

Abstract
This study aims to systematically explore the alkylation effect of 7-OH in silibinin and 2,3-dehydrosilibinin on the antiproliferative potency toward three prostate cancer cell lines. Eight 7-O-alkylsilibinins, eight 7-O-alkyl-2,3-dehydrosilibinins, and eight 3,7-O-dialkyl-2,3-dehydrosilibinins have been synthesized from commercially available silibinin for the in vitro cell-based evaluation. The WST-1 cell proliferation assay indicates that nineteen out of twenty-four silibinin derivatives have significantly improved antiproliferative potency when compared with silibinin. 7-O-Methylsilibinin (2) and 7-O-ethylsilibinin (3) have been identified as the most potent compounds with 98- and 123-fold enhanced potency against LNCaP human androgen-dependent prostate cancer cell line. 7-O-Methyl-2,3-dehydrosilibinin (10) and 7-O-ethyl-2,3-dehydrosilibinin (11) have been identified as the optimal compounds with the highest potency towards both androgen-dependent LNCaP and androgen-independent PC-3 prostate cancer cell lines. 7-O-Ethyl-2,3-dehydrosilibinin (11) was demonstrated to arrest PC-3 cell cycle at the G0/G1 phase and to induce PC-3 cell apoptosis. The findings in this study suggest that antiproliferative potency of silibinin and 2,3-dehydrosilibinin can be appreciably enhanced through suitable chemical modifications on the phenolic hydroxyl group at C-7 and that introduction of a chemical moiety with the potential to improve bioavailability through a linker to 7-OH in silibinin and 2,3-dehydrosilibinin would be a feasible strategy for the development of silibinin derivatives as anti-prostate cancer agents.
Keywords: silibinin derivatives, anti-proliferative activity, prostate cancer, structure-activity relationship, cell apoptosis
Graphical Abstract
1. Introduction
Approximately 300,000 men worldwide, of whom 28,000 are U.S. men, die each year of castration-resistant prostate cancer due to inevitable refractory progression on the first-line treatment with docetaxel [1,2]. As a consequence, prostate cancer has been identified as the fifth and the second leading cause of cancer-related deaths in men worldwide and in American men, respectively. Several new treatments have recently been approved by the US Food and Drug Administration for patients with castration-resistant prostate cancer, but only with very little survival benefit [3,4]. It is thus an urgent need to continue searching for more effective therapeutics for this deadly disease.
Milk thistle (Silybum marianum) and its whole extract silymarin have long been used as chemotherapeutics for hepatotoxicity caused by poisoning mushroom and oxidative xenobiotics, and as dietary supplements for the prevention of hepatotoxicity in Europe and Asia [5]. Silibinin (1, Scheme 1), consisting of a 1:1 mixture of a pair of diastereoisomeric silybin A and silybin B, is the most abundant chemical components of silymarin [6]. Silybin A and silybin B are naturally occurring hybrid molecules of flavonoid and lignan. Several in vitro cell-based and in vivo animal studies have demonstrated that silymarin, silibinin, silybin A, and silybin B possess the potential in treating prostate cancer, but with moderate potency [7,8]. A phase I trial of silibinin-phytosome, a formula of silibinin, at a dose of 13 g/day suggested the safety profiles of silibinin in human but poor oral bioavailability due to first-pass metabolism of glucuronidation and sulfation. The phase II studies in patients with localized prostate cancer revealed that oral administration of high-dose silibinin-phytosome led to high concentration of silibinin in blood but no detectable silibinin in prostate gland [9,10]. Hence, structure modifications of silibinin to engineer new chemical entities with enhanced potency and bioavailability are highly desirable and are the core focus of our research project.
Scheme 1.

Synthesis of silibinin derivatives (2-25). Reactants and conditions: (i) RX, K2CO3, DMF (or acetone); (ii) (a) Air, CH3COOK, DMF, 60 °C, 6 h; (b) RX, K2CO3, DMF
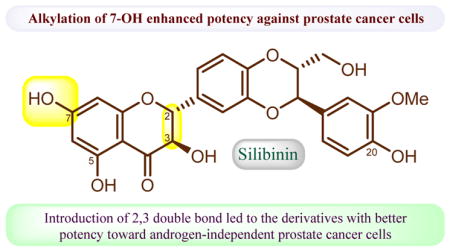
We started this project from exploring the optimal structure moieties of silibinin suitable for modification by designing and synthesizing three groups of silibinin derivatives for in vitro cell-based evaluations against three prostate cancer cell lines. Silibinin was selected as our lead compound because it exhibited similar antiproliferative potency toward prostate cancer cells as each of its two optically pure antipodes (silybin A and silybin B) [11]. 7-O-Methylsilibinin (2) and 2,3-dehydrosilibinin have been demonstrated to possess enhanced antiproliferative potency toward human prostate cancer cells [12,13]. Additionally, 7-O-methylsilibinin (2), 7-O-benzylsilibinin (9), 7-O-methyl-2,3-dehydrosilibinin (10), 7-O-benzyl-2,3-dehydrosilibinin (14), 3,7-O-dimethylsilibinin (18), and 3,7-O-dibenzylsilibinin (25) have been reported by Kren and co-workers to show better inhibitory effects on P-glycoprotein modulatory activity than silibinin [14]. However, only methylated and benzylated derivatives of silibinin have been reported so far [15] while no synthesis and antiproliferative activities of other alkylated derivatives against prostate cancer cell lines have been systematically investigated. In this study, twenty-four alkylated derivatives of silibinin and 2,3-dehydrosilibinin have been synthesized and evaluated as anti-prostate cancer agents. Among them, eighteen new derivatives were prepared from silibinin for the first time. The above-mentioned six known compounds were also synthesized and evaluated for better understanding of the structure-activity relationship data. The findings from this study will pave the way for our further structural manipulations of silibinin to develop potential anti-prostate cancer agents. The design, synthesis, anti-proliferative activity, and structure-activity relationships of these silibinin derivatives were presented in this paper. The cell apoptosis induction and cell cycle regulation by a representative derivative were also described.
2. Results and Discussion
2.1 Chemistry
The challenge for the synthesis of 7-O-alkylsilibinins and 7-O-alkyl-2,3-dehydrosilibinins lies in the competitive reactivity of phenolic hydroxyl groups and the oxidation of silibinin to 2,3-dehydrosilibinin [15]. Selective methylation (53%) and benzylation (81%) of the C-7 phenolic hydroxyl group of silibinin have been achieved by Kren et al. through refluxing with benzyl bromide or methyl iodide in the presence of potassium carbonate and anhydrous acetone [14]. Oberlies and co-workers have also identified the same reaction conditions (MeI, K2CO3, acetone, 30–60°C) for the optimal synthesis of 7-O-methylsilibinin [16]. We have successfully synthesized 7-O-methylsilibinin (2) and 7-O-benzylsilibinin (9) using the above-mentioned procedure in acceptable yields (44% and 54%). However, little to no yields were achieved when we extended this method to prepare other alkylated derivatives. Various conditions were explored by using different bases, solvents, and reactant equivalents. Consequently, as shown in Scheme 1, 7-O-propylsilibinin (4), 7-O-propyl-2,3-dehydrosilibinin (12), 3,7-O-dipropyl-2,3-dehydrosilibinin (20), 7-O-butylsilibinin (5), 3,7-O-dibutyl-2,3-dehydrosilibinin (21), 7-O-pentyl-2,3-dehydrosilibinin (14), 7-O-heptyl-2,3-dehydrosilibinin (16), and 3,7-O-diheptyl-2,3-dehydrosilibinin (24) were prepared from silibinin (1) by treating with 0.75 equivalents of appropriate alkyl halide in the presence of potassium carbonate in DMF (1M) at room temperature. The limiting amount of alkyl halides was used here to prevent multiple alkylation of silibinin. An alternative method for preparation of silibinin derivatives was to dilute the reaction mixture from 1M to 0.23–0.25 M concentration while using the same base (potassium carbonate) and solvent (DMF). Derivatives 3,7-O-dimethyl-2,3-dehydrosilibinin (18), 7-O-ethylsilibinin (3), 7-O-pentylsilibinin (6), 3,7-O-dipentyl-2,3-dehydrosilibinin (22), 7-O-hexylsilibinin (7), 3,7-O-dihexyl-2,3-dehydrosilibinin (23), and 7-O-hexyl-2,3-dehydrosilibinin (15) were prepared from this method. 3,7-O-dimethyl-2,3-dehydrosilibinin (18), 3,7-O-diethyl-2,3-dehydrosilibinin (19) and 3,7-O-dibenzyl-2,3-dehydrosilibinin (25) were prepared through the oxidation of silibinin mediated by potassium acetate in ambient air followed by alkylation at both 7-OH and 3-OH using potassium carbonate as base in DMF. The above-mentioned twenty-two silibinin derivatives were prepared from direct alkylation of silibinin (1). Two other derivatives, 7-O-methyl-2,3-dehydrosilibinin (10) and 7-O-ethyl-2,3-dehydrosilibinin (11) were prepared through the oxidation of 7-O-methylsilibin (2) and 7-O-ethylsilibin (3) in ambient air catalyzed by potassium acetate in DMF.
The structures of the twenty-four silibinin derivatives were characterized by 1D- and 2D NMR analysis in combination with HRMS data. Table 1 summarizes the 1H and 13C NMR data for compounds 4, 13 and 24 that were fully assigned based on the interpretation of their COSY, HMQC, and HMBC data. The signals in the 1H NMR and 13C NMR spectra (δH 3.96-3.92, 1.80, and 1.02; δC 70.4, 22.4, 10.5) of compound 4 and the HRMS data confirmed the addition of a propyl group to 4 when compared with silibinin (1). The propyl group in derivative 4 was assigned to 7-OH based on the key HMBC correlations from the signal at δH 3.96-3.92 (CH2 in propyl) to the signal at δC 168.7 (C-7, Figure 1). Similarly, the butyl group in compound 13 was assigned to 7-OH; and two heptyl groups in compound 24 were assigned to 3-OH and 7-OH (Figure 1 and Table 1). It should be noted that silibinin, serving as the starting material for all syntheses, was a nearly equimolar mixture of the silybin A and silybin B diastereoisomers. Consequently, some NMR signals of 7-O-alkylsilibinins, existing as a pair of diastereoisomers, appear as a pair (Table 1 and experimental section). This issue does not exist with 2,3-dehydrosilybin derivatives as the NMR signals for both enantiomers are undistinguishable.
