

Abstract
One strategy for combating cancer-drug resistance is to deploy rational polytherapy up front that suppresses the survival and emergence of resistant tumor cells. Here we demonstrate in models of lung adenocarcinoma harboring the oncogenic fusion of ALK and EML4 that the GTPase RAS–mitogen-activated protein kinase (MAPK) pathway, but not other known ALK effectors, is required for tumor-cell survival. EML4-ALK activated RAS-MAPK signaling by engaging all three major RAS isoforms through the HELP domain of EML4. Reactivation of the MAPK pathway via either a gain in the number of copies of the gene encoding wild-type K-RAS (KRASWT) or decreased expression of the MAPK phosphatase DUSP6 promoted resistance to ALK inhibitors in vitro, and each was associated with resistance to ALK inhibitors in individuals with EML4-ALK–positive lung adenocarcinoma. Upfront inhibition of both ALK and the kinase MEK enhanced both the magnitude and duration of the initial response in preclinical models of EML4-ALK lung adenocarcinoma. Our findings identify RAS-MAPK dependence as a hallmark of EML4-ALK lung adenocarcinoma and provide a rationale for the upfront inhibition of both ALK and MEK to forestall resistance and improve patient outcomes.
Lung adenocarcinoma cells harboring oncogenic rearrangements of the gene encoding anaplastic lymphoma kinase (ALK), such as EML4-ALK, are typically dependent upon the oncogene for survival. Hence, treatment with an ALK tyrosine-kinase inhibitor (crizotinib or ceritinib) often elicits profound initial anti-tumor responses in ALK fusion–positive (ALK+) patients with lung adenocarcinoma1–3. However, complete, durable responses to ALK-inhibitor monotherapy are rare, and patients invariably succumb to the emergence of drug-resistant disease (acquired resistance). Furthermore, up to 40% of ALK+ patients fail to respond initially to ALK-inhibitor therapy and exhibit innate resistance2,4. Identifying molecular events that limit the response to ALK inhibition is essential for enhancing clinical outcomes.
In most oncogene-driven cancers, including ALK+ lung cancers, efforts to combat resistance have focused on treating acquired resistance after it has emerged2. One alternative strategy for enhancing the initial response and suppressing acquired resistance is to deploy rational upfront polytherapies that target the primary oncoprotein (such as EML4-ALK) and a critical effector of that oncoprotein5,6. This strategy has been successful in patients with BRAFV600E-mutant melanoma, in whom upfront (but not second-line) inhibition of the primary driver BRAFV600E plus its primary effector, MEK1/2, exhibits activity superior to that of RAF- or MEK-inhibitor monotherapy5,7–9.
The optimal upfront co-targeting strategy is less clear in tumors harboring an oncoprotein that engages multiple effector pathways, such as receptor tyrosine kinases (RTKs)10. Mutant EGFR and ALK, ROS1 or RET gene rearrangements are prominent oncogenic RTKs in lung adenocarcinoma11. A rational co-targeting strategy requires understanding of the signaling events that are most critical for survival in tumor cells with a particular oncogenic RTK. We addressed this knowledge gap in EML4-ALK lung adenocarcinoma to provide insight into the oncogenic function of ALK and identify a rational upfront polytherapy strategy to enhance patient survival.
RESULTS
EML4-ALK lung adenocarcinoma cells depend on MAPK
EML4-ALK signals via the PI3K-AKT, MAPK and JAK-STAT pathways3 (Fig. 1a). Which effector is most critical for EML4-ALK–driven cell survival is unclear. We investigated downstream pathway dependencies in EML4-ALK lung adenocarcinoma cells, focusing on the most common ALK fusion variant in lung adenocarcinoma (EML4-ALK E13:A20, variant 1)11. The ALK inhibitors crizotinib or ceritinib decreased cell growth and the abundance of phosphorylated (p-) ALK, p-ERK, p-AKT and p-STAT3 in two patient-derived EML4-ALK (E13:A20) cell lines, H3122 and STE-1 (ref. 12) (Fig. 1b). Inhibition of MAPK (via MEK inhibition), but not of PI3K-AKT or JAK-STAT, suppressed cell growth similar to inhibition of ALK (Fig. 1c and Supplementary Fig. 1a–d). Conversely, constitutive genetic activation of MAPK signaling at the level of the GTPase RAS (KRASG12V/C/D), RAF (BRAFV600E) or (MAP2K1S218D,S222D, MEKDD)13 rescued EML4-ALK lung adenocarcinoma cells from ALK inhibition, whereas genetic activation of AKT signaling (via myristoylated AKT) did not (Fig. 1d–g and Supplementary Figs. 1e–g and 2a–g). K-RASG12V-expressing cells rescued MAPK signaling, but not AKT signaling, in the presence of ALK inhibition (Fig. 1e). Genetic activation of STAT3 partially rescued EML4-ALK lung adenocarcinoma cells from the effects of treatment with an ALK inhibitor, consistent with published work14, although modestly so compared with the activation of MAPK (Supplementary Fig. 2a–h). These data reveal a specific and primary requirement for MAPK signaling in EML4-ALK lung adenocarcinoma cells.
Figure 1.
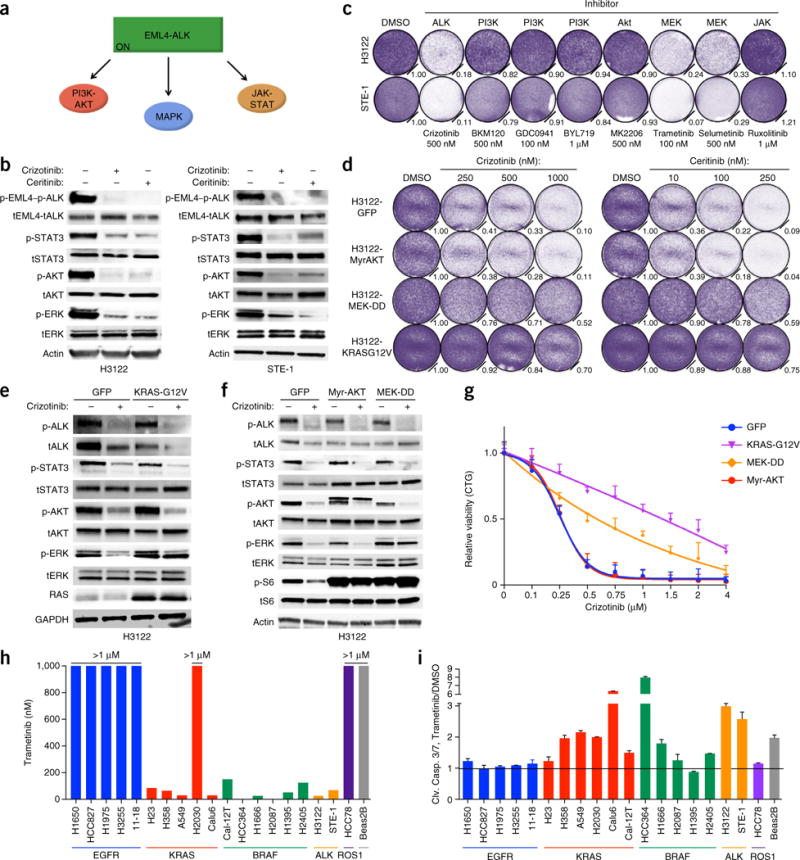
EML4-ALK (E13;A20, variant 1) lung adenocarcinoma cells are specifically dependent upon MAPK signaling. (a) Known EML4-ALK effector pathways. ON, activated. (b) Immunoblot analysis showing inhibition of EML4-ALK effector pathways in H3122 and STE-1 cells treated for 1 h with crizotinib (1 μM) or ceritinib (200 nM) (+) or not (−). Actin (bottom) serves as a loading control; ‘t’ before protein designation indicates total protein. (c) Crystal violet staining and quantification of H3122 and STE-1 cells 5 d after treatment with dimethyl sulfoxide (DMSO) or the indicated inhibitors (target inhibition, Supplementary Fig. 1a,b). Quantification (relative number of stained cells) is shown at bottom right of each plate. (d) Growth-inhibition response to 5 d of treatment with crizotinib or ceritinib in H3122 cells expressing cDNA encoding green fluorescent protein (H3122-GFP) or activated AKT (myristoylated AKT (H3122-MyrAKT)), MEK (H3122-MEK-DD) or RAS (H3122-KRASG12V). (e,f) Immunoblot analysis, with antibodies against the indicated molecules, of lysates of H3122-GFP control cells (e,f) and H3122-K-RAS-G12V cells (e) or H3122-Myr-AKT and H3122-MEK-DD cells (f) treated with 250 nM crizotinib for 30 min (+) or not (−). GAPDH (bottom) serves as a loading control throughout. (g) Growth-inhibition response to 72 h of treatment with crizotinib in STE-1 cells expressing cDNA as in d, relative to untreated controls, presented as ‘CellTiterGlo’ (CTG). (h) Half-maximal inhibition concentration (IC50) values (above bars) for the trametinib dose response in a panel of lung cancer cell lines with the indicated genetic drivers. (i) Induction of cleavage of caspases 3 and 7 (Clv. Casp. 3/7) in the cell lines in h after 24 h of treatment with 100 nM trametinib; values are normalized to those of DMSO-treated cells (Trametinib/DMSO). Data are representative of three experiments (b–i; error bars (g,i), s.e.m.).
