


Abstract
Transposable elements are found throughout the genomes of all organisms. Repressive marks such as DNA methylation and histone H3 lysine 9 (H3K9) methylation silence these elements and maintain genome integrity. However, how silencing mechanisms are themselves regulated to avoid the silencing of genes remains unclear. Here, an anti-silencing factor was identified using a forward genetic screen on a reporter line that harbors a LUCIFERASE (LUC) gene driven by a promoter that undergoes DNA methylation. SUVH1, a Su(var)3–9 homolog, was identified as a factor promoting the expression of the LUC gene. Treatment with a cytosine methylation inhibitor completely suppressed the LUC expression defects of suvh1, indicating that SUVH1 is dispensable for LUC expression in the absence of DNA methylation. SUVH1 also promotes the expression of several endogenous genes with promoter DNA methylation. However, the suvh1 mutation did not alter DNA methylation levels at the LUC transgene or on a genome-wide scale; thus, SUVH1 functions downstream of DNA methylation. Histone H3 lysine 4 (H3K4) trimethylation was reduced in suvh1; in contrast, H3K9 methylation levels remained unchanged. This work has uncovered a novel, anti-silencing function for a member of the Su(var)3–9 family that has previously been associated with silencing through H3K9 methylation.
INTRODUCTION
Chromatin structure, histone modifications and DNA methylation regulate gene expression and influence transposon activity. The model plant Arabidopsis has been used to uncover the molecular framework of DNA methylation, which is critical for the regulation of transposon activity and the maintenance of genome integrity. The RNA-directed DNA methylation (RdDM) pathway is responsible for establishing DNA methylation at CG, CHG and CHH contexts (H = A, C, T) and maintaining asymmetric CHH methylation (1,2). METHYLTRANSFERASE 1 (MET1), the plant homolog of mammalian DNA methyltransferase 1 (DNMT1), maintains CG methylation (3,4). The maintenance of CHG methylation requires the plant-specific methyltransferase CHROMOMETHYLASE 3 (CMT3) (3,4). DNA methylation can also be actively erased through demethylation. Four DNA glycosylases involved in DNA demethylation are known (5,6): DME, which functions primarily in the seed (7), and three DME homologs (ROS1, DML2 and DML3) with broader domains of activity in the plant (8–10).
Histone modifications also influence gene expression. Histone H3 lysine 4 trimethylation (H3K4me3) is a well-recognized active mark and is deposited by the SET domain proteins ATX1, ATX2, ATXR3, and ATXR7 in Arabidopsis (11–15). Histone H3 lysine 9 dimethylation (H3K9me2) is a repressive mark deposited by homologs of Drosophila Su(var)3–9 in various eukaryotes (16). In Arabidopsis, there are ten Su(var)3–9 homologs, which can be divided into four subgroups: SUVH1, SUVH2, SUVH4 and SUVH5 (17). SUVH4, SUVH5 and SUVH6 belonging to the SUVH4 and SUVH5 subgroups are active H3K9me2 methyltransferases (18,19). SUVH2 and SUVH9 in the SUVH2 subgroup are players in RdDM; they are required for the occupancy of NRPE1, the largest subunit of RNA Polymerase V (Pol V) and a major player in RdDM, at regions with DNA methylation (20,21). No functions have been reported for any of the SUVH1 subgroup proteins, which include SUVH1, SUVH3, SUVH7, SUVH8 and SUVH10 (17).
Although DNA methylation and H3K9me2 largely occur in heterochromatic regions, they are also found in euchromatic regions where genes are located. In fact, when such epigenetic modifications are close to genes, the expression of the nearby genes could be repressed (22–24). This raises the question of how genes with nearby transposable elements can overcome the effects of epigenetic silencing to be expressed. With the goal of identifying negative regulators of gene silencing, a forward genetic screen was performed using a reporter line named YJ11–3F (hereafter referred to as YJ), which harbors a luciferase gene (LUC) driven by a double 35S promoter (d35S), which harbors DNA methylation. A mutation causing decreased luciferase activity was mapped to the SUVH1 locus. Treatment of the YJ and YJ suvh1–1 lines with the cytosine methylation inhibitor 5-Aza-2′-deoxycytidine suppressed the effect of the suvh1–1 mutation, indicating that SUVH1 functions in the DNA methylation pathway. In fact, SUVH1 function was found to be dispensable in the nrpe1 mutant background that is defective in CHH methylation. However, analysis of DNA methylation at the LUC locus as well as at the genome-wide level did not reveal any changes in DNA methylation in suvh1–1; thus, SUVH1 likely functions downstream of DNA methylation. The suvh1–1 mutation led to decreased H3K4me3 levels at SUVH1-targeted loci, but did not affect H3K9me2 levels. The present findings suggest that SUVH1 counteracts the repressive effects of CHH DNA methylation to promote gene expression and reveal an unexpected function of a Su(var)3–9 family member, a function that is opposite to the well-known repressive roles of this family in gene expression.
MATERIALS AND METHODS
Plant materials and crosses
All tissues used in the present study were from 8- to 10-day-old seedlings, and all Arabidopsis strains were in the Columbia ecotype. The reporter lines LUCH (25) and YJ are in the rdr6–11 mutant background (26). The transgene in LUCH was genotyped as follows. The primer pair tailcR and 9-7-2 gtF was used to amplify the genomic fragment without LUCH insertion and the primer pair tailcR and R1 was used to amplify the LUCH insertion. The YJ transgene was genotyped as follows. The primer pair YJ11–3F_For and YJ11–3F_Rev was used to amplify the genomic fragment without the YJ insertion and the primer pair YJ11–3F_Rev and R1 was used to amplify the YJ insertion.
The suvh1–1 allele was isolated in the YJ background through a genetic mutagenesis screen. This allele was genotyped by PCR amplification using the primer pair SUVH1-NlaIVF and SUVH1-NlaIVR followed by restriction digestion with NlaIV (NEB, R0126S). Only the PCR fragment amplified from wild type can be digested with this enzyme. The suvh1–2 allele (SAIL_11_D02) carrying a T-DNA insertion in the promoter of SUVH1 was ordered from ABRC. This allele was genotyped as follows. The primer pair SUVH1sailLP and SUVH1sailRP was used to amplify the genomic fragment without the T-DNA insertion and the primer pair SUVH1sailRP and Sail-LB1 was used to amplify the T-DNA insertion.
ros1–5, ago4–6 and drd1–12 were isolated in the LUCH background (25) and subsequently introduced into YJ and YJ suvh1–1 through crosses. nrpe1–1 (drd3-1) was described previously (27) and was crossed into YJ and YJ suvh1–1.
LUCH suvh1–1 was obtained through the cross of YJ suvh1–1 to LUCH. Genotyping was carried out as described above to identify plants that are homozygous for the LUCH transgene and suvh1–1 but lack the YJ transgene. YJ suvh1–2 was obtained through a cross between YJ and suvh1–2 followed by genotyping.
All genotyping primer sequences are provided in Supplemental Table S6.
RT-PCR
Total RNA was extracted from seedlings with Trizol (Invitrogen, 15596-018) then treated with DNase I (Roche, 04716728001). cDNA was synthesized using oligo-dT primers and RevertAid Reverse Transcriptase (Fermentas, EP0442). RT-qPCR was performed with three technical replicates on a Bio-Rad C1000 thermal cycler equipped with a CFX detection module using iQ™ SYBR® (Bio-Rad, 170-8880). To calculate the fold change of gene expression value, an internal control UBQ5 was used. ΔΔCt is calculated as
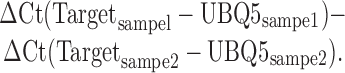 |
The fold change is equal to 2ΔΔCt. The primers used in the study are listed in Supplemental Table S6.
Luciferase live imaging and 5-Aza-2′-deoxycytidine treatment
For luciferase live imaging, 8- to 10-day-old seedlings growing on plates with half-strength Murashige and Skoog (MS) media supplemented with 0.8% agar and 1% sucrose were sprayed with 1 mM luciferin (Promega) in 0.01% Triton X-100. After 5 min incubation in the dark, the plants were placed in a Stanford Photonics Onyx Luminescence Dark Box equipped with a Roper Pixis 1024B camera controlled by the WinView32 software and then imaged with a 1 min exposure time. For 5-Aza-2′-deoxycytodine (Sigma, A3656) treatment, plants were grown in half-strength MS media with 7 μg/ml 5-Aza-2′-deoxycytodine for 2 weeks.
