

Summary
The cyclic adenosine monophosphate–protein kinase A (cAMP–PKA) pathway is a central signalling cascade that transmits extracellular stimuli and governs cell responses through the second messenger cAMP. The importance of cAMP signalling in fungal biology has been well documented and the key conserved components, adenylate cyclase (AC) and the catalytic subunit of PKA (CPKA), have been functionally characterized. However, other genes involved in this signalling pathway and their regulation are not well understood in filamentous fungi. Here, we performed a comparative transcriptomics analysis of AC and CPKA mutants in two closely related fungi: Fusarium graminearum (Fg) and F. verticillioides (Fv). Combining available Fg transcriptomics and phenomics data, we reconstructed the Fg cAMP signalling pathway. We developed a computational program that combines sequence conservation and patterns of orthologous gene expression to facilitate global transcriptomics comparisons between different organisms. We observed highly correlated expression patterns for most orthologues (80%) between Fg and Fv. We also identified a subset of 482 (6%) diverged orthologues, whose expression under all conditions was at least 50% higher in one genome than in the other. This enabled us to dissect the conserved and unique portions of the cAMP–PKA pathway. Although the conserved portions controlled essential functions, such as metabolism, the cell cycle, chromatin remodelling and the oxidative stress response, the diverged portions had species‐specific roles, such as the production and detoxification of secondary metabolites unique to each species. The evolution of the cAMP–PKA signalling pathway seems to have contributed directly to fungal divergence and niche adaptation.
Keywords: cAMP, cell signalling, Fusarium, orthologous, PKA, speciation, transcriptome
Introduction
A hallmark of cellular organisms is their ability to respond to environmental stimuli via sophisticated signal transduction pathways and transcriptional regulation. In most eukaryotic organisms, environmental signals sensed by transmembrane receptors, such as G protein‐coupled receptors (GPCRs), activate adenylate cyclase (AC) via G proteins or Ras proteins to produce a second messenger cyclic adenosine monophosphate (cAMP). After binding with cAMP, the regulatory subunits of protein kinase A (PKA) tetramers (PKR) disassociate from the catalytic subunits (CPKA), which then become active to regulate downstream signalling and gene transcription. Intracellular cAMP levels are balanced by cAMP‐producing AC and cAMP‐scavenging phosphodiesterase (PDE) (Broach, 1991; Kraakman et al., 1999; Ma et al., 1999).
The cAMP–PKA pathway plays a central role in the transduction of environmental signals and in the mediation of diverse cellular functions (Daniel et al., 1998; Seino and Shibasaki, 2005). Its biological functions have been characterized in many organisms, from bacteria to humans. In animals, cAMP signalling regulates many tissue‐ or cell type‐specific functions and processes, including glycogen metabolism (Bollen et al., 1998), heart rate (Zaccolo, 2009) and progesterone secretion (Chin and Abayasekara, 2004). In Saccharomyces cerevisiae, cAMP signalling regulates nutrient sensing and pseudohyphal differentiation (Casperson et al., 1985; Matsumoto et al., 1982). In Schizosaccharomyces pombe, this conserved signalling pathway regulates spore germination and mating under glucose‐ or nitrogen‐limiting conditions (D'Souza and Heitman, 2001; Maeda et al., 1990). In pathogenic fungi, such as Cryptococcus neoformans (D'Souza et al., 2001), Ustilago maydis (Dürrenberger et al., 1998) and Magnaporthe oryzae (Choi and Dean, 1997; Xu et al., 1997), cAMP signalling also regulates various infection processes. For instance, in the opportunistic fungal pathogen C. neoformans, the cAMP–PKA pathway regulates mating and virulence (D'Souza et al., 2001). In the rice blast fungus M. oryzae, cAMP signalling is essential for the formation and function of the appressorium, a specialized structure that facilitates plant penetration (Choi and Dean, 1997; Xu et al., 1997).
The filamentous ascomycetes Fusarium graminearum (Fg) and F. verticillioides (Fv) are two fungal pathogens that threaten cereal crops worldwide. Although Fg causes head blight on Triticum aestivum (wheat) and Hordeum vulgare (barley), and ear and stalk rot on Zea mays (maize; Goswami and Kistler, 2004; Leslie and Summerell, 2006; Sutton, 1982), Fv primarily causes stalk and ear rot in maize and Sorghum bicolor (sorghum) (Leslie and Summerell, 2006). In addition to causing yield losses, both fungi produce species‐specific mycotoxins, such as deoxynivalenol (DON) and aurofusarin in Fg (Desjardins et al., 1993; Goswami and Kistler, 2004) and fumonisin B1 and bikaverin in Fv (Kedera et al., 1999; Lazzaro et al., 2012; Sydenham et al., 1990), which pose serious threats to animal and human health.
Recently, the cAMP signalling pathway has been characterized in these two pathogenic Fusarium species, as well as in their sister species, F. oxysporum (Fo). Choi and Xu (2010) reported that the deletion of the AC gene FAC1 in Fv led to reduced virulence, but that deletion of one catalytic subunit of PKA, CPK1, had little effect on fungal virulence, even though both deletions reduced conidiation and vegetative growth. The FAC1 deletion mutant exhibited normal fumonisin B1 production, but showed increased production of bikaverin and increased resistance to oxidative and heat stresses. In Fg, FAC1 and CPK1 were essential for normal vegetative growth, conidiation, ascospore maturation and release, DON production, and pathogenesis (Hu et al., 2014), but a deletion mutant of a CPK1 paralogue, CPK2, had no detectable phenotype. An FoCPKA (CPK1 homologue) deletion mutant of Fo O‐685, a strain pathogenic to Arabidopsis thaliana, showed normal conidiation, but abnormal microconidium germination, and was non‐pathogenic (Kim et al., 2011). Mutant phenotype analysis suggests that the cAMP–PKA pathway shares some common functions among these three closely related species, but has also diverged to regulate distinct functions, such as secondary metabolite production and sexual development, in each species.
In this study, we performed a comparative transcriptomics analysis among two sets of mutants (Δfac1 and Δcpk1) in two Fusarium species that share 8750 orthologues (Ma et al., 2010). We used Affymetrix fungal multigenome ExonChips to monitor gene expression. We identified genes that were differentially expressed in the mutants compared with their respective wild‐type strains. Based on the differentially expressed genes (DEGs) detected in the mutants and the phenomics data available in the public domain, we reconstructed the Fg cAMP signalling pathway, which regulates important biological processes through key regulators, including protein kinases (PKs) and transcription factors (TFs). By comparing the expression patterns of all 8750 orthologues in three genetic backgrounds (wild‐type, Δfac1 and Δcpk1), we identified 482 orthologues that exhibit different patterns of gene expression, suggesting potential functional divergence between the orthologues, despite high levels of sequence similarity. Our comparative study enabled us to dissect the cAMP signalling pathway in these two Fusarium species into conserved and species‐specific components. In agreement with our phenotypic observations, conserved portions in both species controlled essential functions, such as metabolism, the cell cycle, protein synthesis and the stress response. By contrast, diverged components regulated species‐specific functions, such as the biosynthesis and detoxification of species‐specific secondary metabolites.
Results
Reconstructed Fg cAMP–PKA pathway based on DEGs and phenome data
Key regulators of the cAMP–PKA pathway
We reconstructed the Fg cAMP–PKA signalling pathway based on Δfac1 and Δcpk1 mutant expression data, previously characterized Fg TFs (Son et al., 2011) and kinases (Wang et al., 2011), and information from the Pathogen–Host Interaction Database (Winnenburg et al., 2008). FAC1‐ and/or CPK1‐dependent key regulators were defined as TFs and PKs that were differentially expressed in one or both Δfac1 and Δcpk1 mutants. We identified 65 TFs and 22 PKs with a false discovery rate (FDR) of less than 0.05 (Table S2, see Supporting Information). According to previous phenotypic characterizations of all TF and PK knockout lines (Son et al., 2011; Wang et al., 2011), the mutants of 13 TFs and 15 PKs exhibited aberrant phenotypes, including defects in virulence, DON and zearalenone (ZEA) production, sporulation, sexual reproduction and the stress response (Fig. 1A). To study the transcriptional regulation of this set of key regulators, we further characterized their expression patterns using 60 Fg transcriptomics datasets available at PLEXdb (Dash et al., 2012). These 60 experiments included four datasets that captured: (i) the active pathogen–host interaction during the course of wheat coleoptile infection (Guenther et al., 2009; Zhang et al., 2012); (ii) conidial germination (Seong et al., 2008); (iii) sexual reproduction (Hallen et al., 2007); and (iv) DON induction (Gardiner et al., 2009; Jonkers et al., 2012).
Figure 1.