We explored whether the MAPK dependence noted above was present generally in oncogene-driven lung cancer. Most lung adenocarcinomas harbor a genetic lesion encoding a product capable of hyperactivating MAPK signaling, including oncogenic KRAS, BRAF, EGFR, MEK, ERBB2, MET, ALK, RET and ROS1, among other events in the MAPK pathway11. MEK inhibitors have shown limited efficacy in lung adenocarcinoma patients to date, which indicates the need for better definition of tumor subsets that are most MEK dependent15. We found that the EML4-ALK models were among the most sensitive to MEK inhibition in a panel of oncogene-driven lung adenocarcinoma models (Fig. 1h,i and Supplementary Fig. 3). This MEK-inhibitor sensitivity in EML4-ALK models was similar to that observed in lung adenocarcinoma cells with oncogenic K-RAS or BRAF, but not to that of cells with oncogenic EGFR, which had little response to the MEK inhibitor trametinib (Fig. 1h,i and Supplementary Fig. 3). The data reveal the specificity of dependence on the MAPK pathway in ALK+ lung adenocarcinoma cells.
EML4-ALK cells activate RAS-MAPK via the EML4 HELP domain
Although GTPases of the RAS family canonically link RTKs to MAPK signaling, whether oncogenic ALK fusion proteins activate RAS to promote MAPK signaling is unknown. Using GST–RAS-binding domain (RBD) affinity capture16, we found that activation of RAS was coupled to ALK signaling in both H3122 cells and STE-1 cells (Fig. 2a,b). As three major RAS isoforms (H-RAS, N-RAS and K-RAS) typically function in cancer cells, we explored which isoform(s) was (were) coupled to EML4-ALK in lung adenocarcinoma cells. Using RAS isoform–specific antibodies17, we found that all three RAS isoforms were activated by EML4-ALK in lung adenocarcinoma cells (Fig. 2a,b). Although genetic silencing of each RAS isoform individually had no effect on signaling or cell viability, simultaneous knockdown of all three substantially suppressed cell growth and the abundance of p-ERK but not that of p-STAT3 or p-AKT (Fig. 2c,d and Supplementary Fig. 4a). These results suggest that RAS controls MAPK signaling, but not STAT3 or AKT signaling, downstream of EML4-ALK in lung adenocarcinoma cells.
Figure 2.
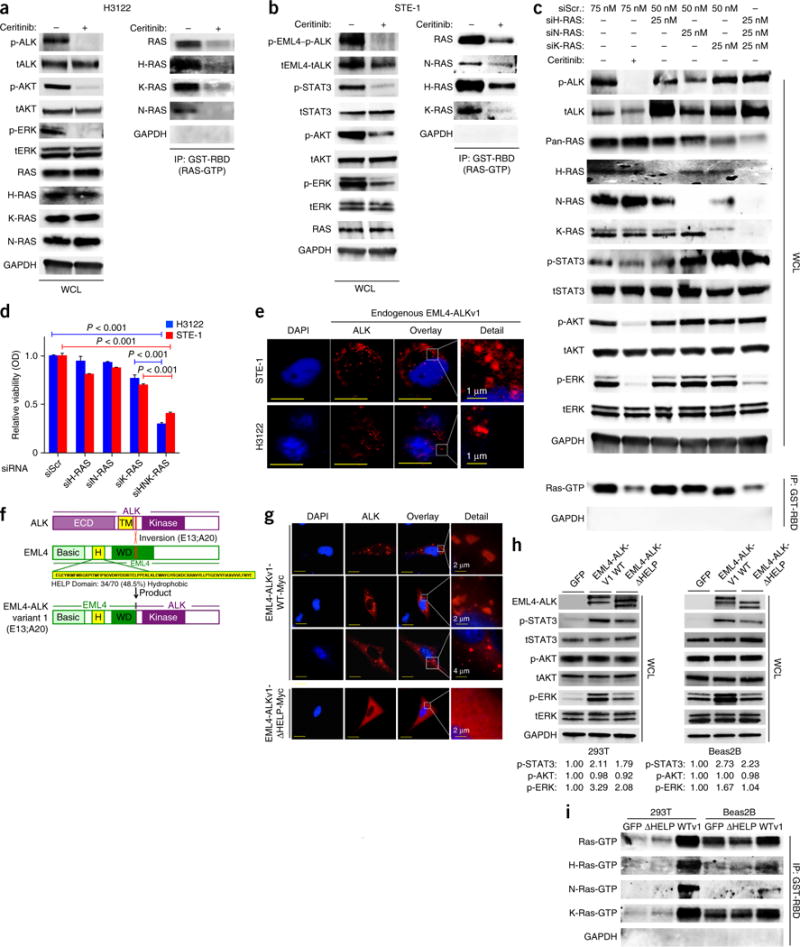
Cells expressing EML4-ALK (E13:A20, variant 1) activate H-, N- and K-RAS to drive MAPK signaling, via the HELP domain of EML4. (a,b) Immunoblot analysis, with antibodies against the indicated molecules, of whole-cell lysates (WCL) or GST-RBD-immunoprecipitated (IP) lysates of H3122 (a) and STE-1 (b) cells treated for 30 min with 200 nM ceritinib (+) or not (−). (c) Immunoblot analysis, with antibodies against the indicated molecules, of WCL or GST-RBD IP lysates of cells transfected with scrambled siRNA (siScr) or siRNA directed against H-RAS (siH-RAS), N-RAS (siN-RAS) or K-RAS (siK-RAS) and treated with 200 nM ceritinib (+) or DMSO (−) for 30 min. (d) Crystal violet assay of cell cultures treated as in c or with siRNA directed against all three RAS isoforms (siHNK-RAS) OD, optical density. P < 0.001 (unpaired t-test). (e) Immunofluorescence staining of endogenous EML4-ALK in STE-1 and H3122 cells with antibody against ALK. Red shows EML4-ALK expression; blue shows DAPI nuclear staining. Inset (far right) shows high magnification of EML4-ALK localization. Representative images from three independent experiments are shown. Scale bars indicate 10 μM. (f) Schematic of ALK (top), showing the extra-cellular domain (ECD) and transmembrane domain (TM), as well as the E13;A20 inversion (below); EML4 (middle), showing the hydrophobic HELP domain (H) and WD-repeat domain (WD); and the EML4-ALK variant 1 (E13:A20) (bottom). (g) Immunofluorescence staining of overexpressed Myc-tagged EML4-ALKWT (EML4-ALKv1-WT-Myc) or ΔHELP EML4-ALK (EML4-ALKv1-ΔHELP-Myc) with antibody against the Myc tag in Beas2B cells. Red shows EML4-ALK expression; blue shows DAPI nuclear staining. Inset (far right) shows high magnification of EML4-ALK localization. Representative images from three independent experiments are shown. Scale bars indicate 15 μm. (h) Immunoblot analysis, with antibodies against the indicated molecules, of lysates of Beas2B and 293T cells transfected with the indicated constructs. Below, expression of each phosphorylated protein relative to that in the GFP-control condition, assessed by densitometry. (i) GST-RBD pulldown assays to isolate Ras-GTP, with immunoblotting using RAS isoform-specific antibodies, in the cells in h. Data shown are n = 3, ±s.e.m., for quantitative assays and for immunoblot assays representative of three independent experiments.