Table 1.
NMR data for derivatives 4, 13, and 24 (1H NMR: 300 MHz; 13C NMR: 75 MHz).
| Position | 4 (CDCl3) | 13 (CDCl3) | 24 (CDCl3) | |||
|---|---|---|---|---|---|---|
|
|
|
|||||
| δC, type | δH, J in Hz) | δC, type | δH, (J in Hz) | δC, type | δH, (J in Hz) | |
|
|
|
|
|
|||
| 2 | 83.2, CH | 5.00, d (12.0) | 145.3, C | - | 155.6, C | - |
| 3 | 72.6 (72.5), CH | 4.55 (4.54), d (12.0) | 136.2, C | - | 138.6, C | - |
| 4 | 195.9, C | - | 175.4, C | - | 179.1, C | - |
| 4a | 100.8, C | - | 104.0, C | - | 106.1, C | - |
| 5 | 163.8, C | - | 160.9, C | - | 162.1, C | - |
| 6 | 96.1, CH | 6.12 (6.11), d (2.1) | 98.6, CH | 6.36, s | 98.5, CH | 6.33, d (2.1) |
| 7 | 168.7, C | - | 165.6, C | - | 165.2, C | - |
| 8 | 95.2, CH | 6.05 (6.04), d (2.1) | 92.7, CH | 6.45, s | 92.6, CH | 6.40, d (2.1) |
| 8a | 163.0, C | - | 157.0, C | 156.9, C | - | |
| 10 | 78.5, CH | 4.03–4.08, m | 78.9, CH | 4.16-4.12, m | 78.9, CH | 4.15-4.10, m |
| 11 | 76.6 (76.5), CH | 4.97, d (8.4) | 76.6, CH | 5.01, d (8.4) | 76.5, CH | 5.01, d (8.4) |
| 12a | 144.12 (144.09), C | - | 144.0, C | - | 143.7, C | - |
| 13 | 117.5 (117.2), CH | 7.08 (7.11), d (1.8) | 116.9, CH | 7.88, s | 117.8, CH | 7.77, d (2.1) |
| 14 | 129.72 (129.67), C | - | 124.5, C | - | 124.3, C | - |
| 15 | 121.3 (121.05), CH | 7.11 (7.08), dd (8.1, 1.8) | 121.9, CH | 7.83, d (8.4) | 122.9, CH | 7.74, dd (8.4, 2.1) |
| 16 | 116.8 (116.6), CH | 7.21, dd (7.2, 1.5) | 117.4, CH | 7.10, d (8.4) | 117.1, CH | 7.06, d (9.0) |
| 16a | 144.32 (144.27), C | - | 145.1 C | - | 145.7 C | - |
| 17 | 128.0, C | - | 127.8, C | - | 127.8, C | - |
| 18 | 109.7, CH | 6.94, t (1.5) | 109.7, CH | 6.98, s | 109.7, CH | 6.98, s |
| 19 | 147.1, C | - | 147.2, C | - | 147.1, C | - |
| 20 | 146.6, C | - | 146.7, C | - | 146.7, C | - |
| 21 | 114.8, CH | 6.96, s | 114.9, CH | 6.99, s | 114.9, CH | 7.00, s |
| 22 | 121.0, CH | 6.96, s | 121.1, CH | 6.99, s | 121.0, CH | 7.00, |
| 23 | 61.9, CH2 | 3.82, dd (12.6, 2.7) | 61.9, CH2 | 3.86, dd (12.6, 3.6) | 61.8, CH2 | 3.86, dd (12.6, 2.1) |
| 3.57, dd (12.6, 4.2) | 3.60, dd (12.6, 3.6) | 3.60, dd (12.6, 3.0) | ||||
|
|
|
|||||
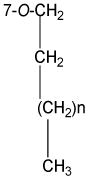
|
70.4, CH2 | 3.96-3.92, overlapped | 68.6, CH2 | 4.03, t (6.6) | 68.9, CH2 | 4.01, t (6.6) |
| 22.4, CH2 | 1.80, Hex (7.5) | 31.1, CH2 | 1.81 quin (7.5) | 31.9 CH2 | 1.85-1.69 m | |
| - | - | 19.3, CH2 | 1.51 Hex (7.5) | 30.2,a 29.1, 26.1, 22.8 (CH2)n | 1.48-1.28 m | |
| 10.5, CH3 | 1.02, t (7.5) | 14.0 CH3 | 1.00, t (7.5) | 14.2 CH3 | 0.90 t(6.6) | |
|
|
|
|||||
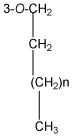
|
- | - | - | - | 73.1, CH2 | 4.01, t (6.6) |
| - | - | - | - | 31.9 CH2 | 1.85-1.69 m | |
| - | - | - | - | 29.2,a 29.1, 26.1, 22.8. (CH2)n | 1.48-1.28 m | |
| - | - | - | - | 14.2 CH3 | 0.88 t(6.9) | |
|
| ||||||
| 19-OMe | 56.2, CH3 | 3.93, s | 56.3, CH3 | 3.95, s | 56.2, CH3 | 3.94, s |
| 3-OH | - | - | - | 6.54, s | - | - |
| 5-OH | - | 11.18 (11.17), s | - | 11.67, s | - | 12.65, s |
| 20-OH | - | 5.75, s | - | 5.79, s | - | 5.75, s |
| 23-OH | - | - | - | - | - | 2.05, s |
These two signals are exchangeable.
Figure 1.

Key HMBC correlations in derivatives 4, 13, and 24
2.2 Anti-proliferative effects towards three prostate cancer cell lines
The effects of various alkyl groups with different chain length and attaching points as well as the C2-C3 double bond on the proliferation of prostate cancer cells were explored. The in vitro anti-proliferative activity of silibinin derivatives was evaluated using WST-1 cell proliferation assay according to the procedure as described in the Experimental Section in both androgen-sensitive (LNCaP) and androgen-insensitive (PC-3 and DU145) human prostate cancer cell lines. Silibinin was used as a positive control for comparison in the parallel experiments and the IC50 values are summarized in Table 2. Nineteen silibinin derivatives exhibited better anti-proliferative potency by comparing their IC50 values with that of silibinin (Table 2). All synthesized 7-O-alkylsilibinins as well as silibinin were more effective in androgen-dependent prostate cancer cells (LNCaP), with IC50 values ranging from 0.35 to 4.66 μM, than in androgen-independent prostate cancer cells (DU145 and PC-3), with IC50 values ranging from 5.29 to 30.33 μM. The 7-O-alkylsilibinins (2-9) are significantly more potent than silibinin toward LNCaP cells. The two most promising derivatives (methyl derivative 2 and ethyl derivative 3) were 98-time and 123-time more potent than silibinin toward the LNCaP cell line based on the comparison of their IC50 values. The antiproliferative activities of all synthesized 7-O-alkyl-2,3-dehydrosilibinins (10-17) were improved by 9 to 30 times when compared with silibinin. This subgroup of 2,3-dehydrosilibinin derivatives demonstrated nearly equivalent potency against both androgen-independent (DU145 and PC-3) and androgen-dependent (LNCaP) prostate cancer cell lines with IC50 values in the range of 2 to 9 μM. The IC50 values of 3,7-O-dialkyl-2,3-dehydrosilibinins (18-25) varied widely from 5 to over 100 μM. The potency of the derivatives significantly decreased with increasing length of the alkyl group, with the ethyl derivative 19 showing the highest potency toward all 3 cell lines (IC50 = 5.57, 10.36, and 8.50 μM for LNCaP, DU145 and PC-3 respectively).
Table 2.