EMS mutagenesis of the YJ line
A 1 ml volume of seeds (∼10 000 seeds) was pre-washed with 0.1% Tween 20 for 15 min then treated with 0.2% EMS for 12 h, followed by three washes with 10 ml water for 1 h with gentle agitation. The seeds were planted in soil to obtain M1 plants, which gave M2 seeds. Mutants with reduced LUC activity, based on LUC live imaging, were isolated in the M2 generation. The mutants were backcrossed to the parental line (YJ) two times prior to further analysis.
Mapping of the suvh1–1 mutation
To identify the mutation responsible for low LUC expression, the mutant was crossed to YJ in the Ler background to generate a mapping population. In the F2 generation, 407 plants showing the low luciferase phenotype were used to narrow the interval that the mutation lies in (see Supplementary Figure S2). The mutation was first positioned close to the marker F7A7 that is located at the top arm of chromosome 5. The mutation was further placed in a 1.5 Mb region between markers F7A7 and MJJ3. A series of five more markers (T32M21, MUK11, MUG13a, MUG13b, K18I23) narrowed down the mutation to a 44 kb region encompassing 11 genes. Sequencing of At5g04940 (SUVH1) revealed a G-to-A mutation resulting in a G-to-E amino acid substitution in the SET domain. Sequences of the mapping primers are provided in Supplemental Table S6.
Plasmid construction
To generate the SUVH1p:SUVH1–3XFLAG transgene, the SUVH1 genomic region including 1.5 kb of the promoter and the coding region lacking the stop codon was amplified from YJ genomic DNA using the primer pair SUVH1smaI and SUVH1claI (Supplemental Table S6) and cloned into the pJL-Blue entry vector (28). The genomic fragment was then introduced into a binary vector containing a pEG301 (29) backbone and a C-terminal 3XFLAG tag using Gateway® LR Clonase® Enzyme mix (Invitrogen, Cat. 11791-019).
McrBC-qPCR
Genomic DNA was extracted using the CTAB method (30), and ribonuclease A (Sigma, R4875-100MG) was used to eliminate RNAs. One hundred nanogram DNA was treated with 2 units of McrBC (New England Biolabs, M0272S) at 37°C for 30 min, and a mix without McrBC was performed in parallel as the control. The mixtures were incubated at 65°C for 20 min to inactivate the McrBC. qPCR was performed using iQ™ SYBR® (Bio-Rad, 170-8880) to quantify the remaining DNA, with the ratio between the McrBC mix and the mix without McrBC as an indicator of the methylation level. The relative DNA levels were equal to the 2ΔCt and ΔCt was equal to the Ct of undigested samples minus the Ct of digested samples.
MethylC-seq library construction
To generate MethylC-seq libraries, genomic DNA was extracted using the DNeasy Plant Mini Kit (Qiagen, 69104) and quantified using a Qubit fluorometer. One microgram of genomic DNA was sonicated into fragments 100 to 300 bp in length using a Diagenode Bioruptor for four cycles with the following parameters: intensity = high, on = 30 s, off = 30 s and time = 15 min. The sonicated DNA fragments were purified using the PureLink PCR Purification Kit (Invitrogen, K3100–01). End repair was performed at room temperature for 45 min using the End-It™ DNA End-Repair Kit (Epicenter, ER0720), with the replacement of dNTP with a mixture of dATP, dGTP and dTTP. Following the incubation, the Agencourt AMPure XP-PCR Purification system (Beckman Coulter, A63881) was used for DNA purification. 3′-end adenylation was performed at 37°C for 30 min using dATP and Klenow Fragment (3′"5′ exo-) (New England Biolabs, M0212), followed by purification using the Agencourt AMPure XP-PCR Purification system. The purified DNA was ligated with methylated adapters from the TruSeq DNA Sample Preparation Kit (Illumina, FC-121-2001) at 16°C overnight using T4 DNA ligase (New England Biolabs, M0202). The ligation products were purified with AMPure XP beads twice. Less than 400 ng ligated product was used for bisulfite conversion using the MethylCode Kit (Invitrogen, MECOV-50) according to the manufacturer's guidelines, except for the addition of 12 μg carrier RNA (Qiagen, 1068337) to the conversion product before column purification. The final conversion product was amplified using Pfu Cx Turbo (Agilent, 600414) under the following PCR conditions: 2 min at 95°C; 9 cycles of 15 s at 98°C, 30 s at 60°C and 4 min at 72°C; and 10 min at 72°C. The PCR product was purified using AMPure XP beads prior to a 101-cycle sequencing run (single end) on an Illumina HiSeq 2000. The methylome data were deposited into the NCBI database under the accession number GSE64600.
Data analysis of the MethylC-seq libraries
The raw reads that passed the Illumina quality control steps were retained, and duplicated reads were removed prior to mapping. The reads were mapped to the TAIR10 genome using BS Seeker (31), and in-house R and Perl scripts were employed to convert the BS Seeker-aligned reads to every cytosine. DMRs (differentially methylated regions) were calculated according to previously described methodology (3). The Arabidopsis genome was divided into 100 bp windows, and the methylation level at each window was calculated separately. The methylation level was defined as the number of methylated cytosines sequenced divided by the total number of cytosines sequenced. To avoid skew caused by few cytosines and low coverage, only windows with at least four cytosines covered by at least four reads each were counted. Windows with an absolute methylation difference greater than 0.4 (CG), 0.2 (CHG) and 0.1 (CHH) and an adjusted P-value (FDR) < 0.01 (Fisher's exact test) were considered DMRs. Only DMRs identified from both replicates of YJ and YJ suvh1–1 were considered suvh1 DMRs.
Chromatin immunoprecipitation (ChIP)
ChIP was performed as previously described (32) using H3K4me3 (abcam, ab8580), H3K4me2 (abcam, ab7766), H3K4me1 (abcam, ab8895) and H3K9me2 (abcam, ab1220) antibodies. qPCR was performed using iQ™ SYBR® (Bio-Rad, 170-8880) to quantify the DNA. The %input was equal to the 2ΔCt times 100 and divided by the dilution of the input. ΔCt was equal to the Ct of input samples minus the Ct of samples with antibodies.
mRNA-seq library construction and data processing
Ten-day-old seedlings from YJ and YJ suvh1–1 were collected for RNA extraction using Trizol (Invitrogen, 15596–018), and the extracted RNA was treated with DNase I (Roche, 04716728001). Two micrograms of the DNase I-treated RNA and the TruSeq RNA Sample Preparation Kit v2 (Illumina, FC-122-1002) were used for library construction. The libraries were sequenced on an Illumina HiSeq 2000 and the data were deposited into the NCBI database under the accession number GSE64600.
The raw reads that passed the Illumina quality control steps were collapsed into a set of non-redundant reads. These non-redundant reads were mapped to the TAIR10 Arabidopsis genome using TopHat v2.0.4 with default settings (33). For the quantification of a given gene or window, reads whose 5′ ends were within the gene or window were counted. The fold change was calculated using the RPKM-normalized read values, and the p-value was calculated based on the Poisson distribution (34).
Phylogenetic analysis of SUVH proteins
The SUVH protein sequences from Arabidopsis thaliana were downloaded from TAIR (35). The SUVH protein sequences from Amborella trichopoda were obtained from the Amborella Genome Database (36) and the sequences from Oryza sativa, Selaginella moellendorffii, and Physcomitrella patens were obtained from the Phytozome website (37). The phylogenetic analysis of SUVH protein sequences was carried out using MEGA 6 with the alignment parameter of Muscle and the tree building parameter of Maximum Likelihood (38).
RESULTS
Two reporter lines with promoter DNA methylation
To identify new factors in DNA methylation and transcriptional gene silencing, particularly negative factors, two reporter lines with a LUC gene driven by the dual cauliflower mosaic virus 35S promoter (d35S) were employed in our lab in forward genetic screens. To avoid the posttranscriptional silencing of the transgenes, both transgenes were introduced into the rdr6–11 background (26). In one reporter line, named LUCH, a single copy of T-DNA was inserted into the 3′ UTR of AT3G07350 (25). In LUCH, high levels of DNA methylation and small RNAs are present at the d35S promoter and LUC expression is strongly de-repressed by decreased DNA methylation in RdDM mutants such as ago4, drd1, nrpe1 and drm2, and further repressed by increased DNA methylation in a ros1 mutant (25).
