

Abstract
Metformin, a drug prescribed to treat type-2 diabetes, exhibits anti-cancer effects in a portion of patients, but the direct molecular and genetic interactions leading to this pleiotropic effect have not yet been fully explored. To repurpose metformin as a precision anti-cancer therapy, we have developed a novel structural systems pharmacology approach to elucidate metformin’s molecular basis and genetic biomarkers of action. We integrated structural proteome-scale drug target identification with network biology analysis by combining structural genomic, functional genomic, and interactomic data. Through searching the human structural proteome, we identified twenty putative metformin binding targets and their interaction models. We experimentally verified the interactions between metformin and our top-ranked kinase targets. Notably, kinases, particularly SGK1 and EGFR were identified as key molecular targets of metformin. Subsequently, we linked these putative binding targets to genes that do not directly bind to metformin but whose expressions are altered by metformin through protein-protein interactions, and identified network biomarkers of phenotypic response of metformin. The molecular targets and the key nodes in genetic networks are largely consistent with the existing experimental evidence. Their interactions can be affected by the observed cancer mutations. This study will shed new light into repurposing metformin for safe, effective, personalized therapies.
Metformin, a drug frequently prescribed for type 2 diabetes, has been shown to decrease the incidence of cancer in diabetic patients by 37% relative to the diabetic patients not taking this drug, making it an important candidate for drug repurposing1. In breast cancer, as well as other cancers, metformin is believed to activate the AMPK signaling pathway, which elicits its anti-cancer effect by inhibiting the mTOR pathway2,3. In addition to the AMPK pathway, several AMPK independent pathways such as RAS4, AKT2, and HIF-1α5 may contribute to the anti-cancer effect of metformin. Metformin has also been proposed to act directly on mitochondria, decreasing mitochondrial respiration and lowering overall energetic efficiency; in cancer cells, this metabolic stress results in a greater increase in aerobic glycolysis than in normal cells, due to the former’s greater metabolic vulnerability6. More recently, Sun et al. attempted to construct a signaling pathway network of the genes through which metformin elicits its effect7. They proposed that seven genes (CDKN1A, ESR1, MAX, MYC, PPARGC1A, SP1, and STK11) and one MYC-centered pathway with CDKN1A, SP1, and STK11 might play important roles in metformin’s therapeutic effect. Larsson et al. examined the differential influence of metformin on the transcriptome and translatome of cancer cells. Their findings indicate that metformin’s primary effect is of inhibition of translation of a subset of mRNAs8. Previously, we proposed that metformin may weakly bind to the AMPKβ subunit, as well as four other proteins which have very similar binding sites (MAP2K2, PDK2, EGFR, and TIAM1)9. In spite of these efforts, the molecular and genetic mechanisms of metformin’s anti-cancer effect have not yet been fully understood. First, the direct molecular targets of metformin and their molecular details of interaction, which lead to its pleiotropic effect, are largely unknown. Such information is critical to understand the personalized therapeutic effect of metformin, as it will allow us to predict the direct impact of amino acid mutations on the drug-target interaction. Second, it is not clear what other molecular and genetic factors that modulate the drug action are responsible for the anti-cancer sensitivity of metformin on a patient-specific basis. It is speculated that genetic modifications in the drug modulation pathway will alter the drug response10. The answers to these questions will facilitate identifying pharmacogenetic biomarkers for the efficacy and side effects of metformin for individual patients, a critical step towards repurposing metformin as a safe and effective precision anti-cancer therapy.
There is evidence that polypharmacology is a common phenomenon. Weak interactions with many targets make a significant contribution to a drug’s effect11. Moreover, cryptic genetic factors may collectively affect drug actions under drug treatments12. It is challenging to determine genome-wide drug-target interactions and their correlations with clinical responses under diverse genetic backgrounds both experimentally and computationally. To address this problem, our innovation here is to extract the key proteomic and genomic factors which explain the drug’s empirically-observed activity on a multi-scale, from molecular details of drug-target interactions to the emergent properties of biological networks. Our strategy creates a start-to-finish picture of the drug’s overall effects, from its directly-binding upstream protein targets to the key down-stream proteins which control the body’s anti-cancer machinery. The strategy also takes instances of congruence as suggestions of polypharmacology, producing multiple binding partners and their modulated pathways of metformin’s interaction network. Using only publically available datasets, our methodology has produced a more complete image of metformin’s biological activity. While we have experimentally verified the interaction between metformin and the majority of the targets we have tested, our results have yielded many additional direct and indirect targets ready for further investigation. Our approach is generalizable to any compound with at least one documented protein receptor for which a crystal structure is available and for which a genome-wide gene expression analysis has been performed, and is particularly suited for identification of polypharmacology.
Specifically, we have developed a structure-based method to identify the targets of a drug molecule on a structural proteome scale, and successfully applied it to drug repurposing13, side effect prediction12, and polypharmacology14. Here, a similar strategy will be applied to identify proteome-wide molecular targets of metformin. Among the top ranked targets, several are kinases. Subsequently, we commissioned a KINOMEscan binding assay between these putative kinase targets and metformin, and validated 5 out of 6 our predictions being correct. After identifying putative interacting proteins, we for the first time developed a novel method to predict the downstream genes and drug modulation pathways by integrating drug targets, protein-protein interaction network, and gene expression using a Prize-Collecting Steiner Tree (PCST) algorithm15. Finally, we analyzed these predicted networks for biological relevance to anti-cancer activity and to determine hidden genetic factors that will impact the drug modulation pathways. In this way, we can correlate drug-target interactions with drug response phenotypes under diverse genetic backgrounds.
Using the multi-scale modeling approach, we have identified twenty putative targets of metformin. Although using different structural templates as a starting point, the two studies resulted in consistent results. Five kinases that we have shown bind directly to metformin (SGK1, EGFR, CDK7, MAPK14, and MAP2K2). Furthermore, we identified genetic factors (TP53, AKT1, and PCNA genes) that do not directly bind to metformin, but are responsible for the anti-cancer effect of metformin. Our predictions are largely consistent with the existing experimental and computational results. They generated abundant testable hypotheses for further in vitro and in vivo validation, and may shed new light into repurposing metformin for safe, effective, personalized anti-cancer therapies. This study also lays the groundwork for high-throughput prediction of pleiotropic drug/target/gene relationships.
Results
Our structural systems pharmacology approach is based on a simplified model of drug action as shown in Fig. 1. We assume that a drug like metformin may bind to multiple proteins (drug targets)12. Functional modifications of targets resulting from the drug binding (inhibition or activation) will propagate to other genes through a protein-protein functional association network, and manifest in the transcriptional, translational, and other observable genome-wide changes. The observed differentially expressed genes upon drug binding can be used as a surrogate of drug response phenotype. Genes without observable changes (cryptic genetic factors) may exist to mediate the information transmission from drug targets to differential genes. A genetic alteration in either a drug target or a cryptic genetic factor will impact the drug’s action. Following this concept, our structural systems pharmacology approach consists of three major steps. First, we apply a previously-proven successful method to identify drug binding targets on a structural genome scale. Then we map the drug targets and differential genes to a protein-protein interaction (PPI) network and identify the drug modulation pathway, a sub-network on the PPI network, which links the drug targets to differentially expressed genes. Subsequently, we detect the critical gene nodes in the drug modulation pathway whose deletion will have significant impacts on information transmission, thereby altering the drug response phenotype. Finally, we map the observed cancer mutations to the binding pockets of experimentally validated molecular targets and identified genetic biomarkers. These analyses provide clues to the alternation of drug-target interaction and network rewiring that may impact the personalized drug response. The method is summarized in Fig. 2, and details are presented in the Supplemental Methods section.