In vitro anti-proliferative activity (IC50, μM)a of the compounds against prostate cancer cell lines
| Comp. No | IC50 (μM) | IC50 (silibinin)/IC50 (derivative) | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| LNCaP b | DU145 c | PC-3d | LNCaP | DU145 | PC-3 | |
| silibinin | 43.03 ± 7.84 | 93.34 ± 13.76 | 72.65 ± 3.15 | 1 | 1 | 1 |
|
| ||||||
| 2 | 0.44 ± 0.03 | 25.09 ± 3.81 | 12.37 ± 2.76 | 98 | 4 | 6 |
| 3 | 0.35 ± 0.16 | 27.72 ± 3.46 | 16.11 ± 1.35 | 123 | 3 | 5 |
| 4 | 4.66 ± 2.95 | 30.33 ± 3.79 | 18.47 ± 1.42 | 9 | 3 | 4 |
| 5 | 2.51 ± 1.64 | 11.96 ± 3.28 | 5.38 ± 0.89 | 17 | 8 | 14 |
| 6 | 1.78 ± 0.47 | 8.93 ± 0.40 | 6.17 ± 0.68 | 24 | 10 | 12 |
| 7 | 1.65 ± 0.42 | 11.35 ± 2.25 | 5.97 ± 0.36 | 26 | 8 | 12 |
| 8 | 4.57 ± 0.40 | 17.35 ± 0.79 | 10.91 ± 1.80 | 9 | 5 | 7 |
| 9 | 2.04 ± 0.19 | 8.77 ± 0.85 | 5.29 ± 0.32 | 21 | 11 | 14 |
|
| ||||||
| 10 | 2.76 ± 0.18 | 7.92 ± 0.55 | 2.39 ± 0.97 | 16 | 12 | 30 |
| 11 | 2.58 ± 0.07 | 7.59 ± 0.66 | 3.25 ± 0.31 | 17 | 12 | 22 |
| 12 | 4.41 ± 2.07 | 5.36 ± 1.18 | 4.69 ± 0.62 | 10 | 17 | 15 |
| 13 | 3.19 ± 0.24 | 5.81 ± 1.03 | 4.55 ± 0.33 | 13 | 16 | 16 |
| 14 | 4.62 ± 0.16 | 6.92 ± 0.19 | 6.05 ± 0.29 | 9 | 14 | 12 |
| 15 | 4.01 ± 1.01 | 8.84 ± 0.87 | 6.94 ± 0.65 | 11 | 11 | 10 |
| 16 | 4.81 ± 0.45 | 7.49 ± 0.92 | 7.09 ± 0.30 | 9 | 12 | 10 |
| 17 | 3.71 ± 0.14 | 8.97 ± 0.98 | 7.98 ± 0.35 | 12 | 10 | 9 |
|
| ||||||
| 18 | 6.88 ± 2.37 | 22.24 ± 2.43 | 15.27 ± 4.44 | 6 | 4 | 5 |
| 19 | 5.57 ± 2.47 | 10.36 ± 1.29 | 8.50 ± 1.47 | 8 | 9 | 9 |
| 20 | 10.95 ± 3.23 | 20.60 ± 1.45 | 17.41 ± 4.63 | 4 | 5 | 5 |
| 21 | 46.57 ± 10.06 | 36.49 ± 2.68 | 45.76 ± 11.61 | 1 | 3 | 2 |
| 22 | 28.74 ± 8.07 | 28.70 ± 1.42 | 52.15 ± 2.38 | 1 | 3 | 1 |
| 23 | >100 | >100 | >100 | <1 | <1 | <1 |
| 24 | >100 | >100 | >100 | <1 | <1 | <1 |
| 25 | >100 | >100 | >100 | <1 | <1 | <1 |
IC50 is the drug concentration effective in inhibiting 50% of the cell viability measured by the WST-1 cell proliferation Assay after 3 days exposure.
Human androgen-sensitive prostate cancer cell line
Human androgen-independent prostate cancer cell line
Human androgen-independent prostate cancer cell line
The structure-antiproliferative activity relationships of silibinin derivatives based on this study can be summarized as below:
Methylation and ethylation at 7-OH of silibinin led to appreciable enhancement of antiproliferative potency (98 and 123 times) against androgen-dependent human prostate cancer cells, but only moderate improvement of potency against androgen-independent prostate cancer cells.
Propylation, butylation, and pentylation at 7-OH of silibinin resulted in significant increase in antiproliferative potency against both androgen-dependent and androgen-independent prostate cancer cell lines.
2,3-dehydrosilibinin derivatives possess better anti-proliferative potency than silibinin derivatives with C2-C3 single bond toward androgen-resistant human prostate cancer cell lines (DU145 and PC-3).
The effect of the length of 7-O-alkyl group in 2,3-dehydrosilibinin derivatives on the antiproliferative potency is not apparent, suggesting that 7-OH of 2,3-dehydrosilibinin can act as one optimal moiety for chemical manipulations.
3,7-O-Dialkyl-2,3-dehydrosilibinins are apparently less potent than the corresponding 7-O-alkyl-2,3-dehydrosilibinin against three prostate cancer cell lines.
The antiproliferative potency of 3,7-O-dialkyl-silibinins against three prostate cancer cell lines varied with the length of the alkyl group. The activity is completely diminished as the alkyl chain reaches 6 carbon atoms.
2.3 Effects of 7-O-ethyl-2,3-dehydrosilibinin (11) on PC-3 cell cycle progression and apoptosis
It has been reported that silibinin can arrest rat (H-7 and I-8) and human prostate cancer cell (LNCaP) cycle at G1 phase [17, 18]. 7-O-Ethyl-2,3-dehydrosilibinin (11) was selected for flow cytometry evaluation of its effect on PC-3 cell cycle regulation because it is one of the derivatives that exhibited optimal cell proliferation inhibition on both androgen-dependent LNCaP and androgen-independent PC-3 prostate cancer cell models. At 20 μM, compound 11 can cause PC-3 cell accumulation in a G0/G1 phase (Figure 3) by increasing the cell population in this phase from 49% (control) to 62% (treated with 11) at 24 hours, and from 55% (control cells) to 61% (treated with 11) at 30 hours. The cell population in G2 phase slightly decreased from 34% in control cells to 23% at 24 hours, and from 30% in control cells to 25% at 30 hours.
Figure 3.

Cell cycle analysis of PC-3 cells. PC-3 cancer cells were untreated or treated with 7-O-ethyl-2,3-dehydrosilibinin (11). Cells were harvested after 24 and 30 hours, fixed, stained, and analyzed for DNA content. The distribution and percentage of cells in G1/G0, and G2 phase of the cell cycle are indicated.
Silibilin was reported by Agarwal and co-workers to induce cell apoptosis and to suppress cell proliferation in PC-3 tumor xenografts in a time-dependent manner [19]. To distinguish the apoptotic PC-3 cells from those undergoing necrosis in response to increasing doses of 7-O-ethyl-2,3-dehydrosilibinin (11), F2N12S and CYTOX AADvanced double staining flow cytometry-based assay was chosen for this study. Staurosporine was used as a specific apoptotic inducer and positive apoptotic control in all these experiments (data not shown). As shown in Figure 4, incubation of the PC-3 cells with compound 11 for 16 h induced considerable levels of apoptotic cell death in the androgen-independent PC-3 prostate cancer cell line in a dose-responsive manner. Specifically, 10 μM of compound 11 could induce detectable early phase of apoptosis in PC-3 cells as compared with control cells; treatment with 30 μM of compound 11 led to 68% early apoptotic cells and 25% late apoptotic/necrotic cells. Both apoptotic and necrotic cell populations increased in response to increasing concentration of compound 11 (0–50 μM final concentration range). The maximal apoptotic cell population was reached when the PC-3 cancer cells were exposed to compound 11 at the concentration of 40 μM.
Figure 4.

Evolution of viable, apoptotic, and necrotic PC-3 cells populations in response to increasing dosages of 7-O-ethyl-2,3-dehydrosilibinin (11).
3. Conclusion
In summary, twenty-four silibinin derivatives including eight 7-O-alkylsilibinins (2-9), eight 7-O-alkyl-2,3-dehydrosilibinins (10-17), and eight 3,7-O-dialkyl-2,3-dehydrosilibinins (18-25) have been synthesized from commercially available slibinin. Their in vitro antiproliferative activities against both androgen-dependent and androgen-independent prostate cancer cell lines have been assessed by WST-1 cell proliferation assay. Nineteen of them are more potent than silibinin. 7-O-Methylsilibinin (2) and 7-O-ethylsilibinin (3) have been identified as the optimal compounds with 98- and 123-fold increase in antiproliferative potency against LNCaP human androgen-dependent prostate cancer cell line. Among 2,3-dehydrosilibinin derivatives, 7-O-methyl-2,3-dehydrosilibinin (10) and 7-O-ethyl-2,3-dehydrosilibinin (11) emerged as the most potent compounds against both androgen-dependent LNCaP and androgen-independent PC-3 prostate cancer cell lines. Similar to silibinin, 7-O-ethyl-2,3-dehydrosilibinin (11) was proved to be capable of inducing PC-3 cell cycle regulation in G0/G1 phase and of activating PC-3 cell apoptosis. Our findings suggest that antiproliferative potency of silibinin and 2,3-dehydrosilibinin can be significantly enhanced through suitable chemical modifications on the phenolic hydroxyl group at C-7 and that introduction of a chemical moiety with the potential to improve bioavailability through a linker to 7-OH in silibinin and 2,3-dehydrosilibinin would be a viable strategy for the development of silibinin derivatives as anti-prostate cancer agents.
4. Experimental
4.1 General synthetic procedures
NMR spectra were obtained on a Bruker Fourier 300 spectrometer in CDCl3, CD3OD, or DMSO-d6. The chemical shifts are given in δ (ppm) referenced to the respective solvent peak, and coupling constants are reported in Hz. Anhydrous THF and dichloromethane were purified by PureSolv MD 7 Solvent Purification System from Innovative Technologies (MB-SPS-800). All other reagents and solvents were purchased from commercial sources and were used without further purification. Silica gel column chromatography was performed using silica gel (32–63 μM). Preparative thin-layer chromatography (PTLC) separations were carried out on 1000 μm AnalTech thin layer chromatography plates (Lot No.13401). Silibinin (> 98.0%) was purchased from Fisher Scientific (TCI America, Cat # 50-014-46874).
4.2 Synthesis of silibinin derivatives
4.2.1 General procedure for Method 1
The solution of sililbinin (1 equiv.) and potassium carbonate (3 equiv.) in acetone (0.1 M) was stirred for 10 min prior to being added an appropriate alkyl iodide or alkyl bromide (1 equiv.) through syringe. The reaction was allowed to proceed with refluxing under argon for 4 h. After cooling down to room temperature, the reaction was quenched with 1M HCl, and the acetone was removed in vacuo. The residue was diluted with ethyl acetate, and the subsequent mixture was rinsed with brine. The organic layer was dried over anhydrous sodium sulfate and concentrated in vacuo to give a crude product, which was purified by column chromatography over silica gel eluting with 50% ethyl acetate in hexane to yield the desired compound.
4.2.2 General procedure for Method 2
The mixture of silibinin (1 equiv.), anhydrous potassium carbonate (2 equiv.), alkyl iodide or alkyl bromide (0.75 equiv.), and DMF (1 M) was stirred under argon at room temperature for 24 h. The reaction was quenched by addition of 1 M HCl followed by diethyl ether and ethyl acetate. The subsequent mixture was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo to give a crude product. PTLC purification of the crude product, using 3% methanol in DCM as eluent, yielded the desired product.