The other line is YJ, which has not been described before. YJ also harbors a LUC transgene driven by a d35S promoter. Genetic segregation analysis showed that the T-DNA in YJ was inserted into a single genomic locus. Through TAIL-PCR, we found that the T-DNA in YJ was inserted into the 3′ UTR of AT1G02740. To determine the copy number of the LUC transgene in YJ, qPCR was carried out to measure the relative LUC DNA levels in YJ and LUCH. As shown in Supplementary Figure S1A, the DNA levels of LUC were similar in YJ and LUCH. Based on the fact that LUCH is a single copy T-DNA insertion (25), we conclude that YJ is also likely a single copy T-DNA insertion. The DNA methylation level at the LUC transgene in YJ was analyzed using a qPCR-based assay. DNA was digested or not by the restriction enzyme McrBC that cleaves methylated DNA, and real-time PCR was performed. Less PCR amplification is expected at hypermethylated regions following McrBC treatment. This analysis showed that DNA methylation was present at the d35S promoter but not at the LUC coding region in YJ (Supplementary Figure S1B).
Despite the presence of DNA methylation at the d35S promoter in YJ, LUC expression levels were much higher in YJ than in LUCH (Supplementary Figure S1C). When YJ and LUCH were grown on media containing the cytosine methylation inhibitor 5-Aza-2′-deoxycytidine, LUC expression was increased by five- and 28-fold, respectively (Supplementary Figures S1D and S1E), suggesting that the LUC transgene in both reporter lines is under repression by DNA methylation.
Isolation of suvh1 mutants
The YJ line was treated with ethyl methanesulfonate (EMS) for a forward genetic screen that aimed to identify negative factors in DNA methylation or transcriptional gene silencing. A mutant exhibiting reduced luciferase luminescence was isolated, and RT-qPCR confirmed the reduced expression of the transgene (Figure 1A, YJ versus YJ suvh1–1). Traditional map-based cloning (Supplementary Figure S2) revealed a G-to-A mutation that caused a G-to-E substitution in the SET domain of SUVH1 (Figure 1B) and this mutation was named suvh1–1. In addition, we ordered a suvh1 mutant with a T-DNA insertion in the promoter of SUVH1 and named it suvh1–2. The expression of SUVH1 was greatly reduced in suvh1–2 as determined by RT-qPCR (Supplementary Figure S1F). The suvh1–2 allele was introduced into the YJ line through crosses to generate YJ suvh1–2. As in YJ suvh1–1, both luciferase luminescence and LUC transcript levels were reduced in YJ suvh1–2 as compared to YJ (Figure 1A). A wild-type SUVH1 genomic fragment introduced into YJ suvh1–1 completely rescued the reduced LUC expression in 19 out of 20 T1 transgenic lines (Figure 1A, data not shown). All these results confirmed that loss or reduction of function in SUVH1 led to a decrease in LUC expression in YJ. The equal expression of SUVH1 in YJ and YJ suvh1–1 (Supplementary Figure S1F) indicated that the suvh1–1 mutation affects SUVH1 function at the protein level. The introduction of the suvh1–1 mutation into LUCH also led to decreased luciferase luminescence and reduced LUC transcript levels (Figure 1C); thus, the suvh1–1 mutation decreased LUC expression in both the YJ and LUCH backgrounds. These studies show that SUVH1 is required for the expression of two transgenes. This was unexpected as three other SUVH genes, SUVH4, SUVH5, and SUVH6 belonging to another subgroup of Arabidopsis Su(var)3–9 homologs, are required for transcriptional gene silencing (18,19,39–41).
Figure 1.
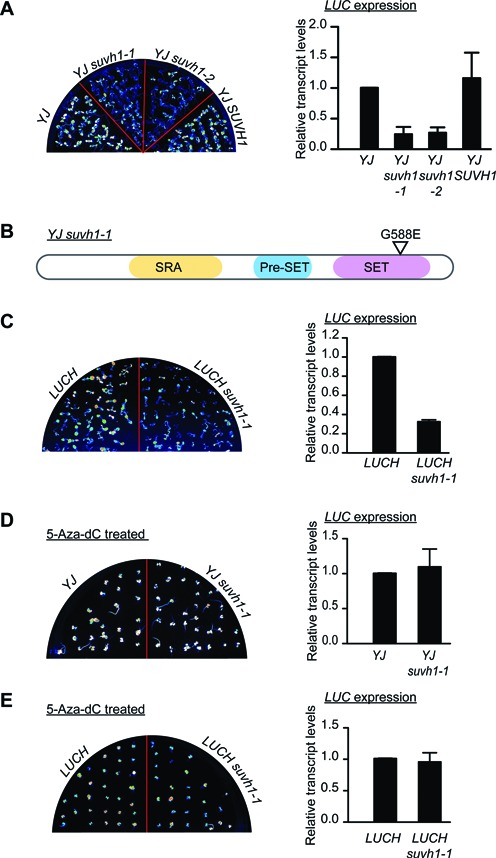
Characterization of suvh1 mutants. (A) The suvh1–1 mutation led to decreased expression of the luciferase gene (LUC) in the YJ background. (Left panel) LUC luminescence of 8-day-old YJ, YJ suvh1–1, YJ suvh1–2 and YJ SUVH1 seedlings grown on MS media. (Right panel) RT-qPCR revealed decreased LUC transcript levels in the suvh1 mutants in the YJ background. ‘YJ SUVH1’ indicates the YJ SUVH1p:SUVH1–3XFLAG suvh1–1 line. In YJ SUVH1, the phenotype of the YJ suvh1–1 mutant was rescued by the transgene containing a wild-type SUVH1 genomic region and a 3XFLAG tag at the C-terminus of SUVH1. (B) A diagram of the SUVH1 protein and the substitution caused by the suvh1–1 mutation. The SUVH1 protein contains an SRA domain, a Pre-SET domain and a SET domain. The G-to-E substitution caused by suvh1–1 occurs in the SET domain. (C) The suvh1–1 mutation led to decreased LUC expression in the LUCH background. (Left panel) LUC luminescence of 8-day-old LUCH and LUCH suvh1–1 seedlings grown on half-strength MS media. (Right panel) RT-qPCR showed decreased LUC expression in the suvh1–1 mutant in the LUCH background. (D–E) The effects of suvh1–1 on LUC expression were suppressed by 5-Aza-2′-deoxycidine treatment in both the YJ (D) and LUCH (E) backgrounds. (Left panels) LUC luminescence of seedlings grown on half-strength MS media containing 7 μg/ml 5-Aza-2′-deoxycytidine for 14 days for YJ and YJ suvh1–1 (D) and LUCH and LUCH suvh1–1 (E). (Right panels) RT-qPCR showed rescued LUC transcript levels in the treated seedlings. Error bars in (A–E) represent standard deviation from three biological replicates.
To determine whether SUVH1 regulates LUC expression through the DNA methylation pathway, LUC expression levels were analyzed in YJ suvh1–1, LUCH suvh1–1 and control plants (YJ and LUCH, respectively) treated with the cytosine methylation inhibitor 5-Aza-2′-deoxycytidine (5-Aza-dC). Luminescence imaging and RT-qPCR revealed that the decreases in LUC expression observed with suvh1–1 were completely eliminated in both the YJ and LUCH backgrounds following the chemical treatment (Figure 1D and E). Therefore, eliminating the DNA methylation of the LUC reporter genes completely suppressed the suvh1–1 molecular phenotype, suggesting that SUVH1 functions through the DNA methylation pathway.
The suvh1–1 mutation does not affect DNA methylation
The next question addressed was whether the suvh1–1 mutation leads to increased DNA methylation. First, the DNA methylation level at the LUC transgene was analyzed using McrBC-qPCR. Surprisingly, despite the drastic decrease in LUC expression in both YJ suvh1–1 and LUCH suvh1–1 (Figure 1A and C), no differences were observed for the methylation levels at the d35S promoter when comparing YJ to YJ suvh1–1 or LUCH to LUCH suvh1–1 (Figure 2A and B). For the LUC coding region, methylation was nearly absent in YJ and low in LUCH, and increased DNA methylation was not observed in the suvh1–1 background (Figure 2A and B). To further assess whether SUVH1 influences DNA methylation levels, MethylC-seq was performed to interrogate the status of DNA methylation on the genomic scale. Two biological replicates were performed for YJ and YJ suvh1–1; the bisulfite conversion efficiency and coverage are listed in Supplemental Tables S1 and S2. No methylation level differences were observed at either the highly methylated d35S promoter or the unmethylated LUC coding region when comparing YJ and YJ suvh1–1 (Figure 2C and D). These results confirmed that the decreased LUC expression observed in the suvh1–1 mutant was not attributable to increased DNA methylation, indicating that SUVH1 functions downstream of DNA methylation.
Figure 2.
