

The dynamic association of specific transcripts with polysomes participates in the regulation of seed germination by dormancy.
Abstract
Dormancy is a complex evolutionary trait that temporally prevents seed germination, thus allowing seedling growth at a favorable season. High-throughput analyses of transcriptomes have led to significant progress in understanding the molecular regulation of this process, but the role of posttranscriptional mechanisms has received little attention. In this work, we have studied the dynamics of messenger RNA association with polysomes and compared the transcriptome with the translatome in dormant and nondormant seeds of Arabidopsis (Arabidopsis thaliana) during their imbibition at 25°C in darkness, a temperature preventing germination of dormant seeds only. DNA microarray analysis revealed that 4,670 and 7,028 transcripts were differentially abundant in dormant and nondormant seeds in the transcriptome and the translatome, respectively. We show that there is no correlation between transcriptome and translatome and that germination regulation is also largely translational, implying a selective and dynamic recruitment of messenger RNAs to polysomes in both dormant and nondormant seeds. The study of 5′