 32.8, 32.5, 28.4, 26.71, 26.67, 19.6, 19.2; HRMS (ESI) m/z calcd for C33H40Cl3NO5SiNa [M + Na]+ 686.1634, found 686.1650.
Fmoc-(S)-Phe-(S)-N-Me-Tyr(TBDPS)-OTce (7)
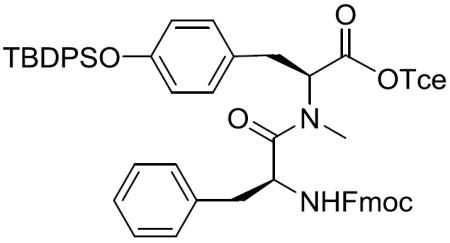
To a stirred solution of 13 (1.77 g, 2.65 mmol) in CH2Cl2 (19.09 mL) was added trifluoroacetic acid (TFA) (8.18 mL, 106.16 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 5 h, the reaction mixture was concentrated in vacuo to give crude deprotected amine as a dark yellow oil, which was used for the next reaction without further purification.
To a stirred solution of deprotected amine in CH2Cl2 (40 mL) was added DIEA (3.70 mL, 21.23 mmol), Fmoc-(S)-Phe-OH (1.34 g, 3.45 mmol) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) (2.02 g, 5.31 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 36 h, the reaction mixture was concentrated in vacuo and diluted with EtOAc. The whole mixture was washed with 0.5 M aqueous KHSO4, saturated aqueous NaHCO3 and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (10% EtOAc in hexane) to give 7 (1.03 g, 42% over two steps) as a white amorphous solid: [α]20D −45.50 (c 0.84, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.71–7.64 (m, 4H), 7.53 (dd, J = 12.3, 7.5 Hz, 2H), 7.44–7.36 (m, 4H), 7.36–7.27 (m, 6H), 7.23–7.11 (m, 5H), 6.72 (d, J = 8.4 Hz, 2H), 6.60 (d, J = 8.4 Hz, 2H), 5.34 (d, J = 8.9 Hz, 1H), 4.83–4.65 (m, 2H), 4.72 (s, 2H), 4.38 (dd, J = 10.4, 7.0 Hz, 1H), 4.23 (dd, J = 10.4, 7.2 Hz, 1H), 4.14 (dd, J = 6.9, 6.9 Hz, 1H), 3.25 (dd, J = 14.5, 5.4 Hz, 1H), 3.04 (dd, J = 13.6, 7.1 Hz, 1H), 2.93 (dd, J = 14.6, 9.5 Hz, 1H), 2.84 (dd, J = 13.6, 6.3 Hz, 1H), 2.72 (s, 3H), 1.06 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 171.5, 168.8, 155.6, 154.7, 144.0, 143.9, 141.5, 141.4, 136.2, 135.7, 135.6, 133.02, 132.97, 130.0, 129.8, 129.7, 129.1, 128.6, 127.9, 127.2, 127.1, 125.3, 125.2, 120.11, 120.07, 94.7, 74.7, 67.1, 61.4, 52.2, 47.3, 39.1, 34.9, 33.9, 29.9, 26.6, 19.6; HRMS (ESI) m/z calcd for C52H51Cl3N2O6SiNa [M + Na]+ 955.2480, found 955.2492.
Benzyl (S)-2-(((allyloxy)carbonyl)amino)-5-hydroxypentanoate (14)
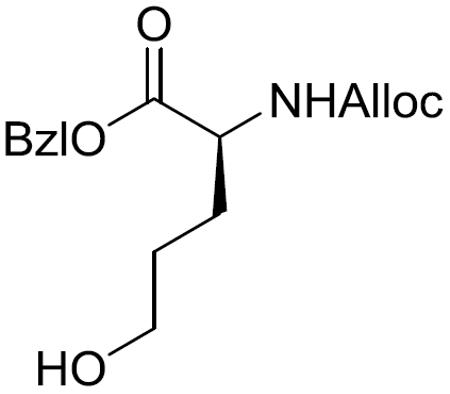
To a stirred solution of (S)-Glu-OBzl (2.50 g, 10.54 mmol) in CH2Cl2 (50 mL) was added 1 M aqueous NaHCO3 (50 mL) and allylchloroformate (2.8 mL, 26.34 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 24 h, the reaction mixture was acidified with 0.5 M aqueous HCl to pH 2–3 and the aqueous layer was extracted with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (25% EtOAc in hexane) to give the N-protected glutamic acid as a colorless oil, which was used for the next reaction without further characterizations.
To a stirred solution of N-protected glutamic acid (2.62 g, 8.15 mmol) in THF (50 mL) was added Et3N(3.4 mL, 24.44 mmol) and ethylchloroformate (2.3 mL, 24.44 mmol) at −10 °C. After being stirred at the same temperature for 4.5 h, NaBH4 solution (1.23 g NaBH4 dissolved in 50 mL water) was added dropwise over 15 min. The reaction mixture was stirred at −10 °C to room temperature for 4 h and then quenched by 0.5 M aqueous KHSO4. The aqueous layer was extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (50% EtOAc in hexane) to give 14 (1.26 g, 39% over 2 steps) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.40–7.31 (m, 5H), 5.90 (ddt, J = 15.7, 10.6, 5.2, Hz, 1H), 5.45 (br d, J = 6.0 Hz, 1H), 5.30 (d, J = 17.1 Hz, 1H), 5.24–5.12 (m, 3H), 4.56 (d, J = 5.4 Hz, 2H), 4.49–4.39 (m, 1H), 3.63 (t, J = 6.1 Hz, 2H), 2.00–1.89 (m, 1H), 1.84–1.72 (m, 1H), 1.72–1.49 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 172.4, 156.1, 135.4, 132.7, 128.8, 128.6, 128.5, 118.0, 67.3, 66.0, 62.1, 53.7, 29.4, 28.2; HRMS (ESI) m/z calcd for C16H21NO5Na [M + Na]+ 330.1317, found 330.1325.
