

Abstract
Internal cavities are important elements in protein structure, dynamics, stability and function. Here we use NMR spectroscopy to investigate the binding of molecular oxygen (O2) to cavities in a well-studied model for ligand binding, the L99A mutant of T4 lysozyme. On increasing the O2 concentration to 8.9 mM, changes in 1H, 15N, and 13C chemical shifts and signal broadening were observed specifically for backbone amide and side chain methyl groups located around the two hydrophobic cavities of the protein. O2-induced longitudinal relaxation enhancements for amide and methyl protons could be adequately accounted for by paramagnetic dipolar relaxation. These data provide the first experimental demonstration that O2 binds specifically to the hydrophobic, and not the hydrophilic cavities, in a protein. Molecular dynamics simulations visualized the rotational and translational motions of O2 in the cavities, as well as the binding and egress of O2, suggesting that the channel consisting of helices D, E, G, H, and J could be the potential gateway for ligand binding to the protein. Due to strong paramagnetic relaxation effects, O2 gas-pressure NMR measurements can detect hydrophobic cavities when populated to as little as 1%, and thereby provide a general and highly sensitive method for detecting oxygen binding in proteins.
Internal cavities in proteins are important structural elements that may produce functional motions1, such as drug and ligand binding2 and conformational transitions into high-energy states3,4,5,6,7. To explore their locations and dynamic aspects, specific binding of noble gases, particularly xenon, into protein cavities has been studied by X-ray crystallography8,9,10. In addition, small organic compounds and paramagnetic agents as well as noble gases have been used as probes in various nuclear magnetic resonance (NMR) studies11,12,13,14,15. The fact that small organic compounds and noble gases can associate with internal cavities indicates that proteins are sufficiently dynamic to enable the access of small molecules and that cavities may function as gateways for them.
Penetration of dissolved oxygen (O2) into proteins was originally investigated by quenching of fluorescence16,17. The paramagnetic effects of O2, such as paramagnetic shifts and paramagnetic relaxation enhancements (PREs), have been used to study protein solvent exposure and topology by NMR spectroscopy18,19,20,21. Although many crystal structures of heme-proteins with O2 ligands and their migration processes inside the proteins have been investigated by X-ray crystallography22,23 and molecular dynamics (MD) simulation24,25, to the best of our knowledge, association of O2 with internal cavities of proteins in solution has been investigated by NMR spectroscopy only for ribonuclease A12,26, deoxymyoglobin13, and the B domain of protein A20. In particular, Teng and Bryant investigated O2-induced PREs for backbone and side chain protons of ribonuclease and showed that structural fluctuations in the protein provide access to the protein interior for O2. However, the O2-induced PREs were not simply correlated with the depth of a buried proton or hydrophobicity indices. Rather, large PREs were correlated with the distance to the closest hydrophobic cavity12,20. These studies suggest that O2 represents a useful paramagnetic NMR probe to explore the surface crevices and cavities of proteins, which have the potential for ligand binding.
By using gas-pressure NMR and MD simulation, we investigate O2 accessibility to the protein interior for the cavity-enlarged L99A mutant of T4 lysozyme, which has two hydrophilic cavities (cavity 1: 50 Å3, cavity 2: 25 Å3) and two hydrophobic cavities (cavity 3: 25 Å3, cavity 4: 150 Å3). Cavity 4 was enlarged from 39 Å3 to 150 Å3 by the Leu → Ala mutation at position 9927. The L99A mutant has been used as a model system for understanding protein dynamics in the ligand binding process. X-ray crystallography found that three xenon atoms are present in cavity 4 under 8 bar of xenon pressure, and cavity 4 has been shown to allow the binding of benzene and substituted benzenes10,28,29. Although X-ray crystallography suggested that the enlarged cavity in L99A is sterically inaccessible to incoming ligands, NMR spin relaxation studies showed the presence of conformational fluctuations around the hydrophobic cavities and the rapid exchange of benzene and indole with the protein interior30,31,32,33,34. Our objective here is to understand the selectivity of O2 to hydrophilic and hydrophobic cavities and the coupling between protein conformational fluctuation and accessibility of O2 to internal cavities of the protein.
Results and Discussion
Reversible association of oxygen
We used on-line gas pressure NMR spectroscopy up to 7 bar absolute pressure to demonstrate gas binding into cavities of the cavity-enlarged L99A mutant of the T4 lysozyme. Time-dependent changes in 1H NMR spectra of the protein were observed, when the concentration of molecular oxygen (O2) decreases from 1.8 mM (corresponding to 1.4 bar absolute pressure) to 0.27 mM (corresponding to atmospheric pressure; 0.2 bar O2 partial pressure) at 298 K. A well-separated peak stemming from L121 Hδ1 changed its frequency (about 0.05 ppm) during 18.7 hours after pressure decreased, and the chemical shift change during the final hour was 0.0009 ppm, which is at the level of indiscernible changes in chemical shifts (1H: ±0.001 ppm). Therefore, we regarded that 18.7 hours is sufficient to reach a new equilibrium of gas dissolution in the NMR tube (see Supplementary Fig. S1 online). All NMR measurements were started more than 18.7 hours after gas pressure was changed. In the present pressure range, spectral changes were perfectly reversible.
Oxygen-induced spectral changes
Figure 1a shows 1H/15N refocused heteronuclear single-quantum coherence (HSQC) spectra of 15N-labeled L99A at O2 concentrations from 0.27 mM to 6.4 mM. O2-induced chemical shift changes were observed for cross-peaks of L84, K85, Y88, D89, A99, I100, L118, and A130. At 6.4 mM of O2, their O2-induced 15N chemical shifts are about 0.1–1.0 ppm. In addition, cross-peaks for Y88, I100, and L118 became weaker or disappeared with increasing O2 concentration. In contrast, N2 and Ar gas did not induce perceptible changes in chemical shifts and cross-peak intensities in the same ranges of gas concentrations (N2 ~3.3 mM, Ar ~7 mM; see Supplementary Fig. S2 online).
Figure 1.

(a) 1H/15N refocused-HSQC spectra of 15N labeled L99A of T4 lysozyme at 298 K at different oxygen concentrations from 0.27 mM to 6.4 mM. Amide groups showing significant changes in 15N chemical shift are indicated. (b) 1H/13C constant time HSQC spectra of 13C/15N labeled L99A of T4 lysozyme at different oxygen concentrations from 0.27 mM to 8.9 mM. Positive and negative crosspeaks are presented by same color. Methyl groups showing significant changes in 1H/13C chemical shift and a loss of crosspeak intensities are indicated.
Figure 1b shows the region for methyl group signals in 1H/13C constant time (CT) HSQC spectra of 15N/13C-labeled L99A. O2-induced chemical shift changes and/or loss of signal intensities are significant for the methyl groups of I78δ1, I78γ2, L84δ1, L84δ2, V87γ2, A99β, M102ε, V103γ2, V111γ1, V111γ2, L118δ1, L118δ2, L121δ1, A129β, A130β, L133δ1, and I150γ2. In contrast, chemical shift changes were not observed when Ar was increased to 5 bar (~7 mM) or N2 was increased to 7 bar (~4.6 mM, see Supplementary Fig. S3 online).
