

Abstract
While N6‐methyladenosine (m6A) is a well‐known epigenetic modification in bacterial DNA, it remained largely unstudied in eukaryotes. Recent studies have brought to fore its potential epigenetic role across diverse eukaryotes with biological consequences, which are distinct and possibly even opposite to the well‐studied 5‐methylcytosine mark. Adenine methyltransferases appear to have been independently acquired by eukaryotes on at least 13 occasions from prokaryotic restriction‐modification and counter‐restriction systems. On at least four to five instances, these methyltransferases were recruited as RNA methylases. Thus, m6A marks in eukaryotic DNA and RNA might be more widespread and diversified than previously believed. Several m6A‐binding protein domains from prokaryotes were also acquired by eukaryotes, facilitating prediction of potential readers for these marks. Further, multiple lineages of the AlkB family of dioxygenases have been recruited as m6A demethylases. Although members of the TET/JBP family of dioxygenases have also been suggested to be m6A demethylases, this proposal needs more careful evaluation.
Also watch the Video Abstract.
Keywords: adenine methylation, chromatin, dioxygenases, methyltransferases, modified DNA, restriction modification, transcription regulation
Introduction
In the early 1950s Luria, Anderson, Ralston and co‐workers uncovered cellular processes regulating the host range of bacteriophages 1, 2, 3. Subsequent investigations of this phenomenon by Arber, Meselson and co‐workers led to the discovery of restriction‐modification (R‐M) systems, a landmark event in the history of molecular biology 4, 5. While much subsequent work focused on characterizing restriction enzymes as tools for recombinant DNA technology, the biology and biochemistry of the R‐M systems proved to be interesting in their own right 6, 7, 8. These systems are the most widespread prokaryotic biological conflict systems facilitating both discrimination of cellular “self” DNA from invasive “non‐self” DNA and destruction of the latter 6, 9, 10, 11. In their most basic form, R‐M systems are linked genes (like other prokaryotic operons), which code for a modification enzyme that covalently modifies DNA and a restriction endonuclease that cuts DNA upon recognizing specific sequence signatures 7, 10. However, R‐M systems often exhibit great diversity, and include other linked genes whose products might perform various accessory functions, such as target site recognition, DNA unwinding, long‐distance DNA‐looping and translocation, and regulation or augmentation of the restriction activity 6, 7, 10, 11, 12, 13, 14.
While the most common modification catalyzed by R‐M systems is methylation of specific bases in DNA, recent studies suggest that there might be others, including incorporation of different modified bases and modification of the DNA‐backbone by replacement of the non‐bridging oxygen atom of the phosphate by a sulfur 15, 16, 17. Modification methylases (MTases) methylate either cytosine or adenine in DNA 18, 19, 20, 21. Cytosine is methylated either on the carbon at the 5 position of the pyrimidine ring (C5) or at the exo‐cyclic NH2 group at the 4 position (N4), whereas adenine is methylated on the exocyclic NH2 group at the 6 position of the purine ring (N6) (Fig. 1A). In classic R‐M systems, modification of DNA serves as the discriminatory tag, which prevents the restriction of self DNA, while allowing the non‐self DNA, which is not modified, to be targeted 8, 10. Several bacteriophages have evolved counter‐strategies against R‐M systems in the form of DNA modifications generated by enzymes encoded in their genomes, which inhibit restriction enzymes 10, 22, 23. These modifications include, N6‐methyladenine (m6A), adenine modified at N6 by glycine (momylation), deazaguanines, 5‐hydroxymethyluracil, hypermodified thymines, 5‐methylcytosine (5mC), and 5‐hydroxymethylcytosine (5hmC) and its glycosylated derivatives 15, 24, 25. As part of the ongoing arms‐race, prokaryotes have in turn evolved several specialized restriction systems targeting invasive DNA with such modifications 15, 26.
Figure 1.
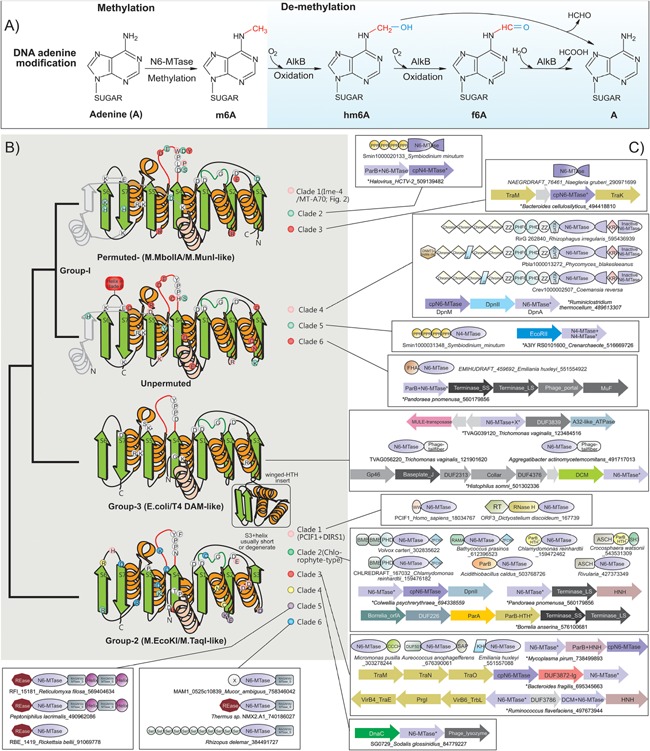
m6A methylation and demethylation reactions, topology, and conserved features of eukaryotic N6A‐MTases. A: Groups modifying the nucleotide are colored red and blue. B: Topology and anatomy of MTase domains. Cartoon representations of principle groups of eukaryotic MTases grouped according to their higher order relationships (shown to the left). Conserved strands are colored green and helices orange. Additionally, lineage‐specific structural elements are shown in gray. Ancestrally conserved residues are shown in gray circles at their structural position, whereas clade specific residues are shown in their respective colors. C: Representative domain architectures and gene neighborhoods for different clades within the three groups are illustrated. Genes in operons are shown with the arrow head pointing to the 3′ direction of the coding strand. Proteins are denoted by their gene name if present, species name, and Genbank identifier (GI) separated by underscores. Proteins from species not available in Genbank are given a temporary id, separated by the species name. The full sequence can be accessed in the Supporting Information. Standard abbreviations are used for domain names. Additional non‐standard names include: X, domains of uncharacterized function; cpN6‐MTase, circularly permuted Group I‐like MTase; RAGNYA, RAGNYA fold domain found in the methylase‐specificity subunit; Helix, α‐helical element that forms coiled coils; ZFCW, PHDX/ZFCW domain; DUF3872‐Ig, an all‐β Ig fold domain.
Following the discovery of R‐M systems, studies pointed to more extensive functions for methyl modifications in prokaryotes and their viruses. m6A was found to mark replication origins of genomic and plasmid replicons, and regulate replication and chromosome segregation 27, 28, 29, 30. Similarly, m6A marks also help distinguishing the DNA strands during mismatch repair 31. Furthermore, in several bacteria, transcription was found to be regulated by specific m6A patterns associated with a given gene 31. m6A and other modified bases were found to regulate transcription and facilitate packaging of a genome length of DNA into the phage head following replication 32. These findings led to the concept that m6A could encode information over and beyond that encoded by the bases of DNA (genetic information) – a form of information termed “epigenetic” 33, 34, 35.
By the 1980s, it had become clear that DNA modifications were not the unique preserve of prokaryotes – several were discovered in eukaryotes, including m6A, 5mC, and the hypermodified thymine (base J) 24, 36, 37, 38. Of these, 5mC was found to be widely distributed across eukaryotes, including humans and other mammals, thereby making it the subject of intense investigation 39, 40. While 5mC in eukaryotes was found to be generated by enzymes having evolutionary links to prokaryotic R‐M and counter R‐M MTases 41, it was found to be an important epigenetic mark with diverse functional consequences in different eukaryotes 42, 43, 44, 45. Recent work has shown that 5mC is not a terminal modification: it is further oxidized by action of the TET/JBP family of 2‐oxoglutarate and Fe (II)‐dependent dioxygenases (2OGFeDOs) to give rise to 5hmC, 5‐formylcytosine (5fC), and 5‐carboxylcytosine (5caC) 46, 47, 48, 49, 50. While functions of these oxidized 5mC derivatives remain to be fully understood, it is becoming apparent that they might be both epigenetic marks of their own right, as well as intermediates in 5mC demethylation 51.
In contrast to 5mC, m6A (the dominant epigenetic mark in prokaryotes) remained largely neglected in eukaryotes 24, 36. This has recently changed: multiple groups have reported conclusive, genome‐wide evidence for m6A modifications from diverse eukaryotes and potential epigenetic roles for this modification 52, 53, 54. Given that these discoveries are likely to elicit much interest and raise several new questions, in this article we attempt to provide an overview of the natural history of the N6A methylation, demethylation, and “reading” apparatus.
Eukaryotic N6A‐MTases belong to three broad groups
Comprehensive genomic analysis revealed that eukaryotes have acquired N6A‐MTase domains (Box 1) from prokaryotic precursors on at least 13 independent occasions in their evolutionary history (Fig. 1B and C), each defining a distinct clade. These clades in turn belong to three major higher‐order groups (groups 1–3), whose primary radiation occurred in bacteria and their phages in R‐M and counter R‐M systems, and epigenetic systems associated with DNA replication and repair (i.e. the classic Dam MTases). We describe below the eukaryotic clades and their provenance.
Box 1. Anatomy of N6A‐MTases‐MTase domains.
The majority of nucleic acid MTases belong to a superfamily of enzymes displaying a classical Rossmann fold catalytic domain, and use AdoMet as the methyl group donor 20, 21, 55, 56, 57. This superfamily additionally includes diverse enzymes catalyzing methylation of a wide array of small molecules and proteins. Structurally, it is characterized by a distinctive connector between the first conserved strand and helix of the Rossmann domain, assuming a “double‐headed loop” conformation, and binding the ribose moiety of AdoMet (Fig. 1B). Additional contacts with ribose and adenine of AdoMet are mediated by residues from the two downstream conserved strand‐helix elements of the domain 19, 57.
Within this superfamily, nucleic acid MTases belong to two distinct clades, one including all nucleic acid C5 MTases 58 and the other uniting enzymes catalyzing methylation of both N4C and N6A, those modifying the N2 position of guanines in RNA, as well as certain protein MTases methylating the amide group of glutamine in proteins such as the ribosomal protein L3 and peptide release factors (HemK family) 59, 60, 61. Members of the latter clade are characterized by a [DNSH]PP[YFW] motif at the C‐terminus of conserved strand‐4 of the Rossmann domain 18, 20, 21, 62 (Fig. 1B). These MTases follow a conserved catalytic mechanism: the target base is held in place by π–π stacking interactions with the aromatic residue [YFW] in the last position of the above motif 62, 63. The target NH2 group is the donor for hydrogen bonds with the polar group of the first residue [DNSH] of their conserved motif, and with the backbone carbonyl of the peptide bond between the next two prolines. Consequently, the NH2 group is primed for a SN2 reaction with the CH3 group from AdoMet, and resultant conformational inversion of the newly formed CH3NH group 63.
In the clade of N6A‐MTases, those acting on DNA had a single origin, probably being derived from the more ancient and nearly universally conserved protein and rRNA MTases. Among N6A‐MTases, one clade is characterized by a circular permutation, bringing strand‐3 of the conserved core of the Rossmann domain to the N‐terminus 59. This group includes several MTases of R‐M systems (e.g. M.MboII and M.MunI; Fig. 1B) 21, 59, 64. N4C‐MTases appear to have been derived on multiple occasions within the wider radiation of N6A‐MTases, and are characterized by a strand‐4 associated motif with serine in the first position 64. Target specificity of N6A (also N4C) MTases is largely determined by specific elements that were traditionally called “target recognition domains (TRDs)” and used to further classify these enzymes 63, 65. We refrain from using the term TRD because they are not evolutionarily related or even functionally equivalent domains, and instead describe them as necessary based on their actual structure.
Group‐1 contains MTases structurally related to prokaryotic M.MboIIA/M.MunI (circularly permuted) and DnpA (unpermuted)
Members of this group were acquired by eukaryotes on at least six distinct occasions (Fig. 2). The most widespread of these, the Ime4‐like (also called MT‐A70) clade 59, 66, with circularly permuted MTase domains, in turn radiated into six distinct eukaryotic sub‐clades 53 (Fig. 2). Of these, the subclades typified by human METTL3 (yeast Ime4) and human METTL14 (yeast Kar4) are most conserved, and are typically in a single copy per genome 59, 66. METTL14 representatives often show disruptions of their active site motifs suggesting that they are inactive versions (Supporting Information). METTL3 and METTL14 cognates are typically subunits of a dimeric enzyme, catalyzing N6A methylation of specific positions in mRNAs 67, 68. Consistent with this, in METTL3 the MTase is fused to N‐terminal ssRNA‐binding CCCH domains (Fig. 2). Of the other four eukaryotic sub‐clades of the Ime4‐like/MT‐A70 clade that prototyped by METTL4 is widely, albeit patchily, distributed (Fig. 3). Recent work in the nematode Caenorhabditis elegans suggests that it is likely to be a DNA MTase 53. The remaining eukaryotic subclades of the Ime4‐like/MT‐A70 clade show even more sporadic phyletic patterns, distantly related microbial eukaryotes being united close together in the phylogenetic tree (Fig. 2), hence indicating extensive lateral transfer of these genes between them. One of these subclades is typified by fusion of the MTase domain to multiple C‐terminal ZZ‐domains 20 (Fig. 2), a treble‐clef fold Zn‐binding domain mediating protein–protein interactions in chromatin 69, 70. The eukaryotic Ime4‐like/MT‐A70 clade is nested within a prokaryotic radiation that includes MTases of the BglII R‐M system 53 (Fig. 2).
Figure 2.
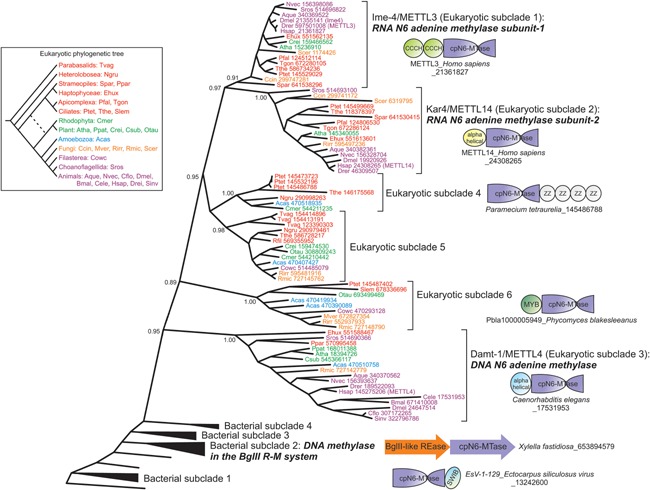
Approximate maximum‐likelihood phylogenetic tree of the Ime‐4/MT‐A70 methylase clade generated using the FastTree and MEGA5 programs. Proteins are labeled using species abbreviations and gi, and colored based on their phylogenetic position in the eukaryotic tree (shown on left). Bootstrap values for major branches of the tree are shown. Related bacterial subclades from which the Ime‐4/MT‐A70 MTases were derived form successive outgroups to the eukaryotic subclades. Species abbreviations for all figures: Aano, Aureococcus anophagefferens; Acas, Acanthamoeba castellanii; Aque, Amphimedon queenslandica; Atha, Arabidopsis thaliana; Bden, Batrachochytrium dendrobatidis; Bnat, Bigellowiella natans; Bmal, Brugia malayi; Ccin, Coprinopsis cinerea; Ccor, Conidiobolus coronatus; Cele, Caenorhabditis elegans; Cflo, Camponotus floridanus; Cmer, Cyanidioschyzon merolae; Cowc, Capsaspora owczarzaki; Crei, Chlamydomonas reinhardtii; Csub, Coccomyxa subellipsoidea; Ddis, Dictyostelium discoideum; Dmel, Drosophila melanogaster; Drer, Danio rerio; Ehux, Emiliania huxleyi; Esil, Ectocarpus siliculosus; Glam, Giardia lamblia; Hsap, Homo sapiens; Lmaj, Leishmania major; Mbre, Monosiga brevicollis; Mver, Mortierella verticillata; Ncra, Neurospora crassa; Ngru, Naegleria gruberi; Nvec, Nematostella vectensis; Otau, Ostreococcus tauri; Otri, Oxytricha trifallax; Pfal, Plasmodium falciparum; Pmar, Perkinsus marinus; Ppal, Polysphondylium pallidum; Ppar, Phytophthora parasitica; Ppat, Physcomitrella patens; Ptet, Paramecium tetraurelia; Rfil, Reticulomyxa filose; Rirr, Rhizophagus irregularis; Rmic, Rhizopus microspores; Scer, Saccharomyces cerevisiae; Sinv, Solenopsis invicta; Spar, Saprolegnia parasitica; Spom, Schizosaccharomyces pombe; Slem, Stylonychia lemnae; Spun, Spizellomyces punctatus; Sros, Salpingoeca rosetta; Tgon, Toxoplasma gondii; Tpse, Thalassiosira pseudonana; Tthe, Tetrahymena thermophila; Tvag, Trichomonas vaginalis; Vcar, Volvox carteri.
Figure 3.
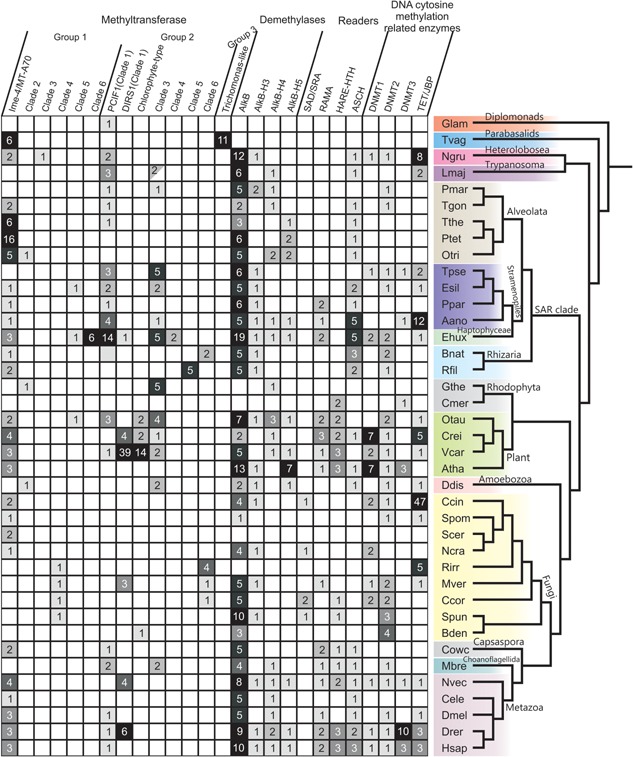
Phyletic patterns of DNA adenine methylases, demethylases, and potential modified DNA‐binding domains (readers) in comparison with key components of the DNA C5 methylation apparatus. Proteins are shown along the x‐axis, whereas organisms are shown along the y‐axis according to their positions in a consensus eukaryotic phylogram. Shaded boxes (with a number) represent the presence and count of representatives in species with multiple paralogs. The blank box represents the absence. The half‐shaded box denotes the presence of the family in Trypanosoma and not Leishmania major. Species abbreviations are as in Fig. 2 legend.
Two further clades of circularly permuted MTases, distinct from the above, representing independent transfers from bacteria, show more restricted distributions. They are characterized by unusual variants of the diagnostic strand‐4 associated motif (Fig. 1B). Of these, the sporadically distributed Clade 2 is related to versions encoded by myxobacteria and archaeal dsDNA viruses (Figs. 1B,C and 3) and often fused to RNA‐binding PPR domains. They present an S in the strand‐4 motif, suggesting that eukaryotic versions might have been recruited for a role in N4C modification in RNA (Fig. 1C). The last of the permuted clades (Clade 3) is currently observed only in the heterolobosean Naegleria 20, and appears to have been derived from potential counter‐restriction MTases of bacterial mobile elements that transfer DNA using Type‐IV secretion systems 71.
The remaining three N6A‐MTase clades in group‐1 (Clades 4–6) display an unpermuted catalytic domain. Of these, Clade 4 is only seen in basal fungi (Fig. 3), suggesting that they were lost on multiple occasions upon early acquisition in fungal evolution. They are fused to Chromo, DNMT3‐like Zn‐finger, ZZ, PHD, GATA, AT‐hook, and KRI domains (Fig. 1C), indicating likely interactions with both DNA and proteins, including methylated histones in chromatin 20, 72, 73. These proteins have a second C‐terminal inactive MTase domain with the KRI domain inserted between the conserved strands‐3 and 4 (Fig. 1C). Prokaryotic versions of this clade are found in DpnII R‐M systems, which code for the DpnII restriction endonuclease and two MTases (Fig. 1C) 74. The first (DpnM) acts as the conventional modification enzyme, which protects self DNA from restriction, while the second MTase (DpnA) is a single‐strand DNA specific MTase, only activated to protect incoming ssDNA during transformation 75. Thus, DpnII systems exempt transforming DNA allowing bacteria to maintain genetic diversity through recombination. Given the specific relationship of fungal versions to DpnA, they too probably act on ssDNA.
Clade 5 is characterized by an S in the strand‐4 motif and is found in distantly related unicellular photosynthetic eukaryotes (Fig. 3). Some of these are fused to the RNA‐binding PPR domains 76, 77, suggesting that they might also modify cytosine at the N4 position in RNA like the aforementioned clade (Figs. 1 and 3). Several of their prokaryotic counterparts are the MTases of the EcoRII‐like R‐M systems. Clade 6 is restricted but lineage‐specifically expanded in the haptophyte algae, like Emiliania (Fig. 3), and are fused to an N‐terminal FHA‐fold domain 78. They are derived from prokaryotic versions encoded by the ParB‐Terminase large subunit (Tls) locus found in several phages and prophages (Fig. 1C), which are predicted to modify phage DNA as part of the DNA‐packaging process 15.
Group‐2 MTases are prototyped by prokaryotic M.EcoKI/M.TaqI
These MTases are characterized by complete or partial degeneration into coils of the helices before and after strand‐3 18, 79. They also display a helix N‐terminal to the core MTase domain with a conserved residue that helps position the asparagine in the strand‐4‐associated motif in the active site (Fig. 1B). Six clades from this group, representing independent transfers from bacteria, are present in eukaryotes (Fig. 3). The first and most widespread clade in this group is defined by the PCIF1 protein, which is traceable to the last eukaryotic common ancestor (Fig. 3). PCIF1 is usually fused to an N‐terminal WW domain (Fig. 1C), which recruits it to the carboxy‐terminal tail (CTD) of the RNA polymerase II (RNAPII) largest subunit 80. The strong conservation of this enzyme, which is typical of RNA‐modification enzymes, and interaction with the CTD, which plays an important role as a scaffold for RNA‐processing 81, raises the possibility that it might methylate mRNA or a CTD‐associated ribonucleoprotein. Also in this clade are MTase domains that are embedded in the polyprotein of DIRS1‐type retrotransposons (Figs. 1C and 3) 82, and which were probably derived from the cellular PCIF1. These elements are highly mobile across species, and are seen in diverse eukaryotes (Fig. 3) 20, 82. However, all copies of the DIRS1 MTase domain are likely inactive because of substitutions affecting catalytic and substrate‐binding residues 20. Hence, they might merely interact with template transcripts of the DIRS1 transposon, or mimic endogenous PCIF1 to regulate transposon polyprotein localization by interacting with RNAPII.
The second clade in this group, the “chlorophyte‐type Dam” clade, contains two families predominantly found in chlorophyte algae 20. The first family usually occurs as a single copy in chlorophytes, and exists as fusions to one or more BMB/PWWP and a ZfCW/PHD‐X domain (Fig. 1C). These domains indicate that they might interact with modified or unmodified histones 73, 83, 84. The second family, present only in certain chlorophytes and chytrid fungi, is characterized by an N‐terminal fusion to a ParB‐type helix‐turn‐helix (HTH) (Supporting Information; Figs. 1C and 3). Prokaryotic members of this clade are found both in phage ParB‐Tls loci and DpnII‐type R‐M systems, where they are the primary modification MTase DpnM (Fig. 1C) 85. Furthermore, both the chlorophyte‐type Dam and the linked ParB‐HTH found in the second family are fused in cyanobacteria to ASCH domains, predicted to bind modified nucleic acids (see below).
The third clade in this group, typified by the Chlamydomonas protein CHLREDRAFT_205675 (gi: 159485216), is broadly distributed in microbial eukaryotes (Fig. 3). They often occur as two paralogs, suggesting that they might form a dimer like METTL3‐METTL14 68. Further, like METTL3, they are often fused to RNA‐binding domains, namely CCCH and KH (Fig. 1) 59. This suggests that at least a subset of this clade is involved in RNA methylation. Their bacterial cognates are encoded by mobile conjugative elements, which they might protect from restriction during DNA‐transfer, and less frequently by R‐M systems. In both cases, they might be found alongside a gene for a DNA C5‐MTase, and in some cases a second N6A‐MTase (Fig. 1C). The fourth clade from this group is represented by paralogous copies seen thus far only in the haptophyte alga Emiliania, and appears to have been derived from a bacteriophage version (Fig. 1C).
MTases of Clades 5 and 6 in this group are restricted to rhizarians and/or basal fungi (Fig. 3). They are fused to the DNA‐binding MTase‐S domain, which contains a RAGNYA fold, seen in diverse nucleic‐acid‐binding contexts where it recognizes specific nucleotide sequences 79, 86, 87. Clade 5 MTases in the rhizarian Reticulomyxa are found in up to five copies, and at least one is fused to an N‐terminal restriction endonuclease domain, thereby retaining the ancestral Type I R‐M system architecture (Figs. 1C and 3). These are also found in bacterial endosymbionts/parasites, pointing to possible lateral acquisition from such organisms.
Group‐3 MTases are prototyped by Dam MTases of Escherichia coli and bacteriophage T4
These are characterized by an additional N‐terminal helix and a winged HTH domain inserted after the second conserved strand‐helix unit, which help in recognition and flipping of the target adenine 62 (Fig. 1). This clade is only seen in the basal eukaryote Trichomonas (up to 10 nearly identical copies), and its members are fused to a bacteriophage tail‐fiber domain 20 (Figs. 1C and 3). They are in the vicinity of transposons coding for an A32‐like packaging ATPase, suggesting that they might have been dispersed by these transposons 20 (Fig. 1C).
These observations indicate that there were multiple origins for N6A‐MTases in eukaryotes involving several independent transitions to RNA‐modification upon acquisition from prokaryotic DNA‐modification systems (Box 2).
Box 2. Evolutionary trends in eukaryotic N6A‐MTases.
N6A‐MTases share several common evolutionary trends with C5‐MTases and DNA‐modifying 2OGFeDOs of the TET/JBP family (Fig. 3). Both types of MTases have been independently transferred on several occasions from prokaryotes, and their viruses to eukaryotes and their viruses: N6A‐MTases and C5‐MTases on 13 and 8 occasions, respectively (Fig. 3) 20. Whereas some transfers occurred in the stem eukaryotes (e.g. PCIF1), others happened only in terminal branches (Fig. 3). Most eukaryotic versions show extensive gene‐loss and are sometimes laterally transferred between lineages (Fig. 2). Since eukaryotes typically lack R‐M systems, the acquired N6A‐MTases are reused in different functional capacities 88. This is often accompanied by fusions to domains, which on the one hand enable specific interactions with methylated histones/other chromatin proteins 83, 84, DNA 73, or both, and on the other hand facilitate interactions with RNA 59. Convergent fusions to the same type of domain are observed in more than one clade (Fig. 1C), suggesting that there are comparable selective pressures acting on independently acquired N6A‐MTases to recruit them in similar functional contexts. A comparable set of multiple, independent fusions to chromatin‐related domains are also observed in eukaryotic C5‐MTases and TET/JBP proteins, suggesting that such fusions represent a common evolutionary mechanism by which DNA‐modifying enzymes of prokaryotic provenance are recruited as generators of epigenetic DNA‐modifications in eukaryotic chromatin 20.
While use of N6A‐MTases as epigenetic DNA‐modifiers can be seen as a functional continuation of their prokaryotic counterparts, a more pronounced functional shift is their repeated recruitment as RNA MTases in eukaryotes. This is known or predicted (based on fusions to RNA‐binding domains) to have happened on at least 4–5 occasions. While a similar shift to RNA specificity has been reported among eukaryotic C5‐MTases, i.e. DNMT2 39, 89 and at least in one clade of TET/JBP enzymes 15, 90, it appears to be more common in N6A‐MTases. This difference might be related to the distinct C‐terminal module in C5‐MTases that predisposes them to preferentially bind dsDNA 20. In contrast, many N6A‐MTases (e.g. DpnA from DpnII‐type systems) were already targeting ssDNA 74. When acquired by eukaryotes, they likely encountered abundant pre‐mRNA in the nucleus, which potentially mimicked their ancestral ssDNA substrate, thereby enabling a functional shift toward RNA methylation. Interestingly, diverse complements of N6A‐MTases are specifically found in phylogenetically distant microbial photosynthetic eukaryotes; in contrast, land plants show few N6A‐MTases (Fig. 3). This suggests that methylation of both DNA and (pre)mRNA, including perhaps chloroplast transcripts, is likely to be important for the regulation of physiology in microbial algae 52.
How do eukaryotic methylomes correlate with the presence of N6A‐MTases?
Since the 1970s, studies have detected and estimated m6A in DNA from diverse eukaryotes 24, 36, 38. Recently, some of these have been reproduced using more sensitive and reliable methods, such as ultra‐high‐performance liquid chromatography coupled with tandem mass spectrometry. For at least a few organisms, methylomes have been directly inferred using technologies, such as single‐molecule, real‐time (SMRT) sequencing and methylated DNA‐immunoprecipitation sequencing (MeDIP‐Seq) 34, 35, 52, 53, 54, 91, 92. This allows us to interrogate the correlation between the detection of m6A in an organism and potential N6A‐MTases coded by a genome.
Notable cases include ciliates, which were reported as having 0.8–2.5% of adenines as methylated 24, 36, 93. All ciliates code for members from 2–3 distinct sub‐clades of the Ime4‐like (MT‐A70) clade (Fig. 2), suggesting that one or more of these enzymes probably generate the observed m6A. The chlorophyte Chlamydomonas was reported as having 0.5% of its adenines methylated 24, 36, 52. A recent study has provided exquisite detail on its methylome 52: the bulk of the m6A is associated with specific motifs centered on an AT dinucleotide (one third of them mapping to motifs CATG and GATC), with a bimodal distribution around the transcription start site. These m6A‐enriched regions show a periodicity of around one per 130–140 bp, being typically localized to inter‐nucleosomal linker regions. Additionally, there are lower abundance m6A methylation sites, lacking periodicity, distributed throughout the gene body; these may be only partially methylated. Chlamydomonas has two MTases from two subclades of the Ime4‐like (MT‐A70) clade (Fig. 2), and multiple chlorophyte‐type Dams (Fig. 3), which could collectively account for the observed methylation. Versions with BMB/PWWP and ZfCW/PHD‐X domains could interact with histones to set up the observed inter‐nucleosomal DNA methylation 52. These MTases are conserved across chlorophytes but not land plants (Fig. 3), suggesting that such N6A methylation patterns were lost during the emergence of the land plants.
Surprisingly, m6A was also identified in C. elegans in which no type of DNA methylation had previously been observed 53. In wild‐type worms, m6A levels are variable (0.01–0.4% of adenines) but consistently elevated in certain mutant backgrounds (see below). Knockdown of the only candidate DNA m6A MTase gene, damt‐1, specifically reduced m6A in genomic DNA and knockouts suppressed mutants with elevated m6A. These results provide strong evidence that in C. elegans damt‐1 is the likely DNA methylase 53. Me‐DIP‐ and SMRT‐sequencing suggest that m6A is enriched at certain motifs, namely AGAA and GAGG, the former being only 10–50% methylated and the latter 50–100%. Interestingly, unlike in Chlamydomonas, the C. elegans motifs are asymmetric in that methylation at these motifs will be necessarily limited to a single strand 53. Recent investigations in Drosophila have revealed that early stage embryos display methylation at ∼0.07% of the adenines, which rapidly fell to ∼0.001% in late stage embryos and adults 54. This is unlike C. elegans, where m6A is present ubiquitously, both in embryos and adults. Again, unlike C. elegans, m6A in Drosophila was found to peak in gene bodies of transposons, but not regions upstream and downstream of them 52. Drosophila has three members of the Ime4‐like (MT‐A70) clade (Fig. 3, Supporting Information): two of these are likely to constitute the conserved mRNA methylating enzyme. The third (CG14906), an ortholog of C. elegans damt‐1, is predicted to be the primary N6A‐MTase in Drosophila.
Thus, in principle, other organisms with a METTL4 representative, such as vertebrates (including humans), land plants, and stramenopiles, might possess m6A in DNA. However, in several organisms, such as the fission yeast Schizosaccharomyces pombe and nematodes, a METTL4 representative is the only Ime4‐like/MT‐A70 clade MTase in the genome (Fig. 3); hence, it might functionally back up the absent METTL3/METTL14 dyad and operate on ssRNA. This is also consistent with the single‐strand biased motifs identified for C. elegans damt‐1 53. Therefore, at least in some organisms, the METTL4 representative could be predominantly an RNA MTase. While phyletic patterns of N6A‐MTases do not suggest the presence of DNA methylation in the last eukaryotic common ancestor, early‐branching eukaryotes, such as Trichomonas and Naegleria, have representatives of one or more clades of N6A‐MTases, implying that they might possess m6A in their genomic DNA (Fig. 3). Similarly, genomes of basal fungi, which possess one or more N6A‐MTase, are predicted to possess a robust m6A signal (Fig. 3).
How are m6A marks reset?
Removal of 5mC marks is part of a key epigenetic resetting mechanism operating at critical developmental junctures in certain eukaryotes 94, 95. The recent discovery of the TET/JBP family of 2OGFeDOs has cast light on how this might happen via a combination of oxidative modification of 5mC and nucleotide excision repair 51. With the earlier discovery of a related family of 2OGFeDOs, prototyped by E. coli AlkB, it became apparent that these enzymes could repair DNA with adenine and cytosine methylated, respectively, at the N1 and N3 positions by mutagenic alkylating agents 96, 97, 98. It was also predicted then that certain members of the AlkB family were likely to demethylate m6A in RNA 96. Subsequently, such demethylation of m6A in RNA was indeed observed to be the mechanism for resetting methyl marks generated by N6A RNA MTases 68, 99. Oxidation of the methyl group by these enzymes results in formation of N6hmA and N6fA, which restore the original base, releasing formaldehyde and formate 100. The recent C. elegans study demonstrated via in vitro and in vivo experiments that nmad‐1, a member of the AlkB family, demethylated m6A in DNA 53. This suggested that in addition to repair of DNA damaged by alkylating adducts, members of the AlkB family are likely to be key players in m6A demethylation in DNA. It remains to be seen whether the metastable intermediates, N6hmA and N6fA, have any independent role in DNA as proposed for modified RNA 100.
While bacteria typically have only a single AlkB representative (Fig. 3), the family underwent explosive radiation in eukaryotes upon being acquired by lateral transfer from bacteria. The basal eukaryotic lineages, the parabasalids and diplomonads, lack members of the AlkB family, suggesting that it was perhaps absent in the ancestral eukaryote (Fig. 3). By the time that other early‐branching eukaryotes – such as euglenozoans and heteroloboseans – split off from the remaining eukaryotes the AlkB family had already radiated into at least five distinct clades (prototyped by human AlkBH1, AlkBH4, AlkBH6, AlkBH7, and AlkBH8; Supporting Information). Subsequently, five further widespread clades emerged along with smaller clades restricted to particular eukaryotic lineages. Of these, the most widespread, AlkBH1, retained the ancestral DNA repair role, while the AlkBH8 and FTO clades specialized in RNA modification 101, 102, 103. C. elegans nmad‐1 belongs to the AlkBH4 clade defined by a N‐terminal Zn‐binding domain and might be primarily involved in DNA m6A demethylation 53. However, an nmad‐1 ortholog is absent in key lineages with confirmed m6A in DNA (Supporting Information). We observed that these organisms instead possess members of the AlkBH5 clade with fusions to domains such as the PHD finger and the AT‐hook DNA‐binding motif. Similarly, AlkBH3 also shows fusions to the PHD finger, and different DNA‐binding domains (SAD/SRA and AT‐hook motif; Fig. 4). Hence, members of the AlkBH5 and AlkBH3 clades conceivably demethylate m6A in DNA (Fig. 4).
Figure 4.
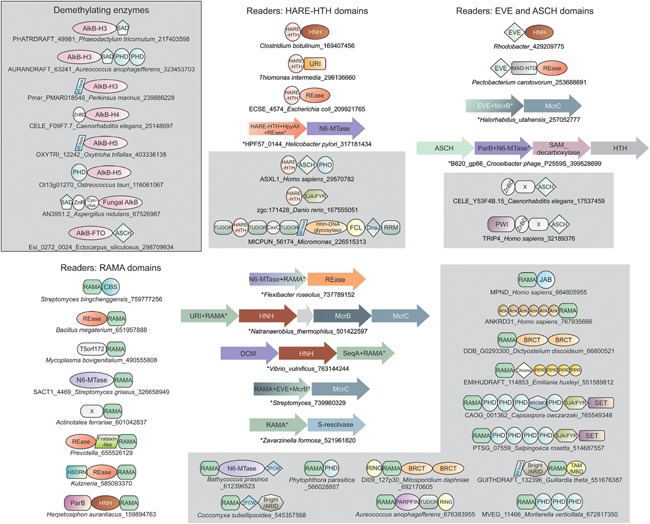
Domain architectures and gene neighborhoods of N6A demethylases and predicted modified DNA‐binding domains. Domains and gene neighborhoods are grouped based on the principal domain of that group. Domains architectures and operons are labeled as in Fig. 1. X refers to uncharacterized domains.
Recently, it was proposed that the Drosophila TET enzyme, DmTET, functions as a m6A demethylase 54. However, this proposal is at odds with other observations. DmTET is closely related to vertebrate TETs, and retains all features of the active site of the latter enzymes which accommodate a pyrimidine rather than a purine 15, 49. A diversity of characterized eukaryotic TET/JBP enzymes have been observed only to modify 5‐methylpyrimidines rather than purines 49, 50, 51, 104, 105. This is further confirmed by contextual analysis of bacterial and phage TET/JBP family members, which are predicted to primarily modify pyrimidines 15, 90. Consistent with these, purified DmTET demonstrated 5mC oxidation capacity in vitro on DNA substrates 54. However, all experiments demonstrating its purported m6A demethylation activity were done with additional Drosophila cell extracts 54. The study did not rule out the presence of the Drosophila nmad‐1 ortholog in these extracts, which could have catalyzed the actual demethylation like its C. elegans ortholog 53. However, a secondary role for DmTET via a pathway involving oxidation of thymines opposite to m6A cannot be ruled out. Hence, until further independent confirmation, the role of TET/JBP enzymes as m6A demethylating enzymes should be treated with caution.
Multiple domains potentially discriminate m6A marks in DNA
In several bacteria, proteins containing a SeqA domain specifically bind DNA with hemimethylated m6A sites, and distinguish parent from newly synthesized duplexes 106. Given the prevalence of m6A modification in eukaryotes, in principle, multiple DNA‐binding proteins might recognize it. We had earlier developed a method to predict modified DNA‐binding domains by using contextual information from domain architectures and conserved gene‐neighborhoods of R‐M and counter‐restriction systems 15. By identifying homologs of domains thus recovered in eukaryotes, we had successfully predicted proteins involved in discrimination of DNA with 5mC and its oxidized derivatives. Applying a similar approach, we were able to predict multiple domains that might be involved in recognition of m6A in eukaryotes 15.
Most prevalent of these is a previously unrecognized domain that was initially observed fused to both N6A‐MTases and nuclease domains belonging to different structural folds: REase, HNH, URI, ParB, serine‐resolvase, and T5orf172, most of which are restriction endonucleases of N6A‐modifying R‐M systems (Fig. 4; Supporting Information) 6, 107. It is also fused to McrB‐like AAA+ domains, involved in DNA translocation/looping in certain R‐M systems 10, 12, and to DNA‐binding domains such as the hemimethylated m6A‐binding SeqA domain (Fig. 4). Accordingly, we named it the RAMA (restriction enzyme adenine methylase associated) domain. We also observe that it has been transferred to eukaryotes, and is found in lineages predicted or known to possess m6A, such as animals, chlorophyte algae, stramenopiles, rhodophytes, and certain nucleocytoplasmic large DNA viruses (Fig. 3). In eukaryotes, RAMA domains are frequently fused to JAB deubiquitinating peptidase (DUB) domains in paralogs of the MYSM1 enzymes that deubiquitinate the monoubiquitinated histone H2A (H2A‐K119u) 108. Less common domain architectures across eukaryotes combine the RAMA domain in a single polypeptide with (Fig. 4): the chlorophyte‐type DAM in the alga Bathycoccus; ankyrin repeats; the histone MTase SET domain; domains recognizing modified histones (PHD finger, Chromo, Tudor, and Bromo domains); a domain recognizing phosphopeptides in chromatin (BRCT); DNA‐binding domains (ARID/BRIGHT, TAM/MBD, PARP‐zinc finger, and AT‐Hook); the ubiquitin E3‐ligase (RING) domain 72, 73, 83, 84.
The HARE‐HTH (for instance found in human ASXL1) is another domain showing similarity to the RAMA domain in its architectural and functional linkages 109. In prokaryotes, it is fused to an array of endonuclease domains, which serve as restriction enzymes in N6A‐modifying R‐M systems, and N6A‐MTase domains 109. In eukaryotes, it is linked to a comparable set of chromatin‐related domains (Fig. 4). Thus, both these domains display architectures that in prokaryotes are suggestive of recognition of modified bases in R‐M systems (Fig. 4), while in eukaryotes they are consistent with a role in recognition of similar epigenetic marks in chromatin. Strikingly, the HARE‐HTH protein AXSL1 is a subunit of the H2A‐K119u MYSM1 DUB in vertebrates (MYSM1 itself is fused to a DNA‐binding MYB domain) 110. Hence, both paralogous H2A‐K119 DUBs are likely to bind DNA, and possibly recognize modified bases in DNA in conjunction with H2A deubiquitination.
PUA‐like domains display a β‐barrel fold, and are the common structural denominator of several families of proteins recognizing modified nucleic acids. These include the SAD/SRA, which recognizes 5mC in DNA; EVE, which binds DNA with 5hmC; and the PUA, which binds modified RNA (including the YTH family which binds m6A containing RNA) 20, 68, 111, 112, 113, 114, 115. Another family displaying this fold, the ASCH domain, was predicted to bind several modified bases, and is found fused to or operonically associated with the N6A‐MTase domain on multiple occasions in bacteria 15, 112 (Fig. 4). This suggests that eukaryotic ASCH domains might also serve as m6A discrimination modules. This proposal is attractive given that C. elegans, with m6A in DNA, lacks both RAMA and HARE‐HTH domains, but has a protein with an ASCH domain. This protein, TRIP4/ASC1, combines the C‐terminal ASCH domain with an N‐terminal RNA‐binding PWI domain, and central Zn‐binding domain, which interacts with specific transcription factors (Fig. 4) 112, 116 and the RNA demethylase FTO 117. TRIP4/ASC1 is prevalent across eukaryotes, and is part of the basal transcription apparatus, where it serves as a co‐activator, suggesting that recognition of m6A marks by TRIP4/ASC1 might have a role in transcription regulation 116. A divergent version of the SAD(SRA) domain is also fused to an AlkB 2OGFeDO domain in several fungi 90 (Fig. 4). While the SAD(SRA) domain typically recognizes single‐strand 5mC sites 118, 119, 120, this version of the domain has distinct features suggesting that it could potentially recognize m6A in RNA or DNA as opposed to 5mC. Hence, it is conceivable that these AlkB‐like proteins also function as m6A demethylases.
What are the roles for m6A in eukaryotic DNA?
While research on m6A function in eukaryotes is still in its infancy, certain consistent features are already seen emerging. Genetic evidence in C. elegans 53, biochemical evidence of m6A in internucleosomal linkers 52, and domain architectures of the N6A‐MTases in fungi, chlorophytes, and ciliates (Fig. 3) suggest that modification of DNA is likely coordinated with recognition of specific histone methylation marks. In Chlamydomonas, genes showing m6A are significantly associated with active transcription as opposed to those with no m6A methylation 52. In C. elegans, loss of function of the LSD1‐like of histone H3 methyl/di‐methyl lysine 4 (H3K4me1/me2) demethylase, spr‐5, causes multi‐generational increase in m6A levels. Conversely, deletion of the potential N6A‐MTase, damt‐1, reduces elevated H3K4me2 levels of spr‐5 mutant worms. Knockdown of the H3K9me binding protein eap‐1 121, which reduces H3K4me2 levels in spr‐5 mutant worms, also reduced levels of m6A in spr‐5 mutant worms. This indicates a tight link between H3K4me2 and m6A marks in C. elegans 53. Given that across eukaryotes H3K4me2 is acquired during active transcription 125, here too m6A might show a link to active transcription. In Drosophila, elevated levels of m6A in gene bodies of transposons were associated with increased levels of transcription of these elements 54. Thus, in three model systems m6A is potentially associated with active transcription. This might in part relate to early observations that m6A in DNA results in lower stability of duplexes, thus favoring stand separation during transcription 122.
In contrast, 5mC shows a conserved association with condensed and transcriptionally silent chromatin across eukaryotes 42, 94, 123. However, while apparently contrary in their functional consequences, 5mC and m6A do not seem to be correlated in eukaryotes. Some eukaryotes, such as nematodes and ciliates, possess only an m6A modification system. Others, such as Naegleria, chlorophytes and several fungal lineages, show co‐occurring systems capable of generating m6A, 5mC, oxidized 5mC, and perhaps oxidized thymines 49, 104, 105. Kinetoplastids lack m6A‐generating systems but possess 5mC and oxidized thymine systems 15, 37, 124. Thus, unlike histone‐methylation, which is universal across eukaryotes, DNA modifications are patchily utilized. Therefore, though they might play critical roles in certain lineages, they have been evolutionarily subordinate to histone modifications, which are an essential feature of eukaryotic life 72.
The m6A methylomes are very different in the three models in which they have been determined 52, 53, 54. Hence, despite common features of m6A modifications in distantly related eukaryotes, there are likely to be functional differences that are currently poorly understood. In Chlamydomonas, extensive modification (84% of genes), a clear TSS‐associated pattern, and symmetric methylation sites, suggest that the modification might play a role in the organization of transcriptionally active chromatin 52. In contrast, in C. elegans there are no strong local patterns of m6A, the levels of m6A are “noisy” even in wild‐type animals, and methylation motifs are single‐strand biased. These observations, along with the remarkable trans‐generational increase coupled to H3K4me2 marks in the spr‐5 mutant, suggest that m6A might reinforce the function of the H3K4me2 mark – the two marks together serving as epigenetic memory for genes that have been actively transcribed 125. Because a large fraction of genes are actively transcribed during germ‐line development, these marks might need to be reset to restore the ground state during meiosis and zygote development. Consistent with this, buildup of m6A and H3K4me2 levels is correlated with infertility that emerges after several generations 53. The asymmetric DNA motifs of m6A in C. elegans are reminiscent of mRNA motifs of methylation catalyzed by enzymes of the related METTL3/METTL14 clades 126. Hence, it is possible that damt‐1 and other METTL4‐like enzymes act on ssDNA generated during transcription.
Conclusions and prospects
Evolutionary analysis of m6A MTases adds further evidence to the recent hypothesis that key players in eukaryotic chromatin first emerged in prokaryotic conflict systems 15, 20. In the latter systems they play a role in self‐nonself discrimination or diversification of secondary metabolites to effectively target rivals with antibiotics and evade stealing of products such as siderophores 88, 107. The origin of the eukaryotic nucleus probably opened niches that selected for prokaryotic modification enzymes and modified DNA readers in new capacities 88. Interestingly, these were often also deployed in RNA‐related roles. This might again relate to the eukaryotic separation of cytoplasmic translation from nuclear transcription allowing for similar enzymes to be utilized in both RNA‐ and DNA‐based regulation 15, 68.
Renewed focus on m6A in eukaryotic DNA has added it to the growing list of epigenetic marks in DNA 15, 37, 49, 51, 105. However, there are still several biochemical questions needing careful consideration in the future: (i) which members of the AlkB family are involved in demethylation of m6A? (ii) Is there a genuine, direct role for TET/JBP enzymes in removal of m6A marks? (iii) Are partners used by DNA N6A‐MTases and demethylases (such as WTAP in the RNA methylation system)? (iv) How does the methylation‐ and demethylation‐apparatus interact with histone modifications and the transcription machinery? (v) What are the readers of m6A marks? (vi) Is binding of transcription factors and chromatin proteins affected by m6A? (vii) Do oxidized metastable derivatives of m6A have any role in dynamics of m6A‐based regulation?
In terms of biology, an important question is how prevalent m6A marks are in eukaryotes. It remains to be established if METTL4 orthologs outside of nematodes are involved in DNA m6A methylation. Further, investigating fungal and basal eukaryotic systems 104, 105 might help understand elements of their functions that are not obvious from mammalian systems.
The authors have declared no conflicts of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Data
Acknowledgment
This work is supported by the funds of the Intramural Research Program of National Library of Medicine at the National Institutes of Health, USA.
References
- 1. Luria SE, Human ML. 1952. A nonhereditary, host‐induced variation of bacterial viruses. J Bacteriol 64: 557–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ralston DJ, Krueger AP. 1952. Phage multiplication on two hosts. Isolation and activity of variants of staphylococcus phage P 1. Proc Soc Exp Biol Med 80: 217–20. [DOI] [PubMed] [Google Scholar]
- 3. Anderson ES, Felix A. 1952. Variation in Vi‐phage II of Salmonella typhi . Nature 170: 492–4. [DOI] [PubMed] [Google Scholar]
- 4. Arber W, Linn S. 1969. DNA modification and restriction. Annu Rev Biochem 38: 467–500. [DOI] [PubMed] [Google Scholar]
- 5. Meselson M, Weigle JJ. 1961. Chromosome breakage accompanying genetic recombination in bacteriophage. Proc Natl Acad Sci USA 47: 857–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Loenen WA, Dryden DT, Raleigh EA, Wilson GG, et al. 2014. Highlights of the DNA cutters: a short history of the restriction enzymes. Nucleic Acids Res 42: 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roberts RJ. 1980. Restriction and modification enzymes and their recognition sequences. Gene 8: 329–43. [DOI] [PubMed] [Google Scholar]
- 8. Roberts RJ, Belfort M, Bestor T, Bhagwat AS, et al. 2003. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31: 1805–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kobayashi I. 2001. Behavior of restriction‐modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res 29: 3742–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bickle TA, Kruger DH. 1993. Biology of DNA restriction. Microbiol Rev 57: 434–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rao DN, Dryden DT, Bheemanaik S. 2014. Type III restriction‐modification enzymes: a historical perspective. Nucleic Acids Res 42: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Anantharaman V, Iyer LM, Aravind L. 2012. Ter‐dependent stress response systems: novel pathways related to metal sensing, production of a nucleoside‐like metabolite, and DNA‐processing. Mol Biosyst 8: 3142–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, et al. 2013. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra‐genomic conflicts, defense, pathogenesis and RNA processing. Biol Direct 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iyer LM, Abhiman S, Aravind L. 2008. MutL homologs in restriction‐modification systems and the origin of eukaryotic MORC ATPases. Biol Direct 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iyer LM, Zhang D, Burroughs AM, Aravind L. 2013. Computational identification of novel biochemical systems involved in oxidation, glycosylation and other complex modifications of bases in DNA. Nucleic Acids Res 41: 7635–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang L, Chen S, Vergin KL, Giovannoni SJ, et al. 2011. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc Natl Acad Sci USA 108: 2963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen S, Wang L, Deng Z. 2010. Twenty years hunting for sulfur in DNA. Protein Cell 1: 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng X. 1995. Structure and function of DNA methyltransferases. Annu Rev Biophys Biomol Struct 24: 293–318. [DOI] [PubMed] [Google Scholar]
- 19. Cheng X, Roberts RJ. 2001. AdoMet‐dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res 29: 3784–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iyer LM, Abhiman S, Aravind L. 2011. Natural history of eukaryotic DNA methylation systems. Prog Mol Biol Transl Sci 101: 25–104. [DOI] [PubMed] [Google Scholar]
- 21. Bujnicki JM. 1999. Comparison of protein structures reveals monophyletic origin of the AdoMet‐dependent methyltransferase family and mechanistic convergence rather than recent differentiation of N4‐cytosine and N6‐adenine DNA methylation. In Silico Biol 1: 175–82. [PubMed] [Google Scholar]
- 22. Warren RA. 1980. Modified bases in bacteriophage DNAs. Annu Rev Microbiol 34: 137–58. [DOI] [PubMed] [Google Scholar]
- 23. Witmer H, Wiatr C. 1985. Polymer‐level synthesis of oxopyrimidine deoxynucleotides by Bacillus subtilis phage S P10: characterization of modification‐defective mutants. J Virol 53: 522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gommers‐Ampt JH, Borst P. 1995. Hypermodified bases in DNA. FASEB J 9: 1034–42. [DOI] [PubMed] [Google Scholar]
- 25. Kaminska KH, Bujnicki JM. 2008. Bacteriophage Mu Mom protein responsible for DNA modification is a new member of the acyltransferase superfamily. Cell Cycle 7: 120–1. [DOI] [PubMed] [Google Scholar]
- 26. Liu G, Ou HY, Wang T, Li L, et al. 2010. Cleavage of phosphorothioated DNA and methylated DNA by the type IV restriction endonuclease ScoMcrA. PLoS Genet 6: e1001253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abeles A, Brendler T, Austin S. 1993. Evidence of two levels of control of P1 oriR and host oriC replication origins by DNA adenine methylation. J Bacteriol 175: 7801–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lobner‐Olesen A, Skovgaard O, Marinus MG. 2005. Dam methylation: coordinating cellular processes. Curr Opin Microbiol 8: 154–60. [DOI] [PubMed] [Google Scholar]
- 29. Rajewska M, Wegrzyn K, Konieczny I. 2012. AT‐rich region and repeated sequences – the essential elements of replication origins of bacterial replicons. FEMS Microbiol Rev 36: 408–34. [DOI] [PubMed] [Google Scholar]
- 30. Mohapatra SS, Fioravanti A, Biondi EG. 2014. DNA methylation in Caulobacter and other Alphaproteobacteria during cell cycle progression. Trends Microbiol 22: 528–35. [DOI] [PubMed] [Google Scholar]
- 31. Casadesus J, Low D. 2006. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev 70: 830–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sternberg N, Coulby J. 1990. Cleavage of the bacteriophage P1 packaging site (pac) is regulated by adenine methylation. Proc Natl Acad Sci USA 87: 8070–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Casadesus J, Low DA. 2013. Programmed heterogeneity: epigenetic mechanisms in bacteria. J Biol Chem 288: 13929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fang G, Munera D, Friedman DI, Mandlik A, et al. 2012. Genome‐wide mapping of methylated adenine residues in pathogenic Escherichia coli using single‐molecule real‐time sequencing. Nat Biotechnol 30: 1232–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murray IA, Clark TA, Morgan RD, Boitano M, et al. 2012. The methylomes of six bacteria. Nucleic Acids Res 40: 11450–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rae PMM, Steele RE. 1978. Modified bases in the DNAs of unicellular eukaryotes: an examination of distributions and possible roles, with emphasis on hydroxymethyluracil in dinoflagellates. BioSystems 10: 37–53. [DOI] [PubMed] [Google Scholar]
- 37. Borst P, Sabatini R. 2008. Base J: discovery, biosynthesis, and possible functions. Annu Rev Microbiol 62: 235–51. [DOI] [PubMed] [Google Scholar]
- 38. Achwal CW, Iyer CA, Chandra HS. 1983. Immunochemical evidence for the presence of 5mC, 6mA and 7mG in human, Drosophila and mealybug DNA. FEBS Lett 158: 353–8. [DOI] [PubMed] [Google Scholar]
- 39. Ooi SK, Bestor TH. 2008. The colorful history of active DNA demethylation. Cell 133: 1145–8. [DOI] [PubMed] [Google Scholar]
- 40. Cheng X, Blumenthal RM. 2008. Mammalian DNA methyltransferases: a structural perspective. Structure 16: 341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bestor TH. 1990. DNA methylation: evolution of a bacterial immune function into a regulator of gene expression and genome structure in higher eukaryotes. Philos Trans R Soc Lond B Biol Sci 326: 179–87. [DOI] [PubMed] [Google Scholar]
- 42. Ooi SK, O'Donnell AH, Bestor TH. 2009. Mammalian cytosine methylation at a glance. J Cell Sci 122: 2787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Furner IJ, Matzke M. 2011. Methylation and demethylation of the Arabidopsis genome. Cur Opin Plant Biol 14: 137–41. [DOI] [PubMed] [Google Scholar]
- 44. Zemach A, McDaniel IE, Silva P, Zilberman D. 2010. Genome‐wide evolutionary analysis of eukaryotic DNA methylation. Science 328: 916–9. [DOI] [PubMed] [Google Scholar]
- 45. Selker EU. 2004. Genome defense and DNA methylation in Neurospora. Cold Spring Harb Symp Quant Biol 69: 119–24. [DOI] [PubMed] [Google Scholar]
- 46. Tahiliani M, Koh KP, Shen Y, Pastor WA, et al. 2009. Conversion of 5‐methylcytosine to 5‐hydroxymethylcytosine in mammalian DNA by MLL partner T ET1. Science 324: 930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. He YF, Li BZ, Li Z, Liu P, et al. 2011. Tet‐mediated formation of 5‐carboxylcytosine and its excision by TDG in mammalian DNA. Science 333: 1303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ito S, Shen L, Dai Q, Wu SC, et al. 2011. Tet proteins can convert 5‐methylcytosine to 5‐formylcytosine and 5‐carboxylcytosine. Science 333: 1300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hashimoto H, Pais JE, Zhang X, Saleh L, et al. 2014. Structure of a Naegleria Tet‐like dioxygenase in complex with 5‐methylcytosine DNA. Nature 506: 391–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang L, Chen W, Iyer LM, Hu J, et al. 2014. A TET homologue protein from Coprinopsis cinerea (CcTET) that biochemically converts 5‐methylcytosine to 5‐hydroxymethylcytosine, 5‐formylcytosine, and 5‐carboxylcytosine. J Am Chem Soc 136: 4801–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pastor WA, Aravind L, Rao A. 2013. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 14: 341–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fu Y, Luo G‐Z, Chen K, Deng X, et al. 2015. N(6)‐methyldeoxyadenosine marks active transcription start sites in chlamydomonas. Cell 161: 879–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Greer EL, Blanco MA, Gu L, Sendinc E, et al. 2015. DNA methylation on N(6)‐adenine in C. elegans . Cell 161: 868–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang G, Huang H, Liu D, Cheng Y, et al. 2015. N(6)‐methyladenine DNA modification in Drosophila. Cell 161: 893–906. [DOI] [PubMed] [Google Scholar]
- 55. Aravind L, Mazumder R, Vasudevan S, Koonin EV. 2002. Trends in protein evolution inferred from sequence and structure analysis. Curr Opin Struct Biol 12: 392–9. [DOI] [PubMed] [Google Scholar]
- 56. Burroughs AM, Iyer LM, Aravind L. 2009. Natural history of the E1‐like superfamily: implication for adenylation, sulfur transfer, and ubiquitin conjugation. Proteins 75: 895–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schubert HL, Blumenthal RM, Cheng X. 2003. Many paths to methyltransfer: a chronicle of convergence. Trends Biochem Sci 28: 329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Foster PG, Nunes CR, Greene P, Moustakas D, et al. 2003. The first structure of an RNA m5C methyltransferase, Fmu, provides insight into catalytic mechanism and specific binding of RNA substrate. Structure 11: 1609–20. [DOI] [PubMed] [Google Scholar]
- 59. Anantharaman V, Koonin EV, Aravind L. 2002. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res 30: 1427–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heurgue‐Hamard V, Champ S, Engstrom A, Ehrenberg M, et al. 2002. The hemK gene in Escherichia coli encodes the N(5)‐glutamine methyltransferase that modifies peptide release factors. EMBO J 21: 769–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nakahigashi K, Kubo N, Narita S, Shimaoka T, et al. 2002. HemK, a class of protein methyl transferase with similarity to DNA methyl transferases, methylates polypeptide chain release factors, and hemK knockout induces defects in translational termination. Proc Natl Acad Sci USA 99: 1473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Horton JR, Liebert K, Bekes M, Jeltsch A, et al. 2006. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J Mol Biol 358: 559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bheemanaik S, Reddy YVR, Rao DN. 2006. Structure, function and mechanism of exocyclic DNA methyltransferases. Biochem J 399: 177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gong W, O'Gara M, Blumenthal RM, Cheng X. 1997. Structure of pvu II DNA‐(cytosine N4) methyltransferase, an example of domain permutation and protein fold assignment. Nucleic Acids Res 25: 2702–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Malone T, Blumenthal RM, Cheng X. 1995. Structure‐guided analysis reveals nine sequence motifs conserved among DNA amino‐methyltransferases, and suggests a catalytic mechanism for these enzymes. J Mol Biol 253: 618–32. [DOI] [PubMed] [Google Scholar]
- 66. Bujnicki JM, Feder M, Radlinska M, Blumenthal RM. 2002. Structure prediction and phylogenetic analysis of a functionally diverse family of proteins homologous to the MT‐A70 subunit of the human mRNA:m(6)A methyltransferase. J Mol Evol 55: 431–44. [DOI] [PubMed] [Google Scholar]
- 67. Liu J, Yue Y, Han D, Wang X, et al. 2014. A METTL3‐METTL14 complex mediates mammalian nuclear RNA N6‐adenosine methylation. Nat Chem Biol 10: 93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet 15: 293–306. [DOI] [PubMed] [Google Scholar]
- 69. Ponting CP, Blake DJ, Davies KE, Kendrick‐Jones J, et al. 1996. ZZ and TAZ: new putative zinc fingers in dystrophin and other proteins. Trends Biochem Sci 21: 11–3. [PubMed] [Google Scholar]
- 70. Aravind L, Iyer LM. 2002. The SWIRM domain: a conserved module found in chromosomal proteins points to novel chromatin‐modifying activities. Genome Biol 3: RESEARCH0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cabezon E, Ripoll‐Rozada J, Pena A, de la Cruz F, et al. 2015. Towards an integrated model of bacterial conjugation. FEMS Microbiol Rev 39: 81–95. [DOI] [PubMed] [Google Scholar]
- 72. Aravind L, Abhiman S, Iyer LM. 2011. Natural history of the eukaryotic chromatin protein methylation system. Prog Mol Biol Transl Sci 101: 105–76. [DOI] [PubMed] [Google Scholar]
- 73. Aravind L, Anantharaman V, Abhiman S, Iyer LM. 2014. Evolution of eukaryotic chromatin proteins and transcription factors In Uversky V, ed; Protein Families: Relating Protein Sequence, Structure, and Function. Hoboken, New Jersey: Wiley. p. 421–502. [Google Scholar]
- 74. Johnston C, Polard P, Claverys J‐P. 2013. The DpnI/DpnII pneumococcal system, defense against foreign attack without compromising genetic exchange. Mob Genet Elements 3: e25582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Johnston C, Martin B, Granadel C, Polard P, et al. 2013. Programmed protection of foreign DNA from restriction allows pathogenicity island exchange during pneumococcal transformation. PLoS Pathog 9: e1003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yin P, Li Q, Yan C, Liu Y, et al. 2013. Structural basis for the modular recognition of single‐stranded RNA by PPR proteins. Nature 504: 168–71. [DOI] [PubMed] [Google Scholar]
- 77. Small ID, Peeters N. 2000. The PPR motif – a TPR‐related motif prevalent in plant organellar proteins. Trends Biochem Sci 25: 46–7. [DOI] [PubMed] [Google Scholar]
- 78. Stavridi ES, Huyen Y, Loreto IR, Scolnick DM, et al. 2002. Crystal structure of the FHA domain of the Chfr mitotic checkpoint protein and its complex with tungstate. Structure 10: 891–9. [DOI] [PubMed] [Google Scholar]
- 79. Goedecke K, Pignot M, Goody RS, Scheidig AJ, et al. 2001. Structure of the N6‐adenine DNA methyltransferase M. TaqI in complex with DNA and a cofactor analog. Nat Struct Biol 8: 121–5. [DOI] [PubMed] [Google Scholar]
- 80. Fan H, Sakuraba K, Komuro A, Kato S, et al. 2003. PCIF1, a novel human WW domain‐containing protein, interacts with the phosphorylated RNA polymerase II. Biochem Biophys Res Commun 301: 378–85. [DOI] [PubMed] [Google Scholar]
- 81. Napolitano G, Lania L, Majello B. 2014. RNA polymerase II CTD modifications: how many tales from a single tail. J Cell Physiol 229: 538–44. [DOI] [PubMed] [Google Scholar]
- 82. Poulter RT, Goodwin TJ. 2005. DIRS‐1 and the other tyrosine recombinase retrotransposons. Cytogenet Genome Res 110: 575–88. [DOI] [PubMed] [Google Scholar]
- 83. Kouzarides T. 2007. Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- 84. Maurer‐Stroh S, Dickens NJ, Hughes‐Davies L, Kouzarides T, et al. 2003. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci 28: 69–74. [DOI] [PubMed] [Google Scholar]
- 85. Cerritelli S, White SW, Lacks SA. 1989. Crystallization of the DpnM methylase from the DpnII restriction system of Streptococcus pneumoniae . J Mol Biol 207: 841–2. [DOI] [PubMed] [Google Scholar]
- 86. Balaji S, Aravind L. 2007. The RAGNYA fold: a novel fold with multiple topological variants found in functionally diverse nucleic acid, nucleotide and peptide‐binding proteins. Nucleic Acids Res 35: 5658–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim JS, DeGiovanni A, Jancarik J, Adams PD, et al. 2005. Crystal structure of DNA sequence specificity subunit of a type I restriction‐modification enzyme and its functional implications. Proc Natl Acad Sci USA 102: 3248–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Aravind L, Burroughs AM, Zhang D, Iyer LM. 2014. Protein and DNA modifications: evolutionary imprints of bacterial biochemical diversification and geochemistry on the provenance of eukaryotic epigenetics. Cold Spring Harb Perspect Biol 6: a016063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Goll MG, Kirpekar F, Maggert KA, Yoder JA, et al. 2006. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311: 395–8. [DOI] [PubMed] [Google Scholar]
- 90. Iyer LM, Tahiliani M, Rao A, Aravind L. 2009. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 8: 1698–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Weber M, Davies JJ, Wittig D, Oakeley EJ, et al. 2005. Chromosome‐wide and promoter‐specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37: 853–62. [DOI] [PubMed] [Google Scholar]
- 92. Eid J, Fehr A, Gray J, Luong K, et al. 2009. Real‐time DNA sequencing from single polymerase molecules. Science 323: 133–8. [DOI] [PubMed] [Google Scholar]
- 93. Gutierrez JC, Callejas S, Borniquel S, Martin‐Gonzalez A. 2000. DNA methylation in ciliates: implications in differentiation processes. Int Microbiol 3: 139–46. [PubMed] [Google Scholar]
- 94. Hajkova P, Ancelin K, Waldmann T, Lacoste N, et al. 2008. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature 452: 877–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Iqbal K, Jin SG, Pfeifer GP, Szabo PE. 2011. Reprogramming of the paternal genome upon fertilization involves genome‐wide oxidation of 5‐methylcytosine. Proc Natl Acad Sci USA 108: 3642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Aravind L, Koonin EV. 2001. The DNA‐repair protein AlkB, EGL‐9, and leprecan define new families of 2‐oxoglutarate‐ and iron‐dependent dioxygenases. Genome Biol 2: RESEARCH0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Falnes PO, Johansen RF, Seeberg E. 2002. AlkB‐mediated oxidative demethylation reverses DNA damage in Escherichia coli . Nature 419: 178–82. [DOI] [PubMed] [Google Scholar]
- 98. Trewick SC, Henshaw TF, Hausinger RP, Lindahl T, et al. 2002. Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419: 174–8. [DOI] [PubMed] [Google Scholar]
- 99. Aas PA, Otterlei M, Falnes PO, Vagbo CB, et al. 2003. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421: 859–63. [DOI] [PubMed] [Google Scholar]
- 100. Fu Y, Jia G, Pang X, Wang RN, et al. 2013. FTO‐mediated formation of N6‐hydroxymethyladenosine and N6‐formyladenosine in mammalian RNA. Nat Commun 4: 1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Fu D, Brophy JA, Chan CT, Atmore KA, et al. 2010. Human AlkB homolog ABH8 Is a tRNA methyltransferase required for wobble uridine modification and DNA damage survival. Mol Cell Biol 30: 2449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Songe‐Moller L, van den Born E, Leihne V, Vagbo CB, et al. 2010. Mammalian ALKBH8 possesses tRNA methyltransferase activity required for the biogenesis of multiple wobble uridine modifications implicated in translational decoding. Mol Cell Biol 30: 1814–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Jia G, Fu Y, Zhao X, Dai Q, et al. 2011. N6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat Chem Biol 7: 885–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Pais JE, Dai N, Tamanaha E, Vaisvila R, et al. 2015. Biochemical characterization of a Naegleria TET‐like oxygenase and its application in single molecule sequencing of 5‐methylcytosine. Proc Natl Acad Sci USA 112: 4316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chavez L, Huang Y, Luong K, Agarwal S, et al. 2014. Simultaneous sequencing of oxidized methylcytosines produced by TET/JBP dioxygenases in Coprinopsis cinerea . Proc Natl Acad Sci USA 111: E5149–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Guarne A, Zhao Q, Ghirlando R, Yang W. 2002. Insights into negative modulation of E. coli replication initiation from the structure of SeqA‐hemimethylated DNA complex. Nat Struct Biol 9: 839–43. [DOI] [PubMed] [Google Scholar]
- 107. Aravind L, Anantharaman V, Zhang D, de Souza RF, et al. 2012. Gene flow and biological conflict systems in the origin and evolution of eukaryotes. Front Cell Infect Microbiol 2: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Zhu P, Zhou W, Wang J, Puc J, et al. 2007. A histone H2A deubiquitinase complex coordinating histone acetylation and H1 dissociation in transcriptional regulation. Mol Cell 27: 609–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Aravind L, Iyer LM. 2012. The HARE‐HTH and associated domains: novel modules in the coordination of epigenetic DNA and protein modifications. Cell Cycle 11: 119–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nijnik A, Clare S, Hale C, Raisen C, et al. 2012. The critical role of histone H2A‐deubiquitinase Mysm1 in hematopoiesis and lymphocyte differentiation. Blood 119: 1370–9. [DOI] [PubMed] [Google Scholar]
- 111. Aravind L, Koonin EV. 1999. Novel predicted RNA‐binding domains associated with the translation machinery. J Mol Evol 48: 291–302. [DOI] [PubMed] [Google Scholar]
- 112. Iyer LM, Burroughs AM, Aravind L. 2006. The ASCH superfamily: novel domains with a fold related to the PUA domain and a potential role in RNA metabolism. Bioinformatics 22: 257–63. [DOI] [PubMed] [Google Scholar]
- 113. Bertonati C, Punta M, Fischer M, Yachdav G, et al. 2009. Structural genomics reveals EVE as a new ASCH/PUA‐related domain. Proteins 75: 760–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, et al. 2013. Dynamic readers for 5‐(hydroxy) methylcytosine and its oxidized derivatives. Cell 152: 1146–59. [DOI] [PubMed] [Google Scholar]
- 115. Stoilov P, Rafalska I, Stamm S. 2002. YTH: a new domain in nuclear proteins. Trends Biochem Sci 27: 495–7. [DOI] [PubMed] [Google Scholar]
- 116. Jung DJ, Sung HS, Goo YW, Lee HM, et al. 2002. Novel transcription coactivator complex containing activating signal cointegrator 1. Mol Cell Biol 22: 5203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Tung YC, Gulati P, Liu CH, Rimmington D, et al. 2015. FTO is necessary for the induction of leptin resistance by high‐fat feeding. Mol Metab 4: 287–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sharif J, Muto M, Takebayashi S, Suetake I, et al. 2007. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 450: 908–12. [DOI] [PubMed] [Google Scholar]
- 119. Bostick M, Kim JK, Esteve PO, Clark A, et al. 2007. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317: 1760–4. [DOI] [PubMed] [Google Scholar]
- 120. Johnson LM, Bostick M, Zhang X, Kraft E, et al. 2007. The SRA methyl‐cytosine‐binding domain links DNA and histone methylation. Curr Biol 17: 379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Greer EL, Beese‐Sims SE, Brookes E, Spadafora R, et al. 2014. A histone methylation network regulates transgenerational epigenetic memory in C. elegans . Cell Rep 7: 113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Guo Q, Lu M, Kallenbach NR. 1995. Effect of hemimethylation and methylation of adenine on the structure and stability of model DNA duplexes. Biochemistry 34: 16359–64. [DOI] [PubMed] [Google Scholar]
- 123. Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21. [DOI] [PubMed] [Google Scholar]
- 124. Bullard W, Lopes da Rosa‐Spiegler J, Liu S, Wang Y, et al. 2014. Identification of the glucosyltransferase that converts hydroxymethyluracil to base J in the trypanosomatid genome. J Biol Chem 289: 20273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Kerr SC, Ruppersburg CC, Francis JW, Katz DJ. 2014. SPR‐5 and MET‐2 function cooperatively to reestablish an epigenetic ground state during passage through the germ line. Proc Natl Acad Sci USA 111: 9509–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Dominissini D, Moshitch‐Moshkovitz S, Schwartz S, Salmon‐Divon M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A‐seq. Nature 485: 201–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Data