Benzyl (S)-2-(((allyloxy)carbonyl)amino)-5-((tert-butyldimethylsilyl)oxy)pentanoate (8)
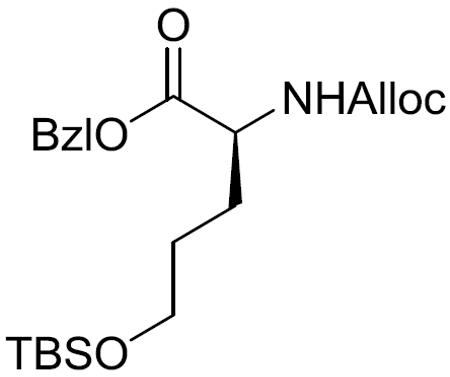
To a stirred solution of 14 (504.4 mg, 1.641 mmol) in DMF (20 mL) was added imidazole (670.4 mg, 9.847 mmol) and tert-butyldimethylsilyl chloride (TBSCl) (742.1 mg, 4.924 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 11 h, the reaction mixture was diluted with EtOAc. The whole mixture was washed with saturated aqueous NaHCO3 and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (15% EtOAc in hexane) to give 8 (419.2 mg, 61%) as a white amorphous solid: 1H NMR (400 MHz, CDCl3) δ 7.39–7.30 (m, 5H), 5.90 (ddt, J = 16.3, 10.9, 5.4 Hz, 1H), 5.46 (br d, J = 7.4 Hz, 1H), 5.30 (d, J = 17.2 Hz, 1H), 5.23–5.10 (m, 3H), 4.56 (d, J = 5.4 Hz, 2H), 4.44–4.36 (m, 1H), 3.59 (t, J = 5.9 Hz, 2H), 1.98–1.87 (m, 1H), 1.81–1.69 (m, 1H), 1.59–1.47 (m, 2H), 0.87 (s, 9H), 0.03 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 172.5, 156.0, 135.5, 132.8, 128.7, 128.5, 128.3, 117.9, 67.2, 65.9, 62.3, 53.8, 29.2, 28.4, 26.1, 18.4, −5.2.; HRMS (ESI) m/z calcd for C22H35NO5SiNa [M + Na]+ 444.2182, found 444.2192.
Fmoc-(S)-Gln(Trt)-OMe (15)
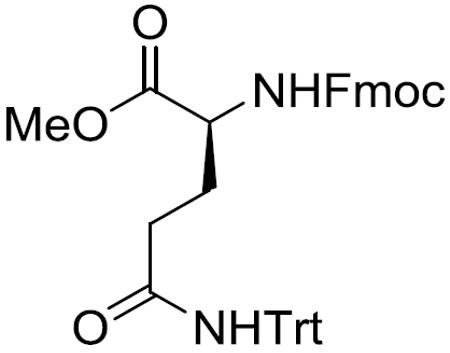
To a stirred solution of Fmoc-(S)-Gln(Trt)-OH (4.7 g, 7.81 mmol) in MeOH (100 mL) was added SOCl2 (2.3 mL, 31.24 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 20 h, the reaction mixture was concentrated in vacuo and the residue was purified by silica gel column chromatography (40% EtOAc in hexane) to give 15 (4.04 g, 83%) as a pale yellow amorphous solid: [α]20D +4.4 (c 1.36, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J = 7.5 Hz, 2H), 7.59 (d, J = 7.2 Hz, 2H), 7.43–7.35 (m, 2H), 7.35–7.18 (m, 17H), 6.89 (s, 1H), 5.59 (br d, J = 7.1 Hz, 1H), 4.45 (dd, J = 10.4, 6.9 Hz, 1H), 4.42–4.31 (m, 2H), 4.22 (dd, J = 6.8, 6.8 Hz, 1H), 3.72 (s, 3H), 2.43–2.27 (m, 2H), 2.27–2.15 (m, 1H), 2.00–1.88 (m, 1H); 13C NMR (100 MHz, CDCl3) δ 172.6, 170.9, 156.4, 144.7, 143.8, 141.4, 128.8, 128.0, 127.8, 127.2, 127.1, 125.2, 120.1, 70.8, 67.1, 53.6, 52.6, 47.3, 33.5, 28.4; HRMS (ESI) m/z calcd for C40H36N2O5Na [M + Na]+ 647.2522, found 647.2533.
N-Hexanoyl-(S)-Gln(Trt)-OMe (4)
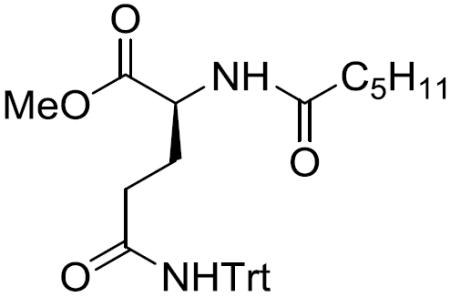
To a stirred solution of 15 (4.04 g) in CH2Cl2 (50 mL) was added Et2NH (25 mL) at 0 °C. After being stirred at 0 °C to room temperature for 5 h, the reaction mixture was concentrated in vacuo and the deprotected amine was used for the next reaction without further purification.