Figure 2 shows the mapping of backbone amide groups and methyl groups showing O2-induced changes in chemical shifts and/or cross-peak intensities. O2-induced changes are specific around the two hydrophobic cavities 3 and 4 in the C-terminal domain of the protein. These results suggest that O2 associated with cavities 3 and 4, and the O2-induced changes resulted from the paramagnetic property of O2 and/or changes in the structure and conformational equilibrium of the protein. Details are discussed further in the sections The paramagnetic effect leads to line broadening and Origin of O2-induced chemical shift changes.
Figure 2. Three-dimensional structure representations of T4 lysozyme L99A, highlighting the locations of amide (red) and methyl (green) groups showing oxygen-induced chemical shift changes or a loss of crosspeak intensity.
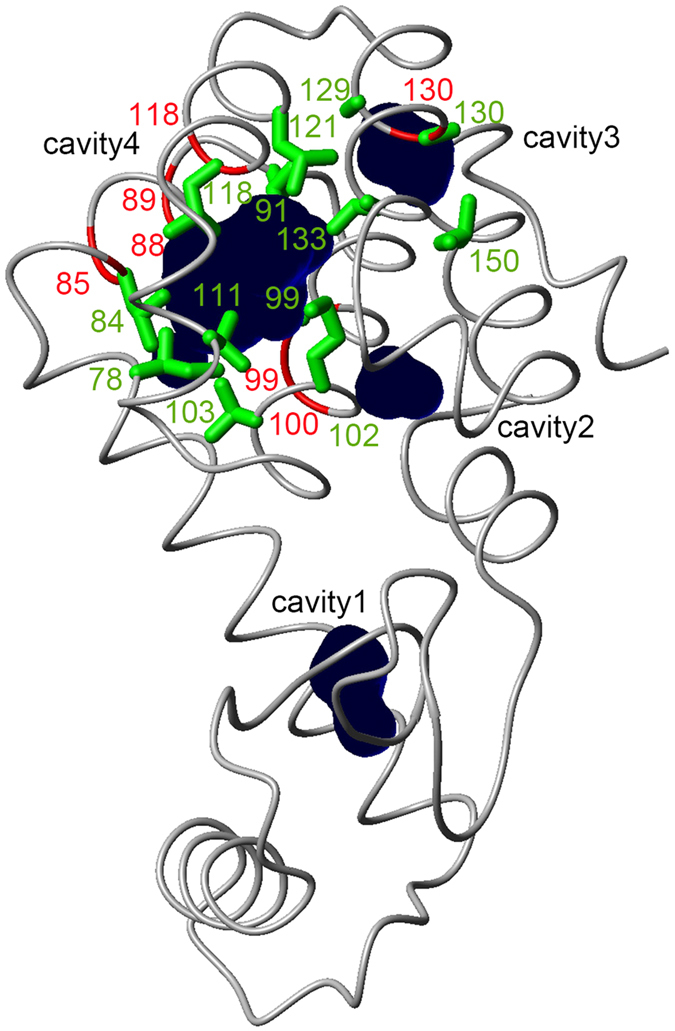
Cavities calculated by the program MOLMOL62 are depicted by dark blue spheres.
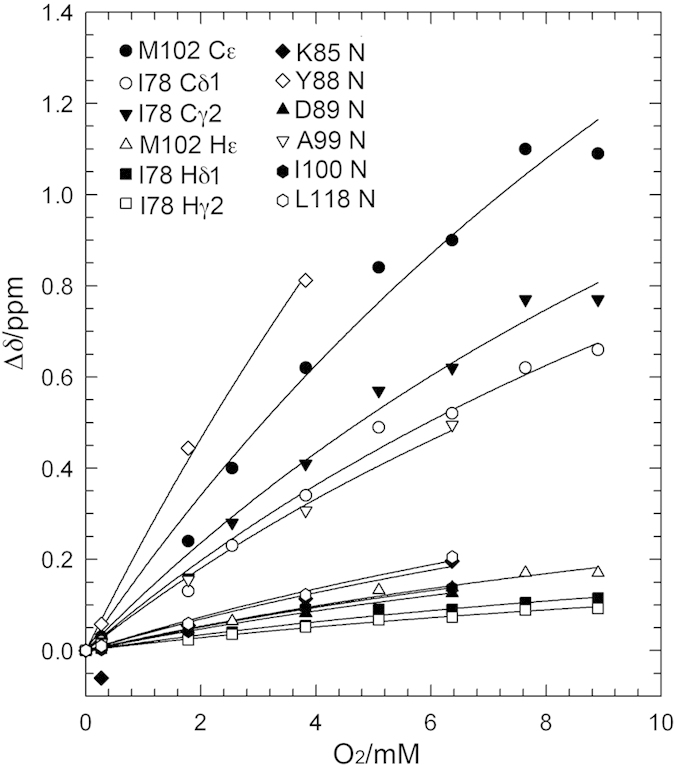
Figure 3 shows the O2-induced chemical shift changes observed for amide nitrogens, methyl carbons, and methyl proton nuclei of the residues around the enlarged hydrophobic cavity (cavity 4). The O2 association constant can be estimated from the concentration dependence of peak positions, assuming exchange between O2-bound and free states. Global fitting to all chemical shift changes was performed, using the following equation (1),
Figure 3. Chemical shift changes of methyl carbons, methyl protons, and amide nitrogens around the enlarged cavity as a function of O2 concentration.
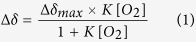 |
where [O2] is the molar concentration of dissolved O2, Δδmax is the residue-specific saturation value of the O2-induced shift, and K is the association constant, which is a global variable in the model fitting to data for all residues simultaneously. The deviations between actual measurements and predicted values from the fit seem to stem from inaccuracies in chemical shift determination. The dissociation constant Kd is the reciprocal of K. K and Kd were 48 ± 7 M−1 and 21 ± 3 mM, respectively. Accordingly, 1.3% of T4 lysozyme L99A was in the O2-bound state at atmospheric pressure (i.e., O2 concentration 0.27 mM). Δδmax for each site is summarized in Table 1. For the other hydrophobic cavity (cavity 3), we did not have sufficient data to estimate K.
Table 1. Chemical shift changes of representative amide nitrogen, methyl protons, and methyl carbons for the oxygen binding to L99A.
| nucleus | Δδ_max/ppma | Std. Error |
|---|---|---|
| K85 N | 0.8 | 0.12 |
| Y88 N | 5.3 | 0.7 |
| D89 N | 0.5 | 0.10 |
| A99 N | 2.1 | 0.2 |
| I100 N | 0.6 | 0.10 |
| L118 N | 0.8 | 0.12 |
| I78 Hδ1 | 0.39 | 0.06 |
| I78 Hγ2 | 0.32 | 0.06 |
| M102 Hε | 0.61 | 0.08 |
| I78 Cδ1 | 2.3 | 0.2 |
| I78 Cγ2 | 2.7 | 0.3 |
| M102 Cε | 3.9 | 0.4 |
aΔδ are obtained by a global fitting for changes in chemical shifts using eq. 1.