Figure 1. A conceptual framework of structural systems pharmacology used in this study.
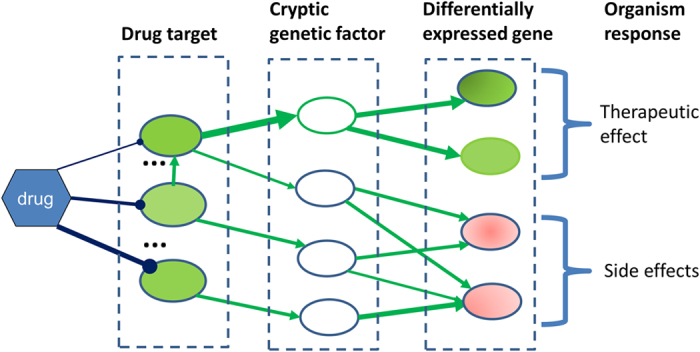
Dark blue lines represent direct molecular interactions between drug and its targets. Green arrows are protein-protein interactions through which the information of drug-target interaction is transmitted.
Figure 2. Methodology Flow Chart.

Bulleted line segments represent datasets and software used; arrows represent the flow of information and the transition from one step to another.
Putative targets of metformin
We proposed that metformin might weakly bind to the AMPKβ subunit9. DPP4 was identified as another direct molecular target of metformin by querying the ChEMBL relational database using SQL16 (Supplemental Methods, Figure S1), and supported by recent studies17,18. Using AMPKβ subunit and DPP4 as templates, we profiled protein structures from the Protein Data Bank (PDB) to identify those with evolutionarily related and similarly shaped binding pockets to that of AMPKβ and DPP4. We identified 19 structures with a binding-site similarity (multiple testing corrected p-value < 0.05) which were not homologs of AMPKβ and DPP4. This set was termed the “putative targets”. Our top-ranked structure, associated with the Fibroblast Activation Protein (FAP), is a member of the prolyl peptidase protein family – the same family as DPP4, explaining its near-identical binding site19. Among the putative targets, six of them are protein kinases.
Binding mode of metformin in putative targets
Docking model experiments between metformin and each of these proteins predicted exothermic binding interactions to take place in all cases, as shown in Table 1. Figure 3A shows an example of the binding pose of metformin on kinase SGK1. Similar to other kinases, the binding site of metformin is adjacent to the ATP binding site9. It implies that metformin may act differently from the common ATP inhibitors of kinases. We further mapped somatic cancer mutations observed in COSMIC (cancer.sanger.ac.uk)20 onto the structures of putative kinase targets. A number of these mutations surround the metformin binding site. Five examples are shown in Fig. 3B–F. These amino acid mutations observed in COSMIC are listed in supplementary Table S1. It is speculated that the mutations of these amino acids, will impact the therapeutic effect of metformin. This finding is potentially important in the context of personalized medicine, as metformin may have increased or decreased drug sensitivity in patients with such mutations.
Table 1. Putative targets of metformin identified from ligand binding site comparison.
| PDB code | Gene symbol | SMAP P-value | Vina binding score Kcal/mol |
|---|---|---|---|
| 1Z0M | PRKAB1 | 0.0 | −4.7 |
| 1Z68 | FAP | 1.0E-10 | −5.0 |
| 3A8N | TIAM1 | 9.22E-04 | −4.8 |
| 1SXJ | RFC1 | 4.79E-03 | −5.1 |
| 3B2V | EGFR | 6.79E-03 | −5.1 |
| 1S9I | MAP2K2 | 6.92E-03 | −5.1 |
| 2BU7 | PDK2 | 9.85E-03 | −4.9 |
| 3DDU | PREP | 1.05E-02 | −4.5 |
| 3NVQ | SEMA7A | 1.63E-02 | −4.6 |
| 1F45 | IL12B | 1.71E-02 | −5.7 |
| 1CJY | PLA2G4A | 1.85E-02 | −4.7 |
| 2ONL | MAPK14 | 1.96E-02 | −5.7 |
| 3O96 | AKT1 | 2.49E-02 | −5.5 |
| 1K8Q | LIPF | 2.81E-02 | −4.5 |
| 3KY9 | VAV1 | 3.01E-02 | −6.4 |
| 1UA2 | CDK7 | 3.21E-02 | −5.5 |
| 3HDN | SGK1 | 3.37E-02 | −4.1 |
| 2OCI | BPHL | 3.50E-02 | −5.4 |
| 2WWW | MMAA | 3.71E-02 | −4.6 |
| 3EAH | NOS3 | 3.77E-02 | −5.7 |
| 1TG6 | CLPP | 4.05E-02 | −2.6 |
Binding between underlined entries and metformin was experimentally tested by KINOMEscan assay. Four putative targets were identified (PDK2, MAP2K2, EGFR and TIAM1) using AMPKβ as a template, and AMPKβ itself was also included (PRKAB1 gene; no p-value is available due to its use as a template).
Figure 3. The predicted binding pose of metformin in the binding pocket of putative kinase targets.

(A) The binding site of metformin (right) is adjacent to the binding site of ATP (left) in SGK1. The interaction patterns of metformin with SGK1 (B), MAP2K2 (C), CDK7 (D), MAPK14 (E), and EGFR (F). The green, labeled residues are those which are known to be involved in carcinogenic mutations.
Binding assay for putative kinase targets
Among our putative targets, six of them are kinases: AKT1, EGFR, MAPK14, MAP2K2, CDK7 and SGK1. We tested the direct binding between metformin and these kinases using
DiscoveRX KINOMEscanTM binding assay. KINOMEscanTM is an active site-directed competition binding assay to quantitatively measure interactions between chemical compounds and kinases. The binding strength is reported by ‘%Control’, which is calculated as shown in equation (1).
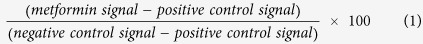 |
DMSO and a pico-molar kinase inhibitor were used as negative and positive controls, respectively. The lower %Control is, the stronger the interaction. As shown in Table 2, our results showed binding between metformin and all the tested six kinases except AKT1. Of the tested kinases, Metformin showed its strongest binding with SGK1 and EGFR. Thus, our predictions are strongly supported by the experimental evidence.
Table 2. Results of KINOMEscan binding assay between metformin and top-ranked kinase targets.
| Gene symbol | % control |
|---|---|
| SGK1 | 59 |
| EGFR | 60 |
| CDK7 | 79 |
| MAP2K2 | 80 |
| MAPK14 | 88 |
| AKT1 | 100 |
‘% control’ is a measure of binding strength. A higher value indicates a weaker interaction; ‘100% control’ indicates no interaction was observed. The results indicate that metformin binds weakly with a large number of kinases.