4.2.3 General procedure for Method 3
The mixture of silibinin (1 equiv.), alkyl iodide or alkyl bromide (1 equiv.), and potassium carbonate (1 equiv.) in DMF (0.23 M) was stirred under argon at room temperature for 48 h prior to being quenched with 1M HCl. After diluting with diethyl ether and ethyl acetate (1:1, v/v), the subsequent mixture was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure. The crude mass obtained was purified by PTLC eluting with 3% methanol in DCM to give the desired product.
4.2.4 General procedure for Method 4
The solution of silibinin (1 equiv.) and potassium acetate (3 equiv.) in DMF (1 M) was stirred at room temperature overnight. To this solution were added potassium carbonate (2 equiv.) and alkyl iodide or alkyl bromide (2 equiv.), and the reaction was allowed to proceed with stirring at room temperature for additional 24 h. The reaction was quenched with 1M HCl, and the reaction mixture was then diluted with diethyl ether and ethyl acetate (1:1, v/v). The subsequent mixture was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated under reduced pressure to furnish a residue, which was purified by PTLC eluting with 3% methanol in DCM to yield the desired derivative.
4.2.5 General procedure for Method 5
To a reaction vial were added 7-O-alkylsilibinin (1 equiv.), potassium acetate (3 equiv.), and DMF (1 M). The reaction was allowed to proceed with stirring at room temperature overnight prior to being quenched with 1 M HCl. After diluting with diethyl ether and ethyl acetate (1:1, v/v), the mixture was rinsed with brine, dried over anhydrous sodium sulfate, and concentrated. Purification of the crude mass by PTLC eluting with 3% methanol in DCM generated the desired derivative.
4.2.6. 7-O-Methysilibin (2)
This derivative was prepared from silibinin by Method 1 as a white powder in 44% yield. m.p. 209.5–210 °C. IR (neat) νmax: 3373, 2981, 2955, 2887, 1639, 1574, 1509 cm−1. 1H NMR (300 MHz, CD3COCD3) δ: 11.67 (s, 1H), 7.82 (s, 1H), 7.15 (d, J = 1.8 Hz, 2H), 7.10 (dd, J = 8.5, 1.8 Hz, 1H), 7.00-6.96 (overlapped, 2H), 6.89 (dd, J = 8.7, 2.3 Hz, 1H), 6.09 (d, J = 2.2 Hz, 1H), 6.07 (d, J = 2.3 Hz, 1H), 5.14 (d, J = 11.6 Hz, 1H), 5.01 (d, J = 7.5 Hz, 1H), 4.86 (br.s, 1H), 4.70 (d, J = 11.6 Hz, 1H), 4.11-4.07 (m, 1H), 4.07-4.06 (m, 1H), 3.880 (3.876) (s, 3H), 3.86 (s, 1H), 3.76 (dd, J = 12.5, 3.3 Hz, 1H), 3.55-3.51 (m, 1H). 13C NMR (75 MHz, DMSO-d6) δ: 198.5, 167.9, 163.3, 162.7, 148.0, 147.2, 144.0, 143.5, 130.18 (130.13), 127.8, 121.7 (121.6), 120.8, 116.93 (116.89), 116.81 (116.77), 115.6, 111.88 (111.84), 101.6, 95.3, 94.1, 83.0, 78.4, 76.1, 71.9 (71.8), 60.5, 56.3, 56.0. HR-MS (ESI) m/z: calcd for C26H25O10 [M+H]: 497.1448; found 497.1448.
4.2.7 7-O-Ethylsilibinin (3)
This derivative was prepared from silibinin by Method 3 as a light yellowish solid in 23%. m.p. 195–196 °C. IR (neat) νmax: 3482, 3447, 2908, 1653, 1576, 1508 cm−1. 1H NMR (300 MHz, CD3COCD3) δ: 11.67 (s, 1H), 7.82 (s, 1H), 7.16-7.15 (overlapped, 2H), 7.10 (dt, J = 8.3, 1.9 Hz, 1H), 6.99 (d, J = 7.0 Hz, 1H), 6.97 (d, J = 7.0 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.07 (d, J = 2.0 Hz, 1H), 6.05 (d, J = 2.0 Hz, 1H), 5.13 (d, J = 11.6 Hz, 1H), 5.01 (d, J = 8.0 Hz, 1H), 4.86-4.84 (m, 1H), 4.72-4.66 (m, 1H), 4.19-4.14 (m, 2H), 4.11 (q, J = 7.0 Hz, 2H), 3.88 (3.87) (s, 3H), 3.79-3.72 (m, 1H), 3.56-3.48 (m, 1H), 1.37 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, DMSO-d6) δ: 198.4, 167.2, 163.3, 162.7, 148.0, 147.3, 143.97 (143.94), 143.5, 130.17 (130.13), 127.8, 121.7 (121.6), 120.8, 116.9, 116.8, 115.6, 111.8, 101.5, 95.6, 94.4, 83.0, 78.4, 76.1, 71.9 (71.8), 64.5, 60.5, 56.0, 14.6. HR-MS (ESI) m/z: calcd for C27H27O10 [M+H]: 511.1604; found 511.1598.
4.2.8 7-O-Propylsilibinin (4)
This derivative was made from silibinin using Method 2 in 3% yield as a light yellow solid. m.p. 175–176 °C. IR (neat) νmax: 3447, 2964, 2925, 2851, 1639, 1571, 1508 cm−1. 1H NMR (300 MHz, CDCl3) and 13C NMR (75 MHz, CDCl3): Table 1. HR-MS (ESI) m/z: calcd for C28H29O10 [M+H]: 525.1760; found 525.1758.
4.2.9 7-O-Butylsilibinin (5)
This compound was prepared from silibinin using Method 2 in 19% yield as a light yellow solid. m.p. 115–116 °C. IR (neat) νmax: 3450, 2957, 2924, 2853, 1639, 1572, 1509 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.192 (11.187) (s, 1H), 7.20 (dd, J = 6.6, 1.5 Hz, 1H), 7.13-7.03 (overlapped, 2H), 6.95 (s, 2H), 6.94 (t, J = 1.5 Hz, 1H), 6.11 (d, J = 1.8 Hz, 1H), 6.04 (6.03) (d, J = 2.4 Hz, 1H), 5.81 (br.s, 1H), 5.00 (d, J = 12.0 Hz, 1H), 4.96 (d, J = 8.1 Hz, 1H), 4.53 (dd, J = 12.0, 4.5 Hz, 1 H), 4.10-4.02 (m, 1H), 3.98 (t, J = 6.6 Hz, 2H), 3.92 (s, 3H), 3.81 (dd, J = 12.6, 2.4 Hz, 1H), 3.56 (dd, J = 12.6, 3.9 Hz, 1H), 2.07 (br.s, 1H), 1.76 (quin, J = 6.6 Hz, 2H), 1.47 (Hex, J = 7.5 Hz, 2H), 0.97 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 195.9, 168.6, 163.7, 163.0, 147.1, 146.6, 144.3 (144.2), 144.08 (144.05), 129.70 (129.66), 128.0, 121.3 (121.05), 121.0, 117.5 (117.4), 116.8 (116.6), 114.8, 109.7, 100.8, 96.1, 95.2, 83.2, 78.49 (78.46), 76.54 (76.49), 72.53 (72.44), 68.6, 61.8, 56.2, 31.0, 19.2, 13.9. HR-MS (ESI) m/z: calcd for C29H31O10 [M+H]: 539.1917; found 539.1918.
4.2.10 7-O-Pentylsilibinin (6)
This derivative was prepared from silibinin by Method 3 in 7% yield as a light yellowish solid. m.p. 112–113 °C. IR (neat) νmax: 3460, 2932, 1636, 1570, 1508, 1464 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.18 (11.19) (s, 1H), 7.20 (dd, J = 6.9, 1.6 Hz, 1H), 7.08 (7.11) (dd, J = 5.6, 1.9 Hz, 1H), 7.07 (7.04) (d, J = 3.0Hz, 1H), 6.96 (s, 2H), 6.94 (t, J = 1.5 Hz, 1H), 6.11 (d, J = 2.1Hz, 1H), 6.05 (6.03) (d, J = 3.5 Hz, 1H), 5.77 (br.s, 1H), 5.00 (d, J = 12.0 Hz, 1H), 4.97 (d, J = 9.0 Hz, 1H), 4.54 (dd, J = 12.0, 4.6 Hz, 1H), 4.08-4.02 (m, 1H), 3.97 (t, J = 6.5 Hz, 2H), 3.93 (s, 3H), 3.82 (dd, J = 12.4, 2.5 Hz, 1H), 3.57 (dd, J = 12.4, 3.6 Hz, 1H), 2.03 (br.s, 1H), 1.78 (quin. J = 6.8 Hz, 2H), 1.41-1.36 (m, 4H), 0.93 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 195.9, 168.7, 163.8, 163.0, 147.1, 146.6, 144.30 (144.26), 144.10 (144.08), 129.72 (129.68), 128.0, 121.3 (121.1), 121.0, 117.5 (117.4), 116.8 (116.6), 114.9, 109.7, 100.8, 96.1, 95.2, 83.2, 78.5, 76.57 (76.52), 72.6 (72.5), 69.0, 61.9, 56.2, 28.7, 28.2, 22.5, 14.1. HR-MS (ESI) m/z: calcd for C30H33O10 [M+H]: 553.2073; found 553.2074.
