

Abstract
Vicinal diamines constitute one the most important functional motif in organic chemistry because of its wide occurrence in a variety of biological and pharmaceutical molecules. We report an efficient metal‐free, highly stereoselective intramolecular diamination using a novel chiral hypervalent iodine reagent together with its application as an efficient catalyst for the synthesis of diamines.
Keywords: cyclization, diamination, hypervalent iodine, organocatalysis, stereoselective synthesis
Abstract
Durch die weite Verbreitung in einer Vielzahl von biologischen und pharmazeutischen Molekülen stellen vicinale Diamine eine der wichtigsten Funktionalitäten in der organischen Chemie dar. Wir berichten über eine effiziente metallfreie, hochstereoselektive intramolekulare Diaminierung unter Verwendung eines neuartigen chiralen hypervalenten Iod‐Reagenz zusammen mit seinem Einsatz als effizienter Katalysator für die Synthese von Diaminen.
Hypervalent iodine reagents have found broad application in organic chemistry and are nowadays frequently used in synthesis.1, 2 It is of great interest to investigate their ability as highly selective oxidants,3 electrophilic reagents,4 to improve known reactions like the α‐functionalizations of ketones5 and to develop new reactions such as rearrangements6 using hypervalent iodine compounds. Oxidative transformations are of particular interest and a great challenge for organocatalytic processes. The requirement of developing new catalytic reactions with metal‐free reagents such as iodine is considerable as efficient procedures of this type are still immature.7
The addition of nitrogen nucleophiles to alkenes using hypervalent iodine reagents is known and aziridinations of alkenes,8 aminohydroxylations9 and aminofluorinations10 have already been carried out using stoichiometric amounts of either achiral or chiral reagents. Bifunctional nucleophiles can lead to interesting building blocks as shown in Scheme 1. After the activation of the double bond in compound 1 with the hypervalent iodine reagent, the first nucleophile attacks to give intermediate 2. The hypervalent iodine moiety in 2 is attached to a sp3‐hybridized carbon atom and is therefore an excellent leaving group, several orders of magnitude more reactive than triflates or tosylates.11 Products of type 3 are formed which can be transformed into 1,2‐diamines 4. There are various metal‐catalyzed methods available to carry out vicinal diamination of alkenes,12 but only a few reports in which stoichiometric amounts of hypervalent iodine reagents have been used for diaminations to generate racemates13 and also enantiomerically enriched products.14 However, there are hardly any methods available to carry out the reaction stereoselectively using chiral metal‐free catalysts.15
Scheme 1.
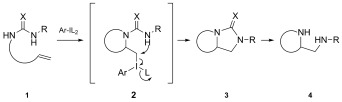
Intramolecular diamination with hypervalent iodine reagents.
Herein, we report the application of novel chiral iodine catalysts for stereoselective intramolecular diamination reactions using various homoallylic guanidine and diaminosulfone derivatives. Initial investigations were carried out using substrate 5 a. It was observed that (diacetoxyiodo)benzene and [bis(trifluoroacetoxy)iodo]benzene led to a sluggish reaction at 0 °C affording a very low yield of product 6 a. As reported in other reactions,16 we tried to activate the reagents with a Lewis acid and observed that, upon addition of BF3 ⋅ OEt2, TMSOTf, or a 1:1 mixture of BF3 ⋅ OEt2 and TMSOTf (Table 1, entries 2–5), the reaction proceeded faster with decent yields for the product. The nature of the solvent and also the reaction temperature have a strong influence on the overall yield as summarized in Table 1.
Table 1.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
For homoallylic guanidine derivatives 7 (Table 2), the nature of the N‐protecting group had a dominant effect on the cyclization. When tert‐butyl oxycarbonate (Boc) was used as the protecting group, only traces of the product 8 were observed (Table 2, entries 1 and 4). However, with carboxybenzoyl (Cbz) or tosylate (Ts) protecting groups, the products were obtained in reasonable yields. Studies have shown that the final carbon–nitrogen bond formation usually proceeds via an SN2‐type transition state with inversion of configuration and, hence, the nucleophilicity of the participating nitrogen moiety has a major influence.17 In addition, methyl substituents on the backbone of the alkene 7 (R1=Me) are less efficient in directing the cyclization as with phenyl substituents in that place (R1=Ph) where the yields are much higher.