Analysis of drug modulated sub-networks
Here, we, for the first time, developed a novel method to predict the drug modulation pathways as protein-protein interaction (PPI) sub-networks, which could lead to experimentally-determined changes in gene expression8, using a Prize-Collecting Steiner Tree (PCST) algorithm15. In this conceptualization, metformin-interacting proteins form the root of a branching tree network, differentially-expressed genes form the leaves of the tree, and confidence-weighted interactions between other proteins connect the root to the leaves. The PCST algorithm attempts to link the root to as many leaves as possible while minimizing edge cost (maximizing confidence), thus predicting the most linked nodes that take part in the network without apparent alteration of expression. Compared with the conventional shortest path method, the PCST will provide a parsimonious solution that jointly optimizes the paths from the root to all the leaf nodes, thus potentially reducing the occurrence of false positives, especially when applied to the noisy PPI network11. In our implementation, protein-protein interactions were obtained from the STRING-DB21 and assigned a cost which was calculated as 1 minus the confidence of the interaction. Figure 4 shows an example of the resulting sub-network structure when MAPK14 is chosen as the root. The leaf nodes of our sub-networks were obtained from a set of genes which are translationally-suppressed in MCF7 cells by metformin – as reported by Larsson et al.8 (see Supplemental Methods). Metformin had been previously demonstrated to inhibit proliferation of MCF7 cells2.
Figure 4. Visualized sub-networks for MAPK14.
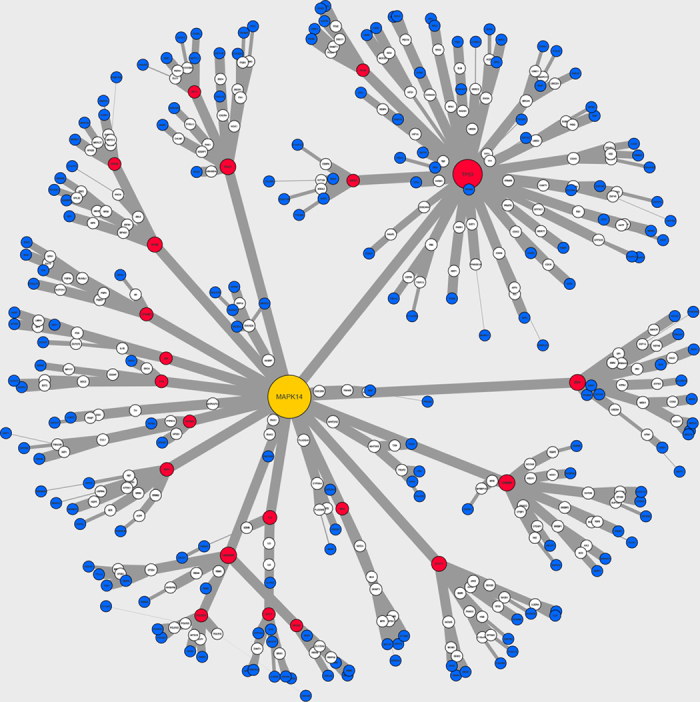
Blue nodes indicate leaf (differentially-expressed genes); for intermediate nodes (cryptic genetic factors) red indicates critical node; yellow indicates root node; all others are white. Node size correlates with betweenness-centrality score. Edge-width correlates with confidence of the interaction. Note that most interactions are of high relative confidence (>0.9 on a scale from 0.0 to 1.0). The arrangement of nodes in space is for ease of comprehension only. Visualized using CytoScape 3.2.154.
We note that the protein-protein interaction scores documented in STRING-DB are noisy. We calculated the distribution of edges both within the STRING-DB human PPI and within our predicted sub-networks. Our results show that the interactions within our networks are consistently of high relative confidence, in contrast to the mainly low relative confidence STRING-DB PPI (Figure S2). This suggests that the PCST algorithm effectively selects high-confidence paths through the STRING-DB PPI and that our predicted sub-networks contain interactions which are likely to occur in vivo.
As a control, we generated three additional sets of sub-networks. The first set consisted of sub-networks with randomly chosen genes at the root (the “R-control” set, n = 16). The second set was generated using a random selection of genes from the leaf nodes (“L-control”, n = 20). Noting that our predicted binding targets and binding assay demonstrate the interaction between metformin and several kinases, our third set of control sub-networks were constructed using a random selection of genes coding for kinases (“K-control” set, n = 19).
We used the GeneTrail tool to identify biological pathways that were enriched in the non-terminal nodes of the experimental and control sub-networks. The experimental sub-networks were enriched for a greater number of overall KEGG pathways than the L-control and R-control sub-networks but not the K-control group (average of 40.4, 33.9, 32.8, and 39.4 KEGG pathways respectively; exp vs. L-control < 0.05; exp vs. R-control p < 0.05; exp vs. K-control p = 0.741). The same pattern was seen when comparing enrichment of cancer-related KEGG pathways (average of 22.7, 20.0, 19.5, and 22.8 KEGG pathways respectively; exp vs. L-control p < 0.01; exp vs. R-control p < 0.01; exp vs. K-control p = 0.944). Cancer-related KEGG pathways were defined as pathways related to specific cancer types, e.g. ‘Melanoma’ or ‘Small cell lung cancer,’ as well as pathways known to be involved in the development or maintenance of cancer, e.g. ‘VEGF signaling pathway’ or ‘Apoptosis.’ A full list of KEGG enriched pathways defined as cancer-related can be found in the Supplemental Methods.
The results yielded enrichment of multiple relevant KEGG pathways22. ‘Apoptosis’, ‘Cell cycle’, ‘Pathways in cancer’, ‘Chronic myeloid leukemia’, ‘Pancreatic cancer’, ‘Glioma’, ‘Non-small cell lung cancer’, ‘Prostate cancer’, and ‘Bladder cancer’ were all enriched in at least 85% of the sub-networks in each set (at least 18 of 21 experimental sub-networks, 17 of 20 L-control sub-networks, 14 of 16 R-control sub-networks, and 17 of 19 K-control sub-networks, FDR corrected p values < 0.05). Other relevant pathways, including ‘MAPK signaling pathway’, ‘ErbB signaling pathway’, ‘Nucleotide excision repair’, ‘DNA replication’, ‘B cell receptor signaling pathway’, ‘mTOR signaling pathway’, and ‘VEGF signaling pathway’ as well as pathways for several more cancers, were enriched in at least 10 experimental sub-networks and control sub-networks.
As the enrichment of relevant pathways was high in experimental and control sub-networks, we sought a finer method to distinguish between the identified sub-networks.
Based on our KEGG results, as well as our knowledge of metformin’s relevance to cancer, insulin, and AMPK signaling, we assembled a set of ‘key pathways’ which represent a profile of metformin’s documented and putative activity (see Supplemental Methods)22. We calculated the number of instances in which nodes of each sub-network were found in a key pathway, and each sub-network was assigned a “participation” score for each key pathway. Although most networks in both the experimental and control groups were enriched for the same pathways, the nodes of the control sub-networks participated less in key pathways. Analysis of participation within key pathways for the sub-networks between the experimental and control groups showed significant differences. On average, the participation of intermediate nodes in key pathways was 117.3 for the L-control, 117.6 for the R-control, 133.7 for the K-control group, and 131.4 for the experimental group. With approximately equal standard deviations of 12.9, 15.6, 13.3, and 18.8, respectively, the difference between experimental and both L-control and R-control is significant (Welch’s T-test for unequal variance, p < 0.01 and p < 0.05 respectively) but the difference between experimental and K-control is not (p = 0.669). Noting that 6 out of 20 of our putative targets were themselves kinases, we calculated that the average participation score for that sub-group (140.8) exceeded that of the K-control, although the difference was not significant (p = 0.416).