4.2.11 7-O-hexylsilibinin (7)
This derivative was prepared by Method 3 in 5.7 % yield as a light yellowish solid. m.p. 104.2–105 °C. IR (neat) νmax: 3447, 2928, 1639, 1571, 1509 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.18 (11.17) (s, 1H), 7.21 (dd, J = 7.0, 1.6 Hz, 1H), 7.13-7.04 (m, 2H), 6.96 (s, 2H), 6.94 (t, J = 1.5 Hz, 1H), 6.11 (d, J = 1.5 Hz, 1H), 6.05 (6.04) (d, J = 2.2 Hz, 1H), 5.76 (br.s, 1H), 5.00 (d, J =12.0 Hz, 1H), 4.97 (d, J = 8.5 Hz, 1H), 4.54 (dd, J =12.0, 4.6 Hz, 1H), 4.08-4.02 (m, 1H), 3.97 (t, J = 6.5 Hz, 2H), 3.93 (s, 3H), 3.84 (dd, J =12.5, 2.5 Hz, 1H), 3.57 (dd, J = 12.5, 3.7 Hz, 1H), 1.77 (quin. J = 6.8 Hz, 2H), 1.45-1.41 (m, 2H), 1.35-1.30 (m, 4H), 0.91 (t, J = 6.4 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 195.9, 168.7, 163.76 (163.75), 163.01 (162.99), 147.1, 146.6, 144.32 (144.27), 144.12 (144.09), 129.73 (129.68), 128.0, 121.3 (121.1), 121.0, 117.5 (117.4), 116.8 (116.6), 114.8, 109.7, 100.8, 96.1, 95.2, 83.2, 78.51 (78.48), 76.58 (76.53), 72.56 (72.47), 69.0, 61.9, 56.2, 31.6, 29.0, 25.7, 22.7, 14.2. HR-MS (ESI) m/z: calcd for C31H35O10 [M+H]: 567.2230; found 567.2236.
4.2.12 7-O-Heptylsilibin (8)
This derivative was prepared from silibinin using Method 3 in 7% yield as a light yellow solid. m.p. 67–70 °C. IR (neat) νmax: 3444, 2925, 2855, 1636, 1571, 1508 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.184 (11.179) (s, 1H), 7.21 (dd, J = 7.2, 1.5 Hz, 1H), 7.13-7.04 (overlapped, 2H), 6.96 (s, 2H), 6.94 (br.s, 1H), 6.11 (d, J = 1.8 Hz, 1H), 6.05 (6.03) (d, J = 2.1 Hz, 1H), 5.75 (br.s, 1H), 5.00 (d, J = 12.0 Hz, 1H), 4.97 (d, J = 8.4 Hz, 1H), 4.54 (dd, J = 12.0, 4.5 Hz, 1 H), 4.08-4.03 (m, 1H), 3.97 (t, J = 6.3 Hz, 2 H), 3.93 (s, 3H), 3.82 (br.d, J = 12.3 Hz, 1H), 3.63-3.57 (m, 1H), 3.54 (s, 1H), 2.00 (br.s, 1H), 1.77 (quin, J = 6.9 Hz, 2H), 1.49-1.21 (m, 8H), 0.90 (t, J = 6.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 195.9, 168.7, 163.8, 163.0, 147.1, 146.6, 144.31 (144.27), 144.12 (144.09), 129.7, 128.0, 121.1, 121.0, 117.5, 116.8, 114.8, 109.7, 100.8, 96.1, 95.2, 83.2, 78.5, 76.6, 72.6 (72.5), 69.0, 61.9, 56.2, 31.9, 29.1, 29.0, 26.0, 22.8, 14.2. HR-MS (ESI) m/z: calcd for C32H37O10 [M+H]: 581.2386; found 581.2386.
4.2.13 7-O-Benzylsilibinin (9)
This derivative was prepared from silibinin using Method 1 in 80% yield as a light yellow solid. m.p. 93–95 °C. IR (neat) νmax: 3432, 2937, 1634, 1571, 1507 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.25 (11.24) (s, 1H), 7.43-7.34 (m, 5H), 7.19 (dd, J = 4.2, 1.5 Hz, 1H), 7.10-7.01 (m, 2H), 6.97-6.88 (overlapped, 3H), 6.21 (d, J = 1.8 Hz, 1H), 6.13 (6.12) (d, J = 2.4 Hz, 1H), 5.96 (br.s, 1H), 5.07 (s, 2H), 4.99 (d, J = 11.7 Hz, 1H), 4.93 (d, J = 8.4 Hz, 1H), 4.52 (dd, J = 11.7, 3.3 Hz, 1H), 4.09-3.99 (m, 1H), 3.89 (s, 3H), 3.80 (dd, J = 12.3, 2.1 Hz, 1H), 3.55 (dd, J = 12.3, 3.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 196.2, 167.8, 163.6, 162.8, 147.0, 146.4, 144.1, 143.9, 135.6, 129.5, 128.8, 128.4, 127.9, 127.5, 121.2 (121.1), 120.8, 117.3 (117.2), 116.6, 114.9, 109.8, 101.1, 96.4, 95.4, 83.0, 78.4, 76.3, 72.4, 70.5, 61.6, 56.1. HR-MS (ESI) m/z: calcd for C32H29O10 [M+H]: 573.1761; found 573.1769.
4.2.14 7-O-Methyl-2,3-dehydrosilibinin (10)
This derivative was prepared from 7-O-methylsilibinin (2) using Method 5 as a light yellowish solid in 36 % yield. m.p. 195.1–195.5 °C. IR (neat) νmax: 3374, 2955, 2917, 2849, 1655, 1592, 1559, 1503 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.70 (s, 1H), 7.90 (d, J = 2.0 Hz, 1H), 7.84 (dd, J =7.8, 2.0 Hz, 1H), 7.12 (d, J = 8.6 Hz, 1H), 6.99 (s, 2H), 6.97 (s, 1H), 6.63 (br.s, 1H), 6.47 (d, J = 2.1 Hz, 1H), 6.34 (d, J = 2.1 Hz, 1H), 5.74 (br.s, 1H), 5.02 (d, J = 8.3 Hz, 1H), 4.16-4.12 (m, 1H), 3.95 (s, 3H), 3.88 (s, 3H), 3.86 (dd, J = 12.4, 2.6 Hz, 1H), 3.61 (dd, J = 12.4, 3.9 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ: 176.4, 165.4, 160.6, 156.5, 148.0, 147.3, 146.4, 145.4, 143.7, 136.9, 127.6, 123.9, 121.7, 120.9, 117.2, 116. 6, 115.7, 111.9, 104.4, 97.9, 92.3, 78.8, 76.2, 60.4, 56.3, 56.01. HR-MS (ESI) m/z: calcd for C26H23O10 [M+H]: 495.1291; found 495.1293.
4.2.15 7-O-Ethyl-2,3-dehydrosilibinin (11)
This derivative was prepared from 7-O-ethylsilibinin (2) using Method 5 as a light yellowish solid in 17 % yield; m.p. 154–155°C. IR (neat) νmax: 3396, 2979, 2927, 2853, 1653, 1627, 1589, 1501 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.68 (s, 1H), 7.88 (s, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.11 (d, J = 8.6 Hz, 1H), 6.99 (s, 2H), 6.97 (s, 1H), 6.65 (br.s, 1H), 6.45 (s, 1H), 6.36 (s, 1H), 5.76 (br.s, 1H), 5.01 (d, J = 8.3 Hz, 1H), 4.15-4.07 (m, 1H), 4.10 (q, J = 7.0 Hz, 2H), 3.95, (s, 3H), 3.86 (dd, J = 13.6, 2.5 Hz, 1H), 3.60 (dd, J = 12.0, 3.2 Hz, 1H), 1.46 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.4, 165.4, 160.9, 157.0, 147.2, 146.7, 145.3, 145.2, 144.0, 136.2, 127.8, 124.5, 121.9, 121.1, 117.4, 116.9, 114.9, 109.7, 98.5, 92.7, 78.9, 76.6, 64.5, 61.8, 56.3, 14.8. HR-MS (ESI) m/z: calcd for C27H25O10 [M+H]: 509.1448; found 509.1453.
4.2.16 7-O-Propyl-2,3-dehydrosilibinin (12)
This derivative was prepared from silibinin by Method 2 in 3% yield as a yellow solid; m.p. 142–144 °C. IR (neat) νmax: 3397, 2926, 1654, 1590, 1502 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.67 (s, 1H), 7.90 (d, J = 2.1 Hz, 1H), 7.83 (dd, J = 8.7, 2.1 Hz, 1H), 7.11 (d, J = 8.7 Hz, 1H), 6.99 (s, 2H), 6.98 (s, 1H), 6.63 (s, 1H), 6.46 (d, J = 2.1 Hz), 6.37 (d, J = 2.1 Hz), 5.75 (br.s, 1H), 5.01 (d, J = 8.4 Hz, 1H), 4.17-4.12 (m, 1H), 3.99 (t, J = 6.6 Hz, 2H), 3.95 (s, 3H), 3.86 (br.d, J = 12.3, 1H), 3.61 (br.d, J = 12.3, 1H), 1.96 (br.s, 1H), 1.85 (Hex, J = 7.2 Hz, 2H), 1.06 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.2, 165.4, 160.7, 156.8, 147.0, 146.5, 145.1, 144.9, 143.8, 136.0, 127.6, 124.3, 121.7, 120.9, 117.2, 116.8, 114.7, 109.5, 103.8, 98.4, 92.6, 78.7, 76.2, 70.2, 61.7, 56.1, 22.3, 10.4. HR-MS (ESI) m/z: calcd for C28H27O10 [M+H]: 523.1604; found 523.1603.
4.2.17 7-O-Butyl-2,3-dehydrosilibinin (13)
This derivative was prepared from silibinin using Method 3 in 5% yield as a yellow solid; m.p. 142.5–143 °C. IR (neat) νmax: 3383, 2957, 2925, 2853, 1654, 1590, 1502 cm−1. 1H NMR (300 MHz, CDCl3) and 13C NMR (75 MHz, CDCl3): Table 1. HR-MS (ESI) m/z: calcd for C29H29O10 [M+H]: 537.1760; found 537.1759.