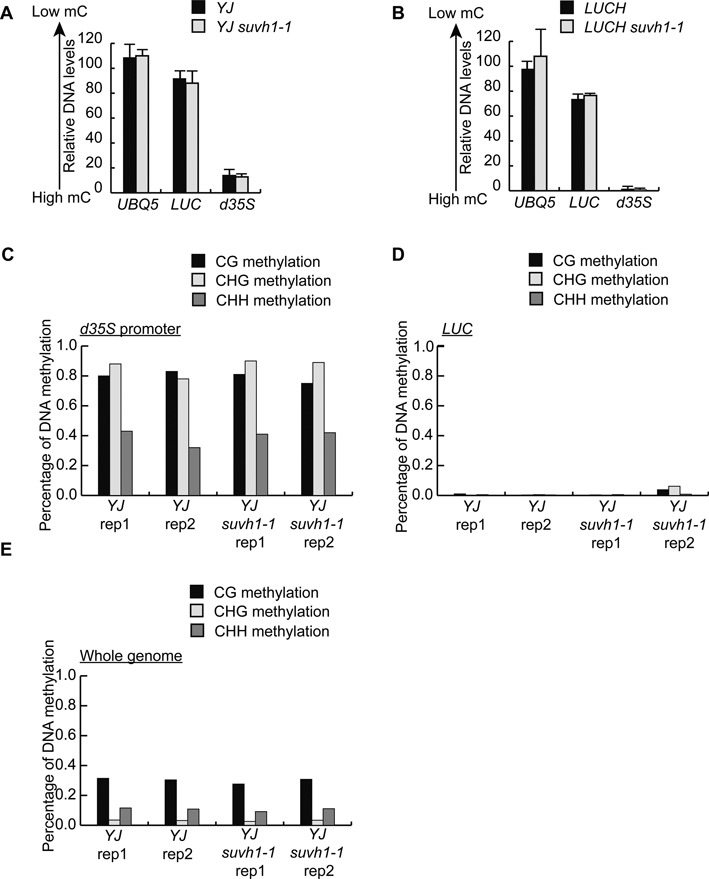
The suvh1–1 mutation does not affect DNA methylation. (A and B) McrBC-qPCR analysis of DNA methylation levels at the d35S promoter and the LUC coding region in YJ (A) and LUCH (B). qPCR was performed using genomic DNA treated with or without McrBC. The relative levels of amplified DNA for UBQ5, LUC and d35S in samples treated with McrBC compared to untreated samples were shown. Error bars were from three technical replicates. Two biological replicates were performed and gave similar results. (C and D) The levels of CG, CHG and CHH DNA methylation at the d35S promoter (C) and LUC coding region (D) in YJ and YJ suvh1–1 as determined through MethylC sequencing. Results from two biological replicates (rep) are shown. (E) Total genomic CG, CHG and CHH DNA methylation in YJ and YJ suvh1–1 as determined through MethylC-seq. Results from two biological replicates (rep) are shown.
We next examined whether SUVH1 influences DNA methylation at endogenous loci. No significant changes in the levels of DNA methylation on the genome-wide scale were found when comparing YJ and YJ suvh1–1 (Figure 2E). To determine whether SUVH1 influences DNA methylation at a subset of genomic loci, differentially methylated regions (DMRs) between YJ and YJ suvh1–1 were identified. There were 144, 4 and 314 CG, CHG and CHH DMRs, respectively, with reduced DNA methylation, and 274, 80 and 276 CG, CHG and CHH DMRs, respectively, with increased DNA methylation in YJ suvh1–1 as compared to YJ. In light of the total number of regions analyzed (1196682 regions, of which 252111, 136201 and 142622 are CG, CHG and CHH methylated regions, respectively), the possibility that the identified DMRs reflected random noise was considered. Specifically, the DMRs obtained in the present study were compared to the DMRs previously reported (3) to identify overlapping DMRs. In the published study, a suvh1 mutant with a T-DNA insertion (SALK_003675) in an exon of SUVH1 was used. The analysis eliminated most of the DMRs identified in the present study, leaving only 12, 1 and 10 hypo CG, CHG and CHH DMRs, respectively, and 10, 16 and 66 hyper CG, CHG and CHH DMRs, respectively, common in the two suvh1 mutants. Moreover, correlation analysis of the methylation levels in YJ and YJ suvh1–1 was performed. As shown in Supplementary Figure S3, there was a tight linear correlation between YJ and YJ suvh1–1 when levels of methylated CG, CHG and CHH were examined. Taken together, the McrBC-qPCR and methylome profiling data indicate that SUVH1 does not affect DNA methylation levels either at the LUC transgene or on a genome-wide scale. Thus, the effect of SUVH1 on LUC expression probably reflects its activity downstream of DNA methylation.
The suvh1–1 mutation causes decreased H3K4me3 levels without affecting H3K9me2 levels at the transgenes
Because SUVH1 encodes a member of the H3K9me2 methyltransferase family, the effect of the suvh1–1 mutation on H3K9me2 levels was analyzed by chromatin immunoprecipitation (ChIP) assays. In YJ, H3K9me2 modifications were detected at the d35S promoter but not at the LUC coding region (Figure 3A). The suvh1–1 mutation did not result in any changes in H3K9me2 levels at the d35S promoter or the LUC coding region (Figure 3A). These findings indicate that SUVH1, unlike its homologs SUVH4, 5 and 6, is not a factor in the H3K9me2 pathway. The active histone methylation mark (H3K4me3) was also analyzed at the LUC transgene. As shown in Figure 3B, no differences were observed in the d35S promoter region, but there was a consistent decrease in the LUC coding region in suvh1–1. In Arabidopsis, there are four genes with known roles in the deposition of H3K4me3, including ATX1 (42), ATX2 (14), ATXR3 (13) and ATXR7 (15). To determine whether the decreased H3K4me3 levels in YJ suvh1–1 were a result of decreased expression of these genes, the expression of the four genes was determined in YJ and YJ suvh1–1. As shown in Figure 3C, the suvh1–1 mutation did not affect the expression of these four genes.
Figure 3.
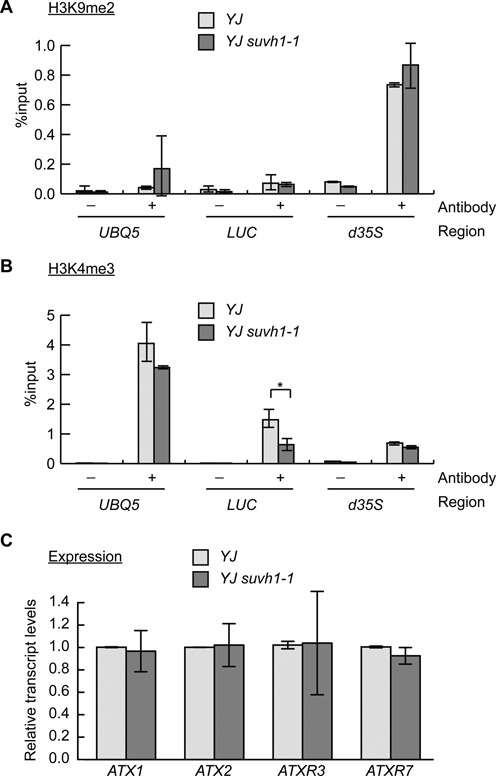
Analysis of histone methylation marks and expression of known H3K4 methyltransferase genes in suvh1–1. (A and B) ChIP-qPCR was performed to measure H3K9me2 (A) and H3K4me3 (B) levels in YJ and YJ suvh1–1. The UBQ5 gene was included as a control. No changes in H3K9me2 levels were observed at the transgene in the two genotypes. Reduced H3K4me3 levels were observed in YJ suvh1–1 at the LUC coding region but not at the d35S promoter. * Significant difference with a P-value <0.05. ‘–’ represents the sample without antibody; ‘+’ represents the sample with H3K4me3 or H3K9me2 antibodies added. Error bars were calculated from three technical replicates. Results were confirmed by three biological replicates. (C) The expression of known genes encoding H3K4 methyltransferases was determined through RT-qPCR. Error bars were calculated from three biological replicates. UBQ5 was used as an internal control.