We investigated how EML4-ALK might engage RAS. Signaling via RAS to its downstream effector pathways typically occurs on a cellular membrane compartment (either the plasma membrane or intracellular membranes)16,18,19. All ALK fusions reported in lung adenocarcinoma contain the kinase domain of ALK but not the native ALK transmembrane domain that enables membrane anchoring20,21. We first examined the subcellular distribution of EML4-ALK using immunofluorescence staining of endogenous ALK in H3122 and STE-1 lung adenocarcinoma cell lines. Endogenous EML4-ALK resided on an intracellular compartment but not the plasma membrane, where many native receptor kinases engage RAS (Fig. 2e). We investigated how a fusion protein with no known membrane-anchoring domain might engage effectors that require a lipid interface to signal, such as RAS, potentially from an intracellular locale. The EML4 portion of EML4-ALKv1 contains Basic, HELP and WD-repeat domains (Fig. 2f). The HELP domain of EML4 consists of approximately 50% hydrophobic residues, which suggests that it might mediate membrane association and access to effectors such as RAS. Although the function of the EML4 HELP domain is poorly understood, removal of it impairs the transforming capacity of EML4-ALK22, and it can regulate EML4’s subcellular localization23,24. We hypothesized that the HELP domain in the EML4 component of the EML4-ALK fusion might be required for proper EML4-ALK localization and RAS-MAPK signaling. We introduced wild-type EML4-ALK (EML4-ALKWT) or a mutant form lacking the HELP domain (∆HELP) into non-transformed lung epithelial (Beas2B) cells and examined EML4-ALK localization and signaling. Overexpressed EML4-ALKWT was present on a distinct intracellular compartment, as assessed by immunofluorescence staining, similar to endogenous EML4-ALK (Fig. 2g). In contrast, the ∆HELP EML4-ALK mutant did not display this discrete intracellular localization but instead exhibited diffuse cytoplasmic expression (Fig. 2g). Furthermore, expression of EML4-ALKWT activated ERK and also STAT3, but not AKT, in both Beas2B cells and 293T cells (Fig. 2h). Moreover, expression of EML4-ALKWT enhanced the GTP loading of each RAS isoform (Fig. 2i). Deletion of the HELP domain impaired the ability of EML4-ALK to activate ERK and RAS in both Beas2B cells and 293T cells (Fig. 2h,i). Moreover, activation of EML4-ALK was uncoupled from RAS activation and MAPK signaling in H2228 lung adenocarcinoma cells that endogenously expressed a rarer EML4-ALK variant (3b) in which EML4 lacks the HELP domain25 (Supplementary Fig. 4b). In these H2228 cells, inhibition of ALK failed to suppress RAS-GTP, p-ERK or cell viability (Supplementary Fig. 4c–f). H2228 cells were less sensitive to MEK inhibition than were H3122 or STE-1 cells (Supplementary Figs. 3 and 4g). Thus, the HELP domain of EML4 in EML4-ALK might regulate EML4-ALK’s subcellular localization and be critical for the activation of RAS-MAPK by EML4-ALK.
Superiority of upfront ALK + MEK polytherapy
Our data indicated that inhibition of ALK was insufficient to fully abrogate MAPK signaling in EML4-ALK lung adenocarcinoma cells (Fig. 1b,e,f). We hypothesized that elimination of this residual MAPK signaling by treatment with sub-maximal doses of MEK inhibitor combined with an ALK inhibitor might enhance the response. The use of a sub-maximal dose of MEK inhibitor is an attractive option, given the clinical toxicity of MEK-inhibitor monotherapy at maximally tolerated doses26,27. The addition of a low dose of trametinib (1 nM) sensitized H3122 and STE-1 cells to inhibition of ALK, with concurrent treatment with trametinib and crizotinib eliciting greater apoptosis than either monotherapy (Fig. 3a–d and Supplementary Fig. 5d). Treatment with the distinct MEK inhibitor selumetinib or with the ERK inhibitor SCH772984 also suppressed cell growth and enhanced the response to the ALK inhibitor alectinib in H3122 cells (Supplementary Fig. 5a). In contrast, trametinib did not sensitize EGFR-mutant lung adenocarcinoma cells to the EGFR inhibitor erlotinib (Supplementary Fig. 5b,c). The superior efficacy of combined inhibition of ALK plus inhibition of the MAPK pathway we observed in vitro was confirmed in vivo with H3122 tumor xenografts, in which substantial tumor regressions occurred only upon treatment with the ALK inhibitor plus trametinib (Fig. 3e and Supplementary Fig. 6a). We observed residual MAPK activity in vivo in the tumors treated with ALK-inhibitor monotherapy (ceritinib, at a dose of 25 mg per kg body weight (mg/kg)), and this residual MAPK signaling was suppressed by the addition of a sub-maximal dose of trametinib (1 mg/kg; Fig. 3f). Whereas mice treated with the maximal tolerated dose of trametinib alone (3 mg/kg) exhibited substantial systemic toxicity, the combination of the ALK inhibitor and a sub-maximal dose of trametinib (1 mg/kg) did not cause significant toxicity (Supplementary Fig. 6b). We similarly observed superior in vivo tumor responses and safety in mice harboring STE-1 xenografts treated with combined crizotinib and (sub-maximal) trametinib, compared with results obtained for each monotherapy (Fig. 3g,h and Supplementary Fig. 6c). Although activated STAT3 modestly decreased sensitivity to an ALK inhibitor (Supplementary Fig. 2), treatment with a JAK inhibitor did not affect tumor growth or response to an ALK inhibitor in EML4-ALK cell lines and tumor xenografts (Supplementary Fig. 6d–f), which suggested specificity of the effects of MEK inhibition on the response to an ALK inhibitor. Our findings show the potential utility, feasibility and specificity of combined ALK inhibitor–MEK inhibitor polytherapy to enhance the initial response in EML4-ALK lung adenocarcinoma.
Figure 3.
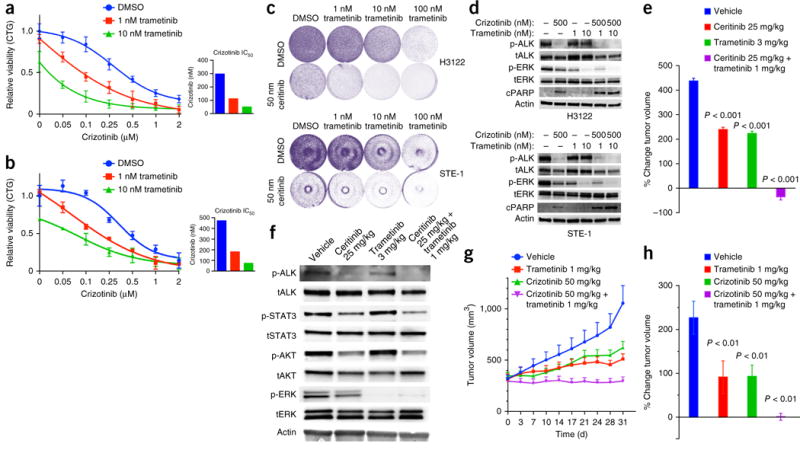
Enhanced therapeutic effect of upfront co-treatment with an ALK inhibitor and a sub-maximal MEK inhibitor. (a,b) Growth-inhibition response (as in Fig. 1g) of H3122 (a) and STE-1 (b) cells treated with crizotinib together with DMSO or trametinib (1 nM or 10 nM). Insets (right) display crizotinib IC50 values. (c) Crystal violet cell-growth assays of H3122 cells (top) and STE-1 cells (bottom) treated as in a,b. (d) Immunoblot analysis, with antibodies against the indicated molecules, of H3122 and STE-1 cells treated for 24 h with crizotinib (500 nM) or trametinib (100 nM) or a combination of those. n ≥ 3, data are shown ±s.e.m. for quantitative assays and for immunoblots representative of three or more independent experiments. (e) Tumor volume (mm3) of H3122 xenografts at 23 d after treatment initiation with ceritinib (25 mg/kg), trametinib (3 mg/kg) or a combination of those at a lower dose of trametinib (1 mg/kg). Values shown are the percentage change in tumor volume from baseline (day 0) ±s.e.m., n = 8 tumors per group. P values, treatment group versus vehicle (unpaired t-test). (f) Immunoblot analysis, with antibodies against the indicated molecules, of lysates from H3122 xenograft tumors 48 h after each treatment. (g,h) Tumor volume (mm3) of STE-1 xenografts during treatment with crizotinib (50 mg/kg), trametinib (1 mg/kg) or a combination of those. Values shown are the tumor volumes over time from baseline (day 0) ±s.e.m., n = 8 tumors per group. P < 0.01, treatment group versus vehicle (unpaired t-test).