Table 2.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
To develop a stereoselective method for diamination reactions, the reaction 5 a → 6 a was performed using lactate‐based chiral hypervalent iodine reagents 9 (Figure 1) under the optimized reaction conditions (Table 1, entry 3). Reagents 9 have been very successful in stereoselective reactions with alkenes9, 10, 14, 18 and also other substrates,19, 20 but here it was observed that only minor stereoselectivities could be obtained (Table 3, entries 1–3). The relatively large conformational flexibility in these reagents causes a weaker interaction of oxygen or nitrogen heteroatoms of the lactate moieties with the iodine which could also potentially be replaced by interactions with nitrogen atoms of the substrate 5 a. Reagent 10 has much less conformational flexibility with a strong interaction of the methoxy oxygen atom with the iodine established by X‐ray analysis.21 With this simple chiral hypervalent iodine reagent 10, much better selectivities (52 % ee) were observed in the cyclization of 5 a to 6 a (Table 3, entry 4).
Figure 1.
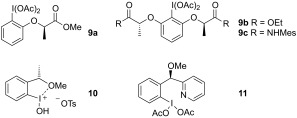
Chiral hypervalent iodine reagents.
Table 3.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
Based on this observation, we designed a novel hypervalent reagent with a pyridine moiety attached to a chiral benzylic center, which should allow an efficient coordination of the pyridine nitrogen to the iodine atom. It is well known that pyridines are good ligands for iodine(III), the close proximity to the stereogenic center should allow high selectivities to be obtained.22 This was indeed observed when the reaction was carried out using the novel hypervalent iodine reagent 11, which led to the product 6 in 72 % ee (Table 3, entry 5). NMR evidence suggests that it is indeed the nitrogen that is coordinating to the iodine, but the instability of the reagent 11 prevents further investigation and we cannot exclude that also the oxygen atom of the methoxy moiety coordinates. Reagent 11 is prepared directly before use and has been investigated with various Lewis acids for its activation, using different solvents and reaction temperatures. It was observed that the highest selectivity (92 % ee) could be obtained with a 1:1 mixture of trimethylsilyl triflate (TMSOTf) and boron trifluoride etherate (BF3 ⋅ OEt2) at −48 °C using acetonitrile as solvent (Table 3, entry 10). Under these reaction conditions BF2OTf ⋅ OEt2 is acting as the Lewis acid.23
The synthesis of reagent 11 is straightforward and is shown in Scheme 2. The aniline derivative 13, obtained by the addition of 2‐lithiopyridine to 2‐cyanoaniline 12, is iodinated to 14 and reduced by using a chiral ruthenium catalyst.24 The alcohol 15, obtained in >99 % ee, is methylated to 16 and oxidized with sodium perborate to reagent 11 in 87 % yield. The absolute configuration of 15 was determined to be (R) by deiodination of 15 and comparison of the optical rotation with a literature reference (see the Supporting Information).25
Scheme 2.
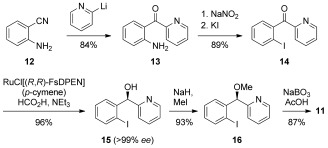
Synthesis of the novel chiral hypervalent iodine(III) reagent 11.
The reagent 11 was then used in the diamination of various substrates. It was again observed that diphenyl‐substituted substrates led to the cyclized products in good yields and with high enantioselectivities. Independent of the linker between the two nitrogen nucleophiles, the cyclization occurred with selectivities at or above 90 % ee using either sulfondiamides 5 (Table 4, entries 5 and 6) or guanidine derivatives 7 (Table 4, entries 2 and 4), whereas dimethyl‐substituted, dimethyl ester‐substituted or unsubstituted substrates showed almost no reaction (Table 4, entries 1, 3, 7, 9–12). Also a substituent R1 on the alkene moiety is tolerated leading to chiral products containing a tetrasubstituted stereocenter with a nitrogen substituent. The absolute stereochemistry of the product 6 a has been determined to be (R) by independent synthesis of 17 a (R=H) (see the Supporting Information). By analogy it can therefore be assumed that the products 6 and 8 (and 17) have (R) absolute stereochemistry for R1=H, Me and (S) for R1=Ph when using reagent 11. The new reagent 11 was also found to show promising selectivities in other stereoselective reactions.26
Table 4.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
All reactions shown in Table 4 were performed with stoichiometric amounts of hypervalent iodine(III) reagent 11 and the optical pure, reduced iodine(I) compound 16 was recovered after the reaction.
We then investigated the possibility of an in situ formation of the hypervalent iodine species by using the iodine(I) catalyst 16 together with stoichiometric amounts of an oxidant. The reaction occurred when 20 mol % of the iodine(I) catalyst 16 was used together with 2.2 equivalents meta‐chloroperbenzoic acid (mCPBA) at −48 °C in acetonitrile. However, the yield was low and the enantioselectivity only moderate. Upon changing the oxidant to sodium perborate and after addition of three equivalents of acetic acid at room temperature, much higher yields and selectivities were obtained. There are reactions reported for the oxidation of alkenes to oxiranes or vicinal acetoxy alcohols using sodium perborate, but these reactions usually take about 24 h for completion and at higher temperatures under highly acidic reaction conditions.27 In the reaction described in Table 5 almost no competing reactions are observed and the desired products 6 can be obtained in reasonable yields with good selectivities. A removal of ‘X’ and the Cbz protecting group is possible by reduction using lithium aluminum hydride, and the free diamines 17 are easily obtained. The Cbz protecting group has to be removed by hydrogenation (Pd/C) prior to reduction.
