

Abstract
Renewable aromatics can be conveniently synthesized from furanics by introducing an intermediate hydrogenation step in the Diels–Alder (DA) aromatization route, to effectively block retro‐DA activity. Aromatization of the hydrogenated DA adducts requires tandem catalysis, using a metal‐based dehydrogenation catalyst and solid acid dehydration catalyst in toluene. Herein it is demonstrated that the hydrogenated DA adducts can instead be conveniently converted into renewable aromatics with up to 80 % selectivity in a solid‐phase reaction with shorter reaction times using only an acidic zeolite, that is, without solvent or dehydrogenation catalyst. Hydrogenated adducts from diene/dienophile combinations of (methylated) furans with maleic anhydride are efficiently converted into renewable aromatics with this new route. The zeolite H‐Y was found to perform the best and can be easily reused after calcination.
Keywords: arenes, biomass, cycloaddition, heterocycles, zeolites
The development of new routes for the production of ‘drop‐in’ aromatic chemicals from renewable resources is currently receiving considerable attention because of the increased demand, finite fossil resources, and the recognized need for more sustainable production routes.1 This attention is further compounded by the challenges associated with the changes in bulk aromatics supply caused by the increased use of shale‐gas‐derived feeds in steam cracker facilities.2 Of the various potential routes to biobased aromatics, Diels–Alder (DA) conversions of sugar‐derived furanic compounds3 are receiving increased attention.4, 5, 6, 7, 8, 9 The DA aromatization strategy typically involves two steps, that is, cycloaddition of a furanic diene with an appropriate dienophile, followed by acid‐catalyzed dehydration of the intermediate DA adduct. A prominent example of this approach is the reaction of 2,5‐dimethylfuran (DMF) and (excess of) ethylene over Brønsted‐acid‐containing BEA‐ and FAU‐type zeolites to produce renewable p‐xylene, a precursor to terephthalic acid (TA) and polyethylene terephthalate (PET), with yields as high as 90 %.4 Wang et al. showed that solid acid oxides, such as WOx‐ZrO2 and niobic acid, also demonstrate high catalytic activities for this reaction.5 Pacheco and Davis later explored DA aromatization of various oxidized derivatives of 5‐hydroxymethylfurfural (HMF) with ethylene over Lewis‐acidic molecular sieves, Sn‐Beta in particular.6
DA aromatization of 2‐methylfuran (MF) and ethylene proved less efficient over either H‐Beta or Sn‐Beta, with toluene selectivities not exceeding 46 %, because MF was consumed by side reactions such as dimerization.7 Indeed, selectivity for aromatization typically decreases as the furan ring becomes less substituted.8 Related to the challenges associated with controlling such furan‐dependent side reactions, careful control over the second catalytic aromatization step is often critical as the intermediate DA adducts are typically unstable and prone to retro‐DA reaction, especially at more elevated temperatures.8, 9 Consequently, one has to run the aromatization reaction at either low temperature10 or, if ethylene is the dienophile, at high pressure.4, 5, 6 Mahmoud et al. addressed the issue of DA adduct instability and furan reactivity by using a mixture of methanesulfonic acid and acetic acid anhydride to synthesize (substituted) phthalic anhydride with high selectivity at 80 °C.8 In an alternative approach, we recently reported on a new three‐step strategy and dealt with this general challenge in DA aromatization by including a mild intermediate hydrogenation step of the oxabicyclic adduct 1.9
This new route, which is run in the liquid phase, is shown in Scheme 1.9a The hydrogenated DA adduct 2 is thermally stable and efficiently aromatized in a tandem catalytic reaction using a mixture of a solid acid dehydration catalyst (e.g., zeolites or resins) and a dehydrogenation catalyst (e.g., metal on a carbon support). For example, catalytic aromatization of 2 in toluene at 200 °C with H‐VUSY (SiO2:Al2O3=11.5) and Pd/C resulted in a combined yield of 84 % for all aromatics.9a Formal catalytic oxidation would then give the desired aromatic di‐ and tricarboxylic acid end products.11 Notably, if this solution‐phase reaction was run with only the zeolite catalyst, very low aromatics yields were obtained, with the γ‐lactone intermediate 3 being the major product instead.9a Surprisingly, we have now discovered that the aromatization reaction also proceeds smoothly without any solvent and dehydrogenation catalyst, by simply mixing the solid reactant and the solid acid catalyst with heating in a standard Kugelrohr oven (see Figure S1 in the Supporting Information).