KRASWT copy-number gain promotes ALK-inhibitor resistance
On the basis of our findings, we hypothesized that the development of acquired resistance to an ALK inhibitor might consistently require re-activation of the RAS-MAPK pathway. We developed multiple in vitro models of acquired resistance to an ALK inhibitor by continuously exposing H3122 cells to either crizotinib (‘crizotinib acquired resistance’ (CAR); n = 3 sub-lines) or ceritinib (‘LDK378 acquired resistance’ (LAR); n = 3 sub-lines) and explored the basis of resistance in the sub-lines derived. Each resistant sub-line was cross-resistant to each ALK inhibitor (Fig. 4a and Supplementary Fig. 7a,b). By DNA-sequencing analysis of ALK, we detected no on-target mutations or alterations in ALK copy number in these sub-lines28 (data not shown). As we found that each in vitro model of ALK-inhibitor resistance showed re-activation of the MAPK pathway during treatment with the ALK inhibitor (Fig. 4b), we investigated whether MAPK signaling was required for resistance. All of the resistant models retained substantial MAPK signaling (MEK) dependence, whereas suppression of JAK-STAT or PI3K-AKT signaling had less impact (Fig. 4c and Supplementary Fig. 7c). Thus, MAPK signaling was rescued and was necessary for acquired resistance to the ALK inhibitor.
Figure 4.
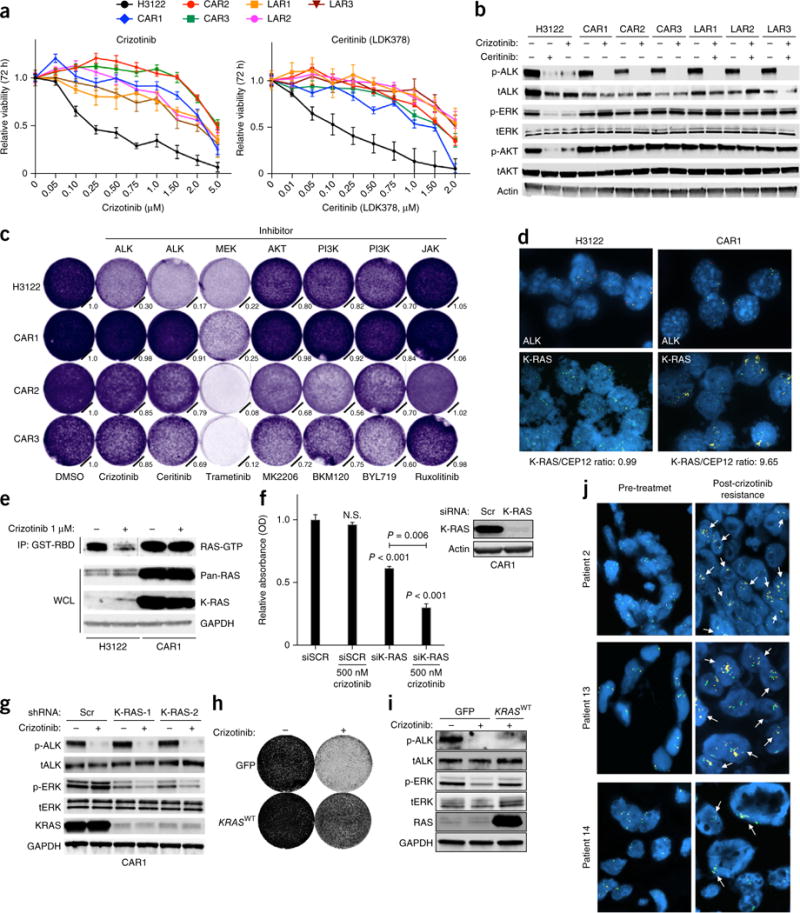
Reactivation of MAPK signaling by KRASWT copy-number gain promotes ALK-inhibitor resistance in EML4-ALK lung adenocarcinoma. (a) Viability of H3122 cells, CAR1–CAR3 cells and ceritinib (LDK378)-resistant cells (LAR1–LAR3) treated (72 h) with the indicated doses of crizotinib or ceritinib; results were normalized to those obtained with DMSO. (b) Immunoblotting of lysates from the indicated cells treated with DMSO or the indicated treatments for 6 h. (c) Growth of H3122 and CAR1–CAR3 cells treated as indicated and stained with crystal violet after 5 d. Normalized cell growth quantification is shown. (d) KRASWT amplification confirmed by FISH in H3122 and CAR1 cells. ALK break-apart (yellow, red), K-RAS (yellow), and CEP12 (control; green) probes are shown, as is the K-RAS/CEP12 ratio. (e) Immunoblotting of WCL and GST-RBD IP lysates of H3122 and CAR1 cells treated with DMSO or 1 μM crizotinib for 30 min (line indicates immunoblot crop). (f) Viability of CAR1 cells transfected with scrambled siRNA or siRNA targeting K-RAS and treated with DMSO or 500 nM crizotinib, assessed by crystal violet staining. N.S., not significant; P < 0.001 or 0.006, for the comparative analysis (unpaired t-test). (g) Immunoblots, with antibodies against the indicated molecules, in lysates of CAR1 cells following the introduction of scrambled shRNA or two independent shRNAs targeting K-RAS and treatment with 1 μM crizotinib or vehicle for 1 h. (h) Crystal-violet growth assay of STE-1 cells expressing GFP or KRASWT and treated with 250 nM crizotinib for 5 d. (i) Immunoblots of GFP- and KRASWT-expressing STE-1 cells treated with 250 nM crizotinib for 1 h. (j) Representative KRAS FISH images showing acquisition of KRASWT amplification in patients #2, #13 and #14. KRAS probes are yellow; CEP12 (control) is green. Arrows indicate representative positive signals. Data are representative of three independent experiments (a–j; error bars (a,f), s.e.m.).
We sought to identify the mechanisms by which the ALK inhibitor–resistant models rescued MAPK signaling, focusing on the crizotinib-resistant models because this ALK inhibitor is currently most widely used. Whole-exome sequencing analysis of the crizotinib-resistant H3122 cell sub-lines CAR1–CAR3 revealed no canonical mutations in ALK, KRAS, NRAS, HRAS, ARAF, BRAF, CRAF, MAP2K1 (encoding MEK1), MAP2K2 (encoding MEK2), MAPK3 (encoding ERK1) or MAPK1 (encoding ERK2) or in genes encoding upstream RTKs that could explain MAPK activation and resistance (data not shown). This exome-sequencing analysis instead revealed focal amplification of the wild-type gene encoding K-RAS (KRASWT) in CAR1 cells (confirmed by fluorescence in situ hybridization (FISH) of KRAS), but not in CAR2 or CAR3 cells (Fig. 4d, Supplementary Fig. 8a and data not shown). CAR1 cells had substantially higher levels of KRAS4A-4B transcripts, K-RAS protein and RAS-GTP than did the parental H3122 cells (Fig. 4e and Supplementary Fig. 8b). Knockdown of K-RAS decreased viability and the abundance of p-ERK and increased apoptosis and sensitivity to crizotinib in CAR1 cells (Fig. 4f,g and Supplementary Fig. 8c–e). Conversely, overexpression of KRASWT promoted resistance to crizotinib in STE-1 and H3122 cells (Fig. 4h,i and Supplementary Fig. 9a,b). These data identify amplification of KRASWT as a mechanism of resistance to an ALK inhibitor.
To explore the clinical relevance of the findings reported above, we obtained tumor biopsies from patients (n = 15) with acquired resistance to ALK-inhibitor therapy and analyzed the biopsies for evidence of KRAS copy-number gain (Supplementary Fig. 8f). We observed focal amplification of KRAS in three patients (3 of 15; 20%) with acquired resistance to the ALK inhibitor, as determined by FISH analysis of KRAS in pre-treatment and post-resistance tumor biopsies (Fig. 4j). The pre-treatment tumor biopsy showed no evidence of KRAS amplification in each case (Fig. 4j), which suggested that this event emerged during the onset of resistance to crizotinib. These resistant tumors with KRAS amplification had no evidence of mutations in ALK, KRAS or BRAF (Supplementary Fig. 8f), which supported the proposal of a primary role for amplification of KRASWT in ALK-inhibitor resistance.