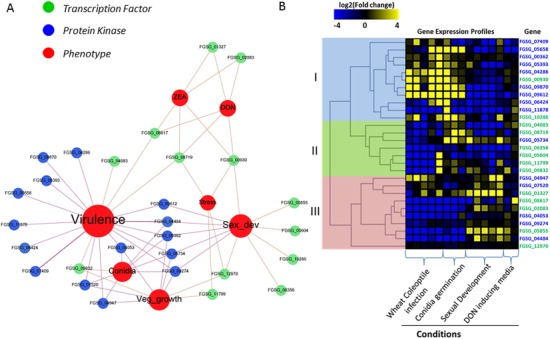
Functional affiliation of the differentially expressed regulators shared in Fusarium graminearum Δfac1 and Δcpk1 mutants. (A) Gene–phenotype networks depict the association of known phenotypes (red nodes) with cyclic adenosine monophosphate (cAMP)‐dependent transcription factors (green nodes) and protein kinases (blue nodes). Genes are differentially expressed in the Δfac1 and/or Δcpk1 mutants. Phenotype information is derived from FgTFPD (Son et al., 2011), a previous kinome analysis (Wang et al., 2011) and the Pathogen–Host Interaction (PHI) Database (Winnenburg et al., 2008). Phenotypes include ‘Conidia’ (conidiation), ‘Stress’ (fungal growth under stress conditions, such as oxidative and osmotic stresses), ‘Veg_growth’ [vegetative growth on potato dextrose agar (PDA) or minimal medium], ‘Sex_dev’ (perithecium and ascospore development), ‘DON’ (deoxynivalenol production), ‘ZEA’ (zearalenone production) and ‘Virulence’ (wheat head blight). (B) Heatmap of gene expression profile (fold changes) of cAMP‐dependent transcription factors (green) and protein kinases (blue) during wheat infection, sexual development, conidial germination and DON‐inducing conditions. Fold changes are log2 values and scale bar shows down‐regulation (blue), up‐regulation (yellow) and no change (black). Genes are divided into three groups (I, II and III) via hierarchical clustering of expression levels.
Using hierarchical clustering, we clustered the regulators of the cAMP–PKA pathway into three groups (Fig. 1B). Interestingly, nine of the 15 PKs (60%) were grouped into Group I, whereas 11 of the 13 TFs were grouped into Groups II and III. In agreement with the mutant data showing that most PKs involved in the cAMP signalling pathway contribute to virulence (Fig. 1A), Group I genes were highly expressed throughout the course of infection and during conidial germination. This finding may reflect the direct contribution of PKs to fungal pathogenesis. The other two groups, and particularly Group III, included most of the regulators controlling sexual reproduction. Mutants in this group of PKs, such as FGSG_04484 (FgSrb10), FGSG_04947 (FgSak1), FGSG_05734 (FgKic1) and FGSG_09274 (FgKin1), had pleiotropic defects in growth, conidiation, sexual reproduction and plant infection, indicating that the corresponding wild‐type genes are involved in basic cellular processes (Wang et al., 2011).
We observed more inconsistencies between gene expression and phenotypes for TFs than for PKs. For example, the only two TFs (FGSG_10286, FGSG_00930) in Group I lacked a direct association with virulence, as observed for the PKs. The FGSG_00930 mutant exhibited defects in the stress response, toxin production and sexual reproduction. Only sexual reproduction was affected in the FGSG_10286 mutant. In agreement with this finding, FGSG_10286 expression was induced during sexual reproduction. Several deletion mutants of genes that are up‐regulated during infection, such as FGSG_11799 and FGSG_05604, showed normal virulence. This suggests that, unlike PKs, which initiate a chain of reactions with direct functional consequences, TFs influence cell function by reprogramming the transcriptional network, which results in pleiotropic effects. Overall, our data suggest that condition‐specific transcriptional regulation of cAMP‐dependent regulators has a significant impact on fungal responses to environmental signals.
The Fg cAMP–PKA pathway
To decipher the gene regulatory networks involved in cAMP signalling in Fg, we measured the expression profiles of two loss‐of‐function mutants, Δfac1 and Δcpk1, which lack key components of the cAMP signalling pathway. We identified genes that were differentially expressed between each mutant and the wild‐type strain PH1 (Table S2). A total of 1239 genes with FDR < 0.05 showed at least a two‐fold change in expression in the Δfac1 mutant, including 1005 genes that were down‐regulated and 234 genes that were up‐regulated. A total of 294 genes were differentially expressed in the Δcpk1 mutant, including 219 that were down‐regulated and 75 that were up‐regulated. Considering the functional properties of the key regulators and the functional enrichment of all DEGs in these two Fg mutants, we reconstructed the Fg cAMP–PKA signalling pathway, which includes portions controlled by both FAC1 and CPK1 (the FAC1–CPK1 subpathway) and portions controlled only by FAC1 (the FAC1‐unique subpathway) or CPK1 (the CPK1‐unique subpathway; Fig. 2).
Figure 2.
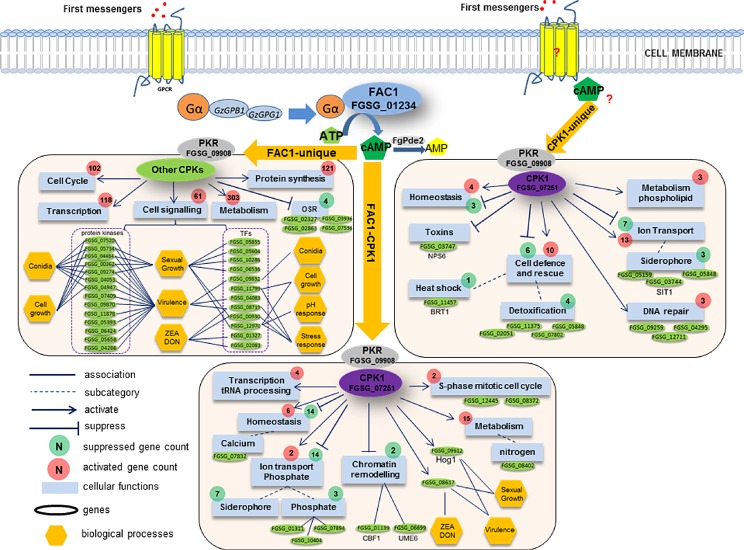
Proposed model for cyclic adenosine monophosphate–protein kinase A (cAMP–PKA)‐mediated transcriptional and signalling pathways in Fusarium graminearum based on transcriptomics analysis of the Δfac1 and Δcpk1 mutants. Environmental cues and first messengers are perceived by G protein‐coupled receptors, and G proteins are activated to generate cAMP through adenylate cyclase FAC1. cAMP then binds to the regulatory subunit of PKA (PKR: FGSG_09908) and releases catalytic subunits of PKA (CPK1, CPK2) that regulate downstream cellular functions. cAMP‐dependent processes can be FAC1 dependent (FAC1‐unique and FAC1–CPK1) or independent (CPK1‐unique), regulating a variety of functions (blue boxes) that are both basic housekeeping and specialized. All genes downstream of and including FAC1 and CPK1 in this network are differentially expressed in the Δfac1 and Δcpk1 mutants based on microarray analysis. The correlation of phenotypes and key regulators (protein kinases and transcription factors) is primarily based on F. graminearum phenome data described in FgTFPD (Son et al., 2011), previous kinome phenotype data (Wang et al., 2011) and the Pathogen–Host Interaction (PHI) Database (Winnenburg et al., 2008). Cellular function association is based on functional enrichment of differentially expressed genes (DEGs) in the Δfac1 and Δcpk1 mutants (Table S2) using FunCat (Munich Information Center for Protein Sequences, MIPS). The F. graminearum gene annotation is derived from the F. graminearum genome annotation database (MIPS) and yeast homology information (Saccharomyces Genome Database). DON, deoxynivalenol; ZEA, zearalenone.
The FAC1–CPK1 subpathway
About 60% of DEGs identified in the Δcpk1 mutant (180) were also identified in the Δfac1 mutant, including 137 and 43 that were down‐regulated and up‐regulated, respectively (Table S2), consistent with the finding that AC and CPKA are two key components of the same cAMP–PKA signalling pathway (D'Souza and Heitman, 2001).
Functional enrichment analysis suggested that the FAC1–CPK1 subpathway positively regulates several essential housekeeping functions, including regulation of the S‐phase of the mitotic cell cycle, tRNA processing, homeostasis and nitrogen metabolism, by modulating the expression of different sets of genes (Fig. 2). By contrast, this subpathway suppressed 16 ion transport genes, including seven siderophore transport genes, three phosphate transport genes and two genes involved in chromatin remodelling (i.e. UME6, a key regulator of meiosis and chromatin remodelling, and CBF1, a centromere‐binding factor) (Fig. 2). In addition, this subpathway regulated mycotoxin production, sexual growth and virulence by controlling a TF (FGSG_08617) and an FgHog1 mitogen‐activated protein kinase (MAPK) (FGSG_09612) (a homologue of the S. cerevisiae osmoregulator HOG1). The link between the FAC1–CPK1 subpathway and the HOG1 homologue suggests that cAMP signalling and the MAPK cascade are interconnected, at least in Fg.