To a stirred solution of the deprotected amine in CH2Cl2 (60 mL) was added Et3N (5.408 mL, 38.801 mmol) and hexanoyl chloride (1.807 mL, 12.934 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 19 h, the reaction mixture was quenched with water and the aqueous layer was filtered. The filtrate was extracted with EtOAc and the combined organic layers were washed with saturated aqueous NaHCO3 and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (25% EtOAc in hexane) to give 4 (2.64 g, 82%) as a pale yellow amorphous solid: [α]20D +4.5 (c 0.48, CH2Cl2); 1H NMR (400 MHz, (CD3)2SO) δ 8.60 (s, 1H), 8.15 (d, J = 6.8 Hz, 1H), 7.30–7.13 (m, 15H), 4.24–4.15 (m, 1H), 3.60 (s, 3H), 2.41–2.25 (m, 2H), 2.10 (t, J = 7.3 Hz, 2H), 1.92–1.80 (m, 1H), 1.75–1.63 (m, 1H), 1.55–1.43 (m, 2H), 1.32–1.17 (m, 4H), 0.85 (t, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 173.6, 172.5, 171.5, 144.6, 128.7, 127.9, 127.0, 70.6, 52.3, 52.1, 36.2, 33.5, 31.4, 27.4, 25.1, 22.4, 14.0; HRMS (ESI) m/z calcd for C31H36N2O4Na [M + Na]+ 523.2573, found 523.2579.
Boc-(S)-Thr[Fmoc-(S)-Phe-(S)-N-Me-Tyr(TBDPS)]-(Z)-Abu-OAllyl (16)

To a stirred solution of 6 (572.3 mg, 0.862 mmol) in CH2Cl2 (8.5 mL) was added Et2NH (8.5 mL) at room temperature. After being stirred at the same temperature for 5 h, the reaction mixture was concentrated in vacuo and the deprotected amine was used without further purification.
To a stirred solution of 7 (1295.1 mg, 1.386 mmol) in THF (15 mL) was added 1 M aqueous NH4OAc (15 mL) and 10% Cd/Pb couple (530.0 mg, 4.348 mmol) at room temperature. After being stirred at the same temperature for 24 h, the reaction mixture was filtered and the filtrate was acidified with 1 M aqueous HCl to pH 2–3. The whole mixture was extracted with CH2Cl2 and the combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (7% MeOH in CH2Cl2) to give the deprotected acid as a white amorphous solid, which was used without further characterization.
To a stirred solution of the deprotected acid (692.3 mg, 0.862 mmol) in CH2Cl2 (17.2 mL) was added HATU (393.4 mg, 1.035 mmol), the deprotected amine and DIEA (300.3 μL, 1.724 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 21 h, the reaction mixture was concentrated in vacuo and diluted with EtOAc. The organic layer was washed with 0.5 M aqueous KHSO4, saturated aqueous NaHCO3 and brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (20% EtOAc in hexane) to give 16 (742.4 mg, 70% over 3 steps) a white amorphous solid: [α]20D −25.9 (c 0.18, CH2Cl2); 1H NMR (600 MHz, CDCl3, a mixture of multiple rotamers, selected peaks are given for the major rotamer) δ 8.29 (br s, 1H), 8.16 (br s, 1H), 7.78–7.70 (m, 2H), 7.67 (br s, 2H), 7.57 (d, J = 7.2 Hz, 2H), 7.53 (d, J = 7.2 Hz, 2H), 7.45–7.22 (m, 13H), 7.01 (d, J = 7.0 Hz, 2H), 6.93 (d, J = 8.0 Hz, 2H), 6.87–6.78 (m, 1H), 6.67 (d, J = 8.2 Hz, 2H), 5.90 (ddt, J = 16.8, 11.2, 5.6 Hz, 1H), 5.65 (br d, J = 5.4 Hz, 1H), 5.43–5.26 (m, 2H), 5.19 (d, J = 10.5 Hz, 1H), 4.95–4.86 (m, 1H), 4.64–4.59 (m, 2H), 4.55–4.41 (m, 2H), 4.39–4.31 (m, 1H), 4.29–4.22 (m, 1H), 4.16–4.07 (m, 2H), 4.07–4.00 (m, 1H), 3.11 (br d, J = 13.7 Hz, 1H), 2.96–2.83 (m, 4H), 2.82–2.68 (m, 1H), 2.47–2.38 (m, 1H), 2.07–1.99 (m, 1H), 1.79–1.73 (m, 3H), 1.43 (s, 9H), 1.06 (br s, 3H), 0.98 (s, 9H), 0.87 (d, J = 6.8 Hz, 3H), 0.82 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3, a mixture of rotamers) δ 172.8, 172.6, 170.7, 170.4, 170.1, 167.6, 167.5, 164.3, 164.1, 163.8, 156.9, 155.7, 155.5, 155.2, 154.9, 154.6, 143.9, 143.7, 143.4, 141.4, 141.30, 141.26, 136.3, 136.1, 135.6, 135.5, 135.4, 135.0, 133.0, 132.8, 132.7, 132.1, 132.0, 130.4, 130.02, 129.95, 129.7, 129.4, 129.3, 129.1, 128.9, 128.6, 127.9, 127.8, 127.23, 127.16, 127.1, 126.8, 126.3, 125.2, 125.0, 120.3, 120.09, 120.05, 118.7, 118.4, 80.4, 80.2, 70.9, 70.3, 67.7, 66.8, 66.1, 65.9, 62.7, 59.9, 58.5, 57.2, 56.4, 52.7, 50.8, 47.3, 47.0, 38.7, 36.6, 33.4, 32.0, 30.2, 29.7, 29.5, 28.4, 26.6, 26.5, 22.8, 19.54, 19.48, 19.1, 19.02, 18.99, 14.9, 14.6, 14.4, 14.2; HRMS (ESI) m/z calcd for C71H83N5O12SiNa [M + Na]+ 1248.5705, found 1248.5718.
Cyclization precursor (5)

To a stirred solution of 16 (742.4 mg, 0.605 mmol) in CH2Cl2 (8 mL) was added Et2NH (8 mL) at room temperature. After being stirred at the same temperature for 5 h, the reaction mixture was concentrated in vacuo and the deprotected amine was used without further purification.
To a stirred solution of 8 (389.6 mg, 0.924 mmol) in THF: MeOH: H2O 2:1:1 (3.7 mL) was added 1 M aqueous LiOH (3.7 mL) at room temperature. After being stirred at the same temperature for 3 h, the reaction mixture was acidified with 1 M aqueous KHSO4 to pH 3–4 and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (8% MeOH in CH2Cl2) to give the deprotected acid as a white amorphous solid, which was used without further characterization.