The participation scores for experimental sub-networks were normalized against the mean participation the R-control group. This Z-score represents the extent to which the sub-network in question participated in key networks beyond what which we would expect by random chance (Table 3).
Table 3. Z-score for participation within key pathways.
| Root | Z-score |
|---|---|
| AKT1 | 2.777 |
| IL12B | 2.585 |
| EGFR | 2.521 |
| MAPK14 | 2.265 |
| TIAM1 | 2.073 |
| SGK1 | 1.498 |
| RFC1 | 1.434 |
| SEMA7A | 1.306 |
| NOS3 | 1.115 |
| VAV1 | 1.051 |
| PLA2G4A | 0.923 |
| CLPP | 0.859 |
| MMAA | 0.795 |
| CDK7 | 0.667 |
| LIPF | 0.411 |
| MAP2K2 | 0.220 |
| PREP | 0.156 |
| FAP | −0.164 |
| PDK2 | −0.867 |
| PRKAB1 | −1.442 |
| BPHL | −1.570 |
Calculated by normalizing participation score against the mean of the R-control set. Higher participation indicates that the intermediate nodes of the network are involved in key pathways to a greater extent.
In summary, sub-networks generated from randomly-selected genes are not different from sub-networks generated from a random sub-set of the differentially-expressed genes in terms of pharmaceutically relevant pathways. Sub-networks based on the putative targets are significantly more closely related to pathways than either of these sub-network sets. However, there is no observed difference between the putative targets and kinases. Considering the fact that a large number of kinases were experimentally validated targets and that kinases share similar ATP binding pockets, it is likely that many other kinases could be potential targets of metformin.
Critical components that may impact drug actions
We further determine hidden genetic factors that would impact the drug modulation pathways. Our working hypothesis is that the node in the network would have a greater impact on the drug’s activity if its deletion significantly reduced the flow of information. Table 4 shows genes which appeared as critical nodes (betweenness-centrality in the 95th percentile, see Supplemental Methods23,24) in at least 20% of experimental sub-networks. We note that several genes were found amongst the critical nodes in at least 50% of sub-networks, in both the set of putative targets and the three control groups. These genes (TP53, PCNA, AKT1, SRC, and INS-IGF2) were apparently selected because of their functional link to significant sub-sets of the genes which were differentially expressed under metformin treatment8, which overwhelmed the choice of root node in the sub-network.
Table 4. Genes that were critical nodes in the sub-network.
| Critical genes | Number of sub-networks where critical |
|---|---|
| TP53* | 20 |
| AKT1* | 20 |
| PCNA* | 17 |
| SRC* | 15 |
| INS-IGF2 | 12 |
| SF3A2 | 10 |
| BIRC5 | 10 |
| DKC1 | 9 |
| ESR1* | 8 |
| BUB1 | 7 |
| BRCA1 | 7 |
| RPL11 | 7 |
| YBX1* | 7 |
| RPS16* | 6 |
| GRB2* | 6 |
| POLR2H | 6 |
| HNRNPK | 5 |
| MDM2 | 5 |
| CASP3 | 5 |
| POLR2A* | 5 |
| CDK5 | 5 |
| SP1 | 5 |
A node is included if the gene appears in at least 20% of experimental sub-networks (5 out of 21). These nodes have a critical role in several sub-networks and may be key cryptic genetic factors in metformin’s signaling network. *indicates genes which have known carcinogenic mutations that could significantly alter the networks.
We proposed that because sub-networks with higher Z-scores could better reflect the documented and proposed activity of metformin, they might be closer to metformin’s true biological activity. Because of this, we suggested that nodes showing a higher degree of betweenness-centrality in networks with higher Z-scores in contrast to networks with lower Z-scores should be more likely to be important factors. We segregated the experimental sub-networks based on Z-score into several overlapping sets: all sub-networks, those with a Z-score greater than 1, and those with a Z-score greater than 2. In Table 5 we recorded the genes that have an especially large discrepancy in their betweenness-centrality in different groups of sub-network. For example, ESR1 is considered critical in 25.0% of R-control sub-networks, 38.10% of all experimental sub-networks, 50.0% of sub-networks with Z > 1, and 80% of sub-networks with a Z > 2. Genes were included in the table if the discrepancy was at least 25 percentage points between highest and lowest percentage.
Table 5. Genes where betweenness-centrality was enriched among one sub-group relative to another.
| Gene | Control (n = 19) | Experimental (n = 21) | Z > 1 (n = 10) | Z > 2 (n = 5) |
|---|---|---|---|---|
| SF3A2 | 37.5% | 47.6% | 60.0% | 100.0% |
| ESR1* | 25.0% | 38.1% | 50.0% | 80.0% |
| BIRC5 | 37.5% | 47.6% | 70.0% | 80.0% |
| MDM2 | 6.3% | 23.8% | 30.0% | 60.0% |
| STAT1* | 0.0% | 14.3% | 30.0% | 60.0% |
| POLR2H | 18.8% | 28.6% | 50.0% | 60.0% |
| CTNNB1 | 6.3% | 9.5% | 20.0% | 40.0% |
| CREBBP | 0.0% | 14.3% | 30.0% | 40.0% |
| RAC1 | 0.0% | 14.3% | 30.0% | 40.0% |
| JUN | 6.3% | 19.0% | 40.0% | 40.0% |
| FOS | 6.3% | 14.3% | 20.0% | 40.0% |
| STAT3 | 6.3% | 19.0% | 20.0% | 40.0% |
| IL8 | 0.0% | 9.5% | 20.0% | 40.0% |
| NFKBIA* | 0.0% | 9.5% | 20.0% | 40.0% |
| BRCA1 | 50.0% | 33.3% | 40.0% | 20.0% |
| HDAC1 | 37.5% | 14.3% | 10.0% | 0.0% |
| CDK5 | 0.0% | 23.8% | 30.0% | 0.0% |
| SDC2* | 18.8% | 19.0% | 30.0% | 0.0% |
For example, SF3A2 was considered critical in 37.50% of sub-networks in the control group, but 100% of experimental networks with Z > 2. Shown here are genes where the disparity was at least 25 percentage points between most-enriched and least-enriched. Many of these nodes are enriched in the highest-scoring networks and may be critical nodes specific to metformin’s anti-cancer activity. *indicates genes which have known carcinogenic mutations that could significantly alter the networks
Cross-validation of critical components
Garnett et al. carried out an elastic-net regression to identify genomic features linked to sensitivity to metformin25. Their analysis identified copy number variation in BRCA1 and BLM, two genes found to be critical nodes in our sub-networks, as factors related to sensitivity to metformin in cancer tissue. Expression levels in several other genes were identified through elastic-net regression whose familial counterparts were predicted in at least one sub-network. These include the IL2RG/IL2, RBM15/RBM5, RPS6KC1/RPS6/RPS16 and UBE2G1/UBE2I sets, as well as MAP4K3 which is related to several predicted MAP and MAPK nodes and direct targets. Finally, the AKT3 gene was identified, pairing with the predicted AKT1 target. Garnett’s results provide a partial cross-validation of our predictions, as shown in Table 6.