Table 5.
The publisher did not receive permission from the copyright owner to include this object in this version of this product. Please refer either to the publisher's own online version of this product or the printed product where one exists.
In summary, we present a highly stereoselective intramolecular diamination of alkenes using a novel, simple hypervalent iodine(III) reagent. This metal‐free method provides rapid access to bicyclic molecules and to diamines in a fast synthetic sequence. Using these results, the first catalytic protocol for a stereoselective diamination of alkenes has been developed.
Experimental Section
All synthetic methods including spectroscopic and analytical data are included in the Supporting Information.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
Acknowledgements
This project was supported by an EU Marie Curie fellowship to P.M. (DIALMEC, No. 298642) and by support from ENCPB to A. L. Support from the School of Chemistry, Cardiff University is also gratefully acknowledged. We thank the EPSRC National Mass Spectrometry Facility, Swansea, for mass spectrometric data.
References
- 1. Hypervalent Iodine Chemistry, (Ed.: T. Wirth), Top. Curr. Chem. ; 2003, 224, Springer, Berlin. [Google Scholar]
- 2. Zhdankin V. V., Hypervalent Iodine Chemistry, Wiley, Chichester: 2014. [Google Scholar]
- 3. Wirth T., Angew. Chem. 2005, 117, 3722–3731; [Google Scholar]; Angew. Chem. Int. Ed. 2005, 44, 3656–3665. [DOI] [PubMed] [Google Scholar]
- 4. Brown M., Farid U., Wirth T., Synlett 2013, 24, 424–431. [Google Scholar]
- 5.Recent review: Merritt E. A., Olofsson B., Synthesis 2011, 517–538. [Google Scholar]
- 6. Singh F. V., Wirth T., Synthesis 2013, 45, 2499–2511. [Google Scholar]
- 7.
- 7a. Richardson R. D., Wirth T., Angew. Chem. 2006, 118, 4510–4512; [Google Scholar]; Angew. Chem. Int. Ed. 2006, 45, 4402–4404; [DOI] [PubMed] [Google Scholar]
- 7b. Singh F. V., Wirth T., Chem. Asian J. 2014, 9, 950–971. [DOI] [PubMed] [Google Scholar]
- 8. Richardson R. D., Desaize M., Wirth T., Chem. Eur. J. 2007, 13, 6745–6754. [DOI] [PubMed] [Google Scholar]
- 9. Farid U., Wirth T., Angew. Chem. 2012, 124, 3518–3522; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012, 51, 3462–3465. [DOI] [PubMed] [Google Scholar]
- 10. Kong W., Feige P., de Haro T., Nevado C., Angew. Chem. 2013, 125, 2529–2533; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013, 52, 2469–2473. [DOI] [PubMed] [Google Scholar]
- 11. Ochiai M., Top. Curr. Chem. 2003, 224, 5–68. [Google Scholar]
- 12.
- 12a. Streuff J., Hövelmann C. H., Nieger M., Muñiz K., J. Am. Chem. Soc. 2005, 127, 14586–14587; [DOI] [PubMed] [Google Scholar]
- 12b. Muñiz K., J. Am. Chem. Soc. 2007, 129, 14542–14543; [DOI] [PubMed] [Google Scholar]
- 12c. Muñiz K., Hövelmann C. H., Streuff J., J. Am. Chem. Soc. 2008, 130, 763–773; [DOI] [PubMed] [Google Scholar]
- 12d. Sibbald P. A., Michael F. E., Org. Lett. 2009, 11, 1147–1149; [DOI] [PubMed] [Google Scholar]
- 12e. Sibbald P. A., Rosewall C. F., Swartz R. D., Michael F. E., J. Am. Chem. Soc. 2009, 131, 15945–15951; [DOI] [PubMed] [Google Scholar]
- 12f. Wen Y., Zhao B., Shi Y., Org. Lett. 2009, 11, 2365–2368; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12g. Cardona F., Goti A., Nat. Chem. 2009, 1, 269–275; [DOI] [PubMed] [Google Scholar]
- 12h. de Figueiredo R. M., Angew. Chem. 2009, 121, 1212–1215; [Google Scholar]; Angew. Chem. Int. Ed. 2009, 48, 1190–1193; [DOI] [PubMed] [Google Scholar]
- 12i. De Jong S., Nosal D. G., Wardrop D. J., Tetrahedron 2012, 68, 4067–4105; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12j. Kong A., Blakey S. B., Synthesis 2012, 44, 1190–1198. [Google Scholar]