In seven other resistant tumor samples (7 of 15; 47%), we discovered KRASWT gene-duplication events in tumor-cell sub-populations (Supplementary Figs. 8f and 9c). Although the function of gene duplication of KRASWT is unknown, gene duplication of BRAFWT or PTPN11WT (the wild-type gene encoding the tyrosine phosphatase SHP-2) can drive MAPK activation and tumor growth29,30. We explored the functional effect of KRASWT gene duplication, which might produce lower levels of KRAS than genomic amplification, by assessing the relationship between low or high levels of KRASWT expression and ALK-inhibitor sensitivity. Expression of high or low levels of KRASWT increased the abundance of p-ERK and promoted resistance to treatment with an ALK inhibitor in H3122 cells (Supplementary Fig. 9a,b). Thus, KRAS gene duplication might promote MAPK signaling and ALK-inhibitor resistance. Our findings reveal KRASWT copy-number gain as a mechanism of ALK-inhibitor resistance in patients with lung adenocarcinoma.
DUSP6 loss promotes ALK-inhibitor resistance
We investigated the mechanism by which CAR2 and CAR3 cells rescued MAPK signaling and became resistant to the ALK inhibitor. CAR2 and CAR3 cells had lower basal levels of RAS-GTP than that of H3122 parental cells, despite having substantial basal phosphorylation of ERK (Fig. 4b and Supplementary Fig. 10a). This finding suggested that re-activation of the MAPK pathway might have occurred via deregulation of components acting downstream of RAS. Phosphatases of the DUSP (‘dual-specificity phosphatase’) family are critical regulators of MEK-ERK signaling and typically control MAPK signaling at a level below RAS31,32. To determine if a specific DUSP might regulate MAPK signaling in EML4-ALK lung adenocarcinoma cells, we examined the levels of members of the DUSP family in transcriptome data sets generated from H3122 parental cells and their ALK inhibitor–resistant derivatives. DUSP6 exhibited higher expression than that of other members of the DUSP family in parental H3122 cells (Supplementary Fig. 10b). Moreover, DUSP6 was significantly down-regulated in the sub-lines CAR2 and CAR3 (Fig. 5a and Supplementary Fig. 10b). We hypothesized that downregulation of DUSP6 might promote reactivation of MAPK and ALK-inhibitor resistance in EML4-ALK lung adenocarcinoma cells. Stable reconstitution of DUSP6 in CAR2 and CAR3 cells restored sensitivity to crizotinib and the ability of crizotinib to suppress MAPK signaling (Fig. 5b,c). Knockdown of DUSP6 also promoted resistance to crizotinib in parental H3122 cells, an effect accompanied by rescue of p-ERK during ALK inhibition (Fig. 5d–f). Thus, loss of DUSP6 drives ALK-inhibitor resistance in EML4-ALK lung adenocarcinoma cells.
Figure 5.
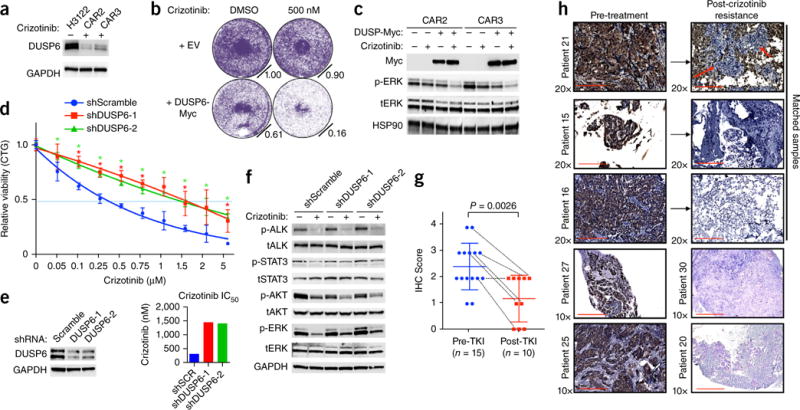
Reactivation of MAPK signaling by suppression of DUSP6 promotes ALK-inhibitor resistance in EML4-ALK lung adenocarcinoma. (a) Immunoblot analysis of DUSP6 expression in H3122 cells and CAR sub-lines. Data shown represent three independent experiments. (b,c) Effects of DUSP6 re-expression in H3122 CAR sub-lines on ALK-inhibitor sensitivity, assessed by crystal violet cell-growth assays (b), and on ALK signaling, assessed by immunoblot analysis (c). Data shown represent three independent experiments. (d) Growth-inhibition response (as in Fig. 1g) to 72 h of treatment with crizotinib in H3122 cells expressing scrambled shRNA or either of two independent shRNAs targeting DUSP6. *P < 0.05, at each concentration (unpaired t-test). (e) Left panel, level of knockdown (by immunoblot analysis), and right panel, IC50 values, of cells in d. (f) Immunoblot analysis in H3122 cells treated with shRNA as in d and treated with 250 nm crizotinib for 30 min. (g,h) Immunohistochemistry analysis (IHC) of DUSP6 expression in pre-treatment (Pre-TKI) and post-resistance (Post-TKI) lung adenocarcinoma tumor biopsies from patients treated with ALK inhibitor (n = 25). TKI, tyrosine kinase inhibitor. Each symbol represents an individual patient tumor; small horizontal lines indicate the mean (±s.d.); dotted lines link results for six individual patients with corresponding paired pre-treatment and after-ALK-inhibitor-resistance tumor samples for analysis. P = 0.0026 between each group (pre-treatment versus post-resistance) (unpaired t-test). Representative images of those used to obtain the immunohistochemistry scores are shown in Supplementary Figure 10d. Red arrows indicate tumor cells. Scale bars indicate 200 μm and 400 μm.
We sought to clinically validate the findings reported above using an immunohistochemistry assay to measure DUSP6 levels in tumors obtained from patients. We conducted a comparative analysis of DUSP6 expression in an additional cohort of treatment-naive and post-ALK-inhibitor-resistance tumor samples from patients (n = 25). DUSP6 was generally lower in samples obtained after they developed resistance to ALK inhibitor than in the pre-treatment tumor specimens (Fig. 5g,h and Supplementary Fig. 10c). In five of six (80%) of the paired tumor biopsies from individual patients in this cohort, DUSP6 expression was lower in the resistant tumor than in the corresponding pre-treatment tumor (Fig. 5g,h). Together our clinical data support the proposal of a critical role for MAPK signaling in EML4-ALK lung adenocarcinoma and identify KRASWT copy-number gain and DUSP6 loss as mechanisms of ALK-inhibitor resistance.
Five patients with resistance to crizotinib in our cohort (#13:KRAS amp +, #14:KRAS amp+, #15:DUSP6 score 0, #18:DUSP6 score 2, #20: DUSP6 score 0) subsequently received either an investigational ALK inhibitor (ASP3026) or ceritinib, and four of the five experienced relatively rapid disease progression (within 2–4 months) (Supplementary Fig. 10c). These data further support the proposal of a potentially important role for increased KRASWT and decreased DUSP6 in limiting the ALK-inhibitor response in patients.
Upfront co-inhibition of ALK-MEK enhances responses
The activation of MAPK can occur via a diverse set of mechanisms in ALK+ lung adenocarcinoma cells, including KRASWT amplification and DUSP6 downregulation or RTK signaling12,28,33–35, or KRAS mutations or loss of NF1 (which encodes a neurofibromatosis protein) (more generally)36. We hypothesized that blocking MAPK signaling upfront would minimize the emergence of resistance in ALK-inhibitor-naive lung adenocarcinoma cells. Treatment with a sub-maximal dose of trametinib (1 nM) plus crizotinib suppressed the development of acquired resistance, and the use of a modestly higher dose of trametinib (10 nM) prevented resistance altogether in both H3122 cells and STE-1 cells in vitro (Fig. 6a,b). Moreover, polytherapy with ceritinib and a sub-maximal dose of trametinib enhanced the magnitude of the initial response and forestalled the development of acquired resistance to ceritinib monotherapy in vivo in the H3122 tumor model (Fig. 6c). Our data provide a rationale for upfront ALK-inhibitor–MEK inhibitor polytherapy to enhance the initial response and eliminate the onset of acquired resistance in ALK+ lung adenocarcinoma.