Scheme 1.
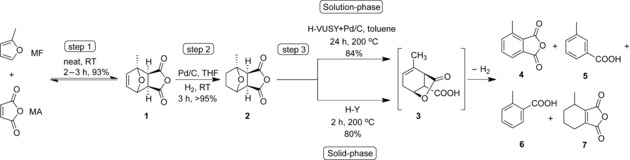
Three‐step catalytic route to renewable aromatic chemicals, exemplified for the synthesis of 4, and 5 and 6. Comparison of the solution‐9a and solid‐phase (this work) conversion of the hydrogenated DA adduct.
Various organic transformations have already been accomplished in the solid state, for example, by using the mechanochemical approach.12 Indeed, such solid‐phase, solventless chemical reactions are very attractive from the point of view of green chemistry and practicality. The facile, new approach reported here for the aromatization of hydrogenated DA adducts furthermore reduces reaction time and allows lower reaction temperatures by using a relatively inexpensive catalyst (Scheme 1, solid‐phase). The reaction requires no input of mechanical energy for it to proceed, except for tumbling the vessel to facilitate mixing. In addition, the omission of the (noble) transition‐metal dehydrogenation catalyst greatly facilitates catalyst recovery and regeneration. Again, the γ‐lactone (e.g., 3) is observed as a primary intermediate and found to be the precursor to all aromatics. Indeed, with 2, 3‐methylphthalic anhydride (4), and the decarboxylation products m‐ and o‐toluic acid (5 and 6) are again obtained. To the best of our knowledge, this is the first report of such a simple, solid‐state approach for the synthesis of biomass‐derived aromatics.
We first explored the influence of selected process parameters and choice of catalyst on the efficiency of this new route, using 2 as the substrate. The large‐pore FAU‐type zeolite Y, containing strong Brønsted acid sites and mesopores (see Table 1 and Figure S2, S3, and S4), proved to be most efficient. The substrate is simply mixed with H‐Y (SiO2/Al2O3=5.2) in a 1:1 w/w ratio in situ by rotating the reaction vessel prior to insertion into the preheated oven. Intimate ex situ mixing of the substrate and catalyst by grinding gave the same catalytic result. Mixing during reaction facilitates mass transfer as well as prevents the creation of hot spots.
Table 1.
Textural and acidic properties of the fresh zeolites and reused[a] H‐Y catalysts.
| Solid acid catalyst (SiO2/Al2O3) | BET surface area [m2 g−1] | Single point adsorption total pore volume [cm3 g−1] | t‐plot micropore volume [cm3 g−1] | Mesopore volume [cm3 g−1] | BJH adsorption average pore diameter [nm] | Total acid sites [mmol NH3 g−1] |
|---|---|---|---|---|---|---|
| H‐Beta (25) | 520 | 0.98 | 0.16 | 0.82 | 12.0 | 0.81 |
| H‐Y (5.2) | 544 | 0.37 | 0.23 | 0.14 | 3.9 | 0.63 |
| Reused H‐Y (5.2)[a] | 556 | 0.40 | 0.24 | 0.16 | 4.4 | 0.63 |
| H‐VUSY (11.5) | 696 | 0.46 | 0.27 | 0.19 | 3.2 | 0.80 |
| H‐SDUSY (32.3) | 799 | 0.53 | 0.27 | 0.26 | 2.8 | 0.30 |
[a] After third reuse and (re)calcination.
Upon heating, the thermally stable substrate melts (melting point of 2 is ca. 98–100 °C)9a and diffuses into the zeolite pores, thus resulting in a dry, free flowing powder (see Movie S1). Monitoring the reaction as a function of time at 200 °C showed rapid, acid‐catalyzed isomerization of 2 into the γ‐lactone 3, with subsequent aromatization being more difficult (Figure 1). Notably, the aromatization reactions are, as expected, accompanied by H2 release (see Movie S2).
Figure 1.
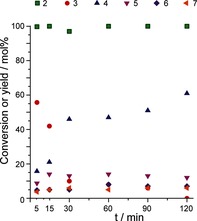
Conversion of 2 and product yields as a function of time at 200 °C over H‐Y at 1:1 w/w ratio.