Relaxation enhancement is due to PRE
The unpaired electrons of the paramagnetic triplet O2 induce relaxation enhancements on longitudinal and transverse spin relaxation rates. We investigated O2-induced longitudinal relaxation enhancements for amide and methyl protons. At 500 MHz we obtained 1H longitudinal relaxation rate constants, R1, for amide protons at 0 mM (Ar, 2 bar) and 6.4 mM (O2, 5 bar) dissolved O2 (see Supplementary Fig. S4 online). Figure 4a shows the paramagnetic relaxation enhancement (PRE) on 1H longitudinal relaxation, ΔR1, for amide protons as a function of residue number, defined as the difference of R1 between 0 mM (Ar, 2 bar) and 6.4 mM of O2. As an example, the relaxation curves for D89 are shown in the inset of Supplementary Fig. S4 online. As illustrated in Fig. 5a, residues exhibiting marked PREs (ΔR1 ≥ 2 s−1) by O2 are selectively observed around the two hydrophobic cavities. In contrast, PREs are small for the rest of the amide protons (ΔR1 = ~1 s−1). We also obtained R1 for methyl protons at different concentration of dissolved O2, at 600 MHz. R1 values at 0 mM (N2, 3 bar), 3.8 mM of dissolved O2 (O2, 3 bar), and their difference, ΔR1, are listed in Supplementary Table S1 online. Figure 6 shows O2-induced ΔR1 for methyl protons of the C-terminal domain (i.e., residues 71–160). Methyl protons exhibiting marked PREs (ΔR1 ≥ 4 s−1) were also located around the two hydrophobic cavities, as shown in Fig. 5b. Note that R1 values of several methyl protons around the two cavities could not be obtained at 3.8 mM of O2 concentration, because their cross-peaks were severely broadened or disappeared (see section: The paramagnetic effect leads to line broadening). These PRE data closely match the O2-induced changes in chemical shifts and peak intensities (Fig. 1).
Figure 4.

(a) Observed and predicted O2-induced 1H longitudinal relaxation enhancements for amide protons against residue number. Difference of longitudinal relaxation rates, ΔR1, for amide protons between 6.4 mM (O2 5 bar) and 0 mM (Ar 2 bar) O2 concentrations. R1 values at each condition are shown in Supplementary Fig. S4 online. Severe line-broadening prohibited quantitative evaluation of ΔR1 for residues 88 and 118 (asterisks). The crystal structure of L99A at 8 atm of xenon pressure possesses three xenon molecules in cavity 4. We added two xenon molecules in cavity 3 and energy minimized. ΔR1 were estimated from 1/r6 weighted distance dependence from each xenon site, using the equation (2). (b) Contributions of each O2-binding site to the predicted 1H longitudinal relaxation enhancements for amide protons. ΔR1 from sites 1–5 were estimated by the following equation: (ΔR1(predict)-f) × (a or b or c or d or e × 105 (1/r1–5)6)/(a × 105 (1/r1)6 + b × 105 (1/r2)6 + c × 105 (1/r3)6 + d × 105 (1/r4)6 + e × 105 (1/r5)6), where r1–5 are distances to each xenon site. The parameters a, b, c, d and e were obtained to be 1.3, 1.1, 1.5, 0.11, 0.10, respectively, by the fitting.
Figure 5.

(a) Mapping of amide groups showing 1H longitudinal relaxation enhancements (ΔR1 ≥ 4 s−1, red; 2 s−1 ≤ ΔR1 < 4 s−1, orange). Data were obtained at 6.4 mM of O2 concentration. Amide groups showing large relaxation enhancements (ΔR1 ≥ 4 s−1) are labeled with residue number. (b) Mapping of methyl groups showing 1H longitudinal relaxation enhancements (ΔR1 ≥ 10 s−1, red; 4 s−1 ≤ ΔR1 < 10 s−1, orange). Data were obtained at 3.8 mM of O2 concentration. Methyl groups showing severe line-broadening are depicted by gray sticks. The picture was prepared using MOLMOL62.
Figure 6. Observed and predicted O2-induced 1H longitudinal relaxation enhancements for methyl protons.

Difference of longitudinal relaxation rates, ΔR1, for methyl protons between 3.8 mM (O2 3 bar) and 0 mM (N2 3 bar) O2 concentrations. R1 values at each condition are listed in Supplementary Table S1. Severe line-broadening prohibited quantitative evaluation of ΔR1 for L84δ2, A99β, L118 δ2, and L121 δ1 (asterisks). The crystal structure of L99A at 8 atm of xenon pressure possesses three xenon molecules in cavity 4. We added two xenon molecules in cavity 3 and energy minimized. ΔR1 were estimated from 1/r6 weighted distance dependence from each xenon site, using equation (2). Identifiers 1–54 have the following assignments: 1:V71γ2, 2:A73β, 3:A74β, 4:V75γ1, 5:V75γ2, 6:I78γ2, 7:I78δ1, 8:L79δ2, 9:A82β, 10:L84δ1, 11:L84δ2, 12:V87γ1, 13:V87γ2, 14:L91δ2, 15:A93β, 16:V94γ1, 17:V94γ2, 18:A97β, 19:A98β, 20:A99β, 21: I100γ2, 22:I100δ1, 23:M102ε, 24:V103γ1, 25:V103γ2, 26:M106ε, 27:T109γ2, 28:V111γ1, 29:V111γ2, 30:A112β, 31:T115γ2, 32:L118δ1, 33:L118δ2, 34:M120ε, 35:L121δ1, 36:L121δ2, 37:A129β, 38:A130β, 39:V131γ1, 40:V131γ2, 41:L133δ1, 42:L133δ2, 43:A134β, 44:T142γ2, 45:A146β, 46:V149γ1, 47:V149γ2, 48:I150γ2, 49:I150δ1, 50:T151γ2, 51:T152γ2, 52:T155γ2, 53:T157γ2, and 54:A160β.
The PRE arises from dipolar interactions between a nucleus and unpaired electrons of the paramagnet and spin relaxation contributions show < r−6 > distance dependence between the paramagnetic center and the nucleus of interest undergoing rotational motion, as described by the Solomon-Bloembergen equation35,36. O2-induced ΔR1 for amide protons predicted by 1/r6-weighted distance analysis is shown in Fig. 4a13. The crystal structure of L99A at 8 atm of xenon pressure (PDB ID, 1c6k) has three xenon atoms in cavity 4 but none in cavity 310. Therefore, we added two xenon molecules (the maximum number of Xe that can be accommodated) to cavity 3 and minimized the total energy. ΔR1 was estimated from 1/r6 distance dependent PRE contribution from each xenon site, using equation (2):
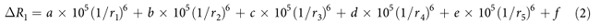 |
where r1–3 are the distances to xenon binding sites 1–3, respectively, in cavity 4 and r4–5 are the distances (Å) to the xenon binding sites 4 and 5, respectively, in cavity 3, and a, b, c, d, e, and f are fitting parameters. Hydrogen atoms were added to the crystal structure by using the WHATIF server37. A linear combination of predicted PREs from the five xenon-binding sites matches the observed ΔR1 pattern well (Fig. 4a). The parameters a, b, c, d, e, and f obtained from the fit were 1.3, 1.1, 1.5, 0.11, 0.10 (Å6/s), and 1.1 (s−1), respectively. Standard error of the estimate (i.e. the square root of the average squared error of prediction) was 0.74 (s−1). The ratio of the parameters a, b, c, d, e, shows the relative O2 occupancy at each xenon site. O2 occupancy in cavity 3 is about 5% of that in cavity 4. In addition, extremely large ΔR1 values were predicted for the amide protons of Y88 and L118, which indeed show severe line broadening with increasing O2 concentration. The R2-value of the correlation between observed and predicted ΔR1 was 0.82.