To a stirred solution of the deprotected acid (195.2 mg, 0.589 mmol) in DMF (12 mL) was added diethyl phosphorocyanidate (DEPC) (134.05 μL, 0.883 mmol), the deprotected amine (607.91 mg, 0.605 mmol) and Et3N (246.27 μL, 1.767 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 24 h, the reaction mixture was diluted with EtOAc and the organic layer was washed with 0.5 N KHSO4, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (50% EtOAc in hexane) to give 5 (602.5 mg, 78%) as a white amorphous solid: [α]20D −16.9 (c 0.43, CH2Cl2); 1H NMR (600 MHz, CDCl3; selected peaks are given because of multiple rotamers) δ 8.25 (br s, 1H), 8.05 (br d, J = 8.0 Hz, 1H), 7.71–7.63 (m, 1H), 7.57 (d, J = 6.8 Hz, 2H), 7.53 (d, J = 6.9 Hz, 2H), 7.43–7.17 (m, 9H), 6.98 (d, J = 6.7 Hz, 2H), 6.95–6.86 (m, 1H), 6.84 (d, J = 8.4 Hz, 2H), 6.61 (d, J = 8.4 Hz, 2H), 6.13 (br d, J = 7.1 Hz, 1H), 5.96 (br d, J = 6.7 Hz, 1H), 5.94–5.83 (m, 1H), 5.83–5.70 (m, 1H), 5.52–5.44 (m, 1H), 5.30 (dd, J = 17.2, 1.3 Hz, 1H), 5.24–5.10 (m, 3H), 4.78 (br s, 1H), 4.70–4.63 (m, 1H), 4.62 (d, J = 5.5 Hz, 2H), 4.57–4.44 (m, 2H), 4.44–4.34 (m, 1H), 4.27–4.15 (m, 1H), 4.15–4.01 (m, 1H), 3.68–3.49 (m, 2H), 3.05 (dd, J = 14.6, 4.2 Hz, 1H), 2.82–2.73 (m, 4H), 2.52 (dd, J = 12.8, 9.5 Hz, 1H), 2.39–2.24 (m, 1H), 2.07–1.95 (m, 1H), 1.92–1.41 (m, 7H), 1.35 (s, 9H), 1.13–1.04 (m, 3H), 0.98 (s, 9H), 0.90–0.78 (m, 15H), 0.04 (d, J = 4.2 Hz, 6H); 13C NMR (150 MHz, CDCl3, a mixture of rotamers) δ 173.9, 171.8, 169.8, 169.5, 168.9, 167.8, 164.5, 156.0, 155.8, 155.6, 154.7, 154.5, 137.0, 135.9, 135.6, 135.5, 133.0, 132.9, 132.8, 132.4, 131.9, 130.2, 130.00, 129.96, 129.7, 129.1, 128.9, 127.8, 127.3, 126.1, 120.1, 118.6, 80.1, 70.0, 66.1, 66.0, 63.1, 62.9, 62.7, 59.7, 57.2, 54.1, 50.0, 37.1, 33.5, 32.0, 30.0, 29.8, 29.6, 29.5, 28.5, 28.40, 28.37, 28.3, 28.1, 26.6, 26.5, 26.1, 25.8, 25.4, 22.8, 19.5, 19.5, 19.1, 18.5, 15.5, 14.6, 14.2, −3.4, −5.2, −5.3; HRMS (MALDI) m/z calcd for C71H100N6O14Si2Na [M + Na]+ 1339.6728, found 1339.6778.
Macrocyclic core (3)

To a stirred solution of 5 (142.4 mg 0.108 mmol) in CH2Cl2 (1.1 mL) was added Me2NH·BH3 (19.10 mg, 0.324 mmol) and Pd(PPh3)4 (24.97 mg, 0.022 mmol) at room temperature. The reaction was protected with aluminum foil from light. After being stirred at the same temperature for 1 h, the reaction mixture was concentrated in vacuo and the deprotected product was used for the next reaction without further purification.
To a stirred solution of the deprotected product in CH2Cl2 (54 mL) was added HATU (49.30 mg, 0.130 mmol) and DIEA (188.22 μL, 1.08 mmol) at room temperature. After being stirred at the same temperature for 12 h, the reaction mixture was concentrated in vacuo and the residue was purified by silica gel column chromatography (40% EtOAc in hexane) to give 3 (62.5 mg, 49%, ≥ 98% de) as a white amorphous solid. Reversed-phase HPLC method for determining the purity of 3: column, Phenomenex Luna 5μ C18(2) 100Å column 250 × 10 mm, 5 μm; flow rate, 2.0 mL/min; elution method, H2O/MeCN=10/90–0/100 linear gradient (0.0–30.0 min), H2O/MeCN =0/100 isocratic (30.0–50.0 min); retention time, 42.1 min. 1H NMR (400 MHz, CDCl3) δ 7.70–7.56 (m, 5H), 7.40 (t, J = 7.0 Hz, 2H), 7.35–7.13 (m, 10H), 6.94 (d, J = 6.9 Hz, 2H), 6.87 (q, J = 7.15 Hz, 1H), 6.80 (d, J = 7.9 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 5.67 (br d, J = 8.0 Hz, 1H), 5.50 (q, J = 6.7 Hz, 1H), 5.01 (dd, J = 8.3, 6.2 Hz, 1H), 4.56–4.48 (m, 1H), 4.39–4.31 (m, 2H), 4.07–3.97 (m, 1H), 3.75–3.62 (m, 2H), 3.06 (dd, J = 14.3, 5.7 Hz, 1H), 2.81–2.70 (m, 4H), 2.56–2.47 (m, 1H), 2.24–2.05 (m, 1H), 1.85–1.58 (m, 7H), 1.47 (s, 9H), 1.35 (d, J = 6.2 Hz, 3H), 1.03 (s, 9H), 0.91–0.85 (m, 15H), 0.08 (s, 3H), 0.04 (s, 3H); 13C NMR (100 MHz, CDCl3, a mixture of rotamers) δ 173.1, 171.7, 171.3, 171.0, 170.1, 170.0, 166.3, 164.2, 154.6, 154.4, 136.4, 135.60, 135.55, 135.52, 135.46, 132.9, 132.8, 132.7, 130.1, 130.03, 129.98, 129.1, 129.0, 128.9, 128.8, 127.84, 127.82, 127.2, 127.1, 120.2, 119.8, 81.0, 71.6, 71.1, 63.4, 62.8, 58.8, 52.3, 52.0, 51.5, 37.6, 33.4, 32.9, 31.0, 30.5, 29.8, 28.9, 28.5, 28.3, 26.60, 26.55, 26.14, 26.06, 19.52, 19.46, 19.1, 18.9, 18.8, 18.7, 18.6, 18.5, 18.4, 14.3, 14.2, 13.1, −5.18, −5.21, −5.23, −5.25; HRMS (ESI) m/z calcd for C64H90N6O11Si2Na [M + Na]+ 1197.6104, found 1197.6054.