- 13. Li H., Widenhoefer R. A., Tetrahedron 2010, 66, 4827–4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Röben C., Souto J. A., González Y., Lishchynskyi A., Muñiz K., Angew. Chem. 2011, 123, 9650–9654; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011, 50, 9478–9482; [DOI] [PubMed] [Google Scholar]
- 14b. Souto J. A., Martínez C., Velilla I., Muñiz K., Angew. Chem. 2013, 125, 1363–1367; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013, 52, 1324–1328. [DOI] [PubMed] [Google Scholar]
- 15. Chávez P., Kirsch J., Hövelmann C. H., Streuff J., Martínez‐Belmonte M., Escudero‐Adán E. C., Martin E., Muñiz K., Chem. Sci. 2012, 3, 2375–2382. [Google Scholar]
- 16. Singh F. V., Rehbein J., Wirth T., ChemistryOpen 2012, 1, 245–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Muñiz K., Martínez C., J. Org. Chem. 2013, 78, 2168–2174. [DOI] [PubMed] [Google Scholar]
- 18. Farid U., Malmedy F., Claveau R., Albers L., Wirth T., Angew. Chem. 2013, 125, 7156–7160; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2013, 52, 7018–7022. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Uyanik M., Yasui T., Ishihara K., Angew. Chem. 2010, 122, 2221–2223; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2010, 49, 2175–2177; [DOI] [PubMed] [Google Scholar]
- 19b. Uyanik M., Yasui T., Ishihara K., Tetrahedron 2010, 66, 5841–5851. [Google Scholar]
- 20.
- 20a. Fujita M., Yoshida Y., Miyata K., Wakisaka A., Sugimura T., Angew. Chem. 2010, 122, 7222–7225; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2010, 49, 7068–7071; [DOI] [PubMed] [Google Scholar]
- 20b. Fujita M., Wakita M., Sugimura T., Chem. Commun. 2011, 47, 3983–3985. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Hirt U. H., Spingler B., Wirth T., J. Org. Chem. 1998, 63, 7674–7679; [Google Scholar]
- 21b. Hirt U. H., Schuster M. F. H., French A. N., Wiest O. G., Wirth T., Eur. J. Org. Chem. 2001, 1569–1579. [Google Scholar]
- 22.
- 22a. Grushin V. V., Shcherbina T. M., Tolstaya T. P., J. Organomet. Chem. 1985, 292, 105–117; [Google Scholar]
- 22b. Kita Y., Okuno T., Tohma H., Akai S., Tetrahedron Lett. 1994, 35, 2717–2720; [Google Scholar]
- 22c. Ochiai M., Suefuji T., Miyamoto K., Shiro M., Chem. Commun. 2003, 1438–1439; [DOI] [PubMed] [Google Scholar]
- 22d. Suefuji T., Shiro M., Yamaguchi K., Ochiai M., Heterocycles 2006, 67, 391–397; [Google Scholar]
- 22e. Pell T. P., Couchman S. A., Ibrahim S., Wilson D. J. D., Smith B. J., Barnard P. J., Dutton J. L., Inorg. Chem. 2012, 51, 13034–13040. [DOI] [PubMed] [Google Scholar]
- 23. Myers E. L., Butts C. P., Aggarwal V. K., Chem. Commun. 2006, 4434–4436. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Wang C., Pettman A., Bacsa J., Xiao J., Angew. Chem. 2010, 122, 7710–7714; [Google Scholar]; Angew. Chem. Int. Ed. 2010, 49, 7548–7552; [DOI] [PubMed] [Google Scholar]
- 24b. Noyori R., Ohkuma T., Angew. Chem. 2001, 113, 40–75; [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001, 40, 40–73; [Google Scholar]
- 24c. Ohkuma T., Ishii D., Takeno H., Noyori R., J. Am. Chem. Soc. 2000, 122, 6510–6511. [Google Scholar]
- 25. Kim H., Nagaki A., Yoshida J., Nat. Commun. 2011, 2, 264. [DOI] [PubMed] [Google Scholar]
- 26.The α‐oxytosylation of propiophenone proceeds with 11 in the presence of p‐TsOH with 42 % ee (78 % yield), the α‐diethylamination of 1‐indanone with 69 % ee (63 % yield) according to a recently published protocol: Mizar P., Wirth T., Angew. Chem. 2014, 126, 6103–6107; [Google Scholar]; Angew. Chem. Int. Ed. 2014, 53, 5993–5997. [DOI] [PubMed] [Google Scholar]
- 27. Xie G., Xu L., Hu J., Ma H., Hou W., Tao F., Tetrahedron Lett. 1988, 29, 2967–2968. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information