Although O2 occupancies at each xenon site are estimated above, contributions of each binding site to ΔR1 of each amide proton are expected to be different, as borne out by Fig. 4b. For instance, the amide protons of residues 129, 130, 153, and 154 exhibit large PREs from site 5 due to their proximity to bound oxygen molecules, even though the O2 occupancy at site 5 is much smaller than that for sites 1–3. The contributions to ΔR1 from the individual binding sites as a function of distance are given in Supplementary Fig. S5 online. The data can be adequately modeled with a 1/r6 distance dependence.
O2-induced ΔR1 for methyl protons of residues 71–160 was also predicted by the 1/r6-weighted distance analysis in Fig. 6. The R2-value of the correlation between observed and predicted ΔR1 was 0.80. These statistically substantial correlations indicate that O2 molecules associate with cavities 3 and 4. The parameters a, b, c, d, e and f obtained were 1.8, 0.0, 3.1, 0.0, 0.046 (Å6/s), and 0.79 (s−1) respectively. Standard error of the estimate was 3.0 (s−1). The predicted O2 occupancy in cavity 3 is much smaller than that of cavity 4, suggesting that O2 binding to cavity 3 is weaker than that to cavity 4. Furthermore, O2 occupancy at site 2 appears to be lower than that at sites 1 and 3. Although these tendencies are consistent with the results of amide protons, the ratios of parameters a–e between amide and methyl protons are different. We considered two explanations. First, the number of the data of methyl protons we used in the fit is smaller than that for amide protons. Even at a few bars of O2 pressure, line broadening by PREs is sufficiently large to prevent a correct estimate of ΔR1 (see section The paramagnetic effect leads to line broadening). Indeed, the standard error of estimate in the case of methyl protons is four times larger than that of amide protons. Alternatively, the values of the predicted ΔR1 increase very steeply, as the distance is less than 3 Å to the xenon site. For instance, even within the same methyl group, the values for each proton are largely different. Thus, in order to obtain a more quantitative prediction, we need to know the exact location and probability of O2 molecules at these binding sites much more precisely to calculate a correct estimate.
According to a calculation by Teng and Bryant, the O2-induced proton relaxation rate constant for a proton in van der Waals contact with O2 is about 6200 s−1 if O2 is bound at all times12. Accordingly, an observed value of 10 s−1 for ΔR1 suggests that the binding probability is ~0.2% or less. However, the O2-bound state probability of the protein seems to be about 5–10 times greater than the predicted binding probability according to our estimation of Kd (i.e., 1.3% at atmospheric pressure). This difference indicates that the effective distance between the O2 and the protein proton is larger than the van der Waals contact. We speculate that O2 does not tightly contact the protein proton and is allowed to move in the cavity space. We discuss the effective distance and dynamics of O2 in hydrophobic cavities in the section: Rotational and translational diffusion of O2 in hydrophobic cavities. Small, but substantial, ΔR1 values were observed at the residues in the N-terminal domain (i.e., residues 1–70), corresponding to the parameter f, and originates from O2 diffusing around the protein surface and proton spin diffusion, as discussed previously38.
The paramagnetic effect leads to line broadening
We investigated the line-widths (Δν1/2) of resonance lines in the 15N and 1H dimensions of the refocused-HSQC as a function of O2 concentration. Because the refocused-HSQC39 provides line widths that are directly proportional to the transverse relaxation rate of in-phase nitrogen coherences, R2N, without contributions from 1H relaxation, it is suitable to estimate the contributions of conformational exchange and PRE contributions to the 15N line-width31. Figure S6 shows 1H and 15N line-widths for residues 85, 88, 89, 99, 100, and 118 as a function of dissolved oxygen concentration. To within experimental error, the 15N line widths with increasing O2 concentration are constant for residues around the hydrophobic cavities, while a strong increase in 1H line widths for the same residues is observed. This observation supports the notion that dipolar PRE is the primary cause of line broadening, as this effect is proportional to the square of the gyromagnetic ratio of the nucleus involved. The observation that all of the amide and methyl protons showing severe line broadening and loss of signal intensities are located less than 6 Å from the closest xenon binding site (Table S1) further supports this. These results show that the PRE to the transverse relaxation rates lead to the line broadening.
Origin of O2-induced chemical shift change
We showed that changes in 15N, 13C, and 1H chemical shifts were specific to O2 and not observed for diamagnetic gases N2 and Ar. We next sought to understand whether O2-induced changes result mainly from the paramagnetic property of O2. In general, localized unpaired electrons of a paramagnet couple to the surrounding nuclei (i.e., hyperfine coupling) and may induce chemical shift changes through spin polarization and delocalization conveyed through the molecular orbitals of the molecule (contact shifts) or through the magnetic field emanating directly from the paramagnetic center (pseudocontact shifts, PCSs). In the case of O2, PCSs result from the anisotropic g-tensor of the unpaired electrons. PCSs depend on the distance between the paramagnet center and the nucleus of interest and the orientation with respect to the principal axes of the magnetic susceptibility tensor (i.e., χ-tensor). If PCSs were significant, the additional magnetic field would be sensed to a similar degree by the nuclei in 15N-1H and 13C-1H bonds and therefore lead to diagonal displacements of signals in 1H/15N and 1H/13C HSQC spectra, where the magnitude of change would be about the same for the bonded nuclei when measured in ppm. Such peak movement was not observed with increasing O2 concentration. To the contrary, 15N and 13C chemical shift changes upon O2 binding were much larger than 1H shift changes. In addition, if oxygen exchanges rapidly with the protein interior without preferred bound orientation or rapidly reorients itself with respect to the protein in the bound state, PCSs would average to zero. Instead, the O2 molecules in the hydrophobic cavities may have frequent collisions with nuclei. The collisions of O2 might allow delocalization of unpaired electron spins to 13C and 15N atoms40,41, causing contact shift. In addition, Bezsonova et al. showed a positive correlation between O2-induced chemical shift changes and increases in a collisionally accessible surface area18. These facts indicate that the collisions of O2 in the cavities could be a reasonable reason for relatively large chemical shift perturbation only for 13C and 15N.