Figure 6.
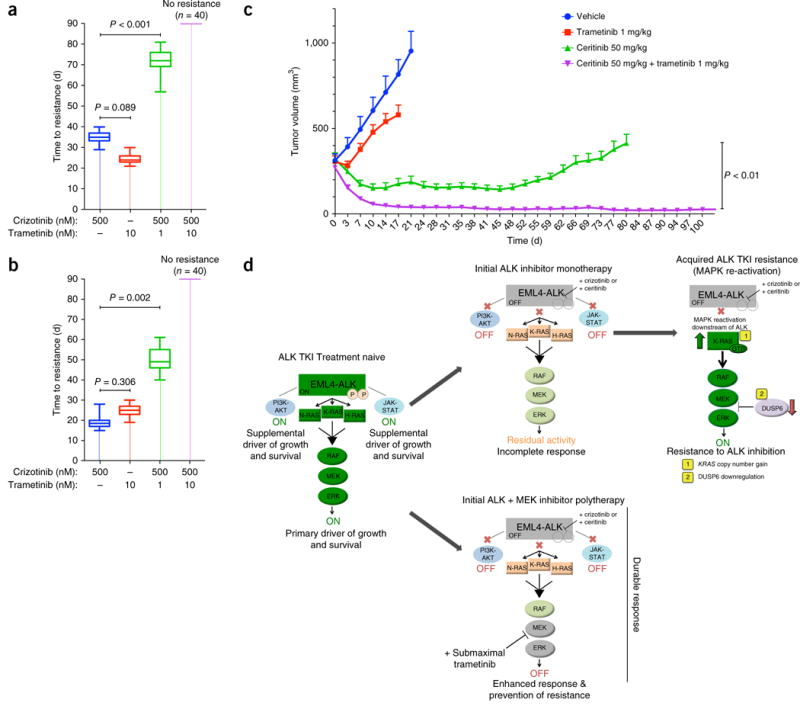
Combined inhibition of ALK and MEK enhances response and eliminates resistance in EML4-ALK lung adenocarcinoma models, in vitro and in vivo. (a,b) Time to acquisition of resistance (defined as days to confluency) in H3122 (a) and STE-1 (b) cells plated in 96-well plates (n = 40) and treated with the indicated drugs. P = 0.002 and P < 0.001, for combination therapy versus control (unpaired t-tests). n = 3, data are presented as box plots, with maximum, minimum and quartile ranges. (c) Tumor volume (mm3) of H3122 xenografts during treatment with ceritinib (50 mg/kg), trametinib (1 mg/kg) or a combination of those, presented as change in tumor volume from baseline (day 0) ±s.e.m., n = 10 tumors per group. P < 0.01, between treatment groups (unpaired t-test). (d) Graphical depiction of the model for EML4-ALK oncogene dependence, in which the tumor cells are dependent primarily on RAS-MAPK signaling. Shown is the mechanism of enhanced efficacy of combined treatment with an ALK inhibitor and a (sub-maximal) MEK inhibitor. EML4-ALK engages RAS-MAPK signaling as the primary downstream effector pathway to drive tumor cell growth and survival (left panel). Upfront ALK monotherapy leads to an incomplete response and tumor-cell survival due to residual MAPK activity (middle top panel). Eventually, these cells acquire resistance to ALK monotherapy by fully rescuing MAPK downstream of EML4-ALK via (1) KRASWT copy number gain or (2) downregulation of the MAPK phosphatase DUSP6 (right top panel). Initial ALK inhibitor–MEK inhibitor polytherapy abrogates this residual MAPK kinase activity to promote greater and more durable upfront responses by minimizing tumor-cell survival and re-activation of MAPK signaling (middle bottom panel).
DISCUSSION
We have established the RAS-MAPK signaling axis as the primary EML4-ALK effector pathway required for the survival of EML4-ALK lung adenocarcinoma cells (Fig. 6d, left panel). EML4-ALK activates all three major RAS isoforms, which leads to substantial RAS-MAPK signaling output that probably explains the specific MAPK dependence of EML4-ALK lung adenocarcinoma models. This RAS-MAPK dependence represents a therapeutic opportunity, as sub-maximal treatment with a MEK inhibitor can enhance the initial anti-tumor effects of ALK-inhibitor therapy by dampening RAS-MAPK output (Fig. 6d, middle panels). This synergistic upfront dual ALK inhibitor–MEK inhibitor polytherapy can also lead to a more durable response by minimizing the opportunity for tumor cells to survive and evolve to develop resistance during initial therapy, thus forestalling acquired resistance (Fig. 6d, middle and right panels).
Our findings highlight reactivation of MAPK signaling as a hallmark feature of acquired ALK-inhibitor resistance in lung adenocarcinoma cells. The mechanisms of resistance to ALK-inhibitor treatment we have reported here and those described previously12,28,33–39 can all activate MAPK signaling. Hence, MAPK-pathway re-activation (via diverse mechanisms) might be a necessary event for the development of ALK-inhibitor resistance. Published work has suggested that inhibition of MEK might be effective in treating ALK-inhibitor resistance after it has occurred (as second-line therapy)35,40. Our study has identified the innate dependence of EML4-ALK lung adenocarcinoma on RAS-MAPK signaling and its underlying mechanistic basis and offers an alternative approach for the treatment of patients with ALK+ lung adenocarcinoma. The upfront ALK inhibitor–MEK inhibitor polytherapy strategy supported by our findings might convert the incomplete and temporary responses obtained with crizotinib, as well as those obtained with more potent ALK inhibitors such as ceritinib and alectinib, into complete, sustained remissions in patients.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Cell culture
All cell lines were maintained in humidified incubators with 5% CO2 at 37 °C. The isogenic crizotinib-resistance cell lines (CAR1–CAR3 and LAR1–LAR3) were maintained in 1 μM crizotinib or 200 nM ceritinib, respectively. The human lung adenocarcinoma cell lines H3122, STE-1, H2228 were maintained in RPMI-1640 medium supplemented with 10% FBS and penicillin-streptomycin 100 μg ml−1. Derivation of STE-1 was described previously12. All cell lines were confirmed to have EML4-ALK via cDNA sequencing and tested negative for mycoplasma contamination.
Compounds
Crizotinib (Selleck Chemicals, Houston, TX, USA), ceritinib (Selleck Chemicals, Houston, TX, USA), trametinib (Selleck Chemicals, Houston, TX, USA), selumetinib (Selleck Chemicals, Houston, TX, USA), ruxolitinib (Selleck Chemicals, Houston, TX, USA), BKM120 (Selleck Chemicals, Houston, TX, USA), BYL719 (Selleck Chemicals, Houston, TX, USA), MK2206 (Selleck Chemicals, Houston, TX, USA), GDC0941 (Selleck Chemicals, Houston, TX, USA) were dissolved in DMSO.
Cell viability and apoptosis assays
For CellTiterGlo (Promega, Madison, WI, USA) viability experiments, cells were seeded in 96-well plates at 5,000 cells/well and were exposed to drugs on the following day. At 72 h after drug addition, CellTiter-Glo reagent was added and luminescence was measured on a Spectramax spectrophotometer (Molecular Devices, Sunnyvale, CA, USA) according the manufacturer’s instructions. All experimental points were set up as octuplicate biological replicates. Data are presented as percentage of viable cells compared with control cells (vehicle treatment). Growth assays were performed via crystal violet staining and quantification. Briefly, cells were seeded in six-well plates and were grown in presence of drug. At 5–7 d, cells were fixed with 4% PFA and stained with crystal violet. Pictures of stained cells were taken using GE Imagequant. For quantification, stained wells were dissolved with 1% SDS solution, and absorbance at 470 nm was measured using a Spectramax spectrophotometer (Molecular Devices, Sunnyvale, CA, USA), and values (denoted as OD) were normalized to those of control (vehicle-treated) cells. All crystal violet images are representative at least three individual experiments. For the apoptosis assay of the lung cancer cell line panel, 10,000 cells were plated in 96-well plates (triplicates) and were exposed to drug the following day. After 24 h, Caspase-Glo reagent (Promega, Madison, WI, USA) was added and luminescence was measured using a Spectramax spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Values were normalized to those of vehicle-treated cells. Statistical significance was assessed by two-sided unpaired t-tests of means calculated by GraphPad and are reported as the P value for significance. Estimated in vitro IC50 values were measured as described previously41.