4.2.18 7-O-Pentyl-2,3-dehydrosilibinin (14)
This derivative was prepared from silibinin by Method 2 in 7% yield as a light yellowish solid. m.p. 124–125 °C. IR (neat) νmax: 3391, 2923, 2853, 1654, 1589, 1500 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.67 (s, 1H), 7.90 (d, J = 1.9 Hz, 1H), 7.83 (dd, J = 8.7, 1.8 Hz, 1H), 7.11 (d, J = 8.7 Hz, 1H), 6.99 (s, 2H), 6.97 (s, 1H), 6.65 (br.s, 1H), 6.45 (d, J =1.9 Hz, 1H), 6.36 (d, J = 1.8 Hz, 1H), 5.76 (br.s, 1H), 5.01 (d, J = 8.3 Hz, 1H), 4.16-4.11 (m, 1H), 4.02 (t, J = 6.5 Hz, 2H), 3.95 (s, 3H), 3.86 (dd, J = 12.4, 2.3 Hz, 1H), 3.60 (dd, J = 12.4, 3.7 Hz, 1H), 1.82 (quin, J = 7.0 Hz, 2H), 1.44-1.38 (m, 4H), 0.95 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.4, 165.6, 160.9, 157.0, 147.2, 146.7, 145.3, 145.1, 144.0, 136.2, 127.8, 124.5, 121.9, 121.1, 117.4, 116.9, 114.9, 109.7, 104.0, 98.6, 92.7, 78.9, 76.6, 68.9, 61.8, 56.3, 28.8, 28.3, 22.6, 14.2. HR-MS (ESI) m/z: calcd for C30H31O10 [M+H]: 551.1917; found 551.1918.
4.2.19 7-O-Hexyl-2,3-dehydrosilibinin (15)
This derivative was prepared from silibinin by Method 3 as a light yellowish solid in 6% yield; m.p. 132–133 °C. IR (neat) νmax: 3383, 2953, 2926, 2871, 1654, 1624, 1589, 1558, 1500 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.67 (s, 1H), 7.90 (d, J= 1.9 Hz, 1H), 7.85 (dd, J= 8.5, 2.1 Hz, 1H), 7.11 (d, J= 8.6 Hz, 1H), 6.99 (s, 2H), 6.98 (s, 1H), 6.63 (s, 1H), 6.46 (d, J = 2.0 Hz, 1H), 6. 37 (d, J = 2.0 Hz, 1H), 5.75 (s, 1H), 5.01 (d, J = 8.3 Hz, 1H), 4.14 (dt, J = 7.6, 3.6 Hz, 1H), 4.02 (t, J= 6.5 Hz, 2H), 3.95 (s, 3H), 3.88 (br.d, J = 12.0 Hz, 1H), 3.61 (br.d, J = 12.0 Hz, 1H), 1.96 (br.s, 1H), 1.82 (quin. J= 6.5 Hz, 2H), 1.49-1.44 (m, 2H), 1.37-1.32 (m, 4H), 0.92 (t, J = 7.0 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.4, 165.6, 160.9, 157.0, 147.2, 146.7, 145.3, 145.1, 144.0, 136.2, 127.8, 124.5, 121.9, 121.1, 117.4, 116.9, 114.9, 109.7, 104.0, 98.6, 92.8, 78.9, 76.6, 67.0, 61.9, 56.3, 31.7, 29.1, 25.8, 22.8, 14.2. HR-MS (ESI) m/z: calcd for C31H33O10 [M+H]: 565.2073; found 565.2069.
4.2.20 7-O-Heptyl-2,3-dehydrosilibinin (16)
This derivative was prepared from silibinin using Method 2 in 8% yield as a yellow solid. m.p. 141–145 °C. IR (neat) νmax: 3391, 2922, 2853, 1651, 1617, 1579, 1497 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.67 (s, 1H), 7.89 (s, 1H), 7.83 (d, J = 7.2 Hz, 1H), 7.11 (d, J = 9.0 Hz, 1H), 6.99 (s, 2H), 6.97 (s, 1H), 6.64 (br.s, 1H), 6.45 (s, 1H), 6.36 (s, 1H), 5.76 (br.s, 1H), 5.01 (d, J = 8.4 Hz, 1H), 4.17-4.10 (m, 1H), 4.02 (t, J = 6.6 Hz, 2H), 3.95 (s, 3H), 3.87 (br.d, J = 13.0 Hz, 1H), 3.60 (br.d, J = 13.0 Hz, 1H), 1.98 (br.s, 1H), 1.83 (quin, J = 6.9 Hz, 2H), 1.51-1.18 (m, 8H), 0.92 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 175.3, 165.6, 160.9, 157.0, 147.2, 146.7, 145.3, 145.1, 144.0, 136.2, 127.8, 124.5, 121.9, 121.1, 117.4, 116.9, 114.9, 109.7, 104.0, 98.6, 92.7, 78.9, 76.6, 69.0, 61.8, 56.3, 31.9, 29.2, 29.1, 26.1, 22.8, 14.3. HR-MS (ESI) m/z: calcd for C32H35O10 [M+H]: 579.2230; found 579.2227.
4.2.21 7-O-Benzyl-2,3-dehydrosilibinin (17)
This derivative was prepared from silibinin by Method 3 in 30% yield as a yellow solid. m.p. 194–196 °C. IR (neat) νmax: 3397, 2926, 2852, 1655, 1592, 1500 cm−1. 1H NMR (300 MHz, CDCl3) δ: 11.71 (s, 1H), 7.89 (d, J = 2.1 Hz, 1H), 7.83 (dd, J = 8.7, 2.1 Hz, 1H), 7.48-7.35 (m, 7H), 7.11 (d, J = 8.7 Hz, 1H), 7.00 (s, 2H), 6.98 (s, 1H), 6.65 (s, 1H), 6.55 (d, J = 2.1 Hz, 1H), 6.46 (d, J = 2.1 Hz, 1H), 5.76 (br.s, 1H), 5.14 (s, 2H), 5.01 (d, J = 8.4 Hz, 1H), 4.19-4.11 (m, 1H), 3.95 (s, 3H), 3.86 (dd, J = 12.3, 2.4 Hz, 1H), 3.61 (dd, J = 12.4, 3.6 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 175.4, 165.0, 161.0, 157.0, 147.2, 146.7, 145.3, 144.0, 136.2, 135.9, 129.0, 128.6, 127.8, 127.7, 124.4, 121.9, 121.1, 117.4, 117.0, 114.9, 109.7, 104.3, 98.9, 93.2, 78.9, 76.6, 70.7, 61.8, 56.3. HR-MS (ESI) m/z: calcd for C32H27O10 [M+H]: 571.1604; found 571.1606.
4.2.22 3,7-O-dimethyl-2,3-dehydrosilibinin (18)
This derivative was prepared from silibinin by Method 4 as a light yellowish solid in 12%. m.p. 203.5–205 °C. IR (neat) νmax: 3408, 2935, 2847, 1654, 1595, 1496 cm−1. 1H NMR (300 MHz, CD3COCD3) δ: 12.69 (s, 1H), 7.76 (dd, J = 8.6, 2.0 Hz, 1H), 7.71 (d, J = 2.0 Hz, 1H), 7.17 (d, J = 1.7 Hz, 1H), 7.08 (d, J = 8.6 Hz, 1H), 7.01 (dd, J = 8.1, 1.7 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 6.72 (d, J = 2.0 Hz, 1H), 6.31 (d, J = 2.0 Hz, 1H), 5.05 (d, J = 8.1 Hz, 1H), 4.29-4.25 (m, 1H), 3.91 (s, 3H), 3.90 (s, 3H), 3.89 (s, 3H), 3.80 (dd, J =12.5, 2.3 Hz, 1H), 3.55 (dd, J = 12.5, 4.0 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ: 178.4, 165.6, 161.1, 156.6, 155.3, 148.0, 147.4, 146.4, 143.8, 138.7, 127.4, 122.8, 122.4, 121.0, 117.5, 117.0, 115.7, 112.0, 105.5, 98.2, 92.7, 78.8, 76.2, 60.4, 60.1, 56.4, 56.0. HR-MS (ESI) m/z: calcd for C27H25O10 [M+H]: 509.1448; found 509.1448.
4.2.23 3,7-O-diethyl-2,3-dehydrosilibinin (19)
This derivative was prepared from silibinin by Method 4 as a light yellowish solid in 26% yield. m.p. 205.5–206.0 °C. IR (neat) νmax: 3403, 2978, 2931, 2895, 1652, 1586, 1496 cm−1. 1H NMR (300 MHz, CDCl3) δ: 12.64 (s, 1H), 7.79-7.76 (overlapped, 2H), 7.07 (d, J = 9.2 Hz, 1H), 6.99 (s, 2H), 6.97 (s, 1H), 6.39 (d, J = 2.1 Hz, 1H), 6.33 (d, J = 2.1 Hz, 1H), 5.82 (s, 1H), 5.01 (d, J = 8.3 Hz, 1H), 4.11 (q, J = 7.3 Hz, 2H), 4.08 (q, J = 7.0 Hz, 2H), 4.18-4.10 (m, 1H), 3.94 (s, 3H), 3.87 (br.d, J = 11.5 Hz, 1H), 3.60 (dd, J = 12.2, 2.8 Hz, 1H), 1.44 (t, J = 7.0 Hz, 3H), 1.36 (t, J = 7.3 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 179.2, 165.0, 162.2, 157.0, 155.7, 147.2, 146.8, 145.8, 143.9, 138.4, 127.8, 124.4, 122.9, 121.0, 117.7, 117.2, 115.0, 109.7, 106.1, 98.4, 92.6, 78.9, 76.6, 68.8, 64.4, 61.8, 56.3, 15.8, 14.8. HR-MS (ESI) m/z: calcd for C29H29O10 [M+H]: 537.1761; found 537.1763.