Do N2 and Ar interact with the cavities of the protein? Similarities of the properties of the gases, such as mole fraction solubility, van der Waals radius and polarizability42,43, imply that N2 and Ar could associate to the hydrophobic cavities of L99A (Further discussions are in Supplementary Information). Indeed, binding of Ar at cavity 4 was observed in crystal structures of L99A at 8 to 32 bar of Ar pressure10. These results indicate that chemical shift changes by binding of noble gases, which can be attributed to changes in structure and conformational equilibrium, are in general much smaller than those by paramagnetic shifts of O2. Based on these observations and considerations, we conclude that O2-induced changes in chemical shifts resulted primarily from changes in contact shifts rather than due to the PCSs of O2 (see following section) and structural changes and conformational equilibria of the protein.
Rotational and translational diffusion of O2 in hydrophobic cavities
In order to investigate rotational and translational diffusion of O2 in cavities 3 and 4, molecular dynamics (MD) simulations of 100 nanoseconds were performed five times. An O2 molecule was inserted in both cavities 3 and 4 of the crystal structure of L99A (PDB ID; 1c6k) from which the pre-existing three xenon molecules were removed. One of the MD simulations is shown in Supplementary Movie S1 online. Note that the movie consists of 500 snapshots taken every 0.2 nanoseconds. O2 frequently moves around each hydrophobic cavity and rotates many times within 100 ns. Similar results were obtained in the four other MD simulations. Figure 7a shows xenon binding sites 1–5 in L99A, and Fig. 7b shows a density map of O2 molecules in cavities 3 and 4, obtained by the 100 nanoseconds MD simulation. While O2 samples almost all spaces in cavity 4, sampling frequencies that were more than 4 times higher than the average were observed only at sites 1 and 3 (Fig. 7b). These results suggest that O2 density is substantially higher at xenon binding sites 1 and 3 than at site 2, which is qualitatively consistent with the prediction by ΔR1 for amide and methyl protons. In the small hydrophobic cavity 3, O2 seems to mostly populate site 5. These results are consistent with the previous one nanosecond MD simulation by Mann and Hermans44.
Figure 7.

(a) Xenon binding sites in T4 lysozyme L99A (PDB ID; 1c6k). Cavity 4 includes xenon binding sites 1–3. We artificially created sites 4 and 5 in cavity 3 to calculate the distance to the cavity. (b) O2 density map calculated by MD simulation of 100 ns. Positions sampled by O2 are depicted by blue wireframe. Positions showing more than 4 times higher probabilities than the average are depicted by purple spheres. Helices D, E, G, H, and J are labeled. The picture was prepared using RasWin Molecular Graphics 2.7.563.
Interestingly, in the case of one of the MD simulations (Supplementary Movie S2 online), the O2 molecule in cavity 3 moved into cavity 4. Such a displacement of O2 from cavity 3 to 4 and vice versa was also observed in 4 of 5 MD trajectories. Then one of the two O2 molecules in cavity 4 egressed from the protein through the cleft between helices D and G. Furthermore, the O2 molecule eventually returned to cavity 4. A series of snapshots of unbinding and binding of O2 are shown in Fig. 8. During the O2 binding process, the O2 molecule binds to the surface near the helices D and G first and then returns to cavity 4 through the center of the channel consisting of helices D, E, G, H, and J. In the other simulation (Supplementary Movie S3 online), we also observed unbinding of O2 from cavity 3 through the cleft between helices H and J, as seen in Supplementary Fig. S7 online. Although it is known that L99A allows xenon and benzene to bind to cavity 4, the pathway of ligand access and egress is unknown. The present results provide the first insights in the potential pathway of ligand binding and unbinding to cavity 4, as well as egress from cavity 3.
Figure 8. A series of snapshots showing unbinding and binding of O2 molecule to T4 lysozyme L99A.

O2 molecules showing unbinding and binding to cavity 4 are indicated by filled triangles. The D, E, G, H, and J helices are labeled. The picture was prepared using VMD 1.9.264.
We separately performed additional one nanosecond MD simulations to understand more details of the rotational diffusion of O2 in cavity 4. A time dependent relaxation of the rotational correlation function was more reasonably fitted to a bi-exponential function than a single-exponential one, as shown in Supplementary Fig. S8 online. Rotational correlation times estimated by a bi-exponential function were 0.164 ± 0.006 ps and 1.41 ± 0.02 ps. Such a bimodal rotational correlation is well known for small molecules in many solvents45,46. The fast and slow components would be related to the inertial characteristics of the small molecule and diffusive solvent motions, respectively45,46. Therefore, we speculate that the fast and slow components in the present case are related to the inertial characteristic of O2 and protein internal dynamics taking place around the cavity, respectively. A different explanation is anisotropy of rotational motion of O2 in the cavity. In any case, it appears that the rotational motions taking place at sub-nanosecond will average the orientation with respect to the principal axes of the magnetic susceptibility tensor and reduce the PCSs. On the other hand, chemical shifts can be changed with an increase in a population of “the O2-bound state”. Because the observed NMR signals are the ensemble average of exchanging conformations, O2-induced ΔR1 of a particular proton would depend on the averaged-distance from the O2 molecule, which would be larger than the van der Waals contact. These results strongly support our discussions on PREs and PCSs.
O2 associates selectively and preferentially to hydrophobic and not to hydrophilic internal protein cavities
Although the size of hydrophilic cavities (cavities 1 and 2) is considered to be enough for O2 binding, we could not detect O2-induced changes in spectral parameters, such as chemical shifts, peak line-widths, and longitudinal relaxation rate constants, for residues around these cavities. Interestingly, X-ray crystallography found electron density in the hydrophobic and hydrophilic cavities (cavities 1, 2, and 4), which are attributed to water molecules. In hydrophilic cavity 1, two well-ordered water molecules were seen, while a single well-ordered water molecule was seen in hydrophilic cavity 2. In hydrophobic cavity 4, weak electron density was distributed around the cavity47. In contrast, no gas molecules or water molecules have previously been detected in cavity 3, as far as we are aware. O2 binding to cavities is generally considered to be in competition with water binding. Water molecules associate to the hydrophilic cavities more than O2 probably due to its dipolar property and higher concentration (i.e., ~55.6 M) in solution. Therefore, the observation of O2 penetration into the hydrophilic cavities of proteins is expected to be difficult. The present data show that O2 penetrates into the protein interior and selectively and preferentially associates with the two hydrophobic cavities of the protein. The preferential partitioning of O2 into hydrophobic regions is consistent with earlier observation of O2 binding to ribonuclease A12 and lipid bilayer18, for example.