SUVH1 has an anti-silencing role at certain endogenous loci
In light of the anti-silencing function of SUVH1 on transgenic LUC expression, its effect on the expression of endogenous loci was also investigated. Specifically, mRNA-seq libraries were constructed to profile the transcriptomes of YJ and YJ suvh1–1. To identify differentially expressed genes, the fold change between YJ and YJ suvh1–1 RPKM-normalized read abundance was calculated (where RPKM indicates reads per kilobase of a gene per million mapped reads), and the p-value was calculated using the Poisson distribution (34). Considering the effect of noise, different combinations of p-values and fold changes were considered when assessing the effect of the suvh1–1 mutation (Supplementary Table S3). Regardless of the cutoff used, the number of genes with decreased transcript levels always exceeded the number of genes with increased transcript levels as a result of the suvh1–1 mutation, suggesting that SUVH1 largely promotes gene expression. To analyze the effect of the suvh1–1 mutation on transcripts located at intergenic regions, the genome was divided into 500 bp static windows, and transcript level comparison was performed for each window. As shown in Supplementary Table S3, the predominant effect of the suvh1–1 mutation was also decreased expression. Lists of differential expressed genes and 500 bp windows are shown in Supplemental Tables S4 and S5, respectively. To validate the library data, eight loci were selected (six genes and two un-annotated transcripts) and analyzed using RT-qPCR. At four of the eight loci (three genes and one un-annotated transcript), decreased transcript levels were consistently detected in YJ suvh1–1 and this decrease was rescued by the SUVH1 construct (Figure 4A). Moreover, these four loci were tested in the LUCH background, and decreased expression was consistently observed with the suvh1–1 mutation (Supplementary Figure S4A). In addition, the expression levels of these four loci were also found to be decreased in YJ suvh1–2 relative to YJ (Supplementary Figure S4B). These results suggest that SUVH1 promotes the expression of certain endogenous genes.
Figure 4.
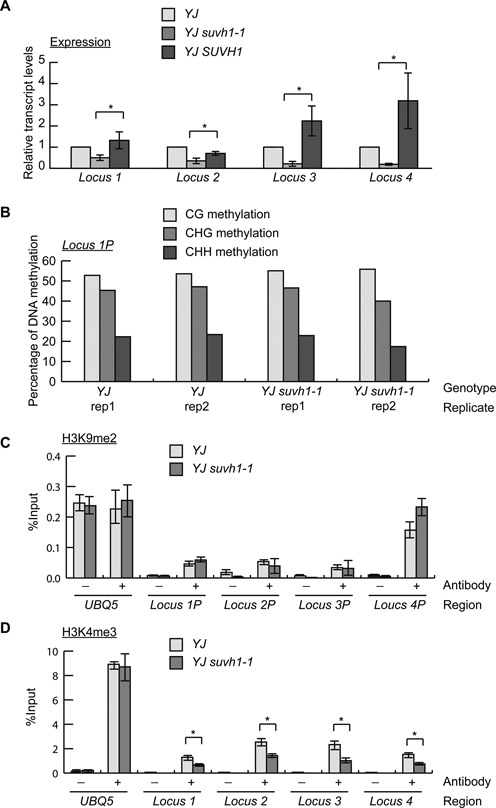
The suvh1–1 mutation leads to the reduced expression of endogenous loci with corresponding reductions in H3K4me3 levels. (A) The expression of four SUVH1-targeted endogenous loci was confirmed by RT-qPCR, and the decreased expression observed in YJ suvh1–1 was rescued in YJ SUVH1 (YJ SUVH1p:SUVH1–3XFLAG suvh1–1) for all four loci. Error bars represent standard deviations calculated from three biological replicates. * Significant difference with a P-value <0.05. (B) The DNA methylation levels of the 1 kb promoter of locus 1 determined from the two biological replicates (rep) of the YJ and YJ suvh1–1 methylome data. In all four MethylC-seq libraries, CG, CHG and CHH methylation was detected, and there were no consistent differences between YJ and YJ suvh1–1. Locus 1P represents the promoter of locus 1. (C) ChIP-qPCR was performed to measure H3K9me2 levels at the promoters of loci 1–4 in YJ and YJ suvh1–1. Locus 1P represents the promoter of locus 1, the same terminology also applies to other loci. No changes in H3K9me2 levels were observed. (D). ChIP-qPCR was performed to measure H3K4me3 levels in the coding (or transcript) regions of the four loci. Reduced H3K4me3 levels were observed in YJ suvh1–1. * Significant difference with a P-value <0.05. UBQ5 was included as a control in (C and D). Error bars representing standard deviations were calculated from three technical replicates in (C and D). Three biological replicates gave similar results. ‘–’ represents the samples without antibody; ‘+’ represents the samples with H3K9me2 or H3K4me3 antibodies added.
SUVH1 promotes H3K4me3 levels at DNA-methylated loci
To follow up on the finding that SUVH1 does not affect DNA methylation but promotes H3K4me3 levels at the LUC transgene (Figure 3B), the DNA and histone methylation status of the four confirmed endogenous loci was also assessed. The MethylC-seq data were used to determine the DNA methylation levels at the four endogenous loci. The promoter regions of the endogenous loci, which we define as 1 kb 5′ of the start of the transcripts, exhibited high levels of DNA methylation (Figure 4B, Supplementary Figure S5). The methylation levels remained unchanged in YJ suvh1–1 (Figure 4B, Supplementary Figure S5), consistent with the observation for the LUC transgene.
H3K9me2 and H3K4me3 ChIP assays were performed to assess the histone methylation levels of the endogenous loci. At coding (or transcript) regions of SUVH1-targeted loci, H3K9me2 was hardly detected, and no difference in H3K9me2 levels was found at these regions between YJ and YJ suvh1–1 (Supplementary Figure S6A). Similarly, no difference in H3K9me2 levels was observed between YJ and YJ suvh1–1 at the promoter regions of these loci (Figure 4C). To test whether SUVH1 affects H3K9me2 at other endogenous loci, H3K9me2 levels were determined at four RdDM loci. No difference was found at these loci between YJ and YJ suvh1–1 (Supplementary Figure S6B). These results indicate that SUVH1 is not likely to affect H3K9me2 levels at endogenous loci.
Like the d35S promoter region (Figure 3B), the promoter regions of SUVH1-targeted endogenous loci had similar levels of H3K4me3 in YJ and YJ suvh1–1 (Supplementary Figure S6C). As for the LUC coding region (Figure 3B), H3K4me3 levels in the coding (or transcript) regions of the endogenous loci were reduced in YJ suvh1–1 (Figure 4D). To determine whether the effects of suvh1–1 on H3K4 modifications are specific to trimethylation, we examined H3K4me1 and H3K4me2 through ChIP assays. For both the promoter regions and coding regions of the LUC transgene as well as the SUVH1-targeted endogenous loci, the suvh1–1 mutation did not affect H3K4me1 or H3K4me2 levels (Supplementary Figure S7).
The genetic relationships between SUVH1 and DNA methylation factors
The findings that SUVH1 functions at genes with DNA-methylated promoters prompted the question of how SUVH1 is related to the RdDM pathway. Pol V produces non-coding scaffold transcripts that recruit siRNAs to chromatin in RdDM (43,44). With mutations in NRPE1 encoding the largest subunit of Pol V, the RdDM pathway is disrupted and CHH methylation cannot be maintained; in contrast, CHG methylation and CG methylation are virtually unaffected (3). To determine whether the SUVH1-targeted loci are regulated by RdDM and whether CHH methylation is required for SUVH1 function, RT-qPCR was performed to detect the transcript levels of LUC and the SUVH1-targeted loci in YJ nrpe1–1 and YJ nrpe1–1 suvh1–1. In YJ nrpe1–1, the expression of three of the four SUVH1-targeted loci (loci 1, 3 and 4) was de-repressed (Figure 5A), indicating that these loci are also under the regulation of RdDM. For LUC and the four endogenous loci, the expression levels were not decreased in YJ suvh1–1 nrpe1–1 relative to YJ nrpe1–1 (Figure 5A). The DNA methylation status at the promoter regions of these loci was determined by McrBC-qPCR. At the d35S promoter and the promoter regions of loci 1–3, a partial loss of DNA methylation was found (Figure 5B). This is consistent with the known role of NRPE1 in the maintenance of CHH methylation. The incomplete loss of DNA methylation at these loci is probably attributable to the fact that these loci contain relatively high levels of CG methylation (Figure 4B, Supplementary Figure S5). These results indicate that the lack of CHH methylation eliminated a need for SUVH1 in the promotion of expression at these loci.
Figure 5.