Compound 17

To a stirred solution of 3 (55.0 mg, 0.048 mmol) in CH2Cl2 (2 mL) was added 2,6-lutidine (340.42 μL, 2.923 mmol) and TMSOTf (423.22 μL, 2.338 mmol) at room temperature. After being stirred at the same temperature for 2h, the reaction mixture was quenched with MeOH and H2O and the aqueous layer was extracted by EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The deprotected amine was used without further purification.
To a stirred solution of 4 (2620.0 mg, 5.233 mmol) in THF:MeOH:H2O 2:1:1 mixture (40 mL) was added 1 N aqueous LiOH (15.7 mL) at room temperature. After being stirred at the same temperature for 6 h, the reaction mixture was acidified with 0.5 M aqueous HCl to pH 2–3 and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The deprotected acid was used without further purification.
To a stirred solution of deprotected amine in DMF (1 mL) was added DIEA (40.72 μL, 0.234 mmol), 3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT) (28.0 mg, 0.094 mmol) and the deprotected acid (34.1 mg, 0.070 mmol) at room temperature. After being stirred at the same temperature for 24 h, the reaction mixture was concentrated in vacuo and the residue was purified by preparative thin layer chromatography (6% MeOH in CH2Cl2) to give 17 (19.2 mg, 29% over 3 steps) as a white amorphous solid: [α]20D −30.9 (c 0.03, CH2Cl2); 1H NMR (600 MHz, (CD3)2SO) δ 9.29 (br s, 1H), 8.90 (br s, 1H), 8.53 (s, 1H), 8.18 (br s, 1H), 8.04 (d, J = 6.7 Hz, 1H), 7.89 (br s, 1H), 7.65–7.61 (m, 1H), 7.56 (d, J = 7.2 Hz, 2H), 7.50 (d, J = 7.2 Hz, 2H), 7.46–7.41 (m, 2H), 7.39–7.31 (m, 4H), 7.28–7.07 (m, 20H), 6.99 (d, J = 8.3 Hz, 2H), 6.61–6.53 (m, 3H), 5.30 (br s, 1H), 5.22–5.14 (m, 1H), 4.64 (d, J = 7.9 Hz, 2H), 4.46–4.40 (m, 1H), 4.34–4.26 (m, 2H), 4.23 (t, J = 7.2 Hz, 1H), 2.90 (dd, J = 14.1, 4.8 Hz, 1H), 2.69–2.64 (m, 1H), 2.63 (s, 3H), 2.41–2.33 (m, 2H), 2.31–2.23 (m, 1H), 2.18–2.01 (m, 3H), 1.87–1.78 (m, 1H), 1.78–1.65 (m, 3H), 1.56–1.43 (m, 6H), 1.39–1.28 (m, 2H), 1.28–1.16 (m, 5H), 1.14 (d, J = 6.3 Hz, 3H), 0.90 (s, 9H), 0.85–0.76 (m, 9H); 13C NMR (150 MHz, (CD3)2SO) δ 172.5, 172.4, 172.2, 171.8, 171.4, 169.5, 168.4, 167.9, 162.8, 153.5, 144.9, 136.9, 134.84, 134.83, 132.1, 132.0, 130.4, 130.3, 130.1, 129.7, 129.0, 128.5, 128.0, 127.93, 127.85, 127.4, 126.3, 119.2, 70.5, 69.2, 61.5, 60.5, 58.3, 54.6, 52.1, 50.9, 49.5, 35.6, 35.2, 32.8, 32.7, 30.9, 30.1, 29.9, 29.1, 28.1, 27.1, 26.1, 25.0, 21.8, 18.8, 18.6, 18.4, 13.8, 13.2; HRMS (MALDI) m/z calcd for C83H100N8O12SiNa [M + Na]+ 1451.7122, found 1451.7187.