O2 association with the hydrophobic cavities and dynamic motion of the protein
Penetration of O2 into the protein interior requires spaces greater than its molecular size, such as a cavity, and transient conformational fluctuations, which provide pathways for penetration. Nucleus-electron dipolar interactions may therefore be modulated by conformational fluctuations of the protein and depend on the solubility of O2 in the protein interior. Our previous work by high-pressure NMR spectroscopy revealed that T4 lysozyme L99A has at least two types of conformational fluctuations6; one takes place within the ground state ensemble, which is limited to the C-terminal domain. These fluctuations occur more rapidly than a millisecond and provide heterogeneous conformations around the hydrophobic cavities. The conformational fluctuations within the ground state ensemble may allow a penetration of gas molecules into the protein interior. Indeed, MD simulations showed that O2 molecules frequently move around cavities 3 and 4 and may go back and forth between the cavities and the outside of the protein during 100 ns. A second motion present for T4 lysozyme L99A is a conformational fluctuation between the ground state and a transiently formed high-energy “excited” state of the protein, which takes place on the millisecond time scale (average 0.7 ms)30. Because the aromatic side chain of F114 flips into the enlarged cavity in the excited state, O2 association will compete with the F114 flip-in in this excited state. Finally, O2-induced changes in chemical shifts and signal intensities were also observed for some methyl groups around cavity 4 (39 Å3) of cysteine-free wild-type (C54T/C97A; WT*) T4 lysozyme (see Supplementary Fig. S9 online). These observations indicate that O2 molecules associated to the cavity 4 of WT* in the absence of a conformational fluctuation between ground and transiently formed excited states. Taken together, these data suggest that O2 association is facilitated by the conformational fluctuation taking place within the ground state ensemble, rather than between the ground and excited states of L99A, and that the transiently formed excited state is not involved in gas binding to the enlarged cavity. The fact that quenching of the tryptophan fluorescence by O2 occurs on nanosecond time scale for several native proteins16,17 agrees with this conclusion.
Conclusion
Gas-pressure NMR spectroscopy using O2 has been used to explore dynamic cavities in T4 lysozyme L99A. We have come to the following conclusions:
O2 preferentially interacts with hydrophobic cavities and induces significant changes in NMR spectra, such as increased peak-widths and longitudinal relaxation rate constants, due to its paramagnetic property.
O2 associates to the two hydrophobic cavities in T4 lysozyme L99A. So far, no gas or water molecules have been detected in cavity 3. The present study provides the first evidence of ligand binding to cavity 3.
O2-induced relaxation enhancements could be adequately accounted for by the paramagnetic dipolar relaxation, assuming 1/r6-weighted contributions from five sites, where r is the distance to the paramagnet.
The dissociation constant for O2 binding to cavity 4 of the protein is 21 mM, indicating that about 1% of the protein contains O2 molecules in the dynamic hydrophobic cavity at ambient pressure.
According to MD simulations, O2 molecules in the hydrophobic cavities of the protein frequently move and rotate on the picosecond to nanosecond time scale. The cleft between helices D and G and the channel consisting of helices D, E, G, H, and J could be the potential gateway for ligand binding to cavity 4. O2 association with the hydrophobic cavities would be facilitated by the conformational fluctuations taking place within the ground state ensemble, rather than between the conformational ground and excited states of the protein.
The rotational and translational motions of O2 in the hydrophobic cavities may effectively reduce potential pseudocontact shift contributions to nuclear shielding.
The combination of NMR and MD simulation provides static and dynamic aspects of O2 binding to hydrophobic cavities. This approach might also be useful to probe the permeation pathway of ions or small molecules, such as channel-blocking molecules in membrane proteins48 and hydrophobic binding pockets for ligands, including drug compounds. Knowledge of protein permeation by oxygen is also highly relevant for optical spectroscopy and microscopy, where O2 dissolved in the protein matrix leads to quenching and bleaching, and knowledge of oxygen association pockets may facilitate the elimination of oxygen-free cavities through protein engineering. This strategy has the potential to greatly improve our understanding of the role played by protein cavities in biologically relevant processes.
Methods
Sample preparation
T4 lysozyme L99A was prepared from the recombinant cysteine-free T4 lysozyme (WT*, C54T/C97A)31. Uniformly 15N-labeled or 15N/13C-labeled L99A was produced in M9 media with 15NH4Cl and 13C6 glucose as the sole nitrogen and carbon sources, following established protocols31. The purified protein sample was dialyzed in 50 mM phosphate buffer including 25 mM NaCl at pH 5.5. Sample concentration was measured by UV absorption at 280 nm and was calculated with a molar extinction coefficient of 25440 M−1cm−1 at 280 nm.
NMR experiments
We used 1H 500 MHz (Bruker BioSpin Co. AVANCE ΙΙΙ) or 600 MHz (Bruker BioSpin Co. AVANCE) NMR spectrometers. In order to study the binding of oxygen (O2), nitrogen (N2), and argon (Ar) to the protein, we used a pressure resistance NMR tube (528-QPV-7, Wilmad-Lab Glass Co.) connected to a gas cylinder by PTFE tubing. Gas pressure was applied to adjust their concentrations in the protein solution. Mole fraction solubility of O2, N2, and Ar in water are 2.3 × 10−5, 1.2 × 10−5, and 2.5 × 10−5, respectively, at 298 K43. In this article, we use absolute pressure (gauge pressure + atmospheric pressure). 1H-NMR, 1H/15N refocused-HSQC39, and 1H/13C CT-HSQC spectra were obtained for 0.50 mM uniformly 15N-labeled or 1.0 mM 15N/13C-labeled T4 lysozyme L99A solution at 298 K at different gas pressures. 1H longitudinal relaxation enhancements for amide and methyl protons were obtained from 1H/15N HSQC and 1H/13C CT HSQC spectra using saturation-recovery, achieved with proton x and y purge pulses followed by a relaxation delay before each scan49. Seven to ten relaxation delays ranging from 0.003 s to 1.5 s were used. Spectral analysis was performed using NMRPipe50 and Sparky51.
Molecular dynamics simulation
Molecular dynamics (MD) simulations of 100 nanoseconds were performed five times using GROMACS 4.6.4 simulator52. The system contained a T4 lysozyme L99A (PDB ID; 1c6k), two O2 molecules, 8 chloride ions, and about 15,000 water molecules. Three xenon molecules in cavity 4 of the protein were removed from the structure, and one O2 molecule was inserted in each hydrophobic cavity (i.e., cavities 3 and 4). The OPLSLL force field53 was used for the protein, and the TIP4P model was used for water54. Potential parameters for O2 and chloride ions were as described in the literature55,56. MD simulations were conducted with the NPT ensemble (300 K, 1 bar) in a truncated dodecahedron box with dimensions of 25.8 Å. Temperature was controlled using a Langevin thermostat with a viscosity of 0.5 ps−1. Pressure was controlled by a Parrinello−Rahman barostat with relaxation times of 2.0 ps57. Electrostatics were treated using the particle mesh Ewald (PME) method with a 10.0 Å cutoff distance58. The van der Waals interactions were expressed using the twin-range cutoff method with 10.0 and 12.0 Å distances. Covalent bonds for hydrogen atoms in the polypeptide were constrained using the linear constraint solver (LINCS)59. Covalent bonds in water were constrained using the SETTLE algorithm60. The integration time step was 2 femtoseconds.
In order to estimate the rotational correlation times of O2 in cavity 4, we performed separate one nanosecond MD simulations. To avoid artifacts from the thermostat and barostat, we carried out the MD simulations in the NVE ensemble with the structure after 100 ns simulation in the NPT ensemble61. Snapshots were recorded every 0.01 picoseconds. Rotational correlation times were calculated using 1 ns trajectories of the O2 molecule in cavity 4. We defined a direction vector between the two oxygen atoms relative to the orientation of the protein.