A blank reaction, without catalyst, was performed at 200 °C under N2 and did not result in conversion of either 2 or 3. Trace metal analyses for potential metal contaminants in zeolite Y samples revealed negligibly low concentrations of Cu, Fe, Ni, V, and Zn (see Table S1), which could potentially catalyze the required dehydrogenation step. This finding indicates that the H2‐evolving step is most likely thermal and noncatalytic. Full conversion of 2 was already seen after 5 minutes at 200 °C, thus yielding 59 % of 3, 16 % of 4, and 9 % of 5, and 5 % of 6. The γ‐lactone 3 is then subsequently gradually aromatized and then not observed anymore after 120 minutes of reaction (see Table S3). At this point, 4 is obtained in 61 % yield, together with 19 % decarboxylation products (5+6). Notably, the increase in 4 observed between 30 and 120 minutes is higher than the amount of 3 left after 30 minutes, thus implying that there may be an intermediate which is not observed, yet contributes to the formation of 4. The yield of the transfer hydrogenation product 7 is as low as 6 %, with 7 being formed mainly at short reaction times.9a
The aromatic product distribution shows that the selectivity for 4 in terms of total aromatics does not change significantly after 30 minutes (73 %). This observation strongly suggests that the rate of the transformation of 3 is directly proportional to the rate of formation of 4. For comparison, the previously reported reactions run with the physical mixture of H‐Y and Pd/C at 200 °C in toluene showed full conversion of 2 only after 24 h at 200 °C, thus giving a similar aromatic product distribution.9a That the extent of decarboxylation is similar for the solid‐phase and solution‐phase reactions, suggests that this selectivity is dictated by the zeolite only.
Variation of the reaction temperature showed that the highest yield of 4 (61 %) and the combined aromatics (80 %) are obtained at 200 °C after 120 minutes (Figure 2). The decarboxylation selectivity increases with temperature at the expense of the formation of 4, with 5+6 making up 12 and 43 % of the total yield of aromatics (4+5+6) at the lowest and highest temperatures, respectively (Figure 2). The use of H‐Beta as the catalyst (SiO2/Al2O3=25) gave a higher yield of 3 (30 %) and a much lower yield of the aromatic products (39 %) under standard reaction conditions (Table 2). As a result of the smaller dimensions of the BEA microstructure compared to that of the FAU‐type H‐Y, not only 2 but also 3 can access the Brønsted acid sites more easily in H‐Y. With conversion of 2 into 3 being more facile, this step could also be catalyzed by weaker acid sites at the external surface of H‐Beta or within its mesopores. Of the zeolite Y catalysts tested (Table 2), H‐Y proved most selective for aromatic product formation. A comparison of the Si/Al ratio and catalyst performance shows that production of the aromatics is not solely related to zeolite acidity. Indeed, the least acidic H‐SDUSY (SiO2/Al2O3=32.3) is more selective than H‐VUSY. These differences in selectivity can be attributed to the degree of mesoporosity, which may enhance mass transport.
Figure 2.
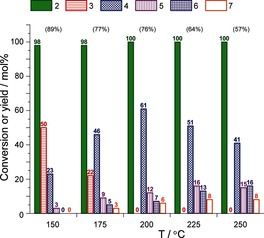
Effect of temperature on the conversion of 2 and product distribution in the aromatization of 2 with H‐Y (1:1 w/w, 120 min). The values within parentheses indicate the amount of 4 formed as a percentage of the total yield of the aromatics (4+5+6).
Table 2.