The FAC1‐unique subpathway
We identified more DEGs in Δfac1 than in Δcpk1, indicating that FAC1 acts upstream of CPK1. Furthermore, this finding suggests the existence of additional cAMP‐dependent signalling components that are independent of CPK1. PKR (FGSG_09908), but not CPK1, was detected among the Δfac1 DEGs. Indeed, no other CPK genes were found among the Δfac1 DEGs, suggesting that the transcription of CPK1 and other potential CPKs was not directly regulated by cAMP. Fg and many other filamentous fungi contain two paralogous PKA catalytic subunit genes: CPK1 and CPK2. Although deletion of CPK2 had no detectable phenotype, the Δcpk1Δcpk2 double mutant exhibited more severe defects than Δcpk1 (Hu et al., 2014). Therefore, it is possible that many downstream targets are co‐regulated by CPK1 and CPK2. Functional enrichment analysis showed that the FAC1‐unique subpathway is involved in a broad spectrum of biological functions, including metabolism, transcription, protein synthesis, cell cycle control and cell signalling (Fig. 2). This observation is consistent with the fact that AC and its product cAMP are important in many fundamental cellular processes. Based on phenomics and transcriptomics data, we identified 17 and 10 regulators that control virulence and sexual reproduction, respectively (Figs 1A and 2). The regulators that contributed most to the phenotype were FGSG_00362, FGSG_05734 (Kic1), FGSG_09274 (FgKin1) and FGSG_08719 (TF), and each of these contributed to different phenotypes. This finding is highly consistent with the phenotypes of Δfac1 in Fg, which included reduced growth, absence of sexual reproduction, decreased DON production and reduced pathogenicity. All of these FAC1‐dependent regulators of key biological processes were co‐regulated in this pathway.
The CPK1‐unique subpathway
The CPK1‐unique subpathway included 114 genes that were only differentially expressed in the Δcpk1 mutant, and these genes were mostly involved in the homeostasis of cations and metal ions, ion transport and cell defence. Both up‐regulated and down‐regulated DEGs were associated with each biological process, suggesting that CPK1 is a dual regulator of these processes (Fig. 2). The detection of the CPK1‐unique subpathway suggests that Fg CPK1 may function independently of FAC1, because intracellular cAMP levels can also be influenced by PDE activities.
In contrast with the other two subpathways of the cAMP–PKA pathway, the CPK1‐unique subpathway was strongly linked to organismal defence. For instance, this subpathway regulated the expression of three genes involved in DNA repair and five genes involved in detoxification. CPK1 suppressed a heat shock response gene, FGSG_11457 (BRT1), probably by contributing to the increased heat tolerance phenotype observed in Δcpk1, but not Δfac1 (Hu et al., 2014). CPK1 also regulated the expression of a non‐ribosomal peptide synthase gene, FGSG_03747 (NPS6). In Alternaria brassicicola and Cochiliobolus heterostrophus (Oide et al., 2006), NPS6 is involved in extracellular siderophore biosynthesis, tolerance to H2O2 stress and fungal virulence. Therefore, NPS6 appears to act downstream of CPK1 in Fg and may regulate iron acquisition, oxidative stress tolerance and virulence.
In summary, our pathway analysis correlated key regulators (TFs and PKs) with many phenotypes (Figs 1 and 2). However, the identification of direct links between genes regulated by the cAMP–PKA signalling pathway and specific biological functions remains challenging, and will require further functional characterization of the genes identified here. Overall, the FAC1‐dependent components control many essential biological functions, including housekeeping functions and host–pathogen interaction processes. By contrast, the CPK1‐dependent components were mostly involved in cell homeostasis, ion transport and cell defence. However, some functions were regulated by both FAC1‐dependent and CPK1‐dependent components. For instance, cAMP was reported to regulate iron transport in S. cerevisiae (Robertson et al., 2000). CPK1 suppressed the expression of 10 genes involved in siderophore transport, including three genes via the CPK1‐unique subpathway and seven genes via the FAC1–CPK1 subpathway.
Conservation of the cAMP–PKA pathway between Fg and Fv
As the cAMP–PKA pathway is a key signalling cascade that regulates cellular systems in response to environmental conditions, we anticipated that this pathway would be functionally conserved in Fg and Fv, two fungal species that diverged less than 40 million years ago (Ma et al., 2013). Based on a total of 8750 orthologues identified between these two phytopathogenic fungi (Ma et al., 2010), we compared the transcriptomics profiles of orthologous genes in the Δfac1 and Δcpk1 mutants of Fg and Fv.
Among the 298 and 153 DEGs detected in the Fv Δfac1 and Δcpk1 mutants (Table S3, see Supporting Information), 215 (72%) and 105 (68%), respectively, have Fg orthologues (Fig. S5, see Supporting Information). Surprisingly, only about one‐third of these orthologues exhibited the same expression pattern in both fungal species when a maximal FDR of 0.05 and a minimal fold change of two were used as thresholds to define DEGs. A total of 63 DEGs, including 39 that were down‐regulated, 21 that were up‐regulated and three that were up‐regulated in one species, but down‐regulated in the other, were shared between the Fg and Fv Δfac1 mutants (Fig. S5, Table S4, see Supporting Information). Thirty‐one DEGs exhibited the same expression patterns in the Fg and Fv Δcpk1 mutants, including seven commonly down‐regulated and 24 commonly up‐regulated genes (Fig. S5, Table S4). The predicted functions of these common DEGs aligned with those of genes that function in the conserved portions of the cAMP–PKA signalling pathway in these two species, including genes involved in key cellular functions, such as primary and secondary metabolism, protein synthesis, mitotic cell cycle control, the stress response, homeostasis and ion transport. Similar to the case in Fg, the DEGs in the conserved cAMP–PKA pathway could be divided into three different categories based on their dependence on FAC1, CPK1 or both FAC1 and CPK1 in Fv (Fig. 3).
Figure 3.
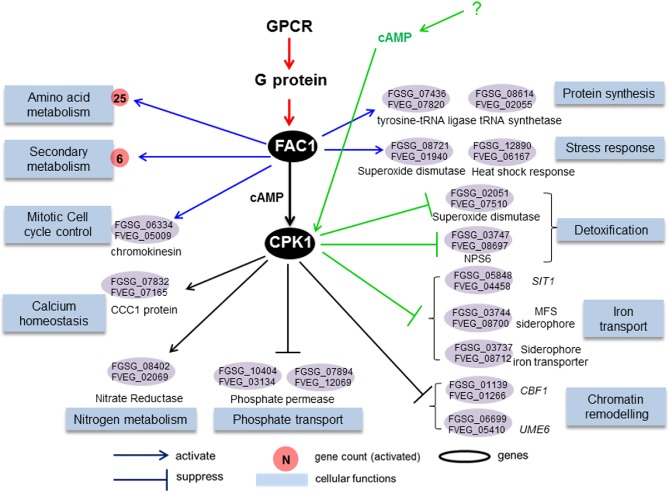
Proposed conserved cyclic adenosine monophosphate–protein kinase A (cAMP‐PKA) pathway for Fusarium graminearum and F. verticillioides. Endogenous cAMP produced by FAC1 activates CPK1 and other CPKs to regulate many conserved biological functions (blue boxes), such as metabolism, protein synthesis, the cell cycle, homeostasis, ion transport and chromatin remodelling via orthologous genes differentially expressed in both species (ovals). These processes are dependent on FAC1 (blue arrows) or FAC1–CPK1 (black arrows), and cAMP can activate CPK1 and regulate detoxification and iron transport, including siderophore transport, independently of FAC1 (green arrows). Cellular function association is based on functional enrichment of differentially expressed genes (DEGs) in the Δfac1 and Δcpk1 mutants (Tables S2 and S3) using FunCat (Munich Information Center for Protein Sequences, MIPS). Fusarium graminearum and F. verticillioides gene annotations are derived from the Fusarium genome annotation database (MIPS) and yeast homology information (Saccharomyces Genome Database). GPCR, G protein‐coupled receptor.
Orthologues regulated by both FAC1 and CPK1 in both Fg and Fv included key regulators of meiosis (FgUME6), chromatin remodelling (FgCBF1), calcium and manganese homeostasis (FGSG_07832, the CCC1 protein), and nitrogen (FGSG_08402) and phosphate (FGSG_10404 and FGSG_07894) metabolism. In the FAC1‐unique pathway in both species, FAC1 positively regulated a kinesin protein (FGSG_06334, FVEG_05009) involved in the mitotic cell cycle, consistent with our current knowledge of the roles of the cAMP–PKA pathway in the regulation of cell cycles. Two orthologues encoding a tRNA ligase (FGSG_07436, FVEG_07820) and tRNA synthetase (FGSG_08614, FVEG_02055) were present in both the Fg and Fv pathways, suggesting that cAMP‐mediated regulation of protein synthesis is conserved in these two species. Orthologues involved in the stress response, such as oxidative and heat stress (FGSG_08721, FGSG_12890), were also detected. Orthologues regulated by the CPK1‐unique subpathway in both Fg and Fv included genes responsible for iron uptake (Fig. 3), such as S. cerevisiae SIT1 (Robertson et al., 2000).