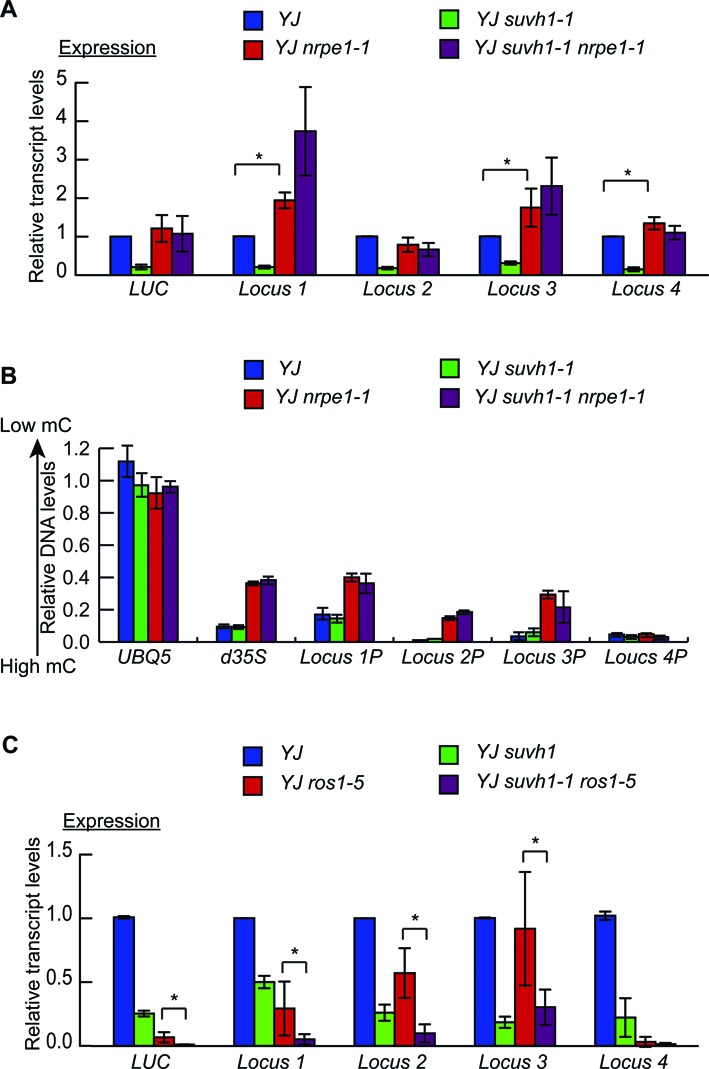
Characterizations of SUVH1-targeted loci in nrpe1 and ros1 mutant backgrounds. (A) The expression of four SUVH1-targeted endogenous loci was detected by RT-qPCR in the nrpe1 mutant background. Error bars representing standard deviations were calculated from three biological replicates. * Significant difference with a P-value <0.05. (B) The DNA methylation levels at the promoter regions of SUVH1-targeted endogenous loci were determined through McrBC-qPCR. Locus 1P represents the promoter of locus 1; the same terminology applies to the other loci. Three biological replicates gave similar results, and those from one biological replicate are shown here. (C) Transcript levels of LUC and the four SUVH1-targeted endogenous loci as determined by RT-qPCR. Error bars representing standard deviations were calculated from three biological replicates. * Significant difference with a P-value <0.05.
The DNA glycosylase/lyase ROS1 is a DNA demethylase (45), and ros1 mutants exhibit increased DNA methylation (at CG, CHG and CHH) (3,9). The transcript levels of the SUVH1-targeted loci were examined in YJ ros1–5 by RT-qPCR to determine whether they are regulated by ROS1. Decreased transcript levels for LUC at the four endogenous loci were found in YJ ros1–5 relative to the YJ control (Figure 5C), indicating that these loci are also regulated by the ROS1 demethylation pathway. Next, the transcript levels of the SUVH1-targeted loci were examined in the YJ suvh1–1 ros1–5 double mutant to determine whether ROS1 and SUVH1 function in the same pathway. Decreased transcript levels were observed for LUC and the SUVH1-targeted loci 1–3 in YJ suvh1–1 ros1–5 compared to YJ ros1–5 (Figure 5C), suggesting that ROS1 and SUVH1 affect these loci independently. At locus 4, the expression in YJ ros1–5 was almost completely diminished, making it impossible to determine whether SUVH1 was functional at this locus in YJ ros1–5 (Figure 5C). These results suggest that ROS1 and SUVH1 are not in the same pathway, which is consistent with the previous finding that the suvh1–1 mutation does not alter DNA methylation levels.
Lack of anti-correlation between promoter DNA methylation and gene expression
DNA methylation is an important mechanism for suppressing the expression of transposable elements and is established through the RdDM pathway. A possible consequence of transposon insertion into the promoter of a gene is suppression of the expression of the gene through DNA methylation. Using the methylome and mRNA-seq data generated in this study, we explored whether there is any anti-correlation between gene expression levels and promoter DNA methylation. We determined the DNA methylation level at 1 kb regions upstream of genes from methylome data and derived the corresponding gene expression levels from mRNA-seq data. As shown in Figure 6, there was no anti-correlation between gene expression and promoter DNA methylation levels. Genes with or without DNA methylation in their promoters were found to have high, medium or low expression levels. Despite the role of DNA methylation in suppressing gene expression, genes with DNA methylation at the promoter region are not necessarily suppressed, indicating that regulatory mechanisms must exist to override this suppressive mark.
Figure 6.
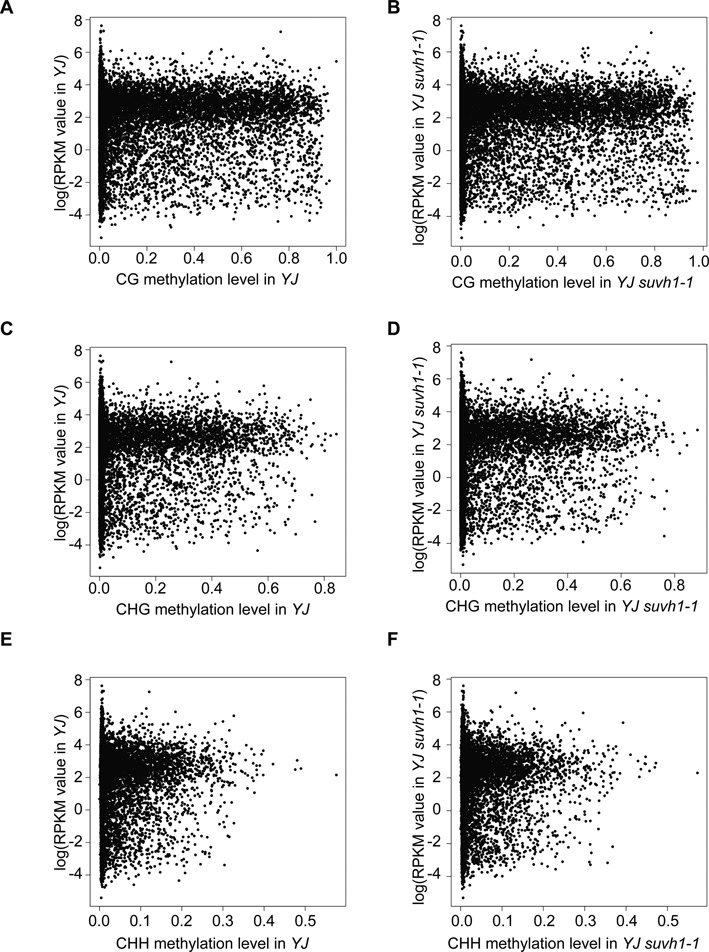
Plots of DNA methylation levels at 1 kb gene promoter regions versus gene expression levels in YJ and YJ suvh1–1. The x-axis represents the level of DNA methylation, and the y-axis represents the natural logarithm of the RPKM (reads per kilobase per million) value for genes from mRNA-seq. (A and B) Correlation plot of CG methylation level with gene expression in YJ (A) and YJ suvh1–1 (B). (C and D) Correlation plot of CHG methylation level with gene expression in YJ (C) and YJ suvh1–1 (D). (E and F) Correlation plot of CHH methylation level with gene expression in YJ (E) and YJ suvh1–1 (F) DNA methylation and gene expression levels were determined from MethylC-seq and mRNA-seq, respectively, in this study.
Evolution of SUVH genes in plants
We examined the phylogeny of SUVH genes from plants (Supplementary Figure S8). Only one SUVH gene exists in the green alga Chlamydomonas reinhardtii, which exhibits DNA methylation in its genome (46). In representative land plants that we examined, such as the moss Physcomitrella patens, the lycophyte Selaginella moellendorffii, the basal angiosperm Amborella trichopoda, the monocot rice and the dicot Arabidopsis, multiple SUVH genes are found. Only the SUVH4 clade genes, which encode H3K9 methyltransferases, are present in all these land plants, suggesting that this clade evolved first. The genes in the SUVH5, SUVH2, and SUVH1 clades are only present in angiosperms, suggesting that they evolved later. These results indicate that the ancient function of SUVH proteins is represented by the SUVH4 clade that represses transcription through H3K9 methylation. The SUVH1 clade, for which our studies implicate an anti-silencing effect, represents a derived function of the SUVH family.