Generation of tyrosine kinase inhibitor–resistant cell lines
To create crizotinib- and ceritinib-resistant cell lines (Supplementary Fig. 5a), H3122 cells were cultured with increasing concentrations of drug, starting with 250 nM crizotinib or 50 nM ceritinib, respectively. When cells began proliferating at normal rates, drug doses were doubled until a concentration of 1 μM crizotinib or 200 nM ceritinib was reached. Fresh drug was added every 72–96 h. Both sets of resistant lines (CAR and LAR) took approximately 4 months to generate. Exome sequencing comparing parental and resistant lines confirmed the cell lines generated were from the same origin. Resistant cell lines (as polyclonal populations) were maintained continuously in the presence of tyrosine kinase inhibitor.
In vitro resistance assay
This assay was adapted from published studies32. Cells were plated in 96-well plates at a density of 7,500 cells per well (~40% confluency), and drug treatments began the following day. Each treatment group had 40 replicates, and drug was replaced every 72–96 h. Resistance was defined as time (in days) for a well to become confluent (90%) in the presence of treatment.
Antibodies and immunoblotting
Antibodies against the following were obtained from Cell Signaling Technology (Danvers, MA, USA) and were used at a dilution of 1:1000: ALK (C26G7), p-Y1604-ALK (#3341), STAT3 123H6 (#9319), p-Y705-STAT3 D3A7 (#9145), AKT (#9272), p-S473-AKT D9E (#4060), ERK1/2 (#3493), p-T202/Y204-ERK1/2 D13.14.4e (#4370), RAS (#3965), DUSP6 (#3058), cleaved (c) PARP 19F4 (#9546), S6 (#2903), pS6 (#2903), BRAFV600E (VE1), cCaspase 3 (#9664), horseradish peroxidase (HRP)-conjugated anti-mouse (#7076) and HRP-conjugated anti-rabbit (#7074). Antibodies to the following were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA, USA): GAPDH (GC5, 1:1000 dilution), actin (I19, 1:1000 dilution;), H-RAS (C-20, 1:200 dilution;), N-RAS (F155, 1:200 dilution;), K-RAS (F234, 1:500 dilution;).
For immunoblotting, cells were washed in PBS and lysed and scraped with 25 mM Tris-HCL (pH 7.6), 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS supplemented with complete protease inhibitors (Roche Diagnostics, Indianapolis, IN, USA) and PhosSTOP phosphatase inhibitors (Roche Diagnostics, Indianapolis, IN, USA). Where indicated, RAS GST-RBD pulldowns were performed on lysates before immunoblot analysis. Lysates were subjected to SDS-PAGE followed by blotting with indicated antibodies and detection via ECL Prime (Amersham Biosciences, Sunnyvale, CA) or Odyssey Licor (Lincoln, NE, USA) with infrared-conjugated secondary antibodies (IR Dye 800 and IR Dye 680).
siRNA experiments
Transfection of siRNA was performed with Dharmafect reagent (Dharmacon, USA). Smartpool siRNA used in experiments were directed against K-RAS, N-RAS and H-RAS and were purchased from Thermo Scientific and used according to the manufacturer’s protocol.
Xenograft studies
H3122 tumor xenografts were generated by injection of 2 × 106 cells in Matrigel into 8-week-old male and female NOD/SCID mice (Mus musculus). Mice were randomized to treatment groups once tumors reached an average size of 150 mm3 (Fig. 3e, n = 8 per treatment group) or 300 mm3 (Fig. 6c, n = 10 per treatment group, and Supplementary Fig. 6c, n = 10 per treatment group). Ceritinib was administered at 25 mg/kg or 50 mg/kg p.o. daily for 5 d. Trametinib was administered at 1 mg/kg or 3 mg/kg p.o. daily for 5 d. Ceritinib (25 mg/kg) or ceritinib (50 mg/kg) and trametinib (1 mg/kg) were administered together p.o. daily for 5 d. STE-1 tumor xenografts were generated by injection of 3 × 106 cells in Matrigel into 8-week-old NOD/SCID mice. Mice were randomized into treatment groups once the tumors reached an average of 300 mm3 (Fig. 3g, n = 8 per treatment group). Crizotinib (50 mg/kg) was administered together with trametinib (1 mg/kg) or vehicle p.o. daily. The vehicle for all drugs was 0.5% methylcellulose and 0.5% Tween-80. Tumor volumes were measured by blinded assessment. All animal experiments were conducted under a UCSF IACUC–approved animal protocol.
Viral transduction
Expression vectors for GFP, Myr-AKT, BRAFV600E, KRAS-G12C/D/V, MEK-DD were in pBABE retroviral expression plasmids obtained from Addgene (Cambridge, MA, USA). The cDNAs encoding KRAS-G12C and KRAS-G12D also encoded the hemagglutinin (HA) tag (Addgene #58901, #58902). EF.STAT3C.Ubc.GFP (CA.STAT3) was obtained from Addgene (Cambridge, MA, USA; plasmid #24983). DUSP6-Myc plasmid was obtained from Addgene (#27975) and inserted into pBABE retroviral expression plasmid. pLenti-KRAS-4B (WT) was a gift from F. McCormick. The viral titration experiments were performed by exposure of H3122 cells to different volumes (concentrations) of viral supernatant (1.0 indicates to 1 mL of viral supernatant in 4 mL media added). EML4-ALKv1-WT-Myc and EML4-ALK-dHELP-Myc were cloned into the pcDNA plasmid backbone and were gifts from H. Mano. 293-GPG viral packaging cells were transfected with pBABE constructs using Lipofectamine-2000 (Life Technologies, Pleasonton, CA) per the manufacturer’s instructions. Virus-containing media was harvested 3 d post transfection and was used to transduce cancer cell lines incubated with 6 μg/ml polybrene for 48 h. Media was changed to standard growth media and puromycin (1 μg/ml for H3122 cells, or 0.5 μ g/ml for STE-1 cells) was added for selection.
shRNA experiments
Lentiviral plasmids (pLKO) encoding shRNA targeting DUSP6 (1,2) or KRAS (1,2) were obtained from Sigma, including DUSP6 [(sh#1), (sh#2)] and KRAS[(sh#1), (sh#2)]. Virus was produced by transfection of 20 μg pLKO.1-shRNA and 20 μg Virapower mixed packaging plasmids. Selection with puromycin was started 48–72 h post viral transduction, after which point experiments were performed.
DNA transfections
Transfection of EML4-ALK plasmids into 293T and Beas2B cells was performed using FuGene 6 (Promega, Madison, WI, USA).
RNA analysis
RNA-seq was performed in triplicate for each cell line on Illumina Hi- Seq 2000 using paired-end 100-bp reads as described41. Reads were aligned and quantified using RSEM. Differential expression analysis between sets of conditions was performed using DESeq and as described41. Quantitative PCR (qPCR) was performed on the QuantStudio 12K Flex Real-Time QPCR System using Taqman probes (Applied Biosystems, Life Technologies) specific to the coding regions of the genes assessed.
RAS activation assay
The RAS GST-RBD activation kit was obtained from Cytoskeleton (Denver, CO, USA; cat #BK008). The protocol was according to the manufacturer’s instructions. Lysis buffer for RAS-GTP pulldowns was 50 mM Tris (ph 7.5), 10 mM MgCl2, 0.5 M NaCl, and 2% Igepal. Snap-freezing of lysates using liquid nitrogen baths was performed directly after lysis.
Immunofluorescence
Endogenous EML4-ALK staining (in H3122 and STE-1 cells) was performed using an antibody against ALK (XP; Cell Signaling Technology). Cells were fixed with 4% PFA and incubated with 0.1% Triton X, and the mounting media used was Prolong Gold + DAPI (Life Technologies). Secondary antibodies (Alexa Fluor 596) were purchased from Thermo Scientific. In Beas2B cells transfected with EML4-ALK, pcDNA-EML4-ALKv1-Myc and pcDNA-EML4-ALK-dHELP-Myc constructs were used. Cells were fixed with 4% PFA and incubated with 0.1% Triton-X. Primary incubation was done with antibodies against Myc (D84C12; #2278) obtained from Cell Signaling Technology, and the mounting media used was Prolong Gold + DAPI (Life Technologies). Secondary antibodies (Alexa Fluor 596) were purchased from Thermo Scientific.