Compound 2

To a stirred solution of 17 (19.2 mg, 0.013 mmol) in CH2Cl2 (1050 μL) was added triisopropylsilane (TIPS) (27.54 μL, 0.134 mmol) and TFA (150 μL) at 0 °C under. After being stirred at 0 °C to room temperature for 2 h, the reaction mixture was concentrated in vacuo and the residue was purified by preparative thin layer chromatography (7% MeOH in CH2Cl2) to give 2 (12.2 mg, 77%) as a white amorphous solid: [α]20D −18.4 (c 0.14, CH2Cl2) 1H NMR (600 MHz, (CD3)2SO) δ 9.30 (br s, 1H), 8.89 (br s, 1H), 8.17 (br s, 1H), 8.08 (d, J = 7.2 Hz, 1H), 7.94 (br s, 1H), 7.66–7.61 (m, 1H), 7.55 (d, J = 7.2 Hz, 2H), 7.50 (d, J = 7.4 Hz, 2H), 7.47–7.42 (m, 2H), 7.39–7.32 (m, 4H), 7.26–7.16 (m, 4H), 7.10 (d, J = 7.0 Hz, 2H), 6.99 (d, J = 8.2 Hz, 2H), 6.72 (s, 1H), 6.61–6.51 (m, 3H), 5.32 (br s, 1H), 5.21 (br s, 1H), 4.64 (d, J = 8.1 Hz, 2H), 4.46–4.40 (m, 1H), 4.37–4.27 (m, 2H), 4.27–4.20 (m, 1H), 2.90 (dd, J = 13.6, 3.5 Hz, 1H), 2.68–2.60 (m, 4H), 2.44–2.37 (m, 1H), 2.16–1.98 (m, 5H), 1.92–1.84 (m, 1H), 1.78–1.65 (m, 3H), 1.53 (d, J = 7.0 Hz, 3H), 1.53–1.44 (m, 3H), 1.38–1.27 (m, 2H), 1.28–1.17 (m, 5H), 1.15 (d, J = 6.2 Hz, 3H), 0.92 (s, 9H), 0.85–0.77 (m, 9H); 13C NMR (150 MHz, (CD3)2SO) δ 173.8, 172.51, 172.49, 172.2, 171.8, 169.5, 168.4, 167.9, 162.9, 153.5, 136.9, 134.9, 134.8, 132.1, 132.0, 130.5, 130.3, 130.1, 129.8, 129.0, 128.00, 127.95, 127.9, 126.4, 119.2, 70.5, 61.5, 60.5, 58.3, 54.7, 52.0, 50.9, 49.4, 35.6, 35.2, 32.9, 31.5, 30.9, 30.2, 29.9, 29.2, 28.1, 27.1, 26.2, 25.0, 21.8, 18.8, 18.6, 18.3, 13.8, 13.2; HRMS (ESI) m/z calcd for C64H86N8O12SiNa [M + Na]+ 1209.6032, found 1209.6008.
Lyngbyastatin 7 (1)

To a stirred solution of 2 (12.2 mg, 0.010 mmol) in DMSO (400 μL) was added IBX (8.6 mg, 0.031 mmol) at room temperature. After being stirred at the same temperature for 17 h, the reaction mixture was concentrated in vacuo and the residue was purified by preparative thin layer chromatography (6% MeOH in CH2Cl2) to give the aldehyde.
To a stirred solution of the aldehyde (3.6 mg, 0.003 mmol) in THF (250 μL) was added 1 M TBAF (3.7 μL, 0.0037 mmol) at 0 °C. After being stirred at 0 °C to room temperature for 2 h, the reaction mixture was diluted with EtOAc. The organic layer was washed with water, dried over MgSO4, filtered, and concentrated in vacuo. The residue was purified by reversed-phase HPLC (column, YMC-Pack ODS-AQ column 250 × 10 mm, 5 μm; flow rate, 2.0 mL/min; elution method, H2O/MeOH=50/50–17/83 linear gradient (0.0–40.0 min), H2O/MeOH=0/100 isocratic (40.0–50.0 min); retention time, 32.8 min) to give 1 (0.41 mg, 14% over 2 steps) as a white amorphous solid: [α]20D −10.0 (c 0.04, MeOH){lit [α]20D −7.4 (c 0.27, MeOH)}; 1H NMR (600 MHz, (CD3)2SO) δ 9.16 (br s, 1H), 8.07 (br s, 1H), 7.88 (br s, 1H), 7.49 (br s, 1H), 7.23 (s, 1H), 7.21–7.13 (m, 4H), 6.99 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 7.1 Hz, 2H), 6.76 (d, J = 8.5 Hz, 2H), 6.73 (br s, 1H), 6.52 (q, J = 7.0 Hz, 1H), 5.47 (br s, 1H), 5.07 (s, 1H), 4.90 (dd, J = 11.5, 1.7 Hz, 1H), 4.78–4.69 (m, 2H), 4.53 (br, 1H), 4.43–4.38 (m, 1H), 3.84–3.76 (m, 1H), 3.08 (d, J = 12.3 Hz, 1H), 2.87 (dd, J = 13.8, 11.7 Hz, 1H), 2.76 (s, 3H), 2.70 (dd, J = 14.0, 11.8 Hz, 1H), 2.43–2.36 (m, 1H), 2.18–2.04 (m, 5H), 1.96–1.88 (m, 1H), 1.85–1.79 (m, 1H), 1.77–1.67 (m, 2H), 1.59–1.47 (m, 7H), 1.30–1.20 (m, 7H), 0.88 (d, J = 6.8 Hz, 3H), 0.85 (t, J = 7.1 Hz, 3H), 0.75 (d, J = 6.9 Hz, 3H); 13C NMR (150 MHz, (CD3)2SO) δ 173.9, 172.70, 172.65, 172.63, 172.3, 170.4, 169.4, 168.9, 162.7, 156.2, 136.7, 131.8, 130.4, 130.0, 129.4, 127.8, 127.4, 126.2, 115.3, 73.7, 71.8, 60.8, 56.1, 55.7, 52.1, 50.3, 48.2, 35.3, 35.1, 32.9, 31.5, 30.9, 30.4, 29.3, 24.9, 21.93, 21.88, 19.3, 17.4, 13.9, 13.1; HRMS (ESI) m/z calcd for C48H66N8O12Na [M + Na]+ 969.4698, found 969.4670.
Validation of the purity of compound 1 and comparison of the retention time between natural lyngbyastatin 7 and compound 1 was done by reversed-phase HPLC (column, Phenomenex Synergi 4μ Hydro-RP 80 Å column 250 × 4.68 mm, 4 μm; flow rate, 1.0 mL/min; elution method, H2O/MeOH=50/50–20/80 linear gradient (0.0–36.0 min), H2O/MeOH=0/100 isocratic (36.0–46.0 min) or H2O/MeCN=80/20–50/50 linear gradient (0.0–30.0 min), H2O/MeCN=50/50–0/100 linear gradient (30.0–45.0 min), H2O/MeCN =0/100 isocratic (45.0–55.0 min)). The HPLC profiles were provided in Supporting Information Figures S4–S7.