Table 6. Genes on the left were independently identified as factors in metformin sensitivity in cancer via the attribute indicated in the central column.
| EN-identified gene | Genomic attribute | Putative target(s) or gene(s) from sub-network |
|---|---|---|
| BRCA1 | Copy number | BRCA1 |
| BLM | Copy number | BLM |
| AKT3 | Expression | AKT1 |
| IL2RG | Expression | IL2 |
| MAP4K3 | Expression | MAP2K2, MAP2K6, MAPKAPK2, MAPK14, MAP3K5, MAPK1 |
| RBM15 | Expression | RBM5 |
| RPS6KC1 | Expression | RPS6, RPS16 |
| UBE2G1 | Expression | UBE2I |
These genes are associated with the factors included on the right, which were part of our set of putative targets or sub-networks.
The ‘drug-specific signaling pathway network’ (DSpathnet)7 for metformin generated recently by Sun et al. produced results which we reinforce here, although the two studies used different data sets and methodology. The genes that we identified as critical components in our predicted sub-networks overlapped significantly with important components of metformin’s DSpathnet. We compared sub-network nodes which were critical (betweenness-centrality in the 95th percentile for a given sub-network) in a given portion of the sub-networks and were also hubs within the DSpathnet (connectivity >14, n = 38). Seven genes were found to be both critical in at least 25% of sub-networks with a Z-score >1 and were also DSpathnet hubs; they were BRCA1, TP53, ESR1, STAT1, JUN, SP1, and AKT1. Their work identified seven critical genes (CDKN1A, ESR1, MAX, MYC, PPARGC1A, STK11, and SP1). All of these genes were identified as critical within at least one predicted sub-network except for STK11 and MYC, which still appeared as nodes within at least one sub-network.
It is interesting to note that the mutations in several genetic biomarkers may lead to the rewiring of the cancer network, as supported by a recent study26. We have marked genes on Tables 4 and 5 which were proposed to be involved in this process.
Discussion
The putative targets of metformin are largely consistent with existing experimental evidence, suggesting that metformin is a polypharmacological agent with multiple weak binding partners
Our results provide evidence for the direct interaction between metformin and several protein targets whose activities contribute directly to the observed pharmacological effects of the drug. These may be considered the upstream nodes of metformin’s predicted network, and were identified independent of context. Several of these predicted targets and functionally-important network nodes have been previously characterized as related to metformin’s effects because of their presence in pathways which are related to AMPK signaling, such as MAPK signaling and RAS signaling, but here for the first time, we have predicted direct interactions between metformin and these proteins using binding site analysis.
There is strong evidence to suggest that many of our putative targets are both directly and functionally linked to metformin. EGFR is a well-known anti-cancer drug target27. Of the kinases we tested for binding with metformin, the strongest direct binding was observed with SGK1. SGK1 participates in many cellular pathways but has gained significant attention in the field of molecular oncology due to its role in several cancer cell pathways28. SGK1 is activated by insulin, growth factors, and steroids – this may explain the increased cancer risk for patients with obesity and insulin resistance29, as well as explain the dual activity of metformin (which we hypothesize to act as an inhibitor of SGK1) in diabetics as well as cancer patients. Earlier, we mentioned that metformin directly acts on the mitochondria6; this result is consistent with our results because SGK1 has been shown to localize in mitochondria in response to cellular stress30. It is possible that interaction with SGK1 allows for metformin’s action on mitochondrial respiration. Other studies28,29 on SGK1 provide evidence of its active role in cancer-related pathways. D’Antona et al. using a specific inhibitor of SGK1 kinase (Si113) tested the proliferation and sensitivity to the antineoplastic agent, Paclitaxel, of colon carcinoma cells. Their results indicate that inhibition of SGK1 enhances apoptotic processes and responsiveness to the Paclitaxel drug and also decreases cell viability29, which in turn, results in a decrease in the proliferation of the carcinoma. They also claim that, given the success of their inhibitor in affecting colon carcinoma, SGK1 inhibitors are excellent candidates for selectively hitting cancer cell pathways without (or with minimal) interference to normal cell pathways. This is because of the reported over-expression of SGK1 in carcinoma cells relative to normal cells29. Wu et al. have elucidated some of the purported pathways and mechanisms by which SGK1 may act in the cancer cell lines. The study by Wu et al. shows that SGK1 expression inhibits chemotherapy induced apoptosis28.
We confirmed binding between metformin and CDK7, a protein involved both in cell cycle control and the activation of RNA-polymerase 2 for transcription. CDK7 phosphorylates many proteins, including p53, a critical regulator of apoptosis and DNA repair31. Our putative sub-networks re-constructed the strong functional relationship between CDK7 and both P53 and RNA polymerase-2.
We also demonstrated the binding of metformin and MAPK14. The precise role of MAPK14 depends upon the tumor subtype and there are cases where inhibition, rather than activation, of MAPK14, has anti-cancer effects32,33,34. For example, in one study, inhibition of MAPK14 phosphorylation (accomplished by using the drug Sorafenib), was shown to suppress non-hodgkins lymphoma32. A different study on Metformin’s effect on human epidermoid carcinoma, shows a similar decrease in the activation of MAPK1433 with a suppressive effect on cancer cell proliferation; another study, shows that Metformin directly decreases the phosphorylation levels of MAPK14 in bovine granulosa cells35, a third study shows that Metformin, used in conjunction with gefitinib, decreased levels of MSH2 protein in human lung squamous cell carcinoma, again, through down regulation of MAPK1434. Finally, a study on metastatic melanomas shows that activation of MAPK14 by urokinase plasminogen activator receptor (uPAR) clustering on cell surfaces (promoted by d-GM3, a ganglioside) results in the activation of matrix metalloproteinase-2 (MMP-2)36 which contributes to cell migration and metastasis in cancer cells. This supports our findings with regards to MAPK14 as a putative target of Metformin, and provides some justification for Metformin’s purported anti-cancer activity. Furthermore, inhibition of MAP2K2 (MEK2), an important signaler in the MAPK signaling pathway, dramatically decreases semaphorin-7a expression in oncogenic Ras cells37. We confirmed the binding of MAP2K2 and metformin and predicted the binding of Semaphorin-7a and metformin. Reynolds described the pathway through which our predicted-target Vav1 activates MEK/ERK signaling via Ras38.
The NOS3 protein, another one of our predicted targets, catalyzes the synthesis of nitric acid, which facilitates the activation of Ras proteins. Knockdown of NOS3 showed that active NOS3 is necessary both for initiation and maintenance of human pancreatic cancer cells. A study by Reynolds et al. suggests that NOS3 acts a link between the different varieties of Ras (HRas, NRas, Kras). It appears that NOS3 activation is increased by the signaling of oncogenic Ras, but the effect of NOS3 comes from subsequent activation of wild-type Ras – for example, oncogenic KRas will lead NOS3 to activate wild-type HRas and NRas39.