The conservation of the cAMP–PKA signalling pathway in the regulation of protein synthesis, ion transport and the stress response can be further extended from the genus Fusarium to Saccharomyces. For example, it is known that S. cerevisiae TPK genes (CPK1 homologues) suppress environmental stress response events by activating the expression of ribosomal protein genes (Gasch, 2003). In Fg Δfac1 and Δcpk1 mutants, down‐regulated genes were highly enriched for ribosomal protein genesis and translation processes, confirming the regulatory role of the cAMP–PKA pathway in protein synthesis in Fg. Similarly, genes involved in protein synthesis were also down‐regulated in the Fv Δfac1 and Δcpk1 mutants. Consistent with the finding that S. cerevisiae TPK regulates SIT1, a siderophore iron transporter (Robertson et al., 2000), SIT1 homologues in Fg and Fv (FGSG_05848 and FVEG_04458) were also negatively regulated by FAC1–CPK1. The other regulatory function in the cAMP–PKA pathway shared by Fg and S. cerevisiae was the organism stress response (Fig. 2). In Fg, a key process regulated by cAMP was the cellular response to oxidative and heat stresses. In S. cerevisiae, a constitutively activated Ras/cAMP pathway resulted in a decreased response to stress conditions, suggesting that there is a negative correlation between endogenous cAMP levels and the cell's tolerance to stress responses (Jones et al., 2003).
Functional divergence of the cAMP–PKA pathway in Fg and Fv
Noise reduction in microarray data
We were concerned that about two‐thirds of the orthologous DEGs identified in the Δfac1 and Δcpk1 mutants in these two closely related species did not show similar expression patterns. This could either reflect the rapid functional divergence of this signalling pathway or errors caused by factors such as the stochastic nature of expression data and the randomness of the hard, arbitrary cut‐off of two‐fold expression changes. In an effort to differentiate true functional divergence of orthologous genes from errors, we developed a program that combined sequence conservation and patterns of orthologous gene expression in two different species, using the ratio of expression level (ROEL) (see Experimental procedures).
In contrast with standard expression analysis, which identifies DEGs on the basis of expression fold changes, this method compares the expression patterns of orthologous genes directly using the level of expression. We compared the expression profiles of 8750 orthologues identified in a comparative genomics study (Ma et al., 2010) in the wild‐type, Δfac1 and Δcpk1 strains in both species. Consistent with their phylogenetic relatedness, we observed a strong correlation in expression level between the orthologues (Pearson's correlation coefficients of 0.80, 0.80 and 0.79 for the wild‐type, Δfac1 and Δcpk1 strains, respectively) (Fig. 4A). We define a pair of orthologues as being functionally diverged if the ROEL score is either above 1.5 or below 0.66, indicating that the expression level is at least 50% higher in one genome than in the other (Table S5, see Supporting Information).
Figure 4.
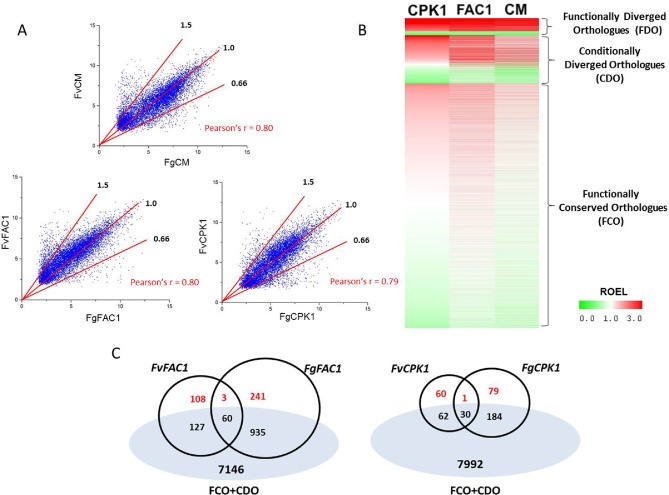
Expression correlation analysis of identified conserved and diverged orthologous gene expression in Fusarium graminearum and F. verticillioides. (A) Scatter plots and Pearson correlation coefficient (r) of orthologous gene expression levels in the two Fusarium species in three genetic backgrounds: wild‐type and Δfac1 and Δcpk1 mutants. Red lines indicate the cut‐offs of the ratio of expression levels (ROELs) (Fg/Fv) of 1.5, 1.0 and 0.66 as indicated. (B) Heatmap of ROELs in the wild‐type (CM) and Δfac1 and Δcpk1 mutants. Functionally diverged orthologues (FDO) have a ROEL of larger than 1.5 or less than 0.66 in all three genetic backgrounds. Conditionally diverged orthologues (CDO) have a ROEL of larger than 1.5 or less than 0.66 in only one or two backgrounds. Functionally conserved orthologues (FCO) have a ROEL of larger than 0.66 and less than 1.5 in all three backgrounds. (C) Venn diagrams showing common differentially expressed genes (DEGs) and distinct DEGs in the two Fusarium species. True diverged DEGs (in red) were identified by removing functionally conserved orthologues (FCO) and conditionally diverged orthologues (CDO).
Combining data from all three genetic backgrounds, we found that most orthologues (79%) had ROEL scores of between 0.66 and 1.5, and were thus defined as functionally conserved orthologues. Approximately 15.5% of the orthologues had diverged expression profiles (ROELs of above 1.5 or below 0.66) under one or two genetic backgrounds, and were considered to be conditionally diverged orthologues (Fig. 4B). The expression levels of about 6% (482) of orthologous genes consistently varied (ROEL of above 1.5 or below 0.66) between these two species among all three tested backgrounds (Fig. 4B), and these genes were defined as functionally diverged orthologues.
Among the DEGs identified in individual mutants of each species, over 70% had orthologous genes in the respective genome and a number were functionally conserved orthologues (Table 1). By removing these conserved orthologues and conditionally diverged orthologues from the distinct DEGs from both species, we defined species‐specific DEGs, which were unique to a species and functionally diverged orthologues (Fig. 4C; Table 1). The diverged orthologue rate was calculated to determine the divergence of each component of the cAMP–PKA pathway between these two species (Table 1). For Fg, the diverged rates were 23% (FAC1) and 40% (CPK1), whereas the diverged rates of Fv were 32% (FAC1) and 49% (CPK1). The consistently higher diverged rates in the Δcpk1 mutant suggested that, compared with FAC1, CPK1 makes a greater contribution to species‐specific functions (Table 1).
Table 1.
Summary of the number and type of differentially expressed genes (DEGs) per mutant per species
| DEGs | Δfvfac1 | Δfgfac1 | Δfvcpk1 | Δfgcpk1 |
|---|---|---|---|---|
| Shareda | 63 | 63 | 31 | 31 |
| Species‐specificb | 100 | 220 | 58 | 71 |
| Functionally diverged orthologues (FDO)b | 11 | 24 | 3 | 9 |
| Conditionally diverged orthologues (CDO) | 54 | 217 | 44 | 81 |
| Functionally conserved orthologues (FCO) | 133 | 778 | 48 | 133 |
| Total DEGs (TD) | 298 | 1239 | 153 | 294 |
| Total orthologues (TO) | 198 | 1019 | 95 | 223 |
| Orthologous DEG ratec (TO/TD) (%) | 76 | 82 | 62 | 75 |
| Diverged orthologue rated (TO − FCO)/TO (%) | 32 | 23 | 49 | 40 |
‘Shared’ DEGs between Δfvfac1 and Δfgfac1, and between Δfvcpk1 and Δfgcpk1.
True diverged DEGs shown in Venn diagrams of Fig. 4C.
Orthologous DEG rate is calculated as the frequency of DEGs that are total orthologues (TO) amongst all DEGs (total DEGs, TD).
Diverged orthologue rate is calculated as the frequency of diverged orthologues (FDO + CDO) amongst total orthologues (TO).
The diverged cAMP–PKA pathway controls species‐specific biological processes
Using the identified Fg‐ or Fv‐specific DEGs (Fig. 4C; Table 1), we reconstructed the unique components of the cAMP–PKA pathway in Fg and Fv, respectively (Fig. 5). In addition to the functional conservation described above, we found that the cAMP–PKA pathway evolved independently in Fg and Fv to control species‐specific functions, such as the production of some species‐specific secondary metabolites, as well as certain primary metabolites used as precursors for these species‐specific secondary metabolites.
Figure 5.
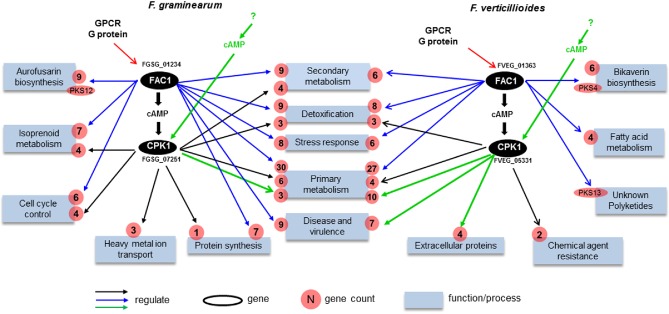
Proposed diverged cAMP–PKA pathways of Fusarium graminearum and F. verticillioides. Blue boxes represent significantly enriched biological processes regulated by the cAMP–PKA pathway. The number of differentially expressed genes (DEGs) involved in different biological processes is indicated in red circles. Blue arrows denote the FAC1‐unique subpathway. Black arrows denote the FAC1–CPK1 subpathway. Green arrows denote the FAC1‐independent activation of CPK1. GPCR, G protein‐coupled receptor.