Additional Information
How to cite this article: Kitahara, R. et al. Detecting O2 binding sites in protein cavities. Sci. Rep. 6, 20534; doi: 10.1038/srep20534 (2016).
Supplementary Material
Acknowledgments
We thank Dr. Marcus Görge Ullisch and Dr. Thomas Breitenbach for help with the gas-pressure setup, Dr. Shun Sakuraba for help with MD simulation, and Mr. Sanshiro Okuda for assistance with the NMR analysis. This work was supported by JSPS KAKENHI Grant Number 25840025 to R. K. Y.Y. was supported by an EMBO Long-Term Fellowship (ALTF 687-2013).
Footnotes
Author Contributions The manuscript was written by R.K. and F.A.A.M. M.X. prepared protein samples, and R.K., Y.Y., M.X. and F.A.A.M. collected and analyzed NMR spectra. T.K. performed MD simulations. All authors reviewed the manuscript.
References
- Hubbard S. J. & Argos P. A functional role for protein cavities in domain:domain motions. J. Mol. Biol. 261, 289–300 (1996). [DOI] [PubMed] [Google Scholar]
- Ogata K. et al. The cavity in the hydrophobic core of Myb DNA-binding domain is reserved for DNA recognition and trans-activation. Nat. Struct. Biol. 3, 178–187 (1996). [DOI] [PubMed] [Google Scholar]
- Kamatari Y. O., Smith L. J., Dobson C. M. & Akasaka K. Cavity hydration as a gateway to unfolding: an NMR study of hen lysozyme at high pressure and low temperature. Biophys. Chem. 156, 24–30 (2011). [DOI] [PubMed] [Google Scholar]
- Kitahara R. et al. A delicate interplay of structure, dynamics, and thermodynamics for function: a high pressure NMR study of outer surface protein A. Biophys. J. 102, 916–926 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche J. et al. Remodeling of the folding free energy landscape of Staphylococcal nuclease by cavity-creating mutations. Biochemistry 51, 9535–9546 (2012). [DOI] [PubMed] [Google Scholar]
- Maeno A. et al. Cavity as a source of conformational fluctuation and high-energy state: High-pressure NMR study of a cavity-enlarged mutant of T4 lysozyme. Biophys. J. 108, 133–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nucci N. V., Fuglestad B., Athanasoula E. A. & Wand A. J. Role of cavities and hydration in the pressure unfolding of T4 lysozyme. Proc. Natl. Acad. Sci. USA 111, 13846–13851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenborn B. P. Binding of xenon to horse haemoglobin. Nature 208, 760–762 (1965). [DOI] [PubMed] [Google Scholar]
- Schoenborn B. P., Watson H. C. & Kendrew J. C. Binding of xenon to sperm whale myoglobin. Nature 207, 28–30 (1965). [DOI] [PubMed] [Google Scholar]
- Quillin M. L., Breyer W. A., Griswold I. J. & Matthews B. W. Size versus polarizability in protein-ligand interactions: Binding of noble gases within engineered cavities in phage T4 lysozyme. J. Mol. Biol. 302, 955–977 (2000). [DOI] [PubMed] [Google Scholar]
- Otting G., Liepinsh E., Halle B. & Frey U. NMR identification of hydrophobic cavities with low water occupancies in protein structures using small gas molecules. Nat. Struct. Biol. 4, 396–404 (1997). [DOI] [PubMed] [Google Scholar]
- Teng C. L. & Bryant R. G. Mapping oxygen accessibility to ribonuclease a using high-resolution NMR relaxation spectroscopy. Biophys. J. 86, 1713–1725 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNaughton L., Hernandez G. & LeMaster D. M. Equilibrium O2 distribution in the Zn2+-protoporphyrin IX deoxymyoglobin mimic: Application to oxygen migration pathway analysis. J. Am. Chem. Soc. 125, 3813–3820 (2003). [DOI] [PubMed] [Google Scholar]
- Nisius L., Stadler M., Kalbitzer H. R. & Brunner E. NMR spectroscopic study of noble gas binding into the engineered cavity of HPr (I14A) from Staphylococcus carnosus. J. Phys. Chem. B 109, 17795–17798 (2005). [DOI] [PubMed] [Google Scholar]
- Desvaux H. et al. Dynamics of xenon binding inside the hydrophobic cavity of pseudo-wild-type bacteriophage T4 lysozyme explored through xenon-based NMR spectroscopy. J. Am. Chem. Soc. 127, 11676–11683 (2005). [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R. & Weber G. Quenching of protein fluorescence by oxygen - Detection of structural fluctuations in proteins on nanosecond time scale. Biochemistry 12, 4171–4179 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun D. B., Vanderkooi J. M., Woodrow G. V. & Englander S. W. Penetration of dioxygen into proteins studied by quenching of phosphorescence and fluorescence. Biochemistry 22, 1526–1532 (1983). [DOI] [PubMed] [Google Scholar]
- Bezsonova I., Forman-Kay J. & Prosser R. S. Molecular oxygen as a paramagnetic NMR probe of protein solvent exposure and topology. Concept Magn. Reson. A 32A, 239–253 (2008). [Google Scholar]
- Ulmer T. S., Campbell I. D. & Boyd J. The effects of dissolved oxygen upon amide proton relaxation and chemical shift in a perdeuterated protein. J. Mag. Reson. 157, 181–189 (2002). [DOI] [PubMed] [Google Scholar]
- Sakakura M., Noba S., Luchette P. A., Shimada I. & Prosser R. S. An NMR method for the determination of protein-binding interfaces using dioxygen-induced spin-lattice relaxation enhancement. J. Am. Chem. Soc. 127, 5826–5832 (2005). [DOI] [PubMed] [Google Scholar]
- Hernandez G., Teng C. L., Bryant R. G. & LeMaster D. M. O2 penetration and proton burial depth in proteins: Applicability to fold family recognition. J. Am. Chem. Soc. 124, 4463–4472 (2002). [DOI] [PubMed] [Google Scholar]
- Springer B. A., Sligar S. G., Olson J. S. & Phillips G. N. Mechanisms of ligand recognition in myoglobin. Chem. Rev. 94, 699–714 (1994). [Google Scholar]
- Phillips S. E. Structure and refinement of oxymyoglobin at 1.6 Å resolution. J. Mol. Biol. 142, 531–554 (1980). [DOI] [PubMed] [Google Scholar]
- Cohen J., Arkhipov A., Braun R. & Schulten K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys. J. 91, 1844–1857 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen J. & Schulten K. O2 migration pathways are not conserved across proteins of a similar fold. Biophys. J. 93, 3591–3600 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng C. L., Hinderliter B. & Bryant R. G. Oxygen accessibility to ribonuclease A: Quantitative interpretation of nuclear spin relaxation induced by a freely diffusing paramagnet. J. Phys. Chem. A 110, 580–588 (2006). [DOI] [PubMed] [Google Scholar]
- Eriksson A. E., Baase W. A. & Matthews B. W. Similar hydrophobic replacements of Leu99 and Phe153 within the core of T4 lysozyme have different structural and thermodynamic consequences. J. Mol. Biol. 229, 747–769 (1993). [DOI] [PubMed] [Google Scholar]