Allegra demonstrated the role of Semaphorin-7a (SEMA7A) in epithelial to mesenchymal transition (EMT)37, an important step in tumorigenesis. The researchers showed that EMT could be prevented by mutating the gene ERF so that it could not be phosphorylated. Semaphorin-7a is normally induced by TGF-b, but when ERF was mutated Semaphorin-7a was expressed at decreased levels and could not be induced by TGF-b. They next demonstrated that Semaphorin-7a is required for TGF-beta-induced EMT.
The identification of several proteins known to be closely related to metformin’s known activity as direct targets lends credence to the idea that the other identified proteins may also directly interact with metformin, although these interactions may less directly lead to metformin’s anti-cancer activity. We have demonstrated the direct binding between metformin and five out of six candidate proteins, and our predictions and evidence strongly support the notion that metformin is a polypharmacological and pleiotropic drug. Where possible, additional ligand binding assays should be performed between metformin and other predicted targets in order to complete the picture of metformin’s biological activity. Our predictions and experimental results suggest that metformin elicits its strong effects by binding weakly with many targets that act in concert to produce a powerful polypharmacological effect. Similar mechanisms have been proposed for the anti-cancer effect of HIV protease inhibitor Nelfinavir40.
Non-metformin-binding genes could be responsible for the therapeutic effect of metformin
In addition to the direct upstream binders of metformin, the predicted protein sub-networks recapitulate many of the pathways we expected to produce metformin’s anti-cancer activity. We note that our network analysis is only based on one single sample, and is context-specific.
Ouchi et al. postulated an “adiponectin-AMPK-PI3-kinase-Akt-eNOS signaling axis” in type 2 diabetes41. Our results are consistent with this model, with our predicted binding between metformin and NOS3, and the inclusion of several genes involved in adipocytokine signaling within predicted interaction sub-networks, including the adipokine Leptin and the transcription factors STAT1 and STAT3.
Our predicted sub-networks have several hub genes (Table 4) which may play critical roles in eliciting metformin’s effects. Several hub genes are enriched in higher-scoring sub-networks (Table 5). Many of them have been previously documented to play a role in cancer, and others may be novel cryptic factors specific to metformin’s modulation pathway. They include the TP53, PCNA, and AKT1 genes, which code for the tumor-suppressor p53, the DNA-repairing PCNA protein42, and the apoptosis-inhibitor AKT1. We see several cancer-relevant genes which are critical specifically within the most participatory sub-networks. Overexpression of RAC1 has been noted as a marker for aggressive breast cancer35. STAT1 expression levels have been used as part of a five-gene signature for non-small-cell lung cancer survival43. These also include the BIRC5 inhibitor of apoptosis, as well as the JUN and FOS genes, coding for c-Jun and c-Fos, which together form the AP-1 transcription factor, and NFKBIA, an inhibitor of the survival-promoting NF-κB complex. Huang et al. showed that down regulation of c-Jun and NF-κB inhibits the invasion of melanoma cells through suppression of MMP-944 None of our predicted networks included the MMP9 gene, however the interactions annotated in the STRING-DB are not all direct, and further investigation may identify a role for MMP-9 in metformin’s anti-cancer activity. The growth-promoting INS-IGF2 gene, and the SRC oncogene were both found to be critical in a large percentage of networks, however their critical role appears to be approximately the same in the experimental and control networks. They also are not critical in two of the most highly-participatory networks, based on MAPK14 and IL12B. These genes may still play a critical role in the overall protein-protein interaction network.
Although our networks identified many critical factors which appear promising for experimental validation, it is important to consider the potentially-large portion of false-negatives within our results. For example, our sub-networks were generated with a maximum depth of 5, limiting the resolution of the final network. This allowed us to perform our analyses computationally-efficiently and revealed many strong functional connections. However, it appears that sub-networks generated using randomly-chosen kinase genes as the root (K-control) are enriched for cancer-related nodes at roughly the same rate as sub-networks based on our putative-targets. This suggest that potentially many of these kinases play significant roles in eliciting metformin’s effect beyond what is apparent in the predicted sub-networks.
Network constraints must also be considered when interpreting the significance of peripheral network nodes. A detail from the publication by Sun et al. illustrates this point7 Their ‘Drug-specific signaling pathway network’ was also applied to metformin. Their methodology attempted to create a complete network of the genes through which metformin elicits its effect, and our work partially recapitulates their results. Their predicted network shares significant overlap with ours; however, they do not speculate on metformin’s direct binding targets. In addition, their network was built using gene expression in response to the metformin treatment from ConnectivityMap (cMAP). While the data is informative, there is also limitation on the direct evidence of those differentially expressed genes in metformin response. Our efforts our distinguished by the fact that our predicted networks are rooted in putative binding targets, some of which we have tested and confirmed.
In metformin’s DSPathNet, the PPARGC1A gene serves as a linker node connecting distinct modules of signaling pathways. Variants of this gene have been linked to breast cancer risk45 and the gene’s product has been shown to play a key role in the metabolism of ERBB2 + breast cancer cells in vivo46. The common location for PPARGC1A within our sub-networks is as a peripheral node downstream of TP53. The motif shows TP53 as a hub node linked to many genes, including SIRT1. SIRT1 is linked to PPARGC1A which connects to MUM1L1, a leaf node. The connection between PPARGC1A and MUM1L1 is notably weak, suggesting that the PCST algorithm connects it to our sub-networks with difficulty. It is plausible that PPARGC1A may play a larger role in eliciting metformin’s anti-cancer effects than our predictions suggest, owing to the constraining set of differentially expressed genes we used when generating the sub-networks. Future applications of the methodology demonstrated here, may wish to limit the number of false negatives by choosing larger sets of network leaf nodes obtained from multiple sources.
Many of the genes in our predicted sub-networks may play an important role in motivating metformin’s anti-cancer activity. We have identified both upstream direct binding targets for metformin, as well as downstream network nodes which transduce its effects to the level of gene expression. We expect that disruption of the putative targets, network hubs, or nodes which play key roles in high-scoring networks (Tables 3, 4, and 5) is likely to inhibit the effects of metformin or cause adverse side effects.
However, as metformin appears to inhibit cancer broadly and through weak binding with several targets, the specific role played by individual network components in a patient is expected to vary based on the individual genetic and homeostatic context. Further experiments, including gene knock-down experiments, may narrow down the predicted biological role for these proteins, paving the way for their adoption as biomarkers for metformin’s efficacy or adverse drug effects.
Towards precision medicine using structural systems pharmacology
Data-driven, network-based association studies provide a promising avenue to realize precision medicine47. However, the power of data-driven approach remains limited if sample sizes are small. Rare mutations may have unexpected consequences on drug response48. To predict drug response de novo under diverse genetic backgrounds, a mechanistic understanding of drug action is required. A drug commonly interacts with multiple targets. Each drug-target interaction modifies the conformational dynamics of the target structure and leads to the change of the functional states. Consequently, the changing conformational and functional states of drug-target interactions affect other molecular components and their interactions through the interplay of complex signal transduction, gene regulation, and metabolic networks that collectively mediate the system-level response to the drug, leading to either therapeutic or adverse effect. A variety of genetic/epigenetic alternations, if involved in the drug interaction and modulation, could impact the drug response. Thus, to associate individual genotype to drug response phenotype, it is essential to model drug actions on multiple scales, from the conformational dynamics of genome-wide drug-target interactions to the emergent property of cellular networks, using a structural systems pharmacology approach49.