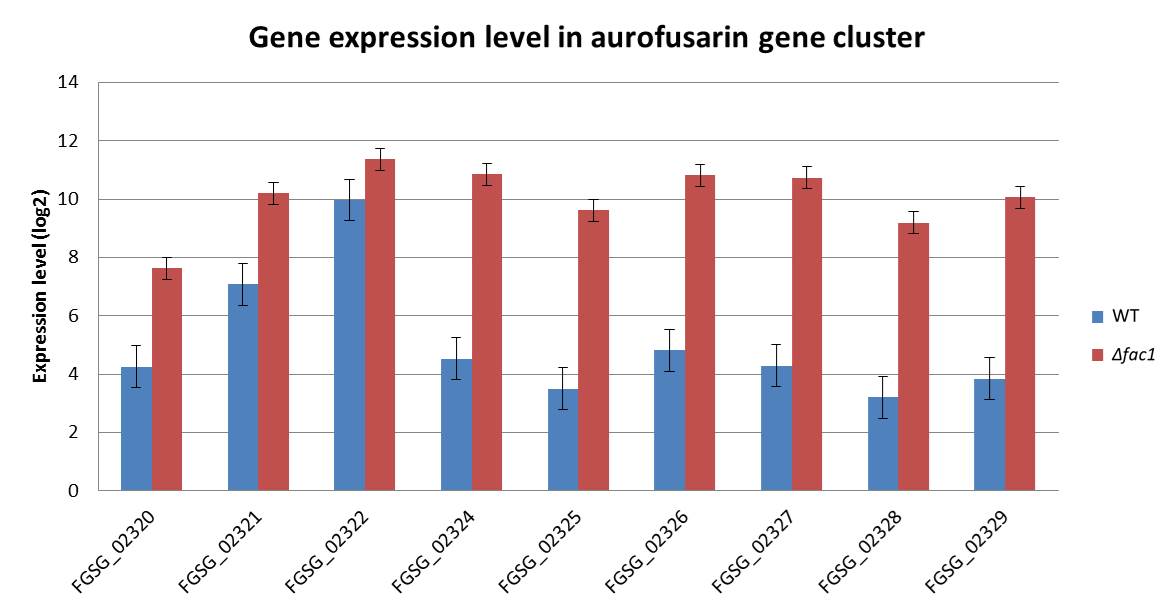
In Fg, the genes responsible for the biosynthesis of aurofusarin, an Fg‐specific secondary metabolite, were upregulated in Δfac1 (Fig. S4, see Supporting Information). In addition, the Fg‐specific component of the cAMP–PKA pathway regulated a primary metabolic pathway that produces isoprenoids used as precursors for the biosynthesis of the Fg‐specific mycotoxin DON (Scott et al., 2004). The isoprenoid metabolic pathway is also regulated by Tri6, a TF that is part of the trichothecene biosynthesis gene cluster (TRI cluster) and regulates DON production (Seong et al., 2009). Indeed, the expression of several TRI genes, including Tri4, Tri5, Tri8 and Tri9, was down‐regulated (Table S2) in Δfac1 and ΔcpkA mutants, under a less stringent cut‐off (P < 0.1). We anticipate that a more significant fold change of the TRI genes may be observed under DON‐inducible conditions. Indeed, DON production was decreased in both Δfac1 and ΔcpkA mutants (Hu et al., 2014), further confirming our prediction.
In Fv, genes responsible for the biosynthesis of bikaverin, a polyketide structurally similar to fatty acids (Hopwood and Sherman, 1990), were up‐regulated in the Δfac1 mutant (Table S2). Bikaverin is one of the main toxic secondary metabolites with antibiotic activity produced by Fv (Lazzaro et al., 2012). Interestingly, the Fv‐unique component of the cAMP–PKA pathway also regulated several genes involved in fatty acid metabolism (Fig. 5). However, no significant difference in expression was observed in the Δfac1 and Δcpk1 mutants for genes involved in the biosynthesis of another key toxic secondary metabolite, fumonisin, even when a less stringent cut‐off was used. The Δfac1 and Δcpk1 mutants were reported to have normal fumonisin production, but increased production of bikaverin (Choi and Xu, 2010), which suggests that fumonisin biosynthesis is independent of the cAMP–PKA pathway.
A few shared annotation terms, such as primary and secondary metabolism, detoxification, stress responses and virulence, were regulated by unique components of the cAMP–PKA pathway in Fg and Fv. Nevertheless, genes involved in these functions were mostly unique for each species and orthologues with diverged functions (Fig. 5). For example, in the ‘detoxification’ category, nine of 12 Fg genes were Fg specific, whereas all 11 Fv genes were unique to Fv. For ‘stress response’, five of six Fg genes and six of eight Fv genes were unique to each species, respectively. Similarly, nine genes in Fg and seven genes in Fv that were annotated as being related to ‘disease and virulence process’ were regulated by unique pathways in each genome. Six of these nine Fg genes were Fg specific, including an integral membrane protein similar to PTH11 (FGSG_07792), a known virulence factor reported in M. oryzae (DeZwaan et al., 1999). Six of the seven Fv genes were unique to Fv, including two pectin lyase genes (FVEG_13545 and 13546), a gene encoding pisatin demethylase (FVEG_12563) and a cell surface ferroxidase gene (FVEG_01690). We also observed the regulatory divergence of orthologous genes. For instance, FvPKS13 (FVEG_10535), which is potentially involved in an unknown polyketide synthesis process, was down‐regulated in the Fv Δfac1 mutant. As a functionally diverged orthologue, the expression of its Fg orthologue (FGSG_03340) was not affected in the Δfac1 and Δcpk1 mutants.
In conclusion, both the genomics and transcriptomics data presented here illustrate that unique components of the cAMP–PKA pathway may have evolved in these two species to regulate virulence and mycotoxin biosynthesis. Functional divergence of orthologues and the acquisition of novel genes occurred during the evolution of cAMP–PKA pathways in both species.
Discussion
The cAMP–PKA signal transduction pathway is a ubiquitous cascade that triggers diverse cellular functions in response to environmental cues. Our study confirmed that this signalling pathway performs conserved functions, as described in many fungal species (D'Souza and Heitman, 2001). However, we also identified DEGs that differed between Fg and Fv for both the Δfac1 and Δcpk1 mutants (Fig. S5). As this pathway interconnects fungal responses with environmental signals, certain portions of the pathway are expected to change when the fungus adapts to different ecological niches. Our study showed that functional divergence of this signalling pathway has directly contributed to Fg and Fv niche adaptation through both the recruitment of novel genes and the establishment of functional divergence of some orthologous genes.
Using comparative genomics and transcriptomics techniques, we investigated the functional conservation and divergence of the cAMP–PKA pathway in Fg and Fv, two evolutionarily related and phytopathogenic ascomycete fungi. Unexpectedly, the number of DEGs identified in this study differed significantly between Fg and Fv. As all hybridizations using Fv RNA samples, including three genetic backgrounds (wild‐type, Δfac1, Δcpk1), published in this work and two unpublished results (Guo et al., unpublished; nitrogen starvation, carbon starvation), resulted in fewer DEGs, we expect that the difference in the number of DEGs identified for Fg and Fv is a result of potential artefacts associated with the microarray experiments. To reduce potential noise inherent in the microarray data, we used the ROEL method to identify strong correlations at the expression level for all of the orthologues.
With this correction, we identified both conserved and species‐specific components of the cAMP–PKA pathway. In both species, genes involved in metabolism, energy and protein binding were down‐regulated in Δfac1, and those responsible for cell defence, the stress response and cell transporters were up‐regulated, suggesting that the biological functions controlled by this essential signalling pathway are largely conserved. Using the shared orthologous DEGs, we reconstructed a conserved cAMP–PKA pathway that controls vegetative growth and the stress response (Fig. 3). This reconstructed pathway is in total agreement with phenotypic studies of the Δfac1 and Δcpk1 mutants, which show reduced hyphal growth and conidiation and increased tolerance to environmental stress (Choi and Xu, 2010; Hu et al., 2014).
The production of species‐specific secondary metabolites governs directly fungal interactions with the host. Interestingly, the species‐specific components of the cAMP–PKA pathway in Fg or Fv reflect such species‐specific adaptation. Not only were these components linked to the production of secondary metabolites and self‐protection against toxicity caused by these toxic secondary metabolites in each species, but they were also linked to the production of their precursors. For instance, the major secondary metabolite associated with Fg during plant infection is DON (McCormick et al., 2010). In addition, the Fg species‐specific cAMP–PKA pathway regulated isoprenoid metabolism (Fig. 5), which synthesizes terpene precursors. However, Fv primarily produces polyketide secondary metabolites, such as bikaverin. Correspondingly, genes linked to the biosynthesis of polyketides and peroxisomal proteins involved in the fatty acid oxidation underlying polyketide synthesis (Maggio‐Hall et al., 2005) were differentially regulated in Fv (Fig. 5). This study further confirmed the importance of species‐specific secondary metabolites in these two mycotoxin producers and suggested that the central cAMP–PKA signalling pathway plays important roles in establishing mycotoxins in these species.
Our study also revealed the power of the functional comparative approach, which not only corrected some system errors inherent in microarray data, but also dissected a complicated signal transduction pathway. Dynamic fungal gene regulatory networks remain to be identified, and the methods developed here have the potential to unveil other signalling pathways in these networks.