- Eriksson A. E., Baase W. A., Wozniak J. A. & Matthews B. W. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature 355, 371–373 (1992). [DOI] [PubMed] [Google Scholar]
- Morton A. & Matthews B. W. Specificity of ligand binding in a buried nonpolar cavity of T4 lysozyme: Linkage of dynamics and structural plasticity. Biochemistry 34, 8576–8588 (1995). [DOI] [PubMed] [Google Scholar]
- Mulder F. A., Mittermaier A., Hon B., Dahlquist F. W. & Kay L. E. Studying excited states of proteins by NMR spectroscopy. Nat. Struct. Biol. 8, 932–935 (2001). [DOI] [PubMed] [Google Scholar]
- Mulder F. A. A., Hon B., Muhandiram D. R., Dahlquist F. W. & Kay L. E. Flexibility and ligand exchange in a buried cavity mutant of T4 lysozyme studied by multinuclear NMR. Biochemistry 39, 12614–12622 (2000). [DOI] [PubMed] [Google Scholar]
- Bouvignies G. et al. Solution structure of a minor and transiently formed state of a T4 lysozyme mutant. Nature 477, 111–114 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder F. A. A., Hon B., Mittermaier A., Dahlquist F. W. & Kay L. E. Slow internal dynamics in proteins: Application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J. Am. Chem. Soc. 124, 1443–1451 (2002). [DOI] [PubMed] [Google Scholar]
- Feher V. A., Baldwin E. P. & Dahlquist F. W. Access of ligands to cavities within the core of a protein is rapid. Nat. Struct. Biol. 3, 516–521 (1996). [DOI] [PubMed] [Google Scholar]
- Solomon I. Relaxation processes in a system of 2 spins. Phys. Rev. 99, 559–565 (1955). [Google Scholar]
- Bloembergen N. & Morgan L. O. Proton relaxation times in paramagnetic solutions. Effects of electron spin relaxation. J. Chem. Phys. 34, 842–850 (1961). [Google Scholar]
- Vriend G. WHAT IF - a molecular modeling and drug design program. J. Mol. Graphics 8, 52–56 (1990). [DOI] [PubMed] [Google Scholar]
- Teng C. L. & Bryant R. G. Experimental measurement of nonuniform dioxygen accessibility to ribonuclease a surface and interior. J. Am. Chem. Soc. 122, 2667–2668 (2000). [Google Scholar]
- Bax A., Ikura M., Kay L. E., Torchia D. A. & Tschudin R. Comparison of different modes of two-dimensional reverse-correlation NMR for the study of proteins. J. Mag. Rson. 86, 304–318 (1990). [Google Scholar]
- Buckingham A. D. & Kollman P. A. Chemical shifts in paramagnetic gas mixtures. Mol. Phys. 23, 65–74 (1972). [Google Scholar]
- Bertini I. & Luchinat C. In Coordination Chem. Reviews, Vol. 150 (eds Lever A. B. P. et al.) Ch. 2, 29–75 (Elsevier, 1996). [Google Scholar]
- Miller T. M. in CRC Handbook of chemistry and physics, 95th edition, Vol. 95 (CRC Press, 2014). [Google Scholar]
- Scharlin P., Battino R., Silla E., Tunon I. & Pascual-Ahuir J. L. Solubility of gases in water: Correlation between solubility and the number of water molecules in the first solvation shell. Pure Appl. Chem. 70, 1895–1904 (1998). [Google Scholar]
- Mann G. & Hermans J. Modeling protein-small molecule interactions: Structure and thermodynamics of noble gases binding in a cavity in mutant phage T4 lysozyme L99A. J. Mol. Biol. 302, 979–989 (2000). [DOI] [PubMed] [Google Scholar]
- Horng M. L., Gardecki J. A. & Maroncelli M. Rotational dynamics of coumarin 153: Time-dependent friction, dielectric friction, and other nonhydrodynamic effects. J. Phys. Chem. A 101, 1030–1047 (1997). [Google Scholar]
- Lin Y. & Jonah C. D. The dynamics of anion solvation in alchohols in Ultrafast dynamics of chemical systems (ed. Simon J. D.) 134–162 (Kluwer Academic Publishers, 1994). [Google Scholar]
- Liu L. J., Quillin M. L. & Matthews B. W. Use of experimental crystallographic phases to examine the hydration of polar and nonpolar cavities in T4 lysozyme. Proc. Natl. Acad. Sci. USA 105, 14406–14411 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray N. W., Zhorov B. S. & Moczydlowski E. G. Interaction of local anesthetics with the K+channel pore domain: KcsA as a model for drug-dependent tetramer stability. Channels 7, 182–193 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion D., Ikura M., Tschudin R. & Bax A. Rapid recording of 2D NMR spectra without phase cycling - Application to the study of hydrogen-exchange in proteins. J. Mag. Rson. 85, 393–399 (1989). [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G. W., Pfeifer J. & Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- Goddard T. D. & Kneller D. G. Sparky-NMR assignment and integradiation software, San Francisco, USA. URL https://www.cgl.ucsf.edu/home/sparky/ (2008).
- Berendsen H. J. C., Vanderspoel D. & Vandrunen R. Gromacs: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 91, 43–56 (1995). [Google Scholar]
- Jorgensen W. L., Maxwell D. S. & TiradoRives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996). [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W. & Klein M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983). [Google Scholar]
- Arora G. & Sandler S. I. Mass transport of O2 and N2 in nanoporous carbon (C168 Schwarzite) using quantum mechanical force field and molecular dynamics simulations. Langmuir 22, 4620–4628 (2006). [DOI] [PubMed] [Google Scholar]
- Chandrasekhar J., Spellmeyer D. C. & Jorgensen W. L. Energy component analysis for dilute aqueous-solutions of Li+, Na+, F−, and Cl− ions. J. Am. Chem. Soc. 106, 903–910 (1984). [Google Scholar]
- Parrinello M. & Rahman A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- Darden T., York D. & Pedersen L. Particle mesh ewald: An Nlog(N) method for ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993). [Google Scholar]
- Hess B., Bekker H., Berendsen H. J. C. & Fraaije J. G. E. M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997). [Google Scholar]
- Miyamoto S. & Kollman P. A. Settle: An analytical version of the shake and rattle algorithm for rigid water models. J. Comput. Chem. 13, 952–962 (1992). [Google Scholar]
- Saito S., Ohmine I. & Bagchi B. Frequency dependence of specific heat in supercooled liquid water and emergence of correlated dynamics. J. Chem. Phys. 138, 094503 (2013). [DOI] [PubMed] [Google Scholar]
- Koradi R., Billeter M. & Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics 14, 51–55 (1996). [DOI] [PubMed] [Google Scholar]
- Bernstein H. J. RasWin Molecular Graphics 2.7.5, New York, USA. URL http://www.rasmol.org/software/RasMol_2.7.5/ (2013).
- Humphrey W., Dalke A. & Schulten K. VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38 (1996). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.