Protease Assays
Porcine pancreatic elastase (Elastin Products Company, Owensville, MO) was dissolved in Tris-HCl (pH 8.0) to give a concentration of 75 μg/mL. N-succinyl-Ala-Ala-Ala-p-nitroanilide (Sigma-Aldrich, St. Louis, MO) was dissolved in Tris-HCl (pH 8.0) to give a 2 mM substrate solution. 1 μL of varying doses of compound 1, symplostatin 5, sivelestat (Sigma-Aldrich) or solvent control (DMSO), 5 μL of elastase solution and 79 μL of Tris-HCl (pH 8.0) were pre-incubated at room temperature for 15 min in a 96-well plate. At the end of the incubation, 15 μL of substrate solution was added to each well by using multichannel pipette, and the reaction was monitored by recording the absorbance at 405 nm every 30 s for 30 min on SpectraMax M5 (Molecular Devices, Sunnyvale, CA).
The inhibitory activity against human neutrophil elastase was also determined using the same procedure with minor modifications. The assay was carried by using 100 μg/mL human neutrophil elastase (Elastin Products Company) and 2 mM N-(OMe-succinyl)-Ala-Ala-Pro-Val-p-nitroanilide (Sigma-Aldrich), both prepared in 0.1 M Tris-0.5 M NaCl (pH 7.5).
Enzyme activity in each well was calculated based on the initial slope of the reaction curve, expressed as a percentage of the initial slope of the uninhibited reaction. IC50 calculations were done by GraphPad Prism 6 based on triplicate experiments.
General Cell Culture Procedure
Human bronchial epithelial cell line (transformed by the adenovirus 12-SV 40 hybrid virus) (BEAS-2B) purchased from ATCC (Manassas, VA) were grown in bronchial epithelial cell growth medium (BEGM, Lonza, Walkersville, MD) under a humidified environment with 5% CO2 at 37 °C. All culture plates and flasks were pre-coated with a mixture of 0.01mg/mL fibronectin (Sigma-Aldrich), 0.03 mg/mL bovine collagen type I (Sigma-Aldrich) and 0.01 mg/mL bovine serum albumin (Sigma-Aldrich) dissolved in bronchial epithelial basal medium (BEBM, Lonza) before use.
Cell Viability Assay (MTT)
BEAS-2B cells were seeded at a density of 7500 cells per well in collagen-coated 96-well plates (Corning, Corning, NY). After 24 h of incubation, the cells were cotreated with varying doses of compound 1, symplostatin 5, sivelestat or solvent control (DMSO), and 100 nM HNE or vehicle (40% [1:1 glycerol: 0.05 M sodium acetate pH 5.0] in BEBM). The cells were incubated for an additional 24 h before the addition of the MTT reagent (Promega, Madison, WI). Cell viability was measured according to the manufacturer's instructions and recorded on SpectraMax M5. IC50 calculations were done by GraphPad Prism 6 based on triplicate experiments.
RNA Extraction, cDNA Synthesis and Quantitative PCR (qPCR) Analysis
BEAS-2B cells were seeded at a density of 3 × 105 cells per well in 6-well plates pre-coated with a mixture of 0.01mg/mL fibronectin, 0.03 mg/mL bovine collagen type I and 0.01 mg/mL bovine serum albumin dissolved in BEBM.
After 24 h of incubation, the medium was replaced with supplement-free BEBM and the cells were further incubated for 24 h. Prior to treatment, the cells were replenished with new supplement-free BEBM and then followed by cotreatment with varying doses of compound 1, symplostatin 5, sivelestat dissolved in DMSO and 100 nM HNE. Control cells were treated with 1% DMSO and 1% vehicle (40% [1:1 glycerol: 0.05 M sodium acetate pH 5.0] in BEBM).
Total RNA was extracted at 3 h post treatment with the aid of RNeasy mini kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. RNA was quantified and qualified using NanoDrop 2000 spectrophotometer (Fisher, Pittsburgh, PA).
From 2 μg of total RNA, cDNA was synthesized by using SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA) and oligo(dT)12–18 primer (Invitrogen).
qPCR was performed with a total reaction volume of 25 μL consisting of 12.5 μL of 2× TaqMan gene expression master mix (Applied Biosystems), 1.25 μL of a 20× TaqMan gene expression assay probe (Applied Biosystems), 0.33 μL of cDNA, and 10.92 μL of RNase-free sterile water. The reactions were dispensed into 96-well optical reaction plates (Applied Biosystems) and detected in an Applied Biosystems 7300 Real-Time PCR system using the thermocycler program: 2 min at 50 °C, 10 min at 95 °C, and 15 s at 95 °C (40 cycles) and 1 min at 60 °C. Each assay was performed in triplicate. IL1A (Hs00174092_m1), IL1B (Hs01555410_m1) and IL8 (Hs00174103_m1) were used as target genes while GAPDH (Hs02758991_g1) was used as endogenous control for normalization. Graphs and data analysis were performed using the Prism software and analyzed using ANOVA followed by Dunnett's t test.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by James and Esther King Biomedical Research Program, TTF Grant 3KF04 and in part by the National Institutes of Health grant R01CA172310. We thank J. Rocca (Advanced Magnetic Resonance Imaging and Spectroscopy) for assistance with NMR data acquisition.