Experimental Procedures
Fungal strains and medium
The wild‐type strain PH‐1 and the Δfac1 and Δcpk1 mutants (Hu et al., 2014) of Fg were routinely incubated on complete medium (CM), as described previously (Wang et al., 2011). Conidia were harvested from 5‐day‐old carboxymethyl cellulose (CMC) cultures (Li et al., 2011). For Fv, the wild‐type strain 7600 and the Δfac1 and Δcpk1 mutants (Choi and Xu, 2010) were maintained on CM plates. All the Fg and Fv strains used in this study were preserved in 15% glycerol at −80 °C.
DNA and RNA extraction
For microarray analysis, RNA was isolated from vegetative hyphae harvested from 36‐h‐old yeast extract–peptone–dextrose (YEPD) cultures with the Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the instructions provided by the manufacturer. Genomic DNA was prepared using the cetyltrimethylammonium bromide (CTAB) method (Brandfass and Karlovsky, 2008).
Chip design, hybridization and quality control
The fungal multigenome ExonChip was generated by Affymetrix and covers up to 120 053 genes (Table S1, see Supporting Information) from nine sequenced phytopathogenic fungal genomes, including seven ascomycete fungi (i.e. F. graminearium, Fo, Fv, F. solani, Verticillium dahliae, M. oryzae and Pyrenophora tritici repentis) and two basidiomycete fungi (i.e. U. maydis and Puccinia graminis). The quality of the fungal ExonChip used in this study was tested by hybridizing the chip with genomic DNA of the nine on‐chip species grown on complete medium. Because all the probes were selected from exons of each gene, a strong hybridization signal should be observed when the probes are hybridized to the genomic DNA. As a result, each fungal DNA hybridization using probes based on native genes yielded high expression values, whereas those based on other species had low values (Fig. S1, see Supporting Information), indicating the reliability of the chip for each species. A few genes that did not yield sufficient numbers of probes in the exon regions (Table S1) failed to produce strong hybridization signals when tested with genomic DNA, and were therefore excluded from the expression analysis. Similarly, expression data per hybridization in the RNA hybridization experiments were checked for quality. Biological replicates of each Fusarium species in the wild‐type, Δfac1 and Δcpk1 genetic backgrounds were used to create scatter plots (Figs S2 and S3, see Supporting Information). The expression values of all replicates are highly correlated, indicating that the hybridization signals are reliable and consistent within biological replicates.
Microarray data analysis
Microarray data (.CEL files) of Fg and Fv wild‐type, Δfac1 and Δcpk1 strains were normalized based on the robust multi‐array average (RMA) algorithm using the affy package from Bioconductor (http://www.bioconductor.org) in R (http://www.r‐project.org/). The R package makecdfenv from Bioconductor was also used to create a probe set environment using an Affymetrix chip description file (CDF). Differential gene expression in Δfac1 and Δcpk1 was identified using the limma package from Bioconductor, with an FDR cut‐off of 0.05. Pearson correlation analysis showed that global gene expression levels for three biological replicates were highly correlated within strains (Figs S2 and S3), indicating the high quality of microarray data. DEGs were identified using the Bioconductor limma package by comparing Δfac1 and Δcpk1 gene expression with wild‐type gene expression. Complete lists of DEGs and fold changes are presented in Tables S2 and S3. Functional enrichment analysis of DEGs was performed in FunCat of the Fg genomics database (FGDB) at the Munich Information Center for Protein Sequences (MIPS) (http://mips.helmholtz‐muenchen.de/proj/funcatDB). Heatmaps of gene expression were created in TIGR Multi‐Experiment Viewer (MEV) version 4.8.1 (http://www.tm4.org/). Origin Pro 9.1 (OriginLab, Northampton, Massachusetts, USA) was used to create scatter plots, column charts and histograms. Gene–phenotype interaction networks (Fig. 1A) were created in Gephi 0.8.2 (Bastian et al., 2009).
Reconstruction of the Fg cAMP–PKA pathway
The proposed cAMP–PKA pathway was built for Fg based on PK and TF genes that were differentially expressed in Δfac1 and Δcpk1. Down‐regulated genes are positively regulated and up‐regulated genes are negatively regulated by FAC1 or CPK1. Genes upstream of FAC1 are orthologous to genes involved in known cAMP–PKA pathways in S. cerevisiae. Three subpathways, i.e. the FAC1–CPK1, FAC1‐unique and CPK1‐unique subpathways, were predicted based on genes that were differentially regulated in both Δfac1 and Δcpk1, in Δfac1 alone or in Δcpk1 alone, respectively. Phenotypic annotation of PKs and TFs was performed using the existing phenome database of Fg TFs (Son et al., 2011) and kinases (Wang et al., 2011), their hypothetical function based on the Fg annotation database (FGDB, MIPS) and their homology with putative orthologues in S. cerevisiae.
Orthologue gene expression analysis
Based on orthologue information and gene expression data for each species, a gene expression matrix for Fg and Fv orthologues was created using in‐house Python scripts. Orthologue expression levels were plotted for the two species in all conditions and the Pearson correlation coefficient was calculated in Origin Pro. For each condition, the ROEL was calculated by dividing the Fv orthologue expression level (mean of three replicates) with that of Fg, and visualized using MEV 4.8.1.
Processing data obtained from PLEXdb
Additional Fg microarray data were downloaded from the Plant Expression Database (PLEXdb) (Dash et al., 2012). The original RMA normalized data file was obtained from PLEXdb. For the first generation chip data (FG1‐FG21), all gene IDs were converted to the FG3 gene ID using a mapping file (ftp://ftpmips.gsf.de/FGDB/v32/FGDB_v32_ProbeSetMapping.tab). Fold changes for all downloaded data were calculated following Student's t‐test on different biological conditions against reference conditions: FG5_H1–H5, FG7_H1–H3, FG14_H5–H8, FG16_H1–H3, FG18_H1–H3 and FG19_H1–H3. Fold changes of all genes were clustered using a hierarchical clustering algorithm and visualized in heatmaps created using MEV 4.8.1.
Data access
All microarray data generated in this study using an Affymetrix multigenome ExonChip were deposited in the Filamentous Fungal Gene Expression database (experiment ID #241 and #242) (http://bioinfo.townsend.yale.edu) (Zhang and Townsend, 2010).
Supporting information
Fig. S1 Quality control of the fungal multigenome ExonChip demonstrated in heatmaps of each fungal genomics DNA hybridization with the ExonChip. Genes are represented in columns and fungal samples in rows (from top to bottom: Puccinia graminis, Magnaporthe oryzae, three Fusarium oxysporum strains Fol 4287, MN25 and Fo47, F. graminearum, F. solani, F. verticillioides andUstilago maydis). Each heatmap shows the signals of all the genes of one species (names below the map) hybridized with all samples. Colour scale: green to red indicates expression values from low to high.
{kind=link}
Fig. S2 Scatter plot matrix of gene expression levels in all hybridization samples of Fusarium graminearum used in this study. Entire gene expression values for all three replicates of all genetic backgrounds, i.e. wild‐type (WT) (FgCM), Δfac1 (FgFAC1) and Δcpk1 (FgCPK1), were plotted in a scatter plot matrix.
{kind=link}
Fig. S3 Scatter plot matrix of gene expression levels in all hybridization samples of Fusarium verticillioides used in this study, including the wild‐type (CM), Δfac1 (mac1) and Δcpk1 (cpka). Entire gene expression values for all three replicates of all genetic backgrounds, i.e. WT (Fv_CM), Δfac1 (Fv_mac1) and Δcpk1 (Fv_cpka), were plotted in a scatter plot matrix.
{kind=link}
Fig. S4 The aurofusarin biosynthesis gene cluster is up‐regulated in the Fusarium graminearum Δfac1 (mac1) mutant compared with the wild‐type (WT).
{kind=link}
Fig. S5 Comparison of differentially expressed genes (DEGs) in Fusarium graminearum (Fg) and F. verticillioides (Fv) Δfac1 and Δcpk1 mutants. The Venn diagram shows the number of DEGs in Fg and Fv Δfac1 mutants and the number of shared DEGs. Green and red represent down‐regulated and up‐regulated genes, respectively. The heat map displays the fold changes (log2‐transformed) of the homologous DEGs compared with the wild‐type (wt) alongside their gene IDs and annotations.
Table S1 Array description: the array includes nine plant‐pathogenic fungal genomes and tiling probes for the whole genomes of Fusarium graminearum and F. oxysporum and partial genome of F. verticillioides.
Table S2 Summary of differentially expressed genes (DEGs) in Fusarium graminearum Δfac1 and Δcpk1 mutants.
Table S3 Summary of differentially expressed genes (DEGs) in Fusarium verticillioides Δfac1 and Δcpk1 mutants.
Table S4 List of shared and distinct differentially expressed genes (DEGs) of Fusarium graminearum and F. verticillioides.
Table S5 Gene expression levels and ratio of expression levels (ROELs) of orthologous genes in Fusarium graminearum and F. verticillioides.
Acknowledgements
This project was supported by the National Research Initiative Competitive Grants Program Grant no. 2008‐35604‐18800 and MASR‐2009‐04374 from the USDA National Institute of Food and Agriculture. LG and L‐JM are also supported by a seed grant from Massachusetts Green High Performance Computing Center (MGHPCC) and the National Research Initiative Hatch Grants Program Grant no. MAS00441. The authors would like to thank Dr Xiaoying Zhou for providing RNA samples for the microarray experiment. The authors would also like to thank the Broad expression platform for conducting hybridization of both DNA and RNA samples to the Affymetrix arrays and processing the microarray results.