Using metformin as a proof-of-concept study, we demonstrate that structural systems pharmacology is able to provide mechanism-based predictive models using a limited number of samples. The power of structural systems pharmacology depends on the quality and coverage of protein structure, molecular interaction, and gene regulatory network, and molecular phenotypic data. In this study, only AMPKβ and DPP4 are used as templates and ~7000 human protein structures are searched through for the potential targets of metformin. It is likely that many targets of metformin remain to be elucidated. With the exponential increase in experimentally solved structures, it is possible for us to construct high-quality 3D drug-target interaction models on a proteome scale. The molecular details of protein-ligand information will allow us to predict the impact of amino acid mutations on the drug action de novo. Moreover, the interaction network applied in this study is incomplete, noisy and is not tissue specific. It may limit the capability to detect novel genetic biomarkers. Recent advance in the proteome-scale mapping of the human interactome network has produced homogenous coverage of the human interactome, significantly expanding the cancer landscape50. The ENCODE project has generated genome-wide gene regulatory networks51. Coupled with molecular phenomics52, we may construct less biased and more complete individual-specific interaction networks53,54. It is expected that the accuracy in the current network analysis will be significantly increased. Putting these together, structural systems pharmacology could be a powerful tool for precision medicine.
Materials and Methods
Overview: a structural systems pharmacology approach to predictive modeling of drug actions under diverse genetic background
As shown in Fig. 2, our methodology begins at the level of molecular structure. We start by ranking the proteins annotated within the PDB by the similarity of their binding sites to that of AMPKβ and dipeptidyl peptidase-IV (DPP4) – two proteins which have been observed to interact directly with metformin9,16. We considered the top hits from this comparison to be our “putative molecular targets”, and simulated the binding between metformin and each putative molecular target to assess the binding pose and interactions between ligand and target. Next, we moved to the larger and more abstract scale of the protein-protein interaction network. We obtained a dataset which represents genes whose expression is perturbed by metformin treatment. Then, for each putative molecular target, we computationally predicted a tree-shaped path (TSP) which functionally linked that putative molecular target to all genes whose expression is perturbed by metformin. We then analyzed the intermediary nodes of each TSP in terms of their association with biological pathways linked to cancer or AMPK signaling, and identified the most critical nodes of each network. Finally, we experimentally validated the binding between metformin and several putative molecular targets. See Supplemental Methods for further details.
Additional Information
How to cite this article: Hart, T. et al. Toward Repurposing Metformin as a Precision Anti-Cancer Therapy Using Structural Systems Pharmacology. Sci. Rep. 6, 20441; doi: 10.1038/srep20441 (2016).
Supplementary Material
Acknowledgments
We sincerely thank editor and the reviewers for their constructive suggestions. This research was supported by the National Library of Medicine of the National Institute of Health under the award number R01LM011177 (ZZ) and R01LM011986 (LX), and the National Institute on Minority Health and Health Disparities of the National Institutes of Health under award number G12MD007599 (LX).
Footnotes
Author Contributions X.L., H.X. and Z.Z. conceived of the project; X.L. designed the experiments; T.H., S.D. and W.H. performed the experiments and analyzed the results; T.H. and X.L. wrote the manuscript; S.D. and Z.Z. edited the manuscript and provided significant additional literature support; all authors reviewed the manuscript.
References
- Libby G. et al. New Users of Metformin Are at Low Risk of Incident Cancer: A Cohort Study among people with type 2 diabetes. Diabetes Care 32(9), 1620–1625 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakikhani M., Dowling R., Fantus I. G., Sonenberg N. & Pollak M., Metformin Is an AMP Kinase-Dependent Growth Inhibitor for Breast Cancer Cells. Cancer Res. 66(21), 10269–10273 (2006). [DOI] [PubMed] [Google Scholar]
- Leclerc G. M., Leclerc G. J., Kuznetsov J. N., DeSalvo J. & Barredo. J. C., Metformin Induces Apoptosis through AMPK-Dependent Inhibition of UPR Signaling in ALL Lymphoblasts. PLoS One 8(8), e74420 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair V. et al. Mechanism of metformin–dependent inhibition of Mammalian target of rapamycin (mTOR) and Ras activity in pancreatic cancer: role of specificity protein (Sp) transcription factors. J. Biol. Chem. 289(40), 27692–27701 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takiyama Y. et al. Tubular injury in a rat model of type 2 diabetes is prevented by metformin. Diabetes 60(3), 981–992 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrzejewski S., Gravel S.-P., Pollak M. & St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2(12), 10.1186/2049-3002-2-12. (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. et al. Deciphering Signaling Pathway Networks to Understand the Molecular Mechanisms of Metformin Action. PLoS Comput. Biol. 11(6), e1004202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson O. et al. Distinct Perturbation of the Translatome by the Antidiabetic Drug Metformin. Proc. Natl. Acad. Sci. 109(23), 8977–8982 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W. & Xie L. Structural basis of pharmacological effects of metformin. 2012 IEEE International Conference on Bioinformatics and Biomedicine: Computational Bioinformatics Workshop, Philadelphia. Proceedings of the 2012 IEEE International Conference on Bioinformatics and Biomedicine Workshops (BIBMW): IEEE Computer Society. 10.1109/BIBMW.2012.6470337 (2012, October). [DOI] [Google Scholar]
- Xie L. et al. Multiscale Modeling of the Causal Functional Roles of nsSNPs in a Genome-Wide Association Study: Application to Hypoxia. BMC Genomics 14(S3), S9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L., Xie L., Kinnings S. L. & Bourne P. E. Novel Computational Approaches to Polypharmacology as a Means to Define Responses to Individual Drugs. Annu. Rev. Pharmacol. Toxicol. 52, 361–79 (2012). [DOI] [PubMed] [Google Scholar]
- Chang R. L., Xie L., Xie L., Bourne P. E. & Palsson B. Ø. Drug Off-Target Effects Predicted Using Structural Analysis in the Context of a Metabolic Network Model. PLoS Comput. Bio. 6(9), e1000938 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Sui S. J. et al. Raloxifene attenuates Pseudomonas aeruginosa pyocyanin production and virulence. Int. J. Antimicrob. Ag. 40 (3), 246–251. (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant J. D. et al. A multidimensional strategy to detect polypharmacological targets in the absence of structural and sequence homology. PLoS Comput. Biol. 6 (1), e1000648 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahid M. J. & Ruan J. A Steiner Tree-Based Method for Biomarker Discovery and Classification in Breast Cancer Metastasis. BMC Genomics 13 (Suppl 6), (S8) (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bento A. P. et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 42 (Database issue), D1083–1090 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay J. R. et al. Inhibition of dipeptidyl peptidase IV activity by oral metformin in Type 2 diabetes. Diabet. Med. 22(5), 654–657 (2005). [DOI] [PubMed] [Google Scholar]
- Cuthbertson J., Patterson S., O’Harte F. P. & Bell P. M. Investigation of the effect of oral metformin on dipeptidylpeptidase-4 (DPP-4) activity in Type 2 diabetes. Diabet. Med. 26 (6), 649–654 (2009). [DOI] [PubMed] [Google Scholar]
- Edosada C. Y. et al. Selective inhibition of fibroblast activation protein protease based on dipeptide substrate specificity. J. Biol. Chem. 281 (11), 7437–7444 (2006). [DOI] [PubMed] [Google Scholar]
- Forbes S. A. et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids. Res. 43 (Database Issue), D805–D811 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen L. J. et al. STRING 8—a Global View on Proteins and Their Functional Interactions in 630 Organisms. Nucleic Acids Res. 37 (Database Issue), D412–416 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M., Goto S., Kawashima S., Okuno Y. & Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32 (suppl 1), D277–D280 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dongen, S. (University of Utrecht, 2000), p. PhD thesis.