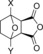
Catalytic aromatization of hydrogenated Diels–Alder adducts using the new solid‐phase route developed in this work.[a]
| Solid acid catalyst (SiO2/Al2O3) | Conversion (mol %)[b] | Molar yield (mol %)[b] | Total aromatics yield [% ] (4+5+6) | Mole balance [%][c] | |||||
|---|---|---|---|---|---|---|---|---|---|
|
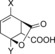
|
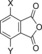
|
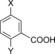
|
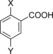
|
|
||||
| H‐Beta (25) | X=CH3; Y=H | 97 | 30 | 27 | 7 | 5 | 2 | 39 | 74 |
| H‐Y (5.2) | X=CH3; Y=H | 100 | –[d] | 61 | 12 | 7 | 6 | 80 | 86 |
| H‐VUSY (11.5) | X=CH3; Y=H | 100 | 15 | 39 | 15 | 7 | 7 | 61 | 83 |
| H‐SDUSY (32.3) | X=CH3; Y=H | 100 | –[d] | 50 | 15 | 5 | 5 | 70 | 75 |
| H‐Y (5.2) | X=Y=H | 100 | 26 | 32 | 25[e] | –[d] | 57 | 83 | |
| H‐Y (5.2) | X=Y=CH3 | 100 | –[d] | 69 | 11[f] | 10[g] | –[d] | 90 | 90 |
[a] Reaction conditions: 0.1 g of hydrogenated adduct, 0.1 g solid‐acid catalyst, 120 min, 200 °C, catalyst is indicated in the Table. [b] Calculated by q‐NMR using 1,4‐dinitrobenzene as the internal standard. [c] Mole balance determined from the total number of moles determined by 1H NMR analysis of the crude reaction mixture. [d] Not observed. [e] Benzoic acid. [f] p‐Xylene. [g] 2,5‐Dimethylbenzoic acid.
The hydrogenated DA adducts of maleic anhydride (MA) with furan or DMF were also investigated (Table 2). As seen before for the liquid‐phase experiments, the DMF‐derived adduct gave the highest cumulative selectivity towards aromatics (90 %). Full decarboxylation was seen to a limited extent for this substrate, with p‐xylene being obtained in 11 % yield. The decarboxylation selectivity for 2,5‐dimethylbenzoic acid is the same as previously seen in solution. Importantly, the reactivity of the furan‐derived adduct is significantly improved in the solid phase (100 % conversion and 57 % total aromatics selectivity), compared to only 24 % conversion and 14 % yield of the aromatics in solution,9a with activity presumably limited by the number of Brønsted acid sites under the latter reaction conditions. Decarboxylation is more severe for this substrate with benzoic acid being obtained in 25 % yield.
Facile catalyst reuse is key for the practical application of such a solid‐phase, zeolite‐only synthesis route, which would be expected to be applied in batch mode. Recovered H‐Y catalyst was therefore first reused directly, that is, after applying only a washing and mild drying step. While activity remained the same, selectivity towards aromatic products was significantly decreased. This decrease was attributed to the deposition of carbonaceous materials, as evidenced by the coloration of the zeolite (see also TGA profiles in Figure S5). Calcination of the recovered, spent H‐Y catalyst at 550 °C in air before reuse proved effective for regeneration, as aromatics yields were fully restored. In fact, the yield of 4 remained almost unchanged after three runs (Figure 3). Importantly, the overall acidity and textural properties of the catalyst were not affected by the reaction or the regeneration process (Table 1).
Figure 3.
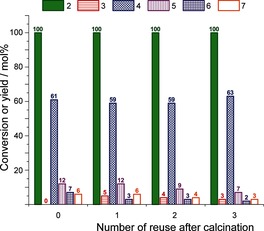
Recyclability tests using H‐Y in the aromatization of 2 (1:1 w/w, 120 min).
In conclusion, our results demonstrate the feasibility of producing renewable aromatic chemicals from biomass‐derived furanics through a zeolite‐only‐catalyzed solid‐phase reaction. The attractive features of this novel approach include shorter reaction times, mild reaction conditions, operational simplicity, and facile reuse of a relatively inexpensive catalyst. We anticipate that this approach can be applied not only to a wide range of dienophile/diene combinations, but also to other types of zeolite‐catalyzed reactions.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary
Acknowledgements
This work is part of research program Technology Areas for Sustainable Chemistry (TASC), which is partly financed by the Netherlands Organization for Scientific Research (NWO). Guus Frissen is acknowledged for some of the NMR analyses.
S. Thiyagarajan, H. C. Genuino, J. C. van der Waal, E. de Jong, B. M. Weckhuysen, J. van Haveren, P. C. A. Bruijnincx, D. S. van Es, Angew. Chem. Int. Ed. 2016, 55, 1368.
Contributor Information
Dr. Ed de Jong, Email: ed.dejong@avantium.com
Dr. Pieter C. A. Bruijnincx, Email: p.c.a.bruijnincx@uu.nl
Dr. Daan S. van Es, Email: daan.vanes@wur.nl
References
- 1.