4.2.24 3,7-O-Dipropyl-2,3-dehydrosilibinin (20)
This derivative was made from silibinin using Method 2 in 9% yield as a light yellow solid. m.p. 157.5–158 °C. IR (neat) νmax: 3420, 2962, 2924, 2853, 1654, 1596, 1496 cm−1. 1H NMR (300 MHz, CDCl3) δ: 12.65 (s, 1H), 7.79 (d, J = 2.1 Hz, 1H), 7.77 (dd, J = 3.0, 1.8 Hz, 1H), 7.08 (d, J = 9.3 Hz, 1H), 6.99 (s, 2H), 6.97 (s, 1H), 6.41 (d, J = 2.4 Hz, 1H), 6.35 (d, J = 2.4 Hz, 1H), 5.75 (br.s, 1H), 5.02 (d, J = 8.4 Hz, 1H), 4.14 (dt, J = 8.4, 2.7 Hz, 1H), 3.99 (t, J = 6.9 Hz, 2H), 3.97 (t, J = 6.6 Hz, 2H), 3.95 (s, 3H), 3.87 (dd, J = 12.6, 2.7 Hz, 1H), 3.61 (dd, J = 12.6, 3.9 Hz, 1H), 1.89-1.71 (m, 4H), 1.05 (t, J = 7.2 Hz, 3H), 0.98 (t, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CD3OCD3) δ: 179.1, 165.2, 162.1, 157.0, 155.6, 147.2, 146.7, 145.7, 143.9, 138.6, 127.8, 124.4, 122.9, 121.0, 117.8, 117.2, 114.9, 109.7, 106.1, 98.5, 92.6, 78.9, 76.6, 74.7, 70.3, 61.8, 56.2, 23.5, 22.5, 10.6. HR-MS (ESI) m/z: calcd for C31H33O10 [M+H]: 565.2073; found 565.2072.
4.2.25 3,7-O-Dibutyl-2,3-dehydrosilibinin (21)
This derivative was prepared from silibinin using Method 2 in 15% yield as a light yellow solid. m.p. 82–85 °C. IR (neat) νmax: 3422, 2956, 2924, 2871, 1654, 1596, 1496 cm−1. 1H NMR (300 MHz, CDCl3) δ: 12.65 (s, 1H), 7.79-7.75 (overlapped, 2H), 7.09 (d, J = 9.3 Hz, 1H), 7.00 (s, 2H), 6.97 (s, 1H), 6.41 (d, J = 2.1 Hz, 1H), 6.34 (d, J = 2.4 Hz, 1H), 5.75 (br.s, 1H), 5.02 (d, J = 8.1 Hz, 1H), 4.18-4.09 (m, 1H), 4.022 (t, J = 6.6 Hz, 2H), 4.017 (t, J = 6.6 Hz, 2H), 3.95 (s, 3H), 3.86 (br.d, J = 12.6 Hz, 1H), 3.61 (br.d, J = 12.6 Hz, 1H), 1.96 (br.s, 1H), 1.84-1.69 (m, 4H), 1.56-1.38 (m, 4H), 0.99 (t, J = 7.5 Hz, 3H), 0.92 (t, J = 7.5 Hz, 3H). 13C NMR (75 MHz, CD3OCD3) δ: 179.1, 165.2, 162.1, 156.9, 155.6, 147.2, 146.7, 145.7, 143.9, 138.6, 127.8, 124.3, 122.9, 121.0, 117.8, 117.2, 114.9, 109.7, 106.1, 98.5, 92.6, 78.9, 76.6, 72.9, 68.6, 61.8, 56.2, 32.3, 31.1, 19.33, 19.30, 14.03, 13.95. HR-MS (ESI) m/z: calcd for C33H37O10 [M+H]: 593.2386; found 593.2381.
4.2.26 3,7-O-Dipentyl-2,3-dehydrosilibinin (22)
This derivative was prepared from silibinin by Method 3 in 6% yield as a light yellowish solid. IR (neat) νmax: 3408, 2953, 2926, 2856, 1652, 1587, 1518, 1494 cm−1. 1H NMR (300 MHz, CDCl3) δ: 12.66 (s, 1H), 7.78-7.74 (overlapped, 2H), 7.08 (d, J = 9.2 Hz, 1H), 6.99 (s, 2H), 6.97 (s, 1H), 6.40 (d, J = 2.2 Hz, 1H), 6.34 (d, J = 2.2 Hz, 1H), 5.79 (br.s, 1H), 5.02 (d, J = 8.3 Hz, 1H), 4.14 (dt, J = 8.0, 3.2 Hz, 1H), 4.02 (t, J = 6.7 Hz, 2H), 4.01 (t, J = 6.5 Hz, 2H), 3.95 (s, 3H), 3.87 (dd, J = 12.5, 2.3 Hz, 1H), 3.61 (dd, J = 12.5, 3.8 Hz, 1H), 1.86-1.70 (m, 4H), 1.50-1.31 (m, 8H), 0.95 (t, J = 7.0 Hz, 3H), 0.89 (t, J = 7.1 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ: 179.1, 165.2, 162.1, 156.9, 155.6, 147.2, 146.7, 145.7, 143.9, 138.6, 127.8, 124.3, 122.9, 121.0, 117.8, 117.1, 114.9, 109.7, 106.1, 98.5, 92.6, 78.9, 76.6, 73.2, 68.9, 61.8, 56.2, 29.9, 28.8, 28.2, 22.62, 22.56, 14.20, 14.16. HR-MS (ESI) m/z: calcd for C35H41O10 [M+H]: 621.2699; found 621.2696.
4.2.27 3,7-O-Dihexyl-2,3-dehydrosilibinin (23)
This derivative was prepared from silibinin by Method 3 as a light yellowish solid in 8% yield. m.p. 109–111 °C. IR (neat) νmax: 3421, 2929, 2858, 1654, 1596, 1518, 1496 cm−1. 1H NMR (300 MHz, CD3OCD3) δ: 12.64 (s, 1H), 7.85 (s, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.70 (s, 1H), 7.16 (s, 1H), 7.02 (d, J = 8.4 Hz, 1H), 7.00 (s, 1H), 6.93 (d, J = 8.1 Hz, 1H), 6.61 (s, 1H), 6.25 (s, 1H), 5.05 (d, J = 8.0 Hz, 1H), 4.24-4.16 (m, 2H), 4.08 (t, J = 6.0 Hz, 2H), 4.06 (t, J = 6.0 Hz, 2H), 3.90 (s, 3H), 3.90-3.78 (m, 1H), 3.59-3.52 (m, 1H), 1.81-1.67 (m, 4H), 1.47-1.18 (m, 12H), 0.91 (t, J = 6.6 Hz, 3H), 0.87 (t, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CD3OCD3) δ: 179.7, 166.1, 162.8, 157.8, 156.3, 148.6, 148.2, 147.2, 144.8, 139.0, 128.9, 124.4, 123.2, 121.7, 118.2, 117.7, 115.8, 1112.0, 106.5, 98.9, 93.3, 80.1, 77.3, 73.2, 69.5, 61.7, 56.4, 32.34, 32.25, 26.45, 26.33, 23.30, 23.27, 14.34, 14.30. HR-MS (ESI) m/z: calcd for C37H45O10 [M+H]: 649.3012; found 649.3017.
4.2.28 3,7-O-diheptyl-2,3-dehydrosilibinin (24)
This derivative was prepared from silibinin by Method 2 in 16% yield as a yellow solid. m.p. 107–108 °C. IR (neat) νmax: 3419, 2926, 2855, 1654, 1597, 1496 cm−1. 1H NMR (300 MHz, CDCl3) and 13C NMR (75 MHz, CDCl3): Table 1. HR-MS (ESI) m/z: calcd for C39H49O10 [M+H]: 677.3325; found 677.3321.
4.2.29 3,7-O-dibenzyl-2,3-dehydrosilibinin (25)
This derivative was prepared from silibinin by Method 4 in 12% yield as a yellow solid. m.p. 196–198 °C. IR (neat) νmax: 3434, 2921, 2851, 1655, 1599, 1496 cm−1. 1H NMR (300 MHz, CDCl3) δ: 12.70 (br.s, 1H), 7.69 (dd, J = 8.4, 2.1 Hz, 1H), 7.68 (s, 1H), 7.50-7.39 (m, 8H), 7.32-7.28 (m, 3H), 7.02 (d, J = 11.1 Hz, 1H), 7.00 (s, 2H), 6.96 (s, 1H), 6.48 (d, J = 2.1 Hz, 1H), 6.45 (d, J = 2.1 Hz, 1H), 5.79 (br.s, 1H), 5.13 (s, 2H), 5.10 (s, 2H), 5.01 (d, J = 8.4 Hz, 1H), 4.15-4.10 (m, 1H), 3.95 (s, 3H), 3.87 (dd, J = 12.6, 2.7 Hz, 1H), 3.61 (dd, J = 12.6, 3.9 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ: 179.0, 164.7, 162.2, 156.9, 156.2, 147.1, 146.7, 145.8, 143.8, 137.8, 136.6, 135.9, 129.0, 128.9, 128.5, 128.52, 128.46, 128.38, 127.8, 127.6, 124.1, 123.1, 121.0, 117.8, 117.1, 114.9, 109.7, 106.4, 98.9, 93.1, 78.9, 76.5, 74.5, 70.6, 61.8, 56.2. HR-MS (ESI) m/z: calcd for C39H33O10 [M+H]: 661.2074; found 661.2078.
4.3 Cell culture
All cell lines were initially purchased from American Type Culture Collection (ATCC™). The PC-3 and LNCaP prostate cancer cell lines were routinely cultured in RPIM-1640 medium supplemented with 10% FBS and 1% penicillin/streptomycin. The DU145 prostate cancer cells were routinely cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% FBS and 1% penicillin/streptomycin. Cultures were maintained in a high humidity environment supplemented with 5% carbon dioxide at a temperature of 37 °C.