- Enright A. J., Van Dongen S. & Ouzounis C. A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30(7), 1575–1584 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett M. J. et al. Systematic Identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlQuraishi M., Koytiger G., Jenney A., MacBeath G. & Sorger P. K. A multiscale statistical mechanical framework integrates biophysical and genomic data to assemble cancer networks. Nat. Genet. 46 (12), 1363–1371 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Yang P. L. & Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 9(1), 28–39 (2009). [DOI] [PubMed] [Google Scholar]
- Wu W. et al. Microarray Analysis Reveals Glucocorticoid-regulated Survival Genes That Are Associated with Inhibition of Apoptosis in Breast Epithelial Cells. Cancer Res. 64(5), 1757–64 (2004). [DOI] [PubMed] [Google Scholar]
- D’Antona A. et al. SI113, a Specific Inhibitor of the Sgk1 Kinase Activity That Counteracts Cancer Cell Proliferation. Cellular Physiology and Biochemistry 35(5), 2006–2018 (2015). [DOI] [PubMed] [Google Scholar]
- O’Keeffe B., Cilia S., Maiyar A., Vaysberg M. & Firestone G. The serum-and glucocorticoid-induced protein kinase-1 (Sgk-1) mitochondria connection: Identification of the IF-1 inhibitor of the F1F0-ATPase as a mitochondria-specific binding target and the stress-induced mitochondrial localization of endogenous Sgk-1. Biochimie 95(6), 1258–1265 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N. et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511(7511), 616–620 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuy B. et al. Multikinase Inhibitor Sorafenib Exerts Cytocidal Efficacy against Non-Hodgkin Lymphomas Associated with Inhibition of MAPK14 and AKT Phosphorylation. Brit. J. Haematol. 152(4), 401–412 (2011). [DOI] [PubMed] [Google Scholar]
- Chaudhary S. C., Kurundkar D., Elmets C. A., Kopelovich L. & Athar M. Metformin, an Antidiabetic Agent Reduces Growth of Cutaneous Squamous Cell Carcinoma by Targeting MTOR Signaling Pathway. Photochem. Photobiol. 88(5), 1149–1156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J. C. et al. Inhibition of P38 MAPK-dependent MutS Homologue-2 (MSH2) Expression by Metformin Enhances Gefitinib-induced Cytotoxicity in Human Squamous Lung Cancer Cells. Lung Cancer 82(3), 397–406 (2013). [DOI] [PubMed] [Google Scholar]
- Schnelzer A. et al. Rac1 in Human Breast Cancer: Overexpression, Mutation Analysis, and Characterization of a New Isoform, Rac1b. Oncogene 19(26), 3013–3020 (2000). [DOI] [PubMed] [Google Scholar]
- Yan Q. et al. Deacetylated GM3 Promotes UPAR-associated Membrane Molecular Complex to Activate P38 MAPK in Metastatic Melanoma. Mol. Cancer 11(6), 665–675 (2013). [DOI] [PubMed] [Google Scholar]
- Allegra M. et al. Semaphorin-7a Reverses the ERF-Induced Inhibition of EMT in Ras-Dependent Mouse Mammary Epithelial Cells. Mol. Biol. Cell 23(19), 3873–3881 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds L. F. et al. Vav1 transduces T cell receptor signals to the activation of the Ras/ERK pathway via LAT, Sos, and RasGRP1. J. Biol. Chem. 279(18), 18239–18246. (2004). [DOI] [PubMed] [Google Scholar]
- Lim K. H., Ancrile B. B., Kashatus D. F. & Counter C. M. Tumour maintenance is mediated by eNOS. Nature 452(7187), 646–649. (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L., Evangelidis T., Xie L. & Bourne P. Drug discovery using chemical systems biology: weak inhibition of multiple kinases may contribute to the anti-cancer effect of Nelfinavir. PLoS Comput. Biol. 7(4), e1002037 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouchi N. et al. Adiponectin Stimulates Angiogenesis by Promoting Cross-Talk Between AMP-Activated Protein Kinase and Akt Signaling in Endothelial Cells. J. Biol. Chem. 279(2), 1304–1309 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. L. et al. Interaction of the P53-Regulated Protein Gadd45 with Proliferating Cell Nuclear Antigen. Science 266 5189), 1376–1380 (1994). [DOI] [PubMed] [Google Scholar]
- Chen H. Y. et al. A Five-Gene Signature and Clinical Outcome in Non-Small-Cell Lung Cancer. N. Engl. J. Med. 356(1), 11–20 (2007). [DOI] [PubMed] [Google Scholar]
- Huang S. C., Ho C. T., Lin-Shiau S. Y. & Lin J. K. Carnosol Inhibits the Invasion of B16/F10 Mouse Melanoma Cells by Suppressing Metalloproteinase-9 through down-Regulating Nuclear Factor-Kappa B and c-Jun. Biochem. Pharmacol. 69(2), 221–232 (2005). [DOI] [PubMed] [Google Scholar]
- Wirtenberger M. et al. Associations of genetic variants in the estrogen receptor coactivators PPARGC1A, PPARGC1B and EP300 with familial breast cancer. Carcinogenesis 27(11), 2201–2208 (2006). [DOI] [PubMed] [Google Scholar]
- McGuirk S. et al. PGC-1α supports glutamine metabolism in breast cancer. Cancer Metab. 1(22), 22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R. & Snyder M. Promise of personalized omics to precision medicine. Wiley Interdiscip. Rev. Syst. Biol. Med. 5(1), 73–82 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N. et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer Discov. 4(5), 546–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L. et al. Towards Structural Systems Pharmacology to Study Complex Diseases and Personalized Medicine. PLoS Comput. Biol. 10(5), e1003554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolland T. et al. A proteome-scale map of the human interactome network. Cell 159(5), 1212–26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidari N. et al. Genome-wide map of regulatory interactions in the human genome. Genome Res., 24(12), 1095–1017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle D., Govindaraju D. R. & Omholt S. Phenomics: the next challenge. Nat. Rev. Genet. 11, 855–66 (2010). [DOI] [PubMed] [Google Scholar]
- Greene C. S. et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 47(6), 569–576 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.