References
- Bastian, M. , Heymann, S. and Jacomy, M. (2009) Gephi: an open source software for exploring and manipulating networks In: ICWSM (Adar E., Hurst M., Finin T., Glance N.S., Nicolov N. and Tseng B.L., eds), The AAAI Press. [Google Scholar]
- Bollen, M. , Keppens, S. and Stalmans, W. (1998) Specific features of glycogen metabolism in the liver. Biochem. J. 336, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandfass, C. and Karlovsky, P. (2008) Upscaled CTAB‐based DNA extraction and real‐time PCR assays for Fusarium culmorum and F. graminearum DNA in plant material with reduced sampling error. Int. J. Mol. Sci. 9, 2306–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broach, J.R. (1991) RAS genes in Saccharomyces cerevisiae: signal transduction in search of a pathway. Trends Genet. 7, 28–33. [DOI] [PubMed] [Google Scholar]
- Casperson, G.F. , Walker, N. and Bourne, H.R. (1985) Isolation of the gene encoding adenylate cyclase in Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. 82, 5060–5063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, E.C. and Abayasekara, D.R.E. (2004) Progesterone secretion by luteinizing human granulosa cells: a possible cAMP‐dependent but PKA‐independent mechanism involved in its regulation. J. Endocrinol. 183, 51–60. [DOI] [PubMed] [Google Scholar]
- Choi, W. and Dean, R.A. (1997) The adenylate cyclase gene MAC1 of Magnaporthe grisea controls appressorium formation and other aspects of growth and development. Plant Cell, 9, 1973–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y.E. and Xu, J.R. (2010) The cAMP signaling pathway in Fusarium verticillioides is important for conidiation, plant infection, and stress responses but not fumonisin production. Mol. Plant–Microbe Interact. 23, 522–533. [DOI] [PubMed] [Google Scholar]
- D'Souza, C.A. and Heitman, J. (2001) Conserved cAMP signaling cascades regulate fungal development and virulence. FEMS Microbiol. Rev. 25, 349–364. [DOI] [PubMed] [Google Scholar]
- D'Souza, C.A. , Alspaugh, J.A. , Yue, C. , Harashima, T. , Cox, G.M. , Perfect, J.R. and Heitman, J. (2001) Cyclic AMP‐dependent protein kinase controls virulence of the fungal pathogen Cryptococcus neoformans . Mol. Cell. Biol. 21, 3179–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel, P.B. , Walker, W.H. and Habener, J.F. (1998) Cyclic AMP signaling and gene regulation. Annu. Rev. Nutr. 18, 353–383. [DOI] [PubMed] [Google Scholar]
- Dash, S. , Van Hemert, J. , Hong, L. , Wise, R.P. and Dickerson, J.A. (2012) PLEXdb: gene expression resources for plants and plant pathogens. Nucleic Acids Res. 40, D1194–D1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins, A.E. , Hohn, T.M. and McCormick, S.P. (1993) Trichothecene biosynthesis in Fusarium species: chemistry, genetics, and significance. Microbiol. Rev. 57, 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeZwaan, T.M. , Carroll, A.M. , Valent, B. and Sweigard, J.A. (1999) Magnaporthe grisea PTH11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. Plant Cell, 11, 2013–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dürrenberger, F. , Wong, K. and Kronstad, J.W. (1998) Identification of a cAMP‐dependent protein kinase catalytic subunit required for virulence and morphogenesis in Ustilago maydis . Proc. Natl. Acad. Sci. 95, 5684–5689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner, D.M. , Kazan, K. and Manners, J.M. (2009) Novel genes of Fusarium graminearum that negatively regulate deoxynivalenol production and virulence. Mol. Plant–Microbe Interact. 22, 1588–1600. [DOI] [PubMed] [Google Scholar]
- Gasch, A. (2003) The environmental stress response: a common yeast response to diverse environmental stresses In: Yeast Stress Responses (Hohmann S. and Mager W., eds), pp. 11–70. Berlin, Heidelberg: Springer. [Google Scholar]
- Goswami, R.S. and Kistler, H.C. (2004) Heading for disaster: Fusarium graminearum on cereal crops. Mol. Plant Pathol. 5, 515–525. [DOI] [PubMed] [Google Scholar]
- Guenther, J.C. , Hallen‐Adams, H.E. , Bucking, H. , Shachar‐Hill, Y. and Trail, F. (2009) Triacylglyceride metabolism by Fusarium graminearum during colonization and sexual development on wheat. Mol. Plant–Microbe Interact. 22, 1492–1503. [DOI] [PubMed] [Google Scholar]
- Hopwood, D.A. and Sherman, D.H. (1990) Molecular genetics of polyketides and its comparison to fatty acid biosynthesis. Annu. Rev. Genet. 24, 37–62. [DOI] [PubMed] [Google Scholar]
- Hu, S. , Zhou, X. , Gu, X. , Cao, S. , Wang, C. and Xu, J.‐R. (2014) The cAMP–PKA pathway regulates growth, sexual and asexual differentiation, and pathogenesis in Fusarium graminearum . Mol. Plant–Microbe Interact. 27, 557–566. [DOI] [PubMed] [Google Scholar]
- Jones, D.L. , Petty, J. , Hoyle, D.C. , Hayes, A. , Ragni, E. , Popolo, L. , Oliver, S.G. and Stateva, L.I. (2003) Transcriptome profiling of a Saccharomyces cerevisiae mutant with a constitutively activated Ras/cAMP pathway. Physiol. Genomics, 16, 107–118. [DOI] [PubMed] [Google Scholar]
- Jonkers, W. , Dong, Y. , Broz, K. and Kistler, H.C. (2012) The Wor1‐like protein Fgp1 regulates pathogenicity, toxin synthesis and reproduction in the phytopathogenic fungus Fusarium graminearum . Plos Pathog. 8, e1002724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedera, C.J. , Plattner, R.D. and Desjardins, A.E. (1999) Incidence of Fusarium spp. and levels of fumonisin B1 in maize in western Kenya. Appl. Environ. Microbiol. 65, 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H.‐S. , Park, S.‐Y. , Lee, S. , Adams, E.L. , Czymmek, K. and Kang, S. (2011) Loss of cAMP‐dependent protein kinase A affects multiple traits important for root pathogenesis by Fusarium oxysporum . Mol. Plant–Microbe Interact. 24, 719–732. [DOI] [PubMed] [Google Scholar]
- Kraakman, L. , Lemaire, K. , Ma, P. , Teunissen, A.W.R.H. , Donaton, M.C.V. , Van Dijck, P. , Winderickx, J. , de Winde, J.H. and Thevelein, J.M. (1999) A Saccharomyces cerevisiae G‐protein coupled receptor, Gpr1, is specifically required for glucose activation of the cAMP pathway during the transition to growth on glucose. Mol. Microbiol. 32, 1002–1012. [DOI] [PubMed] [Google Scholar]
- Lazzaro, I. , Busman, M. , Battilani, P. and Butchko, R.A.E. (2012) FUM and BIK gene expression contribute to describe fumonisin and bikaverin synthesis in Fusarium verticillioides . Int. J. Food Microbiol. 160, 94–98. [DOI] [PubMed] [Google Scholar]
- Leslie, J.F. and Summerell, B.A. (2006) Fusarium Laboratory Manual. Hoboken, New Jersey, USA: Blackwell Publishing. [Google Scholar]
- Li, Y.M. , Wang, C.F. , Liu, W.D. , Wang, G.H. , Kang, Z.S. , Kistler, H.C. and Xu, J.R. (2011) The HDF1 histone deacetylase gene is important for conidiation, sexual reproduction, and pathogenesis in Fusarium graminearum . Mol. Plant–Microbe Interact. 24, 487–496. [DOI] [PubMed] [Google Scholar]
- Ma, L.J. , van der Does, H.C. , Borkovich, K.A. , Coleman, J.J. , Daboussi, M.J. , Di Pietro, A. , Dufresne, M. , Freitag, M. , Grabherr, M. , Henrissat, B. , Houterman, P.M. , Kang, S. , Shim, W.B. , Woloshuk, C. , Xie, X. , Xu, J.R. , Antoniw, J. , Baker, S.E. , Bluhm, B.H. , Breakspear, A. , Brown, D.W. , Butchko, R.A. , Chapman, S. , Coulson, R. , Coutinho, P.M. , Danchin, E.G. , Diener, A. , Gale, L.R. , Gardiner, D.M. , Goff, S. , Hammond‐Kosack, K.E. , Hilburn, K. , Hua‐Van, A. , Jonkers, W. , Kazan, K. , Kodira, C.D. , Koehrsen, M. , Kumar, L. , Lee, Y.H. , Li, L. , Manners, J.M. , Miranda‐Saavedra, D. , Mukherjee, M. , Park, G. , Park, J. , Park, S.Y. , Proctor, R.H. , Regev, A. , Ruiz‐Roldan, M.C. , Sain, D. , Sakthikumar, S. , Sykes, S. , Schwartz, D.C. , Turgeon, B.G. , Wapinski, I. , Yoder, O. , Young, S. , Zeng, Q. , Zhou, S. , Galagan, J. , Cuomo, C.A. , Kistler, H.C. and Rep, M. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature, 464, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, L.J. , Geiser, D.M. , Proctor, R.H. , Rooney, A.P. , O'Donnell, K. , Trail, F. , Gardiner, D.M. , Manners, J.M. and Kazan, K. (2013) Fusarium pathogenomics. Annu. Rev. Microbiol. 67, 399–416. [DOI] [PubMed] [Google Scholar]