4.4 WST-1 cell proliferation assay
PC-3, DU145, and LNCaP cells were plated in 96-well plates at a density of 3,200 each well in 200 μL of culture medium. The cells were then treated with silibinin, or synthesized silibinin derivatives separately at 5 different doses for 3 days, while equal treatment volumes of DMSO (0.25%) were used as vehicle control. The cells were cultured in a CO2 incubator at 37 °C for three days. 10 μL of the premixed WST-1 cell proliferation reagent (Clontech) was added to each well. After mixing gently for one minute on an orbital shaker, the cells were incubated for additional 3 hours at 37 °C. To ensure homogeneous distribution of color, it is important to mix gently on an orbital shaker for one minute. The absorbance of each well was measured using a microplate reader (Synergy HT, BioTek) at a wavelength of 430 nm. The IC50 value is the concentration of each compound that inhibits cell proliferation by 50% under the experimental conditions and is the average from at least triplicate determinations that reproducible and statistically significant. For calculating the IC50 values, a linear proliferative inhibition was made based on at least five dosages for each compound.
4.5 Cell cycle analysis
PC-3 cells were plated in 24-well plates at a density of 200,000 each well in 400 μL of culture medium. After 3 hours of cell attachment, the cells were then treated with compound 11 at 5 μM for 15 hours, while equal treatment volumes of DMSO were used as vehicle control. The cells were cultured in CO2 incubator at 37°C for 24 hours and 30 hours, respectively. Both attached and floating cells were collected in a centrifuge tube by centrifugation at rcf value 450 g for 5 minutes. After discarding the supernatant, the collected cells were re-suspended with 500 μL 80% cold ethanol to fix for 30 minutes in 4°C. The fixed cell could store in −20°C for one week. After fixation, the ethanol was removed after centrifuging and the cells were washed with PBS. The cells were then re-suspend with 100μL of 100mg/mL ribonuclease and were cultured at 37°C for 30 minutes to degrade all RNA. The cells were stained with 200 μL of 50 μg/mL propidium iodide stock solution for 30 minutes at −20°C, and then the fluorescence intensity of PI was detected in individual PC-3 cells using an Attune flow cytometer (Life Technologies) within 0.5 to 1 hour after staining.
4.6 F2N12S and CYTOX AADvanced double staining assay
PC-3 cells were plated in 24-well plates at a density of 200,000 each well in 400 μl of culture medium. After 3 hours of cell attachment, the cells were then treated with the test compound at different concentration for 16 hours, while equal treatment volumes of DMSO were used as vehicle control. The cells were cultured in CO2 incubator at 37°C for 15 hours. Both attached and floating cells were collected in a centrifuge tube by centrifugation at rcf value 450 g for 5 to 6 minutes. The collected cells were re-suspended with 500 μl HBSS to remove proteins which may affect flow signal and centrifuged again. After discarding the supernatant, the collected cells were re-suspended with 300 μl HBSS and stained with 0.3 μl of F2N12S for 3–5 minutes followed by 0.3 μl SytoxAAdvanced for an additional 5 minutes. The fluorescence intensity of the two probes was further measured in individual PC-3 cells using an Attune flow cytometer (Life Technologies) 0.5 to 1 hour after staining.
4.7 Statistical analysis
All data are represented as the mean ± standard deviation (S.D.) for the number of experiments indicated. Other differences between treated and control groups were analyzed using the Student’s t-test. A p-value < 0.05 was considered statistically significant.
Supplementary Material
Highlights.
18 new and 6 known silibinin derivatives were prepared from silibinin.
Nineteen derivatives are more potent than silibinin toward prostate cancer cells.
Alkylation of 7-OH enhanced the antiproliferative effect.
7-O-Ethyl-2,3-dehydrosilibinin induced PC-3 cell G0/G1 arrest and apoptosis.
Modification on 7-OH group is a strategy to search for improved derivatives.
Acknowledgments
This work was financially supported by California State University (CSU)-Fresno. The HRMS data were supported by NIH RCMI program at Xavier University of Louisiana through Grant 2G12MD007595-064 (G.W.) and NIH-NIGMS through Grant 1U54GM104940 (G.W.). We are also grateful to the ASI at CSU-Fresno for a Graduate Research Grant (to S.Z.) and the Graduate Net Initiative at CSU-Fresno for 2015-2016 Graduate Research Fellowships (to S.Z. and X.Z).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.World Cancer Research Fund International. [last accessed on 12.18.15];Prostate Cancer Statistics. http://www.wcrf.org/int/cancer-facts-figures/data-specific-cancers/prostate-cancer-statistics.
- 2.American Cancer Society. [last accessed on 11.7.15];What are the Key Statistics About Prostate Cancer. http://www.cancer.org/cancer/prostatecancer/detailedguide/prostate-cancer-key-statistics.
- 3.Trewartha D, Carter K. Advances in prostate cancer treatment. Nat Rev Drug Discov. 2013;12:823–824. doi: 10.1038/nrd4068. [DOI] [PubMed] [Google Scholar]
- 4.Yap TA, Zivi A, Omlin A, de Bono JS. The changing therapeutic landscape of castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:597–610. doi: 10.1038/nrclinonc.2011.117. [DOI] [PubMed] [Google Scholar]
- 5.Flora K, Hahn M, Rosen H, Benner K. Milk thistle (Silybum marianum) for the therapy of liver disease. Am J Gastroenterol. 1998;93:139–143. doi: 10.1111/j.1572-0241.1998.00139.x. [DOI] [PubMed] [Google Scholar]
- 6.Kim NC, Graf TN, Sparacino CM, Wani MC, Wall ME. Complete isolation and characterization of silybins and isosilybins from milk thistle (Silybum marianum) Org Biomol Chem. 2003;1:1684–1689. doi: 10.1039/b300099k. [DOI] [PubMed] [Google Scholar]
- 7.Vue B, Zhang S, Chen Q-H. Flavonoids with therapeutic potential in prostate cancer. Anticancer Agents Med Chem. 2015;15 doi: 10.2174/1871520615666151008122622. [DOI] [PubMed] [Google Scholar]
- 8.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 2006;26:4457–4498. [PubMed] [Google Scholar]
- 9.Flaig TW, Gustafson DL, Su LJ, Zirrolli JA, Crighton F, Harrison GS, Pierson AS, Agarwal R, Glode LM. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs. 2007;25:139–146. doi: 10.1007/s10637-006-9019-2. [DOI] [PubMed] [Google Scholar]
- 10.Flaig TW, Glode M, Gustafson D, van Bokhoven A, Tao Y, Wilson S, Su LJ, Li Y, Harrison G, Agarwal R, Crawford ED, Lucia MS, Pollak M. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate. 2010;70:848–855. doi: 10.1002/pros.21118. [DOI] [PubMed] [Google Scholar]
- 11.Davis-Searles PR, Nakanishi Y, Kim NC, Oberlies TN, Wani MC, Wall ME, Agarwal R, Kroll DJ. Milk thistle and prostate cancer: differential effects of pure flavonolignans from Silybum marianum on antiproliferative end points in human prostate carcinoma cells. Cancer Res. 2005;65:4448–4457. doi: 10.1158/0008-5472.CAN-04-4662. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal C, Wadhwa R, Deep G, Biedermann D, Gazak R, Kren V, Agarwal R. Anti-cancer efficacy of silybin derivatives --- a structure-activity relationship. PloS One. 2013;8:e60074. doi: 10.1371/journal.pone.0060074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sy-Cordero AA, Graf TN, Runyon SP, Wani MC, Kroll DJ, Agarwal R, Brantley SJ, Polyak MF, Oberlies NH. Enhanced bioactivity of silybin B methylation products. Bioorg Med Chem. 2013;21:742–747. doi: 10.1016/j.bmc.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dzubak P, Hajduch M, Gazak R, Svobodova A, Psotova J, Walterova D, Sedmera P, Kren V. New derivatives of silybin and 2,3-dehydrosilybin and their cytotoxic and P-glycoprotein modulatory activity. Bioorg Med Chem. 2006;14:3793–3810. doi: 10.1016/j.bmc.2006.01.035. [DOI] [PubMed] [Google Scholar]
- 15.Biedermann D, Vavrikova E, Cvak L, Kren V. Chemistry of silybin. Nat Prod Rep. 2014;31:1138–1157. doi: 10.1039/c3np70122k. [DOI] [PubMed] [Google Scholar]
- 16.Althagafy HS, Graf TN, Sy-Cordero AA, Gufford BT, Paine MF, Wagoner J, Polyak SJ, Croatt MP, Oberlies NH. Semisynthesis, cytotoxicity, antiviral activity, and drug interaction liability of 7-O-methylated analogues of flavonolignans from milk thistle. Bioorg Med Chem. 2013;21:3919–3926. doi: 10.1016/j.bmc.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyagi A, Bhatia N, Condon MS, Bosland MC, Agarwal C, Agarwal R. Antiproliferative and apoptotic effects of silibinin in rat prostate cancer cells. Prostate. 2002;53:211–217. doi: 10.1002/pros.10146. [DOI] [PubMed] [Google Scholar]
- 18.Zi X, Agarwal R. Silibinin decreases prostate-specific antigen with cell growth inhibition via G1 arrest leading to differentiation of prostate carcinoma cells: implications for prostate cancer intervention. Proc Natl Acad Sci USA. 1999;96:7490–7495. doi: 10.1073/pnas.96.13.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh RP, Deep G, Blouin MJ, Pllak MN, Agarwal R. Silibinin suppresses in vivo growth of human prostate carcinoma PC-3 tumor xenograft. Carcinogenesis. 2007;28:2567–2574. doi: 10.1093/carcin/bgm218. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.