Immunohistochemistry
10-μm-thick formalin-fixed paraffin-embedded (FFPE) tissue sections were stained with rabbit monoclonal antibody against DUSP6 (AbCam; ab76310; 1:100 dilution). All stained slides were imaged (at 10× and 20×), and tumor cells were identified and scored (by three independent, blinded researchers) from 0–4 in order of increasing staining intensity, where ‘0’ represents no visible DUSP6 staining in tumor cells. All patient tumor samples analyzed were obtained under IRB-approved protocols with informed consent obtained from each subject at each institution.
FISH
Fluorescence in-situ hybridization (FISH) assays were performed as previously described42. Specimens were classified as KRAS-amplification-positive if the KRAS/CEP12 ratio was >2.2 or when >20% tumor cells carried clusters of >15 copies per cell. All called amplifications (patients #2, #13 and #14) met both criteria. All patient tumor samples analyzed were obtained under IRB-approved protocols with informed consent obtained from each subject at each institution.
Statistical analysis
P values were determined with unpaired t-tests between comparator groups using GraphPad software.
Supplementary Material
Acknowledgments
We thank M. McMahon, F. McCormick, K. Shannon and M. Von Zastrow (UCSF) for advice and discussions, and H. Mano (University of Tokyo) for EML4-ALK cDNA constructs and for advice. We acknowledge funding support from the following sources: a US National Institute of Health (NIH) Director’s New Innovator Award, the Howard Hughes Medical Institute, the Doris Duke Charitable Foundation, the American Lung Association, the National Lung Cancer Partnership, the Sidney Kimmel Foundation for Cancer Research and the Searle Scholars Program (T.G.B.); the UCSF Clinical and Translational Science Institute (G.H.); the National Cancer Institute (NCI) of the NIH (R01CA121210 and P01CA129243), a Damon Runyon Clinical Investigator Award and a LUNGevity Career Development Award (C.M.L.); the NIH (NCI5K12CA086913) (R.C.D.); an NIH Paul Calabresi Cancer Development Award in Clinical Oncology (K12CA138464) (J.W.R.); the NIH (NCI-P30CA046934) and Lung Cancer SPORE (NCI-P50CA058187) (M.V.G.); and the La Caixa Foundation and Redes Temáticas de Investigación en Cáncer (RD12/0036/0072) (R.R.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
G.H. contributed to the design, conduct and interpretation of all experiments; V.O. contributed in vivo experiments; E.P., A.T., C.M.B., R.A.O., D.S.N., E.C., A.S. and A.V. contributed cell line experiments and aided in experimental design; S.A. contributed deep-sequencing analysis; L.L. contributed sequencing library preparation; J.F., M.V.-G., D.L.A. and R.C.D. contributed analysis of patient tumor data; S.-H.I.O., P.C.M., N.K., R.R., J.W.R. and R.C.D. contributed patient tumor samples and clinical data; E.A.C. contributed to experimental design and interpretation; E.I. and C.M.L. contributed patient-derived cell lines and conducted experiments; T.G.B. supervised the project and contributed to the design and interpretation of all experiments; and G.H. and T.G.B. wrote the manuscript with input from all co-authors.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Solomon BJ, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371:2167–2177. doi: 10.1056/NEJMoa1408440. [DOI] [PubMed] [Google Scholar]
- 2.Shaw AT, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014;370:1189–1197. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Camidge DR, Doebele RC. Treating ALK-positive lung cancer—early successes and future challenges. Nat Rev Clin Oncol. 2012;9:268–277. doi: 10.1038/nrclinonc.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw AT, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368:2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 5.Robert C, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 6.Chapman PB, Solit DB, Rosen N. Combination of RAF and MEK inhibition for the treatment of BRAF-mutated melanoma: feedback is not encouraged. Cancer Cell. 2014;26:603–604. doi: 10.1016/j.ccell.2014.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Johnson DB, et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600E-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014;32:3697–3704. doi: 10.1200/JCO.2014.57.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flaherty KT, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Long GV, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 10.Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. doi: 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lovly CM, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20:1027–1034. doi: 10.1038/nm.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alessandrini A, et al. Mek1 phosphorylation site mutants activate Raf-1 in NIH 3T3 cells. J Biol Chem. 1996;271:31612–31618. doi: 10.1074/jbc.271.49.31612. [DOI] [PubMed] [Google Scholar]
- 14.Takezawa K, et al. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011;17:2140–2148. doi: 10.1158/1078-0432.CCR-10-2798. [DOI] [PubMed] [Google Scholar]
- 15.Hainsworth JD, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5:1630–1636. doi: 10.1097/JTO.0b013e3181e8b3a3. [DOI] [PubMed] [Google Scholar]
- 16.Bivona TG, et al. Phospholipase Cγ activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 2003;424:694–698. doi: 10.1038/nature01806. [DOI] [PubMed] [Google Scholar]
- 17.Grabocka E, et al. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25:243–256. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2004;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 19.Bivona TG, Philips MR. Ras pathway signaling on endomembranes. Curr Opin Cell Biol. 2003;15:136–142. doi: 10.1016/s0955-0674(03)00016-4. [DOI] [PubMed] [Google Scholar]
- 20.Mano H. ALKoma: a cancer subtype with a shared target. Cancer Discov. 2012;2:495–502. doi: 10.1158/2159-8290.CD-12-0009. [DOI] [PubMed] [Google Scholar]
- 21.Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17:2081–2086. doi: 10.1158/1078-0432.CCR-10-1591. [DOI] [PubMed] [Google Scholar]
- 22.Soda M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 23.Richards MW, et al. Crystal structure of EML1 reveals the basis for Hsp90 dependence of oncogenic EML4-ALK by disruption of an atypical B-propeller domain. Proc Natl Acad Sci USA. 2014;111:5195–5200. doi: 10.1073/pnas.1322892111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pollmann M, et al. Human EML4, a novel member of the EMAP family, is essential for microtubule formation. Exp Cell Res. 2006;312:3241–3251. doi: 10.1016/j.yexcr.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 25.Choi YL, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68:4971–4976. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu T, et al. The clinical effect of the dual-targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res. 2012;18:2316–2325. doi: 10.1158/1078-0432.CCR-11-2381. [DOI] [PubMed] [Google Scholar]
- 27.Zhao YS, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- 28.Katayama R, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra117. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfister S, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest. 2008;118:1739–1749. doi: 10.1172/JCI33656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graham JM, Jr, et al. Genomic duplication of PTPN11 is an uncommon cause of Noonan syndrome. Am J Med Genet. 2009;149A:2122–2128. doi: 10.1002/ajmg.a.32992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caunt CJ, Keyse SM. Dual-specificity MAP kinase phosphatase (MKPs): shaping the outcome of MAP kinase signaling. FEBS J. 2013;280:489–504. doi: 10.1111/j.1742-4658.2012.08716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ercan D, et al. Reactivation of ERK signaling causes resistance to EGFR kinase inhibitors. Cancer Discov. 2012;2:934–947. doi: 10.1158/2159-8290.CD-12-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson TR, et al. Widespread potential for growth factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–509. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang S, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-B signaling. Cell. 2012;151:937–950. doi: 10.1016/j.cell.2012.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crystal AS, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nissan MH, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74:430–2350. doi: 10.1158/0008-5472.CAN-13-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Esfahani K, Agulnik JS, Cohen V. A systemic review of resistance mechanisms and ongoing clinical trials in ALK-rearranged non-small cell lung cancer. Front Oncol. 2014;4:174. doi: 10.3389/fonc.2014.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Doebele RC, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilson FH, et al. A Functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015;27:397–408. doi: 10.1016/j.ccell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanizaki J, et al. Combined effect of ALK and MEK inhibitors in EML4-ALK-positive non-small-cell lung cancer cells. Br J Cancer. 2012;106:763–767. doi: 10.1038/bjc.2011.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin L, et al. Mapping the molecular determinants of BRAF oncogene dependence in human lung cancer. Proc Natl Acad Sci USA. 2014;111:E748–E757. doi: 10.1073/pnas.1320956111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Camidge DR, et al. Optimizing the detection of lung cancer patients harboring anaplastice lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16:5581–5590. doi: 10.1158/1078-0432.CCR-10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.