DISCUSSION
Since the initial discovery of transposons by Barbara McClintock, the regulation of transposons has been widely investigated. DNA methylation is a well-recognized epigenetic mark for the suppression of transposon transcription, and numerous effectors involved in the DNA methylation pathway, from initial establishment to maintenance, have been characterized in plants. However, the understanding of opposing mechanisms that override the effects of DNA methylation is very limited. In the present study, a forward-genetic screening approach was used to identify a factor with an anti-silencing function. SUVH1, which encodes a SET-domain protein, was found to promote the expression of reporter genes only when their promoters harbor DNA methylation.
Although DNA methylation deposition has been well studied, subsequent processes downstream of DNA methylation have not been as thoroughly explored. At present, there are two known types of conserved domains capable of binding methylated DNA: the SET and RING-associated (SRA) domain (47) and the METHYL-CpG-BINDING domain (MBD) (48). In animals, MBD proteins have been implicated in the establishment of repressive chromatin marks through the promotion of histone deacetylase and histone methyltransferase activity (49–52). One family of SRA proteins, the RING-associated VARIANT IN METHYLATION (VIM)/ORTHRUS (ORTH) family and their homologs in animals, Ubiquitin-like PHD and RING finger domain (UHRF1), have all been found to be critical for DNA methylation maintenance through binding methylated CG sites (47). The Su(var)3–9 homologs, which constitute another family of SRA proteins, are associated with the SET domain. Several SRA proteins have been shown to have H3K9me2 methyltransferase activity or to participate in the RdDM pathway (16,17). These DNA-methylation-associated proteins all function in connecting DNA methylation to repressive chromatin marks (namely, H3K9me2 and histone deacetylation). In contrast, SRA proteins have not been associated with active chromatin marks or gene silencing suppression.
Our finding that SUVH1 promotes the expression of LUC and several endogenous loci contradicts the known roles of Arabidopsis SUVH homologs, which have been found to regulate gene expression by promoting silencing (17,20,47). According to the current paradigm, a loss of function suvh mutant would be predicted to exhibit high LUC expression. The low LUC expression in YJ suvh1–1 suggests that SUVH1 has a different role than its homologs with currently known functions. Given that none of the SUVH1 subgroup proteins have been associated with silencing roles, this raises the possibility that this particular subgroup is characterized by anti-silencing functions. ChIP analysis of histone modification levels did not reveal any changes in H3K9me2 abundance in the suvh1–1 mutant, providing a second line of evidence that SUVH1 function may be distinct from those of other SUVH proteins associated with RdDM or H3K9me2.
How SUVH1 promotes the expression of promoter-methylated genes is currently unknown. Based on our finding that SUVH1 function is dispensable in the nrpe1 background and the presence of an SRA domain in SUVH1, we speculate that SUVH1 recognizes CHH methylation. The decreased levels of H3K4me3 in suvh1–1 suggest a possibility that SUVH1 promotes H3K4me3 deposition at target genes. Since SUVH1 was found not to have histone methyltransferase activity (18), SUVH1 probably does so through an H3K4me3 methyltransferase. Alternatively, the reduced levels of H3K4me3 at SUVH1-targeted genes in the suvh1–1 mutant could be an indirect effect of reduced expression of these genes.
Among the SUVH1-targeted endogenous loci, Pol IV-dependent siRNAs were detected at the promoter regions along with CG, CHG and CHH methylation and transposons (Supplementary Figures S9 and S10). We propose the following model for SUVH1 function based on the present findings. With transposons inserting into different positions in the genome over the course of evolution, Pol IV-generated siRNAs function as guides directing DNA methylation at the sites of insertion to inhibit the harmful effects of active transposons. While this is necessary for genome stability, this silencing mechanism could cause a gene to be suppressed if a transposon inserts into its promoter region. To counteract this suppression, however, SUVH1, a protein with a DNA methylation-binding domain, is recruited to these loci by recognizing CHH methylation to promote gene expression.
The above model would predict that SUVH1 function is only necessary in species with CHH DNA methylation near genes. Our phylogenetic analysis of SUVH proteins in plants shows that the SUVH4 clade of H3K9 methyltransferases evolved first and the SUVH1 clade evolved later. Therefore, the SUVH1 clade probably represents a derived function of SUVH proteins, and our present findings suggest this function to be the promotion of the expression of genes with promoter CHH DNA methylation. In a genome-wide methylation analysis of rice (an angiosperm), Selaginella moellendorffii and Physcomitrella patens (two early land plants), and Chlorella sp. NC64A and Volvox carteri (two green algae), CHH methylation was found in promoter regions of genes only in rice (53). The presence of the SUVH1 clade of proteins perhaps coincides with the presence of promoter CHH methylation in genes in angiosperms.
Supplementary Material
Acknowledgments
We thank Drs Markus Schmid and Detlef Weigel for the gift of the pJL-Blue entry vector and Ms. Tianran Jane Jia, Rae Eden Yumul and Drs Wenrong He and Bailong Zhang for comments on the manuscript.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Howard Hughes Medical Institute (HHMI); Gordon and Betty Moore Foundation [GBMF3046]; National Institutes of Health [GM061146]; National Science Foundation of China [91440105 to X.C.]. Funding for open access charge: HHMI.
Conflict of interest statement. None declared.
REFERENCES
- 1.Law J.A., Jacobsen S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010;11:204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matzke M.A., Mosher R.A. RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014;15:394–408. doi: 10.1038/nrg3683. [DOI] [PubMed] [Google Scholar]
- 3.Stroud H., Greenberg M.V., Feng S., Bernatavichute Y.V., Jacobsen S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell. 2013;152:352–364. doi: 10.1016/j.cell.2012.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan S.W., Henderson I.R., Jacobsen S.E. Gardening the genome: DNA methylation in Arabidopsis thaliana. Nat. Rev. Genet. 2005;6:351–360. doi: 10.1038/nrg1601. [DOI] [PubMed] [Google Scholar]
- 5.Morales-Ruiz T., Ortega-Galisteo A.P., Ponferrada-Marin M.I., Martinez-Macias M.I., Ariza R.R., Roldan-Arjona T. DEMETER and REPRESSOR OF SILENCING 1 encode 5-methylcytosine DNA glycosylases. Proc. Natl. Acad. Sci. U S A. 2006;103:6853–6858. doi: 10.1073/pnas.0601109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Penterman J., Zilberman D., Huh J.H., Ballinger T., Henikoff S., Fischer R.L. DNA demethylation in the Arabidopsis genome. Proc. Natl. Acad. Sci. U.S.A. 2007;104:6752–6757. doi: 10.1073/pnas.0701861104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi Y.H., Gehring M., Johnson L., Hannon M., Harada J.J., Goldberg R.B., Jacobsen S.E., Fischer R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell. 2002;110:33–42. doi: 10.1016/s0092-8674(02)00807-3. [DOI] [PubMed] [Google Scholar]
- 8.Gong Z., Morales-Ruiz T., Ariza R.R., Roldan-Arjona T., David L., Zhu J.K. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell. 2002;111:803–814. doi: 10.1016/s0092-8674(02)01133-9. [DOI] [PubMed] [Google Scholar]
- 9.Lister R., O'Malley R.C., Tonti-Filippini J., Gregory B.D., Berry C.C., Millar A.H., Ecker J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ortega-Galisteo A.P., Morales-Ruiz T., Ariza R.R., Roldan-Arjona T. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol. Biol. 2008;67:671–681. doi: 10.1007/s11103-008-9346-0. [DOI] [PubMed] [Google Scholar]