- Ma, P. , Wera, S. , Van Dijck, P. and Thevelein, J.M. (1999) The PDE1‐encoded low‐affinity phosphodiesterase in the yeast Saccharomyces cerevisiae has a specific function in controlling agonist‐induced cAMP signaling. Mol. Biol. Cell, 10, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda, T. , Mochizuki, N. and Yamamoto, M. (1990) Adenylyl cyclase is dispensable for vegetative cell growth in the fission yeast Schizosaccharomyces pombe . Proc. Natl. Acad. Sci. 87, 7814–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggio‐Hall, L.A. , Wilson, R.A. and Keller, N.P. (2005) Fundamental contribution of β‐oxidation to polyketide mycotoxin production in planta . Mol. Plant–Microbe Interact. 18, 783–793. [DOI] [PubMed] [Google Scholar]
- Matsumoto, K. , Uno, I. , Oshima, Y. and Ishikawa, T. (1982) Isolation and characterization of yeast mutants deficient in adenylate cyclase and cAMP‐dependent protein kinase. Proc. Natl. Acad. Sci. 79, 2355–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick, S.P. , Alexander, N.J. and Harris, L.J. (2010) CLM1 of Fusarium graminearum encodes a longiborneol synthase required for culmorin production. Appl. Environ. Microbiol. 76, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oide, S. , Moeder, W. , Krasnoff, S. , Gibson, D. , Haas, H. , Yoshioka, K. and Turgeon, B.G. (2006) NPS6, encoding a nonribosomal peptide synthetase involved in siderophore‐mediated iron metabolism, is a conserved virulence determinant of plant pathogenic Ascomycetes. Plant Cell, 18, 2836–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen, H.E. , Huebner, M. , Shiu, S.‐H. , Güldener, U. and Trail, F. (2007) Gene expression shifts during perithecium development in Gibberella zeae (anamorph Fusarium graminearum), with particular emphasis on ion transport proteins. Fungal Genet. Biol., 44, 1146–1156. [DOI] [PubMed] [Google Scholar]
- Robertson, L.S. , Causton, H.C. , Young, R.A. and Fink, G.R. (2000) The yeast A kinases differentially regulate iron uptake and respiratory function. Proc. Natl. Acad. Sci. 97, 5984–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, D.B. , Jameson, G. and Parker, E. (2004) Isoprenoids: gene clusters and chemical puzzles In: Advances in Fungal Biotechnology for Industry, Agriculture, and Medicine (Tkacz J. and Lange L., eds), pp. 163–198. New York City: Springer US. [Google Scholar]
- Seino, S. and Shibasaki, T. (2005) PKA‐dependent and PKA‐independent pathways for cAMP‐regulated exocytosis. Physiological Reviews, 85, 1303–1342. [DOI] [PubMed] [Google Scholar]
- Seong, K.‐Y. , Zhao, X. , Xu, J.‐R. , Güldener, U. and Kistler, H.C. (2008) Conidial germination in the filamentous fungus Fusarium graminearum . Fungal Genet. Biol. 45, 389–399. [DOI] [PubMed] [Google Scholar]
- Seong, K.Y. , Pasquali, M. , Zhou, X. , Song, J. , Hilburn, K. , McCormick, S. , Dong, Y , Xu, J.R. and Kistler, H.C. (2009) Global gene regulation by Fusarium transcription factors Tri6 and Tri10 reveals adaptations for toxin biosynthesis. Mol. Microbiol. 72, 354–367. [DOI] [PubMed] [Google Scholar]
- Son, H. , Seo, Y.S. , Min, K. , Park, A.R. , Lee, J. , Jin, J.M. , Lin, Y. , Cao, P. , Hong, S.Y. , Kim, E.K. , Lee, S.H. , Cho, A. , Lee, S. , Kim, M.G. , Kim, Y. , Kim, J.E. , Kim, J.C. , Choi, G.J. , Yun, S.H. , Lim, J.Y. , Kim, M. , Lee, Y.H. , Choi, Y.D. and Lee, Y.W. (2011) A phenome‐based functional analysis of transcription factors in the cereal head blight fungus, Fusarium graminearum . Plos Pathog. 7, e1002310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton, J.C. (1982) Epidemiology of wheat head blight and maize ear rot caused by Fusarium graminearum . Can. J. Plant Pathol. 4, 195–209. [Google Scholar]
- Sydenham, E.W. , Gelderblom, W.C.A. , Thiel, P.G. and Marasas, W.F.O. (1990) Evidence for the natural occurrence of fumonisin B1, a mycotoxin produced by Fusarium moniliforme, in corn. J. Agric. Food Chem. 38, 285–290. [Google Scholar]
- Wang, C. , Zhang, S. , Hou, R. , Zhao, Z. , Zheng, Q. , Xu, Q. , Zheng, D. , Wang, G. , Liu, H. , Gao, X. , Ma, J.W. , Kistler, H.C. , Kang, Z. and Xu, J.R. (2011) Functional analysis of the kinome of the wheat scab fungus Fusarium graminearum . Plos Pathog. 7, e1002460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winnenburg, R. , Urban, M. , Beacham, A. , Baldwin, T.K. , Holland, S. , Lindeberg, M. , Hansen, H. , Rawlings C., Hammond‐Kosack, K.E. and Köhler, J. (2008) PHI‐base update: additions to the pathogen host interaction database. Nucleic Acids Res. 36, D572–D576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, J.‐R. , Urban, M. , Sweigard, J.A. and Hamer, J.E. (1997) The CPKA gene of Magnaporthe grisea is essential for appressorial penetration. Mol. Plant–Microbe Interact. 10, 187–194. [Google Scholar]
- Zaccolo, M. (2009) cAMP signal transduction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 158, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.W. , Jia, L.J. , Zhang, Y. , Jiang, G. , Li, X. , Zhang, D. and Tang, W.H. (2012) In planta stage‐specific fungal gene profiling elucidates the molecular strategies of Fusarium graminearum growing inside wheat coleoptiles. Plant Cell, 24, 5159–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. and Townsend, J.P. (2010) The filamentous fungal gene expression database (FFGED). Fungal Genet. Biol. 47, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Quality control of the fungal multigenome ExonChip demonstrated in heatmaps of each fungal genomics DNA hybridization with the ExonChip. Genes are represented in columns and fungal samples in rows (from top to bottom: Puccinia graminis, Magnaporthe oryzae, three Fusarium oxysporum strains Fol 4287, MN25 and Fo47, F. graminearum, F. solani, F. verticillioides andUstilago maydis). Each heatmap shows the signals of all the genes of one species (names below the map) hybridized with all samples. Colour scale: green to red indicates expression values from low to high.
Fig. S2 Scatter plot matrix of gene expression levels in all hybridization samples of Fusarium graminearum used in this study. Entire gene expression values for all three replicates of all genetic backgrounds, i.e. wild‐type (WT) (FgCM), Δfac1 (FgFAC1) and Δcpk1 (FgCPK1), were plotted in a scatter plot matrix.
Fig. S3 Scatter plot matrix of gene expression levels in all hybridization samples of Fusarium verticillioides used in this study, including the wild‐type (CM), Δfac1 (mac1) and Δcpk1 (cpka). Entire gene expression values for all three replicates of all genetic backgrounds, i.e. WT (Fv_CM), Δfac1 (Fv_mac1) and Δcpk1 (Fv_cpka), were plotted in a scatter plot matrix.
Fig. S4 The aurofusarin biosynthesis gene cluster is up‐regulated in the Fusarium graminearum Δfac1 (mac1) mutant compared with the wild‐type (WT).
Fig. S5 Comparison of differentially expressed genes (DEGs) in Fusarium graminearum (Fg) and F. verticillioides (Fv) Δfac1 and Δcpk1 mutants. The Venn diagram shows the number of DEGs in Fg and Fv Δfac1 mutants and the number of shared DEGs. Green and red represent down‐regulated and up‐regulated genes, respectively. The heat map displays the fold changes (log2‐transformed) of the homologous DEGs compared with the wild‐type (wt) alongside their gene IDs and annotations.
Table S1 Array description: the array includes nine plant‐pathogenic fungal genomes and tiling probes for the whole genomes of Fusarium graminearum and F. oxysporum and partial genome of F. verticillioides.
Table S2 Summary of differentially expressed genes (DEGs) in Fusarium graminearum Δfac1 and Δcpk1 mutants.
Table S3 Summary of differentially expressed genes (DEGs) in Fusarium verticillioides Δfac1 and Δcpk1 mutants.
Table S4 List of shared and distinct differentially expressed genes (DEGs) of Fusarium graminearum and F. verticillioides.
Table S5 Gene expression levels and ratio of expression levels (ROELs) of orthologous genes in Fusarium graminearum and F. verticillioides.