- 1a. Christensen C. H., Rass-Hansen J., Marsden C. C., Taarning E., Egeblad K., ChemSusChem 2008, 1, 283–289; [DOI] [PubMed] [Google Scholar]
- 1b. Vennestrøm P. N. R., Osmundsen C. M., Christensen C. H., Taarning E., Angew. Chem. Int. Ed. 2011, 50, 10502–10509; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 10686–10694; [Google Scholar]
- 1c. van Putten R.-J., Dias A. S., de Jong E. in Catalytic Process Development for Renewable Materials (Eds.: P. Imhof, J. C. van der Waal), Wiley-VCH, Weinheim, 2013, pp. 81–117; [Google Scholar]
- 1d. Esposito D., Antonietti M., Chem. Soc. Rev. 2015, 44, 5821–5835. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Bruijnincx P. C. A., Weckhuysen B. M., Angew. Chem. Int. Ed. 2013, 52, 11980–11987; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12198–12206; [Google Scholar]
- 2b. Dauenhauer P. J., Huber G. W., Green Chem. 2014, 16, 382–383. [Google Scholar]
- 3.
- 3a. van Putten R.-J., van der Waal J. C., de Jong E., Rasrendra C. B., Heeres H. J., de Vries J. G., Chem. Rev. 2013, 113, 1499–1597. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Williams C. L., Chang C.-C., Do P., Nikbin N., Caratzoulas S., Vlachos D. G., Lobo R. F., Fan W., Dauenhauer P. J., ACS Catal. 2012, 2, 935–939; [Google Scholar]
- 4b. Chang C.-C., Green S. K., Williams C. L., Dauenhauer P. J., Fan W., Green Chem. 2014, 16, 585–588. [Google Scholar]
- 5. Wang D., Osmundsen C. M., Taarning E., Dumesic J. A., ChemCatChem 2013, 5, 2044–2050. [Google Scholar]
- 6. Pacheco J. J., Davis M. E., Proc. Natl. Acad. Sci. USA 2014, 111, 8363–8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Green S. K., Williams C. L., Dauenhauer J., Fan W., Green Chem. 2014, 16, 585–588; [Google Scholar]
- 7b. Nikbin N., Caratzoulas S., Vlachos D. G., Appl. Catal. A 2014, 485, 118–122; [Google Scholar]
- 7c. Green S. K., Patet R. E., Nikbin N., Williams C. L., Chang C.-C., Yu J., Gorte R. J., Caratzoulas S., Fan W., Vlachos D. G., Dauenhauer P. J., Appl. Catal. B 2016, 180, 487–496. [Google Scholar]
- 8. Mahmoud E., Watson D. A., Lobo R. F., Green Chem. 2014, 16, 167–175. [Google Scholar]
- 9.
- 9a. Thiyagarajan S., Genuino H. C., Śliwa M., van der Waal J. C., de Jong E., van Haveren J., Weckhuysen B. M., Bruijnincx P. C. A., van Es D. S., ChemSusChem 2015, 8, 3052–3056; [DOI] [PubMed] [Google Scholar]
- 9b. van der Waal J. C., de Jong E., van Haveren J., Thiyagarajan S., NL2014023, 2014.
- 10. Shiramizu M., Toste F. D., Chem. Eur. J. 2011, 17, 12452–12457. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Morgan J. A., Lu Z., Clark D. S., J. Mol. Catal. B 2002, 18, 147–154; [Google Scholar]
- 11b. Zaks A., Dodds D. R., J. Am. Chem. Soc. 1995, 117, 10419–10424. [Google Scholar]
- 12.
- 12a. James S. L., Adams C. J., Bolm C., Braga D., Collier P., Friščić T., Grepioni F., Harris K. D. M., Hyett G., Jones W., Krebs A., Mack J., Maini L., Orpen A. G., Parkin I. P., Shearouse W. C., Steed J. W., Waddell D. C., Chem. Soc. Rev. 2012, 41, 413–447; [DOI] [PubMed] [Google Scholar]
- 12b. Wang G. W., Chem. Soc. Rev. 2013, 42, 7668–7700; [DOI] [PubMed] [Google Scholar]
- 12c. Curran D. P., Nat. Chem. 2012, 4, 958; [DOI] [PubMed] [Google Scholar]
- 12d. Rodríguez B., Rantanen T., Bolm C., Angew. Chem. Int. Ed. 2006, 45, 6924–6926; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7078–7080. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Supplementary
Supplementary