- 11.Alvarez-Venegas R., Avramova Z. Methylation patterns of histone H3 Lys 4, Lys 9 and Lys 27 in transcriptionally active and inactive Arabidopsis genes and in atx1 mutants. Nucleic Acids Res. 2005;33:5199–5207. doi: 10.1093/nar/gki830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berr A., McCallum E.J., Menard R., Meyer D., Fuchs J., Dong A., Shen W.H. Arabidopsis SET DOMAIN GROUP2 is required for H3K4 trimethylation and is crucial for both sporophyte and gametophyte development. Plant cell. 2010;22:3232–3248. doi: 10.1105/tpc.110.079962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo L., Yu Y.C., Law J.A., Zhang X.Y. SET DOMAIN GROUP2 is the major histone H3 lysie 4 trimethyltransferase in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18557–18562. doi: 10.1073/pnas.1010478107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saleh A., Alvarez-Venegas R., Yilmaz M., Le O., Hou G., Sadder M., Al-Abdallat A., Xia Y., Lu G., Ladunga I., et al. The highly similar Arabidopsis homologs of trithorax ATX1 and ATX2 encode proteins with divergent biochemical functions. Plant cell. 2008;20:568–579. doi: 10.1105/tpc.107.056614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamada Y., Yun J.Y., Woo S.C., Amasino R.M. ARABIDOPSIS TRITHORAX-RELATED7 is required for methylation of lysine 4 of histone H3 and for transcriptional activation of FLOWERING LOCUS C. Plant cell. 2009;21:3257–3269. doi: 10.1105/tpc.109.070060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rea S., Eisenhaber F., O'Carroll D., Strahl B.D., Sun Z.W., Schmid M., Opravil S., Mechtler K., Ponting C.P., Allis C.D., et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 17.Naumann K., Fischer A., Hofmann I., Krauss V., Phalke S., Irmler K., Hause G., Aurich A.C., Dorn R., Jenuwein T., et al. Pivotal role of AtSUVH2 in heterochromatic histone methylation and gene silencing in Arabidopsis. EMBO J. 2005;24:1418–1429. doi: 10.1038/sj.emboj.7600604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ebbs M.L., Bender J. Locus-specific control of DNA methylation by the Arabidopsis SUVH5 histone methyltransferase. Plant Cell. 2006;18:1166–1176. doi: 10.1105/tpc.106.041400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jackson J.P., Lindroth A.M., Cao X., Jacobsen S.E. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416:556–560. doi: 10.1038/nature731. [DOI] [PubMed] [Google Scholar]
- 20.Johnson L.M., Du J., Hale C.J., Bischof S., Feng S., Chodavarapu R.K., Zhong X., Marson G., Pellegrini M., Segal D.J., et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature. 2014;507:124–128. doi: 10.1038/nature12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Z.W., Shao C.R., Zhang C.J., Zhou J.X., Zhang S.W., Li L., Chen S., Huang H.W., Cai T., He X.J. The SET domain proteins SUVH2 and SUVH9 are required for Pol V occupancy at RNA-directed DNA methylation loci. PLoS Genet. 2014;10:e1003948. doi: 10.1371/journal.pgen.1003948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Henderson I.R., Jacobsen S.E. Tandem repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA methylation and initiate siRNA spreading. Genes Dev. 2008;22:1597–1606. doi: 10.1101/gad.1667808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J., He Y., Amasino R., Chen X. siRNAs targeting an intronic transposon in the regulation of natural flowering behavior in Arabidopsis. Genes Dev. 2004;18:2873–2878. doi: 10.1101/gad.1217304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soppe W.J., Jacobsen S.E., Alonso-Blanco C., Jackson J.P., Kakutani T., Koornneef M., Peeters A.J. The late flowering phenotype of fwa mutants is caused by gain-of-function epigenetic alleles of a homeodomain gene. Mol. Cell. 2000;6:791–802. doi: 10.1016/s1097-2765(05)00090-0. [DOI] [PubMed] [Google Scholar]
- 25.Won S.Y., Li S., Zheng B., Zhao Y., Li D., Zhao X., Yi H., Gao L., Dinh T.T., Chen X. Development of a luciferase-based reporter of transcriptional gene silencing that enables bidirectional mutant screening in Arabidopsis thaliana. Silence. 2012;3:6. doi: 10.1186/1758-907X-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peragine A., Yoshikawa M., Wu G., Albrecht H.L., Poethig R.S. SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans-acting siRNAs in Arabidopsis. Genes Dev. 2004;18:2368–2379. doi: 10.1101/gad.1231804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kanno T., Huettel B., Mette M.F., Aufsatz W., Jaligot E., Daxinger L., Kreil D.P., Matzke M., Matzke A.J. Atypical RNA polymerase subunits required for RNA-directed DNA methylation. Nat. Genet. 2005;37:761–765. doi: 10.1038/ng1580. [DOI] [PubMed] [Google Scholar]
- 28.Yant L., Mathieu J., Dinh T.T., Ott F., Lanz C., Wollmann H., Chen X., Schmid M. Orchestration of the floral transition and floral development in Arabidopsis by the bifunctional transcription factor APETALA2. The Plant cell. 2010;22:2156–2170. doi: 10.1105/tpc.110.075606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Earley K.W., Haag J.R., Pontes O., Opper K., Juehne T., Song K., Pikaard C.S. Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 2006;45:616–629. doi: 10.1111/j.1365-313X.2005.02617.x. [DOI] [PubMed] [Google Scholar]
- 30.Rogers S.O., Bendich A.J. Extraction of DNA from milligram amounts of fresh, herbarium and mummified plant tissues. Plant Mol. Biol. 1985;5:69–76. doi: 10.1007/BF00020088. [DOI] [PubMed] [Google Scholar]
- 31.Chen P.Y., Cokus S.J., Pellegrini M. BS Seeker: precise mapping for bisulfite sequencing. BMC Bioinformatics. 2010;11:203. doi: 10.1186/1471-2105-11-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gendrel A.V., Lippman Z., Martienssen R., Colot V. Profiling histone modification patterns in plants using genomic tiling microarrays. Nat Methods. 2005;2:213–218. doi: 10.1038/nmeth0305-213. [DOI] [PubMed] [Google Scholar]
- 33.Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marioni J.C., Mason C.E., Mane S.M., Stephens M., Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamesch P., Berardini T.Z., Li D., Swarbreck D., Wilks C., Sasidharan R., Muller R., Dreher K., Alexander D.L., Garcia-Hernandez M., et al. The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 2012;40:D1202–D1210. doi: 10.1093/nar/gkr1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amborella Genome Project. The Amborella genome and the evolution of flowering plants. Science. 2013;342:1241089. doi: 10.1126/science.1241089. [DOI] [PubMed] [Google Scholar]
- 37.Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N., et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:D1178–D1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebbs M.L., Bartee L., Bender J. H3 lysine 9 methylation is maintained on a transcribed inverted repeat by combined action of SUVH6 and SUVH4 methyltransferases. Mol. Cell. Biol. 2005;25:10507–10515. doi: 10.1128/MCB.25.23.10507-10515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malagnac F., Bartee L., Bender J. An Arabidopsis SET domain protein required for maintenance but not establishment of DNA methylation. EMBO J. 2002;21:6842–6852. doi: 10.1093/emboj/cdf687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stroud H., Do T., Du J., Zhong X., Feng S., Johnson L., Patel D.J., Jacobsen S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014;21:64–72. doi: 10.1038/nsmb.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alvarez-Venegas R., Pien S., Sadder M., Witmer X., Grossniklaus U., Avramova Z. ATX-1, an Arabidopsis homolog of Trithorax, activates flower homeotic genes. Curr. Biol. 2003;13:627–637. doi: 10.1016/s0960-9822(03)00243-4. [DOI] [PubMed] [Google Scholar]
- 43.Wierzbicki A.T., Ream T.S., Haag J.R., Pikaard C.S. RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat. Genet. 2009;41:630–634. doi: 10.1038/ng.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wierzbicki A.T., Haag J.R., Pikaard C.S. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell. 2008;135:635–648. doi: 10.1016/j.cell.2008.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Agius F., Kapoor A., Zhu J.K. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Proc. Natl. Acad. Sci. U.S.A. 2006;103:11796–11801. doi: 10.1073/pnas.0603563103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng S., Cokus S.J., Zhang X., Chen P.Y., Bostick M., Goll M.G., Hetzel J., Jain J., Strauss S.H., Halpern M.E., et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. U.S.A. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rajakumara E., Law J.A., Simanshu D.K., Voigt P., Johnson L.M., Reinberg D., Patel D.J., Jacobsen S.E. A dual flip-out mechanism for 5mC recognition by the Arabidopsis SUVH5 SRA domain and its impact on DNA methylation and H3K9 dimethylation in vivo. Genes Dev. 2011;25:137–152. doi: 10.1101/gad.1980311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fournier A., Sasai N., Nakao M., Defossez P.A. The role of methyl-binding proteins in chromatin organization and epigenome maintenance. Brief. Funct. Genomics. 2012;11:251–264. doi: 10.1093/bfgp/elr040. [DOI] [PubMed] [Google Scholar]
- 49.Jones P.L., Jan Veenstra G.C., Wade P.A., Vermaak D., Kass S.U., Landsberger N., Strouboulis J., Wolffe A.P. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 50.Nan X., Ng H.-H., Johnson C.A., Laherty C.D., Turner B.M., Eisenman R.N., Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 51.Fuks F., Hurd P.J., Wolf D., Nan X., Bird A.P., Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y., Ng H.-H., Erdjument-Bromage H., Tempst P., Bird A., Reinberg D. Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 1999;13:1924–1935. doi: 10.1101/gad.13.15.1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zemach A., McDaniel I.E., Silva P., Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.