Footnotes
Supporting Information Comparison of the 1H NMR spectrum of natural lyngbyastatin 7 and synthetic compound 1.The HPLC profiles of natural lyngbyastatin 7 and synthetic compound 1 on Phenomenex Synergi 4μ Hydro-RP 80 Å column (250 × 4.68 mm, 4 μm) with two different solvent systems. 1H and 13C NMR spectra of all the synthetic compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Hendrik Luesch is co-founder of Oceanyx Pharmaceuticals, Inc., which is negotiating licenses for patent applications related to the subject matter.
REFERENCES
- (1).Lucas SD, Costa E, Guedes RC, Moreira R. Med. Res. Rev. 2013;33:73–101. doi: 10.1002/med.20247. [DOI] [PubMed] [Google Scholar]
- (2).Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Pharmacol. Rev. 2010;62:726–759. doi: 10.1124/pr.110.002733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Korkmaz B, Moreau T, Gauthier F. Biochimie. 2008;90:227–242. doi: 10.1016/j.biochi.2007.10.009. [DOI] [PubMed] [Google Scholar]
- (4).Von Nussbaum F, Li VM-J. Bioorg. Med. Chem. Lett. 2015;25:4370–4381. doi: 10.1016/j.bmcl.2015.08.049. [DOI] [PubMed] [Google Scholar]
- (5).Chughtai B, O'Riordan TG. J. Aerosol Med. 2004;17:289–298. doi: 10.1089/jam.2004.17.289. [DOI] [PubMed] [Google Scholar]
- (6).Kawabata K, Suzuki M, Sugitani M, Imaki K, Toda S, Miyamoto T. Biochem. Biophys. Res. Commun. 1991;177:814–820. doi: 10.1016/0006-291x(91)91862-7. [DOI] [PubMed] [Google Scholar]
- (7).Aikawa N, Ishizaka A, Hirasawa H, Shimazaki S, Yamamoto Y, Sugimoto H, Shinozaki M, Taenaka N, Endo S, Ikeda T, Kawasaki Y. Pulm. Pharmacol. Ther. 2011;24:549–554. doi: 10.1016/j.pupt.2011.03.001. [DOI] [PubMed] [Google Scholar]
- (8).Aikawa N, Kawasaki Y. Ther. Clin. Risk Manag. 2014;10:621–629. doi: 10.2147/TCRM.S65066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Iwata K, Doi A, Ohji G, Oka H, Oba Y, Takimoto K, Igarashi W, Gremillion DH, Shimada T. Intern. Med. 2010;49:2423–2432. doi: 10.2169/internalmedicine.49.4010. [DOI] [PubMed] [Google Scholar]
- (10).Zeiher BG, Artigas A, Vincent J-L, Dmitrienko A, Jackson K, Thompson BT, Bernard G. Crit. Care Med. 2004;32:1695–1702. doi: 10.1097/01.ccm.0000133332.48386.85. [DOI] [PubMed] [Google Scholar]
- (11).Vegman M, Carmeli S. Tetrahedron. 2013;69(47):10108–10115. [Google Scholar]
- (12).Nagarajan M, Maruthanayagam V, Sundararaman M. J. Appl. Toxicol. 2013;33:313–349. doi: 10.1002/jat.2833. [DOI] [PubMed] [Google Scholar]
- (13).Taori K, Matthew S, Rocca JR, Paul VJ, Luesch H. J. Nat. Prod. 2007;70:1593–1600. doi: 10.1021/np0702436. [DOI] [PubMed] [Google Scholar]
- (14).Salvador LA, Taori K, Biggs JS, Jakoncic J, Ostrov DA, Paul VJ, Luesch H. J. Med. Chem. 2013;56:1276–1290. doi: 10.1021/jm3017305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Yudin AK. Chem. Sci. 2015;6:30–49. doi: 10.1039/c4sc03089c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Giordanetto F, Kihlberg J. J. Med. Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- (17).Yokokawa F, Inaizumi A, Shioiri T. Tetrahedron. 2005;61:1459–1480. [Google Scholar]
- (18).Yokokawa F, Shioiri T. Tetrahedron Lett. 2002;43:8673–8677. [Google Scholar]
- (19).White CJ, Yudin AK. Nat. Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- (20).Yokokawa F, Shioiri T. Tetrahedron Lett. 2002;43:8679–8682. [Google Scholar]
- (21).Gomez-Martinez P, Dessolin M, Guibé F, Albericio F. J. Chem. Soc. Perkin Trans. 1999;1:2871–2874. [Google Scholar]
- (22).Guibé F. Tetrahedron. 1998;54:2967–3042. [Google Scholar]
- (23).El-Faham A, Albericio F. Chem. Rev. 2011;111:6557–6602. doi: 10.1021/cr100048w. [DOI] [PubMed] [Google Scholar]
- (24).Ye Y, Li H, Jiang X. Pept. Sci. 2005;80:172–178. doi: 10.1002/bip.20201. [DOI] [PubMed] [Google Scholar]
- (25).Reddel RR, Ke Y, Gerwin BI, McMenamin MG, Lechner JF, Su RT, Brash DE, Park J-B, Rhim JS, Harris CC. Cancer Res. 1988;48:1904–1909. [PubMed] [Google Scholar]
- (26).Caramori G, Adcock IM, Stefano AD, Chung K. Int. J. COPD. 2014;9:397–412. doi: 10.2147/COPD.S42544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Yoshimoto T, Yoshimoto T, editors. Cytokine Frontiers: Regulation of Immune Responses in Health and Disease. Springer; Tokyo, Japan: 2014. [Google Scholar]
- (28).Bhatia M, Moochhala S. J. Pathol. 2004;202:145–156. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- (29).Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. J. Clin. Invest. 2001;107:1529–1536. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pittet JF, Mackersie RC, Martin TR, Matthay MA. Am. J. Respir. Crit. Care Med. 1997;155:1187–1205. doi: 10.1164/ajrccm.155.4.9105054. [DOI] [PubMed] [Google Scholar]
- (31).Armarego WLF, Chai CLL, editors. Purifcation of Laboratory Chemicals. 7th ed. Butterworth-Heinemann; Oxford, U.K.: 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.