

Abstract
Synapse disruption takes place in many neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS). However, the mechanistic understanding of this process is still limited. We set out to study a possible role for dynein in synapse integrity. Cytoplasmic dynein is a multisubunit intracellular molecule responsible for diverse cellular functions, including long-distance transport of vesicles, organelles, and signaling factors toward the cell center. A less well-characterized role dynein may play is the spatial clustering and anchoring of various factors including mRNAs in distinct cellular domains such as the neuronal synapse. Here, in order to gain insight into dynein functions in synapse integrity and disruption, we performed a screen for novel dynein interactors at the synapse. Dynein immunoprecipitation from synaptic fractions of the ALS model mSOD1G93A and wild-type controls, followed by mass spectrometry analysis on synaptic fractions of the ALS model mSOD1G93A and wild-type controls, was performed. Using advanced network analysis, we identified Staufen1, an RNA-binding protein required for the transport and localization of neuronal RNAs, as a major mediator of dynein interactions via its interaction with protein phosphatase 1–beta (PP1B). Both in vitro and in vivo validation assays demonstrate the interactions of Staufen1 and PP1B with dynein, and their colocalization with synaptic markers was altered as a result of two separate ALS-linked mutations: mSOD1G93A and TDP43A315T. Taken together, we suggest a model in which dynein's interaction with Staufen1 regulates mRNA localization along the axon and the synapses, and alterations in this process may correlate with synapse disruption and ALS toxicity.
Amyotrophic lateral sclerosis (ALS)1 is an adult-onset progressive neurodegenerative disease that targets both upper and lower motor neurons via an unknown mechanism, leading to paralysis and eventually death. Pathological changes affecting synapses in both the primary motor cortex and the peripheral neuromuscular junctions (NMJs) are considered an early occurrence in ALS, often preceding the degeneration of the axons and clinical symptomatic onset (1). Although synapse disruption is common to many neurodegenerative diseases and the molecular mechanisms underlying synapse stabilization and maintenance are of keen interest, the exact mechanisms governing synapse disruption have yet to be understood.
Both upper and lower motor neurons are highly polarized cells, with axons that are several orders of magnitude longer than the diameter of their cell bodies. To survive and maintain proper function, these neurons depend on active intracellular transport (2). The molecular motor kinesin drives transport from the cell body to the nerve periphery, supplying proteins, lipids, RNAs, and other essential materials to the synapse. The dynein/dynactin protein complex drives retrograde transport, moving damaged proteins for degradation, as well as critical signaling molecules such as neurotrophins, to the cell body (3). Dynein is a pleiotropic cellular motor, whose function in numerous cellular pathways may be regulated by specific interactions with different binding partners (4, 5). In addition to its canonical role as a motor protein, dynein has been shown to have an anchoring role as well. For example, the interaction of dynein with microtubule binding nuclear mitotic apparatus protein (NuMA)-protein coupled receptor 1 (LGN) allows dynein to be cortically anchored in order to function in the spindle-positioning process during cell division (4, 6). In neurons, dynein interacts with the neuronal adhesion molecule neural-cell-adhesion-molecule-180, which leads to the specific recruitment of dynein to the cell cortex for synapse stabilization (7). Another example, best characterized in the oocyte, is mRNA anchoring at specific cellular locations (8). Thus, dynein can serve as a motor conducting long-distance signaling, as well as an anchoring agent at distinct domains like the synapse. The switch between dynein's different capacities may be regulated by its phosphorylation state, which may be mediated by protein phosphatase 1 (PP1) (9, 10).
Transport deficits are common in many neurodegenerative disorders (3, 11, 12). In the ALS mouse model SOD1G93A, transport dysfunction can be observed as early as at the embryonic stage (13). Although mutations in dynein or its activator dynactin were demonstrated to lead to synapse disruption and neurodegeneration (14–16), the effect of the mutations in slowing down dynein-mediated transport is not sufficient to create the harsh neurodegeneration observed in ALS (17, 18), suggesting an additional mechanism. One possibility is a switch in the nature of the retrogradely transported cargo from survival signals to stress signals (19). Hence, a change in the composition of dynein complexes may underlie neurodegenerative and synapse elimination mechanisms.
General proteomic screens of protein complexes at the synapse have presented high complexity of both protein composition and signaling network architecture (20–23). Proteomics following immunoprecipitation of receptors such as α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid and N-methyl-D-aspartate from synaptosomes reveal large protein complexes, up to 3000 kDa that can incorporate up to 185 proteins (21, 24). Notably, many of these proteins are involved in localized protein synthesis (25).
Interestingly, dynein was found to be one of the proteins identified from synaptosome proteomics (26), suggesting that dynein plays a role in maintaining synaptic function. Although synapse disruption is one of the early events occurring in many neurodegenerative diseases, the identity of dynein complexes in the synapse and molecular mechanisms of synapse protection are still largely unknown.
Here, we sought to characterize synaptic dynein complexes using a differential proteomic screen of the SOD1G93A mouse model for ALS. The SOD1G93A mouse model is the most studied model for ALS, manifesting many ALS phenotypes, including upper and lower motoneuron degeneration, synaptic disruption, and alterations in dynein functions. Here, we purified synaptosomes from brains of SOD1G93A and control mice, followed by dynein-intermediate chain immunoprecipitation and mass spectrometry analysis to identify changes in dynein interactors. We further utilized the Advanced Network Analysis Tool (ANAT) (27) to predict potential pathways connecting dynein to the immunoprecipitated proteins in the ALS model and control mice. Our results demonstrate distinct populations of dynein-interacting proteins in ALS and in control mice, in addition to several common interactors. In both networks, the RNA-binding protein Staufen1 appeared as a predicted central node linking dynein to PP1B, a component of the catalytic subunit of PP1. In vitro and in vivo validation of the interaction and synaptic colocalization of both Staufen1 and PP1B with dynein, together with altered localization caused by ALS-linked mutations, suggest a role for dynein in the localization of Staufen1 ribonucleoproteins (RNPs) in neurodegenerative diseases such as ALS.
EXPERIMENTAL PROCEDURES
Mice
SOD1G93A mice (28) were originally obtained from Jackson Laboratories, and the colony was maintained by breeding with C57BL mice. DIC-GFP mice were generated as previously described (29) and bred with C57BL mice. SOD1G93A and DIC-GFP mice were crossed to give DIC-GFP/mSOD1G93A progeny. Pregnant ICR mice were obtained from the Institute of Animal Science, Harlan. All animal experimentations were approved by the Animal Ethics Committee of Tel-Aviv University. Primary cultures were harvested from mice embryos of either sex.
Synaptosome Enrichment
Brain tissue from three mice (∼1.2 g total) was homogenized in 12 ml cold homogenization buffer (5 mm Hepes/NaOH, pH 7.4, 0.32 m sucrose, 1X protease inhibitors (Roche, West Grove, PA)) at 400 rpm, and then centrifuged at 1000 g for 10 min at 4 °C. The supernatant was then centrifuged at 160,000 g (30,000 rpm SW41Ti) for 2 h. The pellet was resuspended in resuspension buffer (25 mm Hepes/NaOH, pH 7.4, 150 mm NaCl). Protein concentration was determined (Bio-Rad Protein Assay) and adjusted to a concentration of 10 μg/μl. The samples were then mixed with 2x extraction buffer (2x Triton X-100 (Sigma Aldrich, St. Louis, MO), 25 mm Hepes, 150 mm NaCl, pH 7.4, 2XPI) at a 1:1 volume ratio and incubated on ice for 20 min. The samples were centrifuged at maximum speed (∼17,000 g) in an Eppendorf table centrifuge at 4 °C for 20 min, and the supernatant was transferred into a new tube. The pellet was resuspended in 1x extraction buffer (1xTriton X-100, 25 mm Hepes, 150 mm NaCl, pH 7.4) using twice the volume of the pellet, incubated on ice for 20 min, and centrifuged at 17,000 g at 4 °C for 20 min. The supernatant was collected, added to the previous supernatant, and centrifuged at 17,000 g at 4 °C for 20 min.
Dynein Immunoprecipitation
Synaptosome preparation or brain extracts were precleared with protein A agarose beads (Roche). Following overnight incubation with primary antibody, complexes were incubated with protein A agarose beads for 2 h at 4 °C and then precipitated and washed with PBS with 0.1% Triton X-100 (Sigma). Proteins were eluted by boiling in sample buffer and then 5 μl of 30% acrylamide/Bis solution (Bio-Rad) added to each sample. The samples were then subjected to Western blot analysis or Coomassie Blue staining (Invitrogen LC6025). Precipitation with dynein intermediate chain (DIC) antibody (a kind gift from K. Kevin Pfister, University of Virginia) was carried out using 10 μg of dynein antibody for 10 mg of synaptosome sample and 2 μg of dynein antibody for 2 mg of total brain extract.
Sample Preparation for Mass Spectrometry
In-gel digestion procedure, MALDI MS, and protein identification procedure were performed as previously described (23). The peptides were redissolved in 20 μl 0.1% TFA and separated on an analytical capillary C18 column (150 mm × 100 μm inner diameter column) at 400 nl/min with a linearly increasing concentration of acetonitrile. The eluent was mixed with matrix (7 mg α-cyano-hydroxycinnaminic acid in 1 ml 50% acetonitrile, 0.1% TFA, 10 mm dicitrate ammonium) delivered at a flow rate of 1.5 μl/min and deposited off-line to the Applied Biosystems MALDI metal target every 15 s using the Probot (Dionex, Bannockburn, IL).
Mass Spectrometry
Peptides deposited on the MALDI plates were analyzed on a 4800 proteomics analyzer (Applied Biosystems, Foster City, CA). Collision-induced dissociation was performed at 2 kV with air as the collision gas. MS/MS spectra were collected from 2500 laser shots. Peptides with signal-to-noise ratio above 50 at the MS mode were selected for MS/MS, at a maximum of 20 MS/MS per spot. The precursor mass window was set at 200 relative resolution (full width at half maximum (FWHM)). Mass spectrometry data were searched in ProteinPilot (Applied Biosystems) to identify the proteins.
Protein Identification
ProteinPilot (version 4.5, Sciex) was used for peaklist generation and the embedded Paragon in ProteinPilot for database searches against a mouse Uniprot-Swiss-Prot database (2014/03/21) with all the entries contained in this database (24,650 protein entries). MS/MS spectra were searched with trypsin specificity. False discovery rate analysis was activated, which calculates the false discovery rate corresponding to the Unused ProtScore. The Unused ProtScore should be better than a critical false discovery rate of 1.0% from global false discovery rate from fit. The search effort was thorough ID with biological modifications activated. In this mode, the Paragon first identifies the native peptides, followed by an extra search focusing on the remaining modified peptides from the identified proteins. This second search interrogates the tryptic peptides with more than 100 modifications, including the miss-cleavages. Paragon recalibrates globally the peptide masses without user-defined mass tolerance for precursor and fragment ions. Thus, there were no prior settings for the search with the number of missed and/or nonspecific cleavages, no prior fixed/variable modifications, and no prior mass tolerance for fragment ions. In order to create Supplemental Tables S1 and S2, we filtered for an unused value of >2, with >99% confidence and included proteins that appeared in all repeats from the dynein immunoprecipitated samples but not in any of the IgG control samples. IgG and keratin proteins were omitted. Proteins identified by a single peptide are detailed in Supplemental Table S3, and the corresponding identification spectra detailed in Fig. S3.
Computational Network Inference
To infer key protein–protein interactions (PPIs) we applied the network reconstruction tool ANAT (27). This tool receives as input an anchor protein, a set of terminal proteins, and a set of potential protein–protein interactions with their experimental reliabilities and computes a compact subnetwork that connects the anchor to the terminals via high likelihood pathways. We executed the ANAT algorithm using DYNC1I1 (dynein intermediate chain-DIC1) as the anchor and the high-confidence dynein-bound proteins identified from either the mSOD1 or wtSOD1 as terminals. We defined the centrality score of a protein p in the subnetwork output by ANAT as the percentage of shortest paths going from the anchor to some terminal through p. We used the centrality scores of proteins in the suggested subnetworks to predict key proteins that are likely to be related to dynein's role in the disease. As PPI data in human are much more abundant compared with mouse (by an order of magnitude), we used as input the PPI network of HIPPIE (30) filtered for high-confidence interactions and converted the mouse anchor and terminal proteins into their human orthologs using bioDBnet (31). Cytoscape was used to illustrate the subnetworks (32).
Western Blotting
Brain tissues were excised and homogenized in lysis buffer containing PBS, 1% Triton X-100 (Sigma), and protease inhibitors (Roche), followed by centrifugation and collection of the supernatant. Protein concentration was determined using Bio-Rad Protein Assay. Protein samples were denatured by boiling in SDS sample buffer and then electrophoresed in 10% or 12% polyacrylamide gels. Proteins were transferred to a nitrocellulose membrane then immunoblotted with appropriate primary antibodies (1 μg/ml anti-dynein, 0.44 μg/ml anti-Staufen1, 1.67 μg/ml anti-PP1B), followed by HRP-conjugated secondary antibodies (Jackson Laboratories) and visualized using myECL imager (Thermo), according to manufacturer's instructions. Quantification was performed using the Image J software.
Antibodies
Antibodies used were mouse anti-dynein intermediate chain (Millipore MAB1618), mouse anti-postsynaptic density 95 (Millipore MAB1596), rabbit anti-synapsin (Millipore MAB1543P), rabbit anti-general ERK (Sigma m5670), mouse anti-glyceraldehyde 3-phosphate dehydrogenase (Abcam ab9484), rabbit anti-Staufen1 (Abcam ab137100), rabbit anti-PP1B (Abcam ab154600), mouse anti-dynactin p50 (TL610474) and p150 (Glued) (TL611003) (both from BD Transduction Labs), mouse anti-synaptophysin (Millipore MAB-5258), chicken anti-GFP (Abcam ab 13970), and chicken anti-neurofilament heavy chain (Abcam ab72996).
Cell Culture
Primary spinal cord neurons were cultured from E12.5 mouse embryos as previously described (33, 34). Briefly, spinal cords were excised, trypsinized, and triturated. Supernatant was collected and centrifuged through a BSA cushion. The pellet was resuspended and centrifuged through an Optiprep gradient (10.4% Optiprep (Sigma), 10 mm Tricine, 4% glucose) for 20 min at 760 g with brake turned off. Cells were collected from the interface, washed once in complete medium, and then plated in coated growth chambers. Cells were maintained in Complete Neurobasal Medium (Gibco) containing B27, 10% (v/v) horse serum, 25 μm beta-mercaptoethanol, 100 U/ml penicillin, 100 μg/ml streptomycin (Biological Industries), and Glutamax (Gibco) supplemented with 1 ng/ml glia-derived neurotrophic factor, 0.5 ng/ml ciliary neurotrophic factor, and 1 ng/ml brain-derived neurotrophic factor (all from Alomone Labs). Prior to seeding, growth chambers were coated with 1.5 μg/ml poly dl-ornithine (Sigma) overnight at 37 °C and 3 μg/ml laminin (Sigma) for 2 h at 37 °C. For immunofluorescence staining, 5 × 104 or 104 cells were seeded on cover slides in 24-well plates. Cells were grown at 37 °C in 5% CO2.
RNA Immunoprecipitation
Total protein was harvested from brains of p60 female control and mSOD1 mice in extraction buffer (50 mm Tris-HCl, pH 7.5, 15 mm EGTA, 100 mm NaCl and 0.1% Triton X-100 supplemented with 1x protease inhibitors mixture (Roche Applied Sciences, Pensberg, Germany) and 10 unit/ml of RNAsin (Promega, Madison, WI)). Following a 10-min centrifugation at 17,000 g, the total lysate was incubated with 10 μl of RNase-free DNase I (Sigma) and then incubated at room temperature for 20 min. Ten percent of the extract was used as loading control, and the rest was precleared with protein A agarose beads (Roche Applied Sciences) for 1 h and then incubated overnight with 4 μg antibody, or with no antibody as a control. Agarose beads were then added for 2 h and then rotated at 4 °C. Beads only were used as a control. The precipitate was washed four times in extraction buffer supplemented with 0.5% Triton then washed once in extraction buffer with 1% Triton. RNA was extracted from the precipitate using TRI Reagent (Sigma). cDNA was prepared using the Super Script II Kit (Life Technologies).
Lentiviral Vectors
Genes of interest were cloned into a third-generation lentiviral pLL3.7 backbone (purchased from Addgene). Lentiviruses were generated by transfecting HEK293T cells with a mixture of the vector of interest, vesicular stomatitis virus glycoprotein, and group antigens–polymerase (reverse transcriptase) using the calcium phosphate method. Medium was replaced after 5–8 h, and supernatant was collected 48 h later. 50 mm Hepes were added prior to freezing to maintain neutral pH for long-term storage. When necessary, lentiviruses were concentrated using a PEG Virus Precipitation Kit (Abcam). TDP-43A315T-GFP and mSOD1G93A-GFP were used at an multiplicity of infection (MOI) of 5, and GFP was used at a MOI of 10.
Immunofluorescence Staining
Staining for total RNA was performed using SYTO-14 (500 nm, Life Technologies), according to manufacturer's instructions. When SYTO-14 was used, cells were fixed in 100% methanol for 20 min at −20 °C. Otherwise, cells were fixed in 4% paraformaldehyde for 10 min at room temperature. Cover slides were then washed in PBS and permeabilized with 0.5% Triton in blocking solution containing 5% donkey serum (Jackson Laboratories) and 1 mg/ml BSA (Sigma) for 5–30 min. After three washes, cells were incubated with appropriate antibodies overnight at 4 °C in blocking solution. Antibodies were used at the following concentrations: anti-dynactin, 2.5 μg/ml; anti-synaptophysin, 2 μg/ml; anti-Staufen1, 4.5 μg/ml; anti-PP1B, 2.5 μg/ml. After washing, secondary antibodies (2 μg/ml) in blocking solution were added for 2 h at room temperature. Cells were incubated for 45 min with Alexa Fluor 594/405-conjugated phalloidin (1x, Abcam) diluted in PBS, then washed and mounted with ProLong Gold (Life Technologies, Foster City, CA).
Neuromuscular Junction Staining
Gastrocnemius was excised from adult mice and cleared of connective tissue, washed in PBS and fixed in 4% paraformaldehyde, washed again, and incubated with 1 μg/ml Rhodamine Red-Conjugated Bungarotoxin (Sigma T0195). Tissues were washed and then treated with methanol at −20 °C for 5 min, washed, and blocked in blocking solution for 1 h. Tissues were then rocked with appropriate primary antibodies diluted in blocking solution at room temperature overnight. Antibodies were used at the following concentrations: anti-neurofilament heavy chain, 10 μg/ml; anti-Staufen1, 9 μg/ml; anti-PP1B, 2.5 μg/ml; anti-GFP, 2.5 μg/ml. After washing, secondary antibodies (DyLight 405 anti-chicken 30 μg/ml, AlexaFluor 488 anti-rabbit 4 μg/ml, AlexaFluor 488 anti-chicken 8 μg/ml, AlexaFluor 647 anti-rabbit 3 μg/ml) were added for 4 h at room temperature. Muscle fibers were spread into monolayers under a stereomicroscope and affixed to slides using VectaShield (Vector Laboratories). Cover slides were sealed with clear nail polish.
Image Analysis
Colocalization measurements were obtained from three-dimensional image stacks, using Imaris Coloc (Bitplane Inc.). After background removal, the image was masked for the neurite of interest using either the GFP channel or the phalloidin one and colocalization values reflecting the percentage of colocalized volume per channel. The values from several images were averaged and were conveyed as the percentage of channel A localized to channel B. For quantification of dynactin colocalization, a coloc channel was built as described above and then analyzed with Image J Advanced Profile Plot. Peaks corresponding to colocalized punctae were analyzed for density and intensity.
Statistical Analysis
Statistical analysis was performed using IBM SPSS Statistics v.22. For two-group comparisons a Student's t test was used. For multiple comparisons, analysis of variance (ANOVA) with Dunnet's test was applied. Significance was defined at p < .05.
RESULTS
Immunoprecipitation of Dynein Complexes from Synaptosomes
In order to identify dynein interactors in the synapse and to study possible alterations of dynein complexes in synapses of the mSOD1G93A mouse model for ALS, we performed a differential proteomic analysis (Fig. 1A). ALS targets both upper and lower motor neurons in the motor cortex as well as in the spinal cord. From a technical point of view, synaptosome purification from brains is easier, more specific, and provides more biological material than purification from lower motoneneuron synapses from the spinal cord or neuromuscular junction. Hence, we decided to purify synaptosomes from the brain and perform subsequent validations on NMJs. First, synaptosome preps from two mutant and three wild-type mice brains at symptomatic stage (p120), compared with wild-type SOD1 transgenic mice controls were isolated using a serial centrifugation purification process (Supplemental Fig. S1). In order to validate enrichment of the synaptic fractions in our synaptosome preps, we performed Western blot analysis to examine synaptic markers (Fig. 1B). Indeed, we could see an increase in the ratio of the synaptic marker synapsin to the soluble loading control glyceraldehyde 3-phosphate dehydrogenase. After confirming the enrichment of the synaptic fraction, we immunoprecipitated protein complexes using an antibody against the dynein intermediate chain (DIC) from the synaptosome preparations of mSOD1G93A mice and wtSOD1 controls. The dynein intermediate chain has a scaffolding role in the dynein complex, and its different isoforms and differential phosphorylation patterns regulate specific cargo binding and transport in neurons (reviewed in (35)). As a negative control we used a nonspecific IgG antibody. To test the efficiency and specificity of the immunoprecipitation (IP), we blotted for dynein (Fig. 1C). After verifying the specificity of dynein-complex enrichment from the synaptic pull-downs, the extracts were run on 10% polyacrylamide gel, stained with Coomassie Blue and segmented into five slices (Figs. 1D and 1E). Two different biological sets of dynein pull-downs compared with IgG controls from mSOD1G93A and three sets from wtSOD1 control synaptosomes all compared with IgG negative controls were run on gels, and cut into a total of 50 slices. These gel slices were subjected to mass spectrometry analysis for identification of the synaptic dynein-immunoprecipitated proteins.
Fig. 1.

Preparation of samples for mass spectrometry. (A) Diagram depicting the workflow as described in this paper: Brains were harvested from mSOD1 and wtSOD1 mice, and synaptosomes were then isolated. Next, dynein was immunoprecipitated and the immunoprecipitates run on gels, followed by MS analysis. Identified proteins were subjected to filtration and used for reconstruction of an interaction subnetwork. Predicted interactions of interest were validated by biochemical and imaging techniques. (B) The synaptic preparation, along with complete brain extract, was subjected to Western blot analysis. Synaptosome preparations show enrichment for synapsin; glyceraldehyde 3-phosphate dehydrogenase serves as a loading control. (C) Synaptosome preparation was immunoprecipitated with either dynein antibodies or nonspecific IgG, followed by Western blot analysis for dynein. (D, E) Dynein precipitates from the synaptosomes were run on gel and stained with Coomassie Blue. The gel was then segmented into five slices at approximate molecular weight 121, 65, 45, and 32 KDa for mass spectrometry analysis.
Mass Spectrometry Reveals Distinct Dynein-Bound Populations in ALS
Tryptic digests of the gel slices followed by MS analysis resulted in the identification of 44 total dynein synaptic interactors in the WT control and 24 in the mSOD1G93A sample. We took into account only peptides that appeared with 99% confidence in all replicates of the dynein-immunoprecipitated fractions but did not appear in the controls at this confidence level, and Ig proteins and keratin were removed (Supplemental Tables S1 and S2). We chose this prudent approach at the risk of losing data. Possible interactors that most likely have been filtered out include proteins expressed at lower levels, proteins whose interactions with dynein are relatively weak, or RNA-dependent interactors, as our conditions were not RNA-preserving (Fig. 2A). It is important to note that while several dynactin subunits were detected, several known dynein regulators such as BICD2, LIS1, and NDE1/NDEL1 (also called NudE/Nudel) were not found. Thus, they are unlikely to be enriched at the synapse. Alternatively, the dynein intermediate chain antibody we used may have disrupted dynein's interactions with these proteins (36, 37).
Fig. 2.

Analysis of alterations in dynein-bound synapse fractions in mSOD1 mice and wild-type controls. The mass spectrometry data of dynein interactors in the synapse were filtered for reproducibility and confidence. Proteins that appeared in all separate biological repeats and not in the control with >99% confidentiality were chosen. (A) The list of identified proteins after the filtration process, segregated into correlating groups. (B) Venn diagram depicting distribution of the identified proteins, divided in to the same groups as in A.
Of the identified proteins, 14 appeared in both the WT and mSOD1 samples (Figs. 2A and 2B). These included dynactin subunits, CNOT1 that positively regulates the JAK/STAT pathway; HCN4, a member of the hyperpolarization-activated cyclic nucleotide-gated potassium channels; the ion channels subunits Ryr2 and Kcnh2, as well as Tnks1bp1, which is involved in RNA decay; and CapZ2a, a member of the dynactin complex that regulates actin capping (38). Unique to the wild-type population were neurofilament proteins neurofilament light chain and neurofilament medium chain, calcium/calmodulin-dependent protein kinase II subunit alpha, and members of the solute-carrier family. The mSOD1-specific dynein-bound milieu was smaller and included myosin MYO18A and the protein phosphatase (catalytic subunit C) beta isoform PP1B. Oddly, glial fibrillary acidic protein, a highly specific marker for glia, was also featured in the bound population of both groups, suggesting some glia contamination in our samples, which requires further validation steps.
Staufen1 Is Predicted to Be a Central Node in the Dynein-Bound Protein Network
Mass spectrometry analysis yielded a large number of proteins that interact with dynein in synaptic fractions. In order to better understand how these proteins interact, we applied the ANAT network reconstruction tool (27) to infer high likelihood subnetworks of protein interactions connecting the results of the MS analysis, the “terminal interactors,” to dynein, the network anchor. Each connection is based on previously published data. ANAT analysis provided us with a more comprehensive perspective of the dynein interactome, including proteins that may have eluded detection in the MS due to low abundance and weak interactions. We then looked for central proteins in each of the resulting subnetworks; intuitively, these are internal proteins (i.e. not an anchor or a terminal) that span a large number of terminals below them.
In both the wild-type and mutant networks (Figs. 3A and 3B), we identified the RNA-binding protein Staufen1 as a predicted primary node, suggested to directly interact with dynein and facilitate its downstream interaction with the predicted proteins eukaryotic translation initiation factor kinase, polyadenylate-binding protein 1 and protein phosphatase A, catalytic subunit C. These three predicted interactors play a part in regulating gene expression, suggesting a role for dynein-bound Staufen1 complexes in translational regulation of local expression. Additional nodes are predicted in the wild-type, but not in the mSOD1, network. One of these predicted nodes is asp-glu-ala-his box helicase 9 that mediates the interaction with the mitochondrial isocitrate dehydrogenase 3 beta subunit. Furthermore, ANAT predicted a few other RNA-binding proteins (Supplemental Table S4), which could be the basis for further studies into the cellular pathology of ALS. Among them are the RNA-methylating protein Mettl3 and the RNA-activated translation-initiating factor Eukaryotic translation initiation factor 2-alpha kinase 2, shown to be downregulated in skeletal muscle in ALS mice (39). The fact that most of the RNA-binding proteins including Staufen1 were not identified by MS analysis but only predicted by ANAT supports our assumption that some protein interactions might have been lost due to the non-RNA-preserving conditions during the immunoprecipitation experiments.
Fig. 3.

Predicted networks of dynein interactors in the synapse. Subnetworks connecting the dynein intermediate chain-DYNC1I1 anchor to the genes transcribing the immunoprecipitated proteins identified by MS after filtration (terminals) in wtSOD1 (A) and mSOD1 (B). These subnetworks were reconstructed using the ANAT tool over the HIPPIE high-confidence PPI network and rendered using Cytoscape. Square nodes represent terminals (red—mSOD1 only; green—wtSOD1 only; purple—both). Due to the conversion of protein names to gene symbols, PP1B appears as PP1CB. Predicted nodes are in pink, with size increasing in accordance with centrality in the subnetworks.
Interestingly, of the proteins identified by mass spectrometry, only dynactin (DCTN1), which was identified in the controls, is known to be a direct interactor of dynein. A number of genes (four common, two only in mSOD1 and six only in wtSOD1) were found to be unreachable from the anchor in the PPI network we used, suggesting them to be novel interactors (Table I). Another possible explanation for this could be related to our filtering off low-scoring interactions or the conditions used in the IP experiments, which may decrease forms of protein interactions.
Table I. Proteins identified by MS that remained unmapped by ANAT.
| IPI | UniProt | Gene Symbol | Description |
|---|---|---|---|
| wtSOD1 | |||
| IPI00282186 | F8SLP9 | Pex5l | Isoform 1 of PEX5-related protein |
| IPI00458723 | Q9D8X2 | Ccdc124 | Coiled-coil domain-containing protein 124 |
| IPI00853932 | Q8BPN8 | Dmxl2 | Isoform 1 of DmX-like protein 2 |
| IPI00135965 | P46660;Q3UMG4 | Ina | Alpha-internexin |
| IPI00114279 | P56564;Q543U3;Q8C3T7;Q8C7W8;Q99NG1 | Slc1a3 | Excitatory amino acid transporter 1 |
| IPI00222697 | O55100;Q3U6D7 | Syngr1 | Isoform 1B of Synaptogyrin-1 |
| mSOD1 | |||
| IPI00649326 | Q9JMH9 | Myo18a | Isoform 6 of Myosin-XVIIIa |
| IPI00124564 | Q3UIK4 | Mettl14 | Isoform 1 of Methyltransferase-like protein 14 |
| Common | |||
| IPI00138098 | Q80VP9 | Asphd2 | Aspartate beta-hydroxylase domain-containing protein 2 |
| IPI00469799 | P55937 | Golga3 | Isoform 2 of Golgin subfamily A member 3 |
| IPI00353056 | O70507;B2RY58 | Hcn4 | MCG22630 |
| IPI00319214 | Q8C3P7;Q3U6J4 | Mettl3 | Isoform 1 of N6-adenosine-methyltransferase 70 kDa subunit |
The use of ANAT provided us with a larger assortment of possible players, as well as insight into the centrality of each protein in the dynein-interacting network. Here, the centrality of Staufen1 in both networks, together with its well-characterized, conserved role in mRNP transport and localization, made it an attractive target for further validation and exploration. Thus, we went on to confirm this predicted interaction both in vitro and in vivo.
Validation of Dynein Interactions with Staufen1 in Culture
In vitro experiments were performed on spinal cord cultures enriched for motor neurons from the ventral part of the spinal cord. We chose to look at PP1B, which was one of the proteins identified by MS, despite its being a tertiary node from dynein. PP1B, one of the catalytic subunits of protein phosphatase 1, has been suggested to dephosphorylate dynein to regulate its function, perhaps switching its role from transport to anchoring (9, 10). The subunits differ in their subcellular localization via scaffolding proteins, suggesting access to different substrates within the cell. PP1B was shown to localize to the microtubule-rich dendritic shaft in cortical neurons (40), while PP1A and PP1C1 were enriched in dendritic spines.
In order to validate dynein interaction with Staufen1 and PP1B, we first studied the distribution of Staufen1 and PP1B along neurites and their colocalization with the dynein/dynactin complex and with the synaptic marker synaptophysin. To this end, we cultured spinal cord neurons for 10 days to enable visualization of synaptic markers. Cells were then subjected to immunofluorescent staining with antibodies for Staufen1, PP1B, dynactin (DCTN), an essential dynein cofactor, and the synaptic marker synaptophysin (SYP). We looked at dynactin instead of dynein in immunofluorescent stainings as our anti-dynein antibody proved inefficient in staining. Dynactin, Staufen1, and PP1B are distributed throughout the neurite in a diffuse manner. We used the Imaris software (Bitplane) to quantify the percent of colocalized volume. Over 70% of Staufen1 and nearly 80% of PP1B colocalized with dynactin (Fig. 4A), while 20 and 25% of dynactin colocalized with Staufen1 and PP1B, respectively. As antibodies against both STAU1 and PP1B were raised in rabbits, and in our hands, no reliable signal was detected using other antibodies raised in a different animal host, we could not directly demonstrate colocalization of STAU1 and PP1B. Nonetheless, the similarities in their colocalization with dynactin, together with the note that a direct physical interaction between Staufen1 and PP1B was previously identified using a yeast two-hybrid system (41), suggest that Staufen1 and PP1B may be found together in the same protein complex, possibly in an mRNP granule along the axon and at the synapse.
Fig. 4.
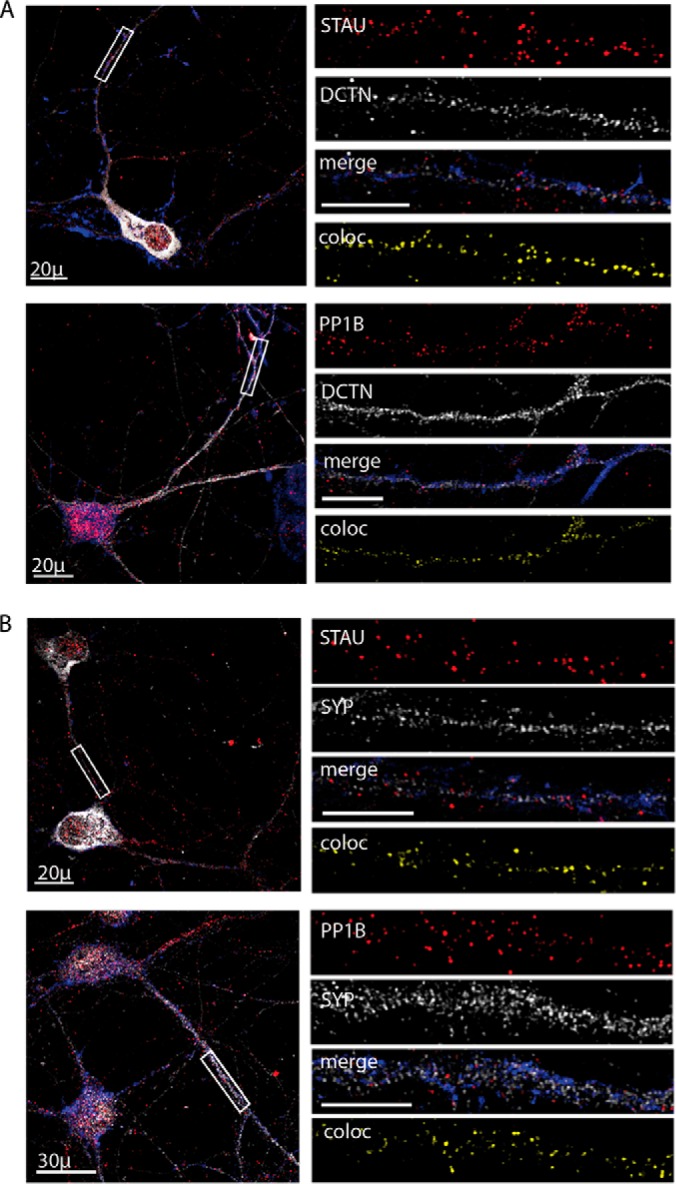
Colocalization of Staufen1 and PP1B with dynactin and synaptic markers in spinal cord cultures. Primary neurons were seeded on cover slides and grown for 10 days and then fixed and immunostained for STAU1, PP1B, DCTN, and SYP. Phalloidin was used to visualize neurites. Representative images of the neurons were taken using a confocal microscope with a 40x objective, with high-magnification close-ups of the axon taken with a 100x objective. High-magnification images were used to analyze volume colocalization along the neurites using the Imaris software, represented in the Coloc channel. (A) Immunostaining for DCTN with STAU and PP1B. (B) Immunostaining for SYP with STAU and PP1B. Unlabeled scale bars represent 10 μm, n = 3, 5–8 images per experimental repetition.
About 32.3 ± 5.8% of Staufen1 protein and 52.6 ± 9.1% of PP1B were colocalized with SYP (Fig. 4B), while 28.8 ± 7.3% and 25.7 ± 9% of SYP colocalized with Staufen and PP1B. This suggests that only a fraction of the Staufen1 complex is found in the synapse. Given Staufen1's known role in the creation and transport of mRNPs, we propose that this fraction contains a subset of mRNAs bound by Staufen1.
Validation of Staufen1 and Dynein Interactions In Vivo
We next sought to study the interaction of Staufen1 with dynein in vivo. In order to test the putative interaction between Staufen1 and dynein, we performed dynein intermediate chain immunoprecipitation on mouse brain extracts (Fig. 5A), followed by Western blot analysis for Staufen1, which indeed precipitated with dynein. We then validated this interaction in the synapse by performing dynein coimmunoprecipitation in synaptosome preps (Fig. 5B).
Fig. 5.

In vivo analysis of dynein–Staufen1 interaction. (A) Protein extracts from p120 adult brains were subjected to immunoprecipitation with dynein antibody. As a control, we used nonspecific IgG. The immunoprecipitate was then subjected to Western blot analysis using antibodies against Stau1 (arrow) and dynein. (B) Synaptosomes were isolated from p120 adult brains, and then IP-ed for dynein and blotted for Staufen1 and dynein. The lower band (∼55kDa), visible in the IP and not in the extract, is the heavy chain of the anti-dynein IgG. (C–E) Gastrocnemius muscle was excised from p120 adult mice, fixed, and treated with bungarotoxin to stain the NMJ and then immunostained. Images were collected with a confocal microscope with a 100x objective. Representative images of NMJs showing enrichment of STAU1 (C), PP1B (D), and dynein intermediate chain (DIC)-GFP (E) at the NMJ, and colocalization with neurofilament (neurofilament heavy chain) are shown. Scale bars represent 10 μm.
After biochemically demonstrating in vivo association of dynein with Staufen1 at the synapse, we wanted to demonstrate the in vivo colocalization of these proteins at the synapse. In order to do so, we examined the distribution of Staufen1 and PP1B at the neuromuscular junction (NMJ), a specialized synapse structure comprised of the motoneuron axon, the muscle, and terminal Schwann cells. Whole mount staining of the gastrocnemius leg muscle was performed, using either anti-Staufen1 or anti-PP1B antibodies, as well as fluorescently labeled bungarotoxin that binds to the postsynaptic acetylcholine receptor and by the neurofilament that stains the neuron and presynaptic side. The results showed that at the NMJ, Staufen1 is enriched along the neuron and around the NMJ (Fig. 5C). A similar distribution was observed for PP1B (Fig. 5D). Enrichment of both Staufen homologs at the NMJ was previously reported, describing this enrichment at the postsynaptic side (42). However, our data suggest that there is also Staufen1 enrichment at the presynaptic side. As our anti-dynein antibody proved incompatible with staining, in order to view dynein localization in the presynaptic NMJ, we took advantage of the DIC-GFP knock-in mice (29). These mice express the neuronal specific dynein intermediate chain 1 tagged by GFP, and their NMJs show prominent GFP signal along the most distal part of their axons (Fig. 5E). Taken together, our data suggest that dynein may interact with Staufen1 at both brain synapses and NMJs.
Staufen1 and PP1B Colocalize with RNA along Neurites
As Staufen1 is a well-known RNA-binding protein, we sought to determine whether Staufen1 colocalizes with RNA along neurites and at synapses in cultured neurons. To this end, we added the SYTO-14 nucleic acid stain to live cells, followed by standard immunofluorescent staining techniques to show that RNA in neurites colocalizes with Staufen1 and PP1B, as well as with the p150 (Glued) subunit of dynactin (DCTN), and the synaptic protein synaptophysin (SYP). We used the Imaris software (Bitplane) to analyze the colocalization. Along the neurite, we observed a high correlation between the localization of RNA and Staufen1 (Figs. 6A and 6C), as well as PP1B (Figs. 6B and 6D). The correlation between RNA and DCTN, when colocalized with either Staufen1 or PP1B (Figs. 6A and 6B), as well as with SYP (Fig. 6D), reinforces our suggestion that an RNP complex of dynein/dynactin, Staufen1, PP1B, and RNA exists and localizes along the neurites and in synapse. In order to test the in vivo interaction of dynein with Staufen1-bound mRNAs, we performed an RNA-IP by immunoprecipitating dynein from control and mSOD1 mice brain extracts, followed by RT-PCR for probing established Staufen1-associated mRNAs, namely CamK2a (43) and Arc (44) (Fig. 6E). Both mRNAs were immunoprecipiated with dynein, while the control mRNA polymerase B (PolB) was not (Fig. 6E), demonstrating a specific interaction of the Staufen1-associated mRNAs with the dynein complexes. As most of the knowledge of Staufen1 mRNA interactors comes from work done on dendrites and both CamK2a and Arc are well-characterized dendritic mRNAs, further work will be needed to identify axonal Staufen1 mRNA interactors.
Fig. 6.
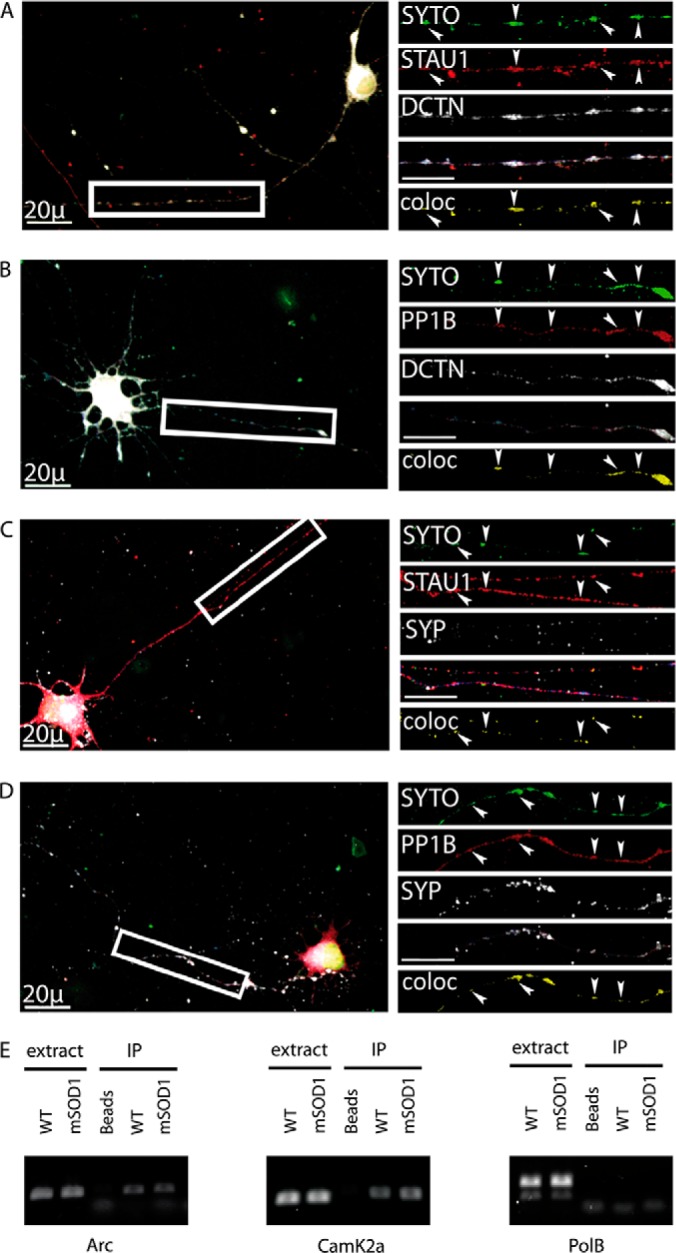
Colocalization of Staufen1 and PP1B with mRNA in neurites. Primary spinal cord neurons were seeded on cover slides and grown for 10 days. They were treated with SYTO-14 for total RNA detection, fixed, and immunostained for STAU1, PP1B, DCTN, and SYP. Images were collected with a confocal microscope. Representative images of the neurons were taken with a 40x objective and high-magnification close-ups of the axon taken with a 100x objective. High-magnification images were used to analyze volume colocalization along the neurites using the Imaris software, represented in the Coloc channel. (A) Colocalization of RNA with STAU1 and DCTN in neurites. (B) Colocalization of RNA with PP1B and DCTN. (C) Colocalization of RNA with STAU1 and SYP. (D) Colocalization of RNA with PP1B and SYP. Unlabeled scale bars represent 10 μm. (E) RNA was harvested from dynein immunoprecipitated from mSOD1 mouse and littermate control brains. Following cDNA preparation, standard RT-PCR was carried out for CamK2a, Arc, and polymerase B (PolB).
ALS-Linked Mutations Affect the Synaptic Localization of Staufen1 and PP1B
Research over the past several years has pointed to an association between RNA metabolism and ALS (reviewed in (45)). ALS-linked mutations in the RNA-binding proteins fused in sarcoma and TDP-43 were discovered, and the repeat expansions in C9orf72, whose mechanism is yet unknown, was also suggested to contribute to ALS-associated neurodegeneration through RNA toxicity (46). ANAT predictions pointed to Staufen1 as a central node in both the mutant SOD1 and wild-type networks. As alterations in both transport and local protein synthesis are hallmarks of neurodegenerative diseases, we examined how ALS-linked mutations affected the distribution of Staufen1 and PP1B along the neurite and the synapse.
First, we examined in vitro cultured spinal cord neurons that were infected with lentiviruses containing GFP-tagged mSOD1G93A and TDP43A315T, two well-characterized ALS-causing mutations. As a control, we used a GFP-expressing lentivirus. Cells were fixed after 10 days in culture and immunostained for Staufen1 and PP1B together with DCTN and SYP. We used Image J to assess changes in the colocalization of Staufen1 and PP1B to SYP and DCTN (Figs. 7A and 7B).
Fig. 7.

ALS-linked mutations alter the association of STAU1 and PP1B with DCTN. (A and B) Primary spinal cord neurons were seeded on cover slides and infected with lentiviruses expressing GFP, GFP-tagged mSOD1G93A and TDP43A315T. After 10 days in vitro (DIV), cells were fixed and immunostained. Images were collected with a confocal microscope. Representative images of the neurons were taken with a 40x objective, with high-magnification close-ups of the axon taken with a 100x objective. DCTN, STAU, and PP1B localization along the axon are not altered as a result of ALS-linked mutations. (C and D) DCTN and STAU1 levels remain constant along the neurite for the GFP control and mSOD1 but are both increased in TDP43-infected neurons. (E) PP1B levels remain constant for all three groups. (F and G) The number of colocalized puncta (viewed in the Coloc channel) of DCTN with STAU1 is constant (F), with an increased intensity in ALS models (G). (H and I) Colocalization of DCTN with PP1B (shown in the Coloc channel) is similarly increased in ALS models. Statistical analysis was done with ANOVA, with Dunnet's post-hoc test, *p < .05. Unlabeled scale bars represent 10 μm, n = 3.
In neurons infected with the ALS-causing mutation mSOD1G93A, the levels of dynactin, Staufen1. and PP1B were not significantly changed (Figs. 7C–7E). However, infection with TDP43A315T appears to cause some increase in the neuritic levels of dynactin and Staufen1. In vivo, however, the levels of dynein are constant in brains and sciatic axoplasm of symptomatic (p120) mSOD1 mice (Supplemental Fig. S2). Interestingly, while Staufen1 levels in the brain remain steady, they are highly variable in the sciatic axoplasm (Supplemental Fig. S2). To assess colocalization in infected cells, we built a coloc channel using Imaris and then analyzed the plot profile of this channel using Image J, looking at the number and intensity of colocalized puncta along the axon. The coloc channel takes into account the intensity of each of the original channels. The number of puncta/μm of colocalized DCTN and STAU1 remained constant in infected neurons (Fig. 7F), but the intensity of the colocalized puncta was increased in the mutants, with a slight increase in the mSOD1 model and a significant increase in the TDP-43 model (Fig. 7G). For PP1B, an increase was observed in both the number and intensity of colocalized puncta for TDP43, and in the intensity of colocalized puncta of PP1B and DCTN in the mSOD1 neurons (Figs. 7H and 7I). Altogether, these changes in the number of colocalized signals and their intensity may suggest that in ALS models there is an increased binding of DCTN to STAU1-containing mRNPs and PP1B along the axons. This increase may mediate dynein binding and possibly enhance retrograde motility of the mRNP.
Next, we tested the colocalization of Staufen1 and PP1B complexes with synaptic markers. We observed that in cells with the ALS-linked mutations the localization of both Staufen1 and PP1B to SYP decreased. In GFP-infected controls, 42% of total Staufen1 in the neurite colocalized to SYP. This was reduced to 27% and 21% in mSOD1 and TDP43-infected neurons, respectively (Figs. 8A and 8C). A similar decrease was observed for PP1B, with mSOD1 and TDP43 infection reducing the colocalized fraction from 40% in the control to 25 and 21%, respectively (Figs. 8B and 8D). The number of synapses per length in the neurites remained constant (Fig. 8E). Thus, while protein levels remain constant, less of the Staufen1 complex is localized to synaptic sites in mSOD1 and TDP43-infected neurons. This is consistent with previous work demonstrating that mutated TDP-43 localization in distal parts of the axon and at the NMJ is altered when mutated (47).
Fig. 8.

ALS-linked mutations decrease the association of STAU1 and PP1B with synaptic markers. Primary spinal cord neurons were seeded on cover slides and infected with lentiviruses expressing GFP, GFP-tagged mSOD1G93A, and TDP43A315T. After 10 DIV, cells were fixed and immunostained. Images were collected with a confocal microscope. Representative images of the neurons were taken with a 40x objective, with high-magnification close-ups of the proximal and distal axon taken with a 100x objective. High-magnification images were used to analyze volume colocalization along the neurites using the Imaris software, represented in the Coloc channel. The percentage of colocalized volume is shown. (A) Colocalization of SYP with STAU in infected neurons. (B) Colocalization of SYP with PP1B in infected neurons. (C) Quantification of SYP and STAU colocalization a ∼15% decrease in colocalized volume in ALS-infected neurons. (D) Quantification of SYP and PP1B colocalization showing a decrease of ∼15% in colocalized volume as a result of ALS-linked mutations. (E) The total number of synapses was quantified using the Image J software and normalized to neurite length. Statistical analysis was done with ANOVA, with Dunnet's post-hoc test, *p < .05. Unlabeled scale bars represent 10 μm, n = 3.
Staufen1 Is Depleted from mSOD1 NMJs as Its Association with Dynein Increases
In order to study dynein-Staufen1 interactions in vivo, we harvested brains from p120 mSOD1 male mice and their control littermates and immunoprecipitated using the anti-dynein antibody followed by Western blot analysis for Staufen1 (Fig. 9A). Although the results were somewhat variable and therefore not statistically significant, we observed a general increase in the relative amount of Staufen1 that precipitated with dynein in these mutant mice (Fig. 9B), suggesting that in ALS models there is alteration in this protein complex.
Fig. 9.

Expression of Staufen1 at the NMJ is depleted in adult mSOD1 mice. (A) Whole brain extracts from mSOD1 mice and healthy littermates were subjected to dynein immunoprecipitation, with nonspecific IgG antibody as a control. The immunoprecipitate was then subjected to Western blot analysis and blotted for Staufen1 (arrow) and dynein. The lower band (∼55 kDa), visible in the IP lanes, but not in the extract, is the heavy chain of either the anti-dynein or the IgG. (B) Densitometry analysis using Image J software shows a consistent but variable increase in the amount of immunoprecipitated Staufen1 in mSOD1 extracts, n = 3. (C-E) Gastrocnemius muscles from DIC-GFP p120 healthy littermates (D) and mSOD1 mice (E) were excised, fixed, and treated with bungarotoxin to stain the postsynaptic NMJ and then immunostained for STAU1 and neurofilament heavy chain. Images were acquired with a confocal microscope with a 100x objective. Analysis of >150 NMJs on three biological repeats show STAU1 localization at the synapse in 19% of NMJs in mSOD1, compared with 64% in the littermate. Statistical analysis was done with Student's t test, *p < .005, n = 3. Scale bar represents 10 μm.
As we demonstrated a decrease in Staufen1 complexes in the synapse of ALS models in vitro, we next asked whether a depletion of Staufen1 from the synapse also occurs in vivo. To test this, we stained gastrocnemius muscle NMJs from control and mSOD1 DIC-GFP mice at a symptomatic stage (p120) for Staufen1 and tested their colocalization with bungarotoxin (Figs. 9C-9E). As expected, muscles from mSOD1 showed NMJ disruption and neuronal denervation from NMJs. Interestingly, the levels of Staufen1 at NMJs that were still intact demonstrated a significant decrease in the mutants (Figs. 9D and 9E). Staufen1 could be detected in over 63% of NMJs in the littermate, compared with 19% in the mSOD1 mouse (Fig. 9C). This may imply that the depletion of Staufen1 is an early occurrence in the NMJ destabilization that occurs in the mSOD1 mouse model for ALS and suggests that Staufen1 may play an important role in the local protein synthesis process at the presynaptic side of the synapse, thus enabling proper synaptic function and structural maintenance.
Taken together with the observation that Staufen1 localization to the synapse was decreased in ALS models (Figs. 8A and 8C), we suggest that the depletion of Staufen1 from the synapse in ALS models may be a result of an active retrograde transport mechanism away from the synapse along the axons.
DISCUSSION
We chose a high-throughput proteomic approach to identify novel dynein interactors in the synapse that may play a role in mechanisms of synapse disruption and ALS pathology. Analysis of the proteomic data using ANAT produced two putative networks of dynein interactors in healthy and in diseased mice. In the mSOD1 mouse model for ALS, mass spectrometry identified the phosphatase PP1B, whose connection to dynein was predicted based on the interaction of both proteins with the RNA-binding protein Staufen1. We validated the interaction between dynein and Staufen1 both in vitro and in vivo and provided evidence to suggest that Staufen1's association and colocalization with dynein is altered in ALS. Both in whole brain extracts and in the neurites of cultured spinal cord neurons, the interaction between dynein and Staufen1 increases, while in cultured cells we could see a reduction in the colocalized fraction of both Staufen1 and PP1B to synaptic markers in two models of ALS, mSOD1G93A and TDP43A315T, similar to the reduction of Staufen1 in mSOD1 NMJs. Altogether, these data may point to an anchoring role for dynein/dynactin at the synapse, leading RNP structures of Staufen1 to localize to the synapse and regulate essential local synthesis events. However, in the ALS model, where we observed a reduction in Staufen1 at the NMJ but an increase in the amount of coimmunoprecipitated Staufen1, as well as increased association of dynactin (Fig. 7) with Staufen1 in cultured neurons, the function of dynein in anchoring Staufen1-RNP may be abrogated, thereby leading to a reduction in the synaptic localization of Staufen1 and possibly impairing the local protein synthesis process, which may lead to neurotoxicity (Fig. 9). The role of dynein as an anchor, in addition to its canonical role as a retrograde motor protein, has been reported in various cellular processes (48–50), including a microtubule-tethering role that supports synaptic maintenance (7). As we do not yet have evidence that Staufen1 physically links PP1B to dynein, we base our suggestion on previous work that has shown a direct interaction between Staufen1 and PP1B (41).
Correct spatiotemporal localization of local protein synthesis is essential for a neuron's health and facilitates the neuron's ability to respond to external stimuli in a precise spatial and temporal manner. It is a highly regulated event that requires the coordination of different processes, including the transport, targeting, anchoring, and on-site translation of mRNA (51). We suggest a model in which dynein plays a dual role in its interaction with Staufen1: in retrograde transport, from the periphery to the soma, and in anchoring at specific sites (Fig. 10). In healthy neurons, Staufen1 is anchored by dynein/dynactin at sites of increased protein translation, such as synapses or axonal branching points. On the other hand, in neurons undergoing neurodegeneration like in ALS, dynein shifts from an anchoring role to a transport role, clearing the Staufen1 mRNP granules from areas of increased local translation back to the cell body. As Staufen1 granules contain mRNAs essential for maintaining the axonal cytoskeleton and synaptic function, their clearance may lead to reduced local translation of these mRNAs, which may accelerate neurodegeneration.
Fig. 10.

Suggested model: Dynein plays a dual role in the synaptic localization of Staufen1. We propose a model in which dynein performs a dual role: in retrograde transport and in anchoring. In healthy neurons, the majority of Staufen1-containing mRNPs bound to dynein are anchored at sites of local protein synthesis, i.e. synapses or axonal branching points. As a result of neurodegeneration, dynein may switch its role to retrograde transport, clearing Staufen1 mRNPs from the distal synapse toward the cell body. Clearance of mRNPs from the distal axon contributes to reduced axonal protein synthesis and may serve to advance neurodegenerative processes.
Staufen has been shown to participate in RNA granule formation, transport, anchoring, and regulation of translational state (reviewed in (52, 53)). In the D. melanogaster oocyte where it was originally described, Staufen is essential for both the spatial and temporal localization of bicoid mRNA to the anterior pole (54) and of oskar mRNA to the posterior pole (55) by associating with dynein and kinesin, respectively, to transport mRNA to distinct locations (56). Anchoring at the poles is thought to be regulated by changes to the cortical actin cytoskeleton facilitated by Swallow (56), although Staufen may also play a role in anchoring additional components at the posterior pole (55, 57). Staufen-mediated RNA transport is microtubule dependent (58) and relies on direct interaction between a dsRNA recognition domain and a stem-loop structure in the mRNA (59), often in the 3′UTR (58, 60). In neurons, Staufen1 has been linked to numerous functions during neurogenesis, as well as in the adult brain, such as synaptic plasticity during memory formation, a process that heavily involves local protein synthesis (61, 62). Indeed, the dendritically localized mRNA CamKIIα, whose translation has been shown to be activity dependent, is found in Staufen1-containing mRNP granules (61, 63). Further, Staufen has been shown to preferentially accumulate at the postsynaptic NMJ, and its levels increase as a result of muscle denervation following injury (42). In Drosophila, Staufen regulates the localization and translation of cora mRNA, which participates in the regulation of bouton number and synapse strength (64). TDP-43 and Staufen1 physically interact, together with Fragile-X mental retardation protein to form a functionally coordinated complex that targets functionally important mRNAs, such as SIRT1, which encodes a protein with neuroprotective functions. In dendrites, this colocalization is increased upon activation (65), while dysregulation of the TDP-43/STAU1/Fragile-X mental retardation protein complex sensitizes neurons to DNA damage and apoptosis (66).
A number of proteomic screens have also attempted to identify novel players in the pathology of ALS (67), and to provide detectable biomarkers in biological fluids, such as plasma (68), cerebrospinal fluid, and brain extracellular fluid (69) of human patients. Transport defects are common to the various genetic etiologies in ALS (70), pointing to ALS as a spatiotemporal mislocalization disease, including mislocalization of mRNA (reviewed in (12)). Transport of specific mRNA species can occur as a response to extrinsic growth stimuli (71) or axonal insult (72). Interestingly, during transport, mRNA remains in a translationally repressed state. This translational repression has recently been suggested to be maintained in Staufen1 mRNP granules by microRNAs (miRNAs). Specifically, neuronal Staufen1 granules have been shown to contain the neuron-specific miRNAs miR-9 and miR-124, together with their target mRNAs (73). Thus, anchoring may be an essential step preceding the release from translational repression.
One possibility we cannot exclude is that in ALS models the observed reduction in Staufen1 and PP1B in the synapse is due to a dysfunction in kinesin-mediated anterograde transport, and further research is required to address this issue. Still, the increased intensity of the p150 dynactin subunit in puncta along the axons (Fig. 7), together with recent studies demonstrating a role for p150 in initiating retrograde transport (14, 74, 75), suggest that indeed alteration in the retrograde transport may lead to the reduction of the Staufen complex in the synapse.
Both Staufen isoforms have been implicated in mRNA degradation, contributing to a process known as Staufen-mediated mRNA decay, a significant posttranscriptional regulatory pathway (76). Staufen recognizes Staufen-binding sites and mediates the recruitment of the Upf1 helicase to 3′ UTRs of specific mRNAs, thereby triggering decay (76, 77). Conversely, Staufen1 binding to trans-activation response (TAR) sequences at the 5′ UTR facilitates initiation of translation, possibly by translocating these mRNAs to polysomes (78). Upf1 was not identified in either our MS data or by ANAT. Interestingly, Upf1 was recently shown to improve neuronal survival in cases of TDP-43 or fused in sarcoma but not SOD1 toxicity (79).
In summary, our work points to changes in Staufen1 localization as a result of ALS mutations and supports the hypothesis that ALS is also an RNA mislocalization disease. Further research is required to characterize the nature of dynein–Staufen1 transport, its bound mRNAs and the functional downstream effects that occur as a result of the shift between anchored and transported Staufen1.
We declare no conflict of interest.
Supplementary Material
Footnotes
Author contributions: N.G., A.M., T.G., K.L., R.S., and E.P. designed the research; N.G., A.M., T.G., K.L., R.S., and E.P. performed the research; X.X. and E.P. contributed new reagents or analytic tools; N.G., A.M., M.C., T.G., K.L., R.S., and E.P. analyzed data; and N.G., A.M., M.C., T.G., X.X., K.L., R.S., and E.P. wrote the paper.
* This work was supported by the Israel Science Foundation (ISF) and the European Research Council (ERC) grants, 561/11, and 309377, respectively, to E.P. ERA-NET E-rare grant (for Research Programs on Rare Diseases, 0601165051) supports E.P. and R.S. X.X. was supported by the Center for Neuroscience and Regenerative Medicine (CNRM)-Uniformed Services University of the Health Sciences. Travel support for N.G.E. was granted by the Constantiner Institute. A.M. was supported in part by a fellowship from the Edmond J. Safra Center for Bioinformatics at Tel-Aviv University.
 This article contains supplemental material Supplemental Tables S1-S4 and Supplemental Figs. S1-S3.
This article contains supplemental material Supplemental Tables S1-S4 and Supplemental Figs. S1-S3.
1 The abbreviations used are:
- ALS
- amyotrophic lateral sclerosis
- ANAT
- Advanced Network Analysis Tool
- DCTN
- dynactin
- DIC
- dynein intermediate chain
- ERK
- extracellular signal-regulated kinases
- GFP
- green fluorescent protein
- NMJ
- neuromuscular junction
- PP1
- protein phosphatase 1
- PPI
- protein–protein interactions
- RNP
- ribonucleotide-protein
- SOD1
- superoxide dismutase 1
- STAU
- Staufen
- SYP
- synaptosphysin
- TDP43
- TAR DNA binding protein 43.
REFERENCES
- 1.Moloney E. B., de Winter F., and Verhaagen J. (2014) ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Frontiers Neurosci 8, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Vos K. J., Grierson A. J., Ackerley S., and Miller C. C. (2008) Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 31, 151–173 [DOI] [PubMed] [Google Scholar]
- 3.Perlson E., Maday S., Fu M. M., Moughamian A. J., and Holzbaur E. L. (2010) Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kardon J. R., and Vale R. D. (2009) Regulators of the cytoplasmic dynein motor. Nat. Rev. Mol. Cell Biol. 10, 854–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu M. M., and Holzbaur E. L. (2014) Integrated regulation of motor-driven organelle transport by scaffolding proteins. Trends Cell Biol. 24, 564–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kiyomitsu T., and Cheeseman I. M. (2012) Chromosome- and spindle-pole-derived signals generate an intrinsic code for spindle position and orientation. Nature Cell Biol. 14, 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perlson E., Hendricks A. G., Lazarus J. E., Ben-Yaakov K., Gradus T., Tokito M., and Holzbaur E. L. (2013) Dynein interacts with the neural cell adhesion molecule (NCAM180) to tether dynamic microtubules and maintain synaptic density in cortical neurons. J. Biolog. Chem. 288, 27812–27824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meignin C., and Davis I. (2010) Transmitting the message: Intracellular mRNA localization. Curr. Opin. Cell Biol. 22, 112–119 [DOI] [PubMed] [Google Scholar]
- 9.Whyte J., Bader J. R., Tauhata S. B., Raycroft M., Hornick J., Pfister K. K., Lane W. S., Chan G. K., Hinchcliffe E. H., Vaughan P. S., and Vaughan K. T. (2008) Phosphorylation regulates targeting of cytoplasmic dynein to kinetochores during mitosis. J. Cell Biol. 183, 819–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allan V. (1995) Protein phosphatase 1 regulates the cytoplasmic dynein-driven formation of endoplasmic reticulum networks in vitro. J. Cell Biol. 128, 879–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinckelmann M. V., Zala D., and Saudou F. (2013) Releasing the brake: Restoring fast axonal transport in neurodegenerative disorders. Trends Cell Biol. 23, 634–643 [DOI] [PubMed] [Google Scholar]
- 12.Gershoni-Emek N., Chein M., Gluska S., and Perlson E. (2015) Amyotrophic lateral sclerosis as a spatiotemporal mislocalization disease: Location, location, location. Int. Rev. Cell Mol. Biol. 315, 23–71 [DOI] [PubMed] [Google Scholar]
- 13.Kieran D., Hafezparast M., Bohnert S., Dick J. R., Martin J., Schiavo G., Fisher E. M., and Greensmith L. (2005) A mutation in dynein rescues axonal transport defects and extends the life span of ALS mice. J. Cell Biol. 169, 561–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lloyd T. E., Machamer J., O'Hara K., Kim J. H., Collins S. E., Wong M. Y., Sahin B., Imlach W., Yang Y., Levitan E. S., McCabe B. D., and Kolodkin A. L. (2012) The p150(Glued) CAP-Gly domain regulates initiation of retrograde transport at synaptic termini. Neuron 74, 344–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikenaka K., Kawai K., Katsuno M., Huang Z., Jiang Y. M., Iguchi Y., Kobayashi K., Kimata T., Waza M., Tanaka F., Mori I., and Sobue G. (2013) dnc-1/dynactin 1 knockdown disrupts transport of autophagosomes and induces motor neuron degeneration. PloS One 8, e54511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eschbach J., and Dupuis L. (2011) Cytoplasmic dynein in neurodegeneration. Pharmacol. Therapeut. 130, 348–363 [DOI] [PubMed] [Google Scholar]
- 17.Zetterström P., Stewart H. G., Bergemalm D., Jonsson P. A., Graffmo K. S., Andersen P. M., Brännström T., Oliveberg M., and Marklund S. L. (2007) Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc. Natl. Acad. Sci. U.S.A. 104, 14157–14162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bergemalm P. O., Hennerdal S., Persson B., Lyxell B., and Borg E. (2009) Perception of the acoustic environment and neuroimaging findings: a report of six cases with a history of closed head injury. Acta Oto-Laryngolog. 129, 801–808 [DOI] [PubMed] [Google Scholar]
- 19.Ekegren T., Hanrieder J., Aquilonius S. M., and Bergquist J. (2006) Focused proteomics in post-mortem human spinal cord. J. Proteome Res. 5, 2364–2371 [DOI] [PubMed] [Google Scholar]
- 20.Grant S. G. (2003) Synapse signalling complexes and networks: Machines underlying cognition. BioEssays 25, 1229–1235 [DOI] [PubMed] [Google Scholar]
- 21.Husi H., Ward M. A., Choudhary J. S., Blackstock W. P., and Grant S. G. (2000) Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nature Neurosci. 3, 661–669 [DOI] [PubMed] [Google Scholar]
- 22.Li K. W., Klemmer P., and Smit A. B. (2010) Interaction proteomics of synapse protein complexes. Anal. Bioanal. Chem. 397, 3195–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klemmer P., Smit A. B., and Li K. W. (2009) Proteomics analysis of immuno-precipitated synaptic protein complexes. J. Proteomics 72, 82–90 [DOI] [PubMed] [Google Scholar]
- 24.Westphal R. S., Tavalin S. J., Lin J. W., Alto N. M., Fraser I. D., Langeberg L. K., Sheng M., and Scott J. D. (1999) Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science 285, 93–96 [DOI] [PubMed] [Google Scholar]
- 25.Jiménez C. R., Eyman M., Lavina Z. S., Gioio A., Li K. W., van der Schors R. C., Geraerts W. P., Giuditta A., Kaplan B. B., and van Minnen J. (2002) Protein synthesis in synaptosomes: a proteomics analysis. J. Neurochem. 81, 735–744 [DOI] [PubMed] [Google Scholar]
- 26.Khanna R., Zougman A., and Stanley E. F. (2007) A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. J. Biochem. Mol. Biol. 40, 302–314 [DOI] [PubMed] [Google Scholar]
- 27.Yosef N., Zalckvar E., Rubinstein A. D., Homilius M., Atias N., Vardi L., Berman I., Zur H., Kimchi A., Ruppin E., and Sharan R. (2011) ANAT: A tool for constructing and analyzing functional protein networks. Science Signal. 4, pl1. [DOI] [PubMed] [Google Scholar]
- 28.Gurney M. E., Pu H., Chiu A. Y., Dal Canto M. C., Polchow C. Y., Alexander D. D., Caliendo J., Hentati A., Kwon Y. W., Deng H. X., and et al. (1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264, 1772–1775 [DOI] [PubMed] [Google Scholar]
- 29.Zhang J., Twelvetrees A. E., Lazarus J. E., Blasier K. R., Yao X., Inamdar N. A., Holzbaur E. L., Pfister K. K., and Xiang X. (2013) Establishing a novel knock-in mouse line for studying neuronal cytoplasmic dynein under normal and pathologic conditions. Cytoskeleton 70, 215–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schaefer M. H., Fontaine J. F., Vinayagam A., Porras P., Wanker E. E., and Andrade-Navarro M. A. (2012) HIPPIE: Integrating protein interaction networks with experiment based quality scores. PloS One 7, e31826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mudunuri U., Che A., Yi M., and Stephens R. M. (2009) bioDBnet: The biological database network. Bioinformatics 25, 555–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., and Ideker T. (2003) Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalmar B., and Greensmith L. (2009) Activation of the heat shock response in a primary cellular model of motoneuron neurodegeneration-evidence for neuroprotective and neurotoxic effects. Cell. Mol. Biol. Lett. 14, 319–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Camu W., and Henderson C. E. (1994) Rapid purification of embryonic rat motoneurons: An in vitro model for studying MND/ALS pathogenesis. J. Neurolog. Sci. 124, 73–74 [DOI] [PubMed] [Google Scholar]
- 35.Pfister K. K. (2015) Distinct functional roles of cytoplasmic dynein defined by the intermediate chain isoforms. Experiment. Cell Res. 334, 54–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang S., Ketcham S. A., Schön A., Goodman B., Wang Y., Yates J. 3rd, Freire E., Schroer T. A., and Zheng Y. (2013) Nudel/NudE and Lis1 promote dynein and dynactin interaction in the context of spindle morphogenesis. Mol. Biol. Cell 24, 3522–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKenney R. J., Weil S. J., Scherer J., and Vallee R. B. (2011) Mutually exclusive cytoplasmic dynein regulation by NudE-Lis1 and dynactin. J. Biolog. Chem. 286, 39615–39622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schafer D. A., Gill S. R., Cooper J. A., Heuser J. E., and Schroer T. A. (1994) Ultrastructural analysis of the dynactin complex: An actin-related protein is a component of a filament that resembles F-actin. J. Cell Biol. 126, 403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soo K. Y., Halloran M., Sundaramoorthy V., Parakh S., Toth R. P., Southam K. A., McLean C. A., Lock P., King A., Farg M. A., and Atkin J. D. (2015) Rab1-dependent ER-Golgi transport dysfunction is a common pathogenic mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropatholog. [DOI] [PubMed] [Google Scholar]
- 40.Bordelon J. R., Smith Y., Nairn A. C., Colbran R. J., Greengard P., and Muly E. C. (2005) Differential localization of protein phosphatase-1alpha, beta and gamma1 isoforms in primate prefrontal cortex. Cereb Cortex 15, 1928–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monshausen M., Rehbein M., Richter D., and Kindler S. (2002) The RNA-binding protein Staufen from rat brain interacts with protein phosphatase-1. J. Neurochem. 81, 557–564 [DOI] [PubMed] [Google Scholar]
- 42.Bélanger G., Stocksley M. A., Vandromme M., Schaeffer L., Furic L., Des Groseillers L., and Jasmin B. J. (2003) Localization of the RNA-binding proteins Staufen1 and Staufen2 at the mammalian neuromuscular junction. J. Neurochem. 86, 669–677 [DOI] [PubMed] [Google Scholar]
- 43.Tübing F., Vendra G., Mikl M., Macchi P., Thomas S., and Kiebler M. A. (2010) Dendritically localized transcripts are sorted into distinct ribonucleoprotein particles that display fast directional motility along dendrites of hippocampal neurons. J. Neurosci. 30, 4160–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kanai Y., Dohmae N., and Hirokawa N. (2004) Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513–525 [DOI] [PubMed] [Google Scholar]
- 45.Droppelmann C. A., Campos-Melo D., Ishtiaq M., Volkening K., and Strong M. J. (2014) RNA metabolism in ALS: When normal processes become pathological. ALS Frontotemporal Degen. 15, 321–336 [DOI] [PubMed] [Google Scholar]
- 46.Rohrer J. D., Isaacs A. M., Mizlienska S., Mead S., Lashley T., Wray S., Sidle K., Fratta P., Orrell R. W., Hardy J., Holton J., Revesz T., Rossor M. N., and Warren J. D. (2015) C9orf72 expansions in frontotemporal dementia and amyotrophic lateral sclerosis. The Lancet. Neurology [DOI] [PubMed] [Google Scholar]
- 47.Alami N. H., Smith R. B., Carrasco M. A., Williams L. A., Winborn C. S., Han S. S., Kiskinis E., Winborn B., Freibaum B. D., Kanagaraj A., Clare A. J., Badders N. M., Bilican B., Chaum E., Chandran S., Shaw C. E., Eggan K. C., Maniatis T., and Taylor J. P. (2014) Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delanoue R., Herpers B., Soetaert J., Davis I., and Rabouille C. (2007) Drosophila Squid/hnRNP helps dynein switch from a gurken mRNA transport motor to an ultrastructural static anchor in sponge bodies. Developmental Cell 13, 523–538 [DOI] [PubMed] [Google Scholar]
- 49.Kotak S., Busso C., and Gönczy P. (2012) Cortical dynein is critical for proper spindle positioning in human cells. J. Cell Biol. 199, 97–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ligon L. A., and Holzbaur E. L. (2007) Microtubules tethered at epithelial cell junctions by dynein facilitate efficient junction assembly. Traffic 8, 808–819 [DOI] [PubMed] [Google Scholar]
- 51.Holt C. E., and Bullock S. L. (2009) Subcellular mRNA localization in animal cells and why it matters. Science 326, 1212–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heraud-Farlow J. E., and Kiebler M. A. (2014) The multifunctional Staufen proteins: Conserved roles from neurogenesis to synaptic plasticity. Trends Neurosci. 37, 470–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tosar L. J., Thomas M. G., Baez M. V., Ibanez I., Chernomoretz A., and Boccaccio G. L. (2012) Staufen: From embryo polarity to cellular stress and neurodegeneration. Front. Biosci. 4, 432–452 [DOI] [PubMed] [Google Scholar]
- 54.Goldstein L. H., Atkins L., Landau S., Brown R. G., and Leigh P. N. (2006) Longitudinal predictors of psychological distress and self-esteem in people with ALS. Neurology 67, 1652–1658 [DOI] [PubMed] [Google Scholar]
- 55.St. Johnston D., Beuchle D., and Nüsslein-Volhard C. (1991) Staufen, a gene required to localize maternal RNAs in the Drosophila egg. Cell 66, 51–63 [DOI] [PubMed] [Google Scholar]
- 56.Breuer A. C., and Atkinson M. B. (1988) Fast axonal transport alterations in amyotrophic lateral sclerosis (ALS) and in parathyroid hormone (PTH)-treated axons. Cell Motility Cytoskeleton 10, 321–330 [DOI] [PubMed] [Google Scholar]
- 57.Allison R., Czaplinski K., Git A., Adegbenro E., Stennard F., Houliston E., and Standart N. (2004) Two distinct Staufen isoforms in Xenopus are vegetally localized during oogenesis. RNA 10, 1751–1763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferrandon D., Elphick L., Nüsslein-Volhard C., and St. Johnston D. (1994) Staufen protein associates with the 3′UTR of bicoid mRNA to form particles that move in a microtubule-dependent manner. Cell 79, 1221–1232 [DOI] [PubMed] [Google Scholar]
- 59.Ramos A., Grünert S., Adams J., Micklem D. R., Proctor M. R., Freund S., Bycroft M., St Johnston D., and Varani G. (2000) RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 19, 997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim Y. K., Furic L., Parisien M., Major F., Des Groseillers L., and Maquat L. E. (2007) Staufen1 regulates diverse classes of mammalian transcripts. EMBO J. 26, 2670–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lebeau G., Maher-Laporte M., Topolnik L., Laurent C. E., Sossin W., Desgroseillers L., and Lacaille J. C. (2008) Staufen1 regulation of protein synthesis-dependent long-term potentiation and synaptic function in hippocampal pyramidal cells. Mol. Cell. Biol. 28, 2896–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dubnau J., Chiang A. S., Grady L., Barditch J., Gossweiler S., McNeil J., Smith P., Buldoc F., Scott R., Certa U., Broger C., and Tully T. (2003) The staufen/pumilio pathway is involved in Drosophila long-term memory. Current Biol. 13, 286–296 [DOI] [PubMed] [Google Scholar]
- 63.Mallardo M., Deitinghoff A., Müller J., Goetze B., Macchi P., Peters C., and Kiebler M. A. (2003) Isolation and characterization of Staufen-containing ribonucleoprotein particles from rat brain. Proc. Natl. Acad. Sci. U.S.A. 100, 2100–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gardiol A., and St Johnston D. (2014) Staufen targets coracle mRNA to Drosophila neuromuscular junctions and regulates GluRIIA synaptic accumulation and bouton number. Developmental Biol. 392, 153–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang I. F., Wu L. S., Chang H. Y., and Shen C. K. (2008) TDP-43, the signature protein of FTLD-U, is a neuronal activity-responsive factor. J. Neurochem. 105, 797–806 [DOI] [PubMed] [Google Scholar]
- 66.Yu Z., Fan D., Gui B., Shi L., Xuan C., Shan L., Wang Q., Shang Y., and Wang Y. (2012) Neurodegeneration-associated TDP-43 interacts with Fragile X mental retardation protein (FMRP)/Staufen (STAU1) and regulates SIRT1 expression in neuronal cells. J. Biolog. Chem. 287, 22560–22572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Massignan T., Casoni F., Basso M., Stefanazzi P., Biasini E., Tortarolo M., Salmona M., Gianazza E., Bendotti C., and Bonetto V. (2007) Proteomic analysis of spinal cord of presymptomatic amyotrophic lateral sclerosis G93A SOD1 mouse. Biochem. Biophys. Res. Commun. 353, 719–725 [DOI] [PubMed] [Google Scholar]
- 68.Palma A. S., De Carvalho M., Grammel N., Pinto S., Barata N., Conradt H. S., and Costa J. (2008) Proteomic analysis of plasma from Portuguese patients with familial amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis 9, 339–349 [DOI] [PubMed] [Google Scholar]
- 69.Maurer M. H., Haux D., Unterberg A. W., and Sakowitz O. W. (2008) Proteomics of human cerebral microdialysate: From detection of biomarkers to clinical application. Proteomics Clin. Applications 2, 437–443 [DOI] [PubMed] [Google Scholar]
- 70.Nardo G., Pozzi S., Mantovani S., Garbelli S., Marinou K., Basso M., Mora G., Bendotti C., and Bonetto V. (2009) Nitroproteomics of peripheral blood mononuclear cells from patients and a rat model of ALS. Antioxidants Redox Signal. 11, 1559–1567 [DOI] [PubMed] [Google Scholar]
- 71.Willis D., Li K. W., Zheng J. Q., Chang J. H., Smit A. B., Kelly T., Merianda T. T., Sylvester J., van Minnen J., and Twiss J. L. (2005) Differential transport and local translation of cytoskeletal, injury-response, and neurodegeneration protein mRNAs in axons. J. Neurosci. 25, 778–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michaelevski I., Medzihradszky K. F., Lynn A., Burlingame A. L., and Fainzilber M. (2010) Axonal transport proteomics reveals mobilization of translation machinery to the lesion site in injured sciatic nerve. Mol. Cell. Proteomics 9, 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peredo J., Villacé P., Ortin J., and de Lucas S. (2014) Human Staufen1 associates to miRNAs involved in neuronal cell differentiation and is required for correct dendritic formation. PloS One 9, e113704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moughamian A. J., and Holzbaur E. L. (2012) Dynactin is required for transport initiation from the distal axon. Neuron 74, 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Moughamian A. J., Osborn G. E., Lazarus J. E., Maday S., and Holzbaur E. L. (2013) Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J. Neurosci. 33, 13190–13203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sorolla M. A., Reverter-Branchat G., Tamarit J., Ferrer I., Ros J., and Cabiscol E. (2008) Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radical Biol. 45, 667–678 [DOI] [PubMed] [Google Scholar]
- 77.Chen S., Lu F. F., Seeman P., and Liu F. (2012) Quantitative proteomic analysis of human substantia nigra in Alzheimer's disease, Huntington's disease and multiple sclerosis. Neurochem. Res. 37, 2805–2813 [DOI] [PubMed] [Google Scholar]
- 78.Zabel C., Mao L., Woodman B., Rohe M., Wacker M. A., Kläre Y., Koppelstätter A., Nebrich G., Klein O., Grams S., Strand A., Luthi-Carter R., Hartl D., Klose J., and Bates G. P. (2009) A large number of protein expression changes occur early in life and precede phenotype onset in a mouse model for Huntington disease. Mol. Cell. Proteomics 8, 720–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Basso M., Samengo G., Nardo G., Massignan T., D'Alessandro G., Tartari S., Cantoni L., Marino M., Cheroni C., De Biasi S., Giordana M. T., Strong M. J., Estevez A. G., Salmona M., Bendotti C., and Bonetto V. (2009) Characterization of detergent-insoluble proteins in ALS indicates a causal link between nitrative stress and aggregation in pathogenesis. PloS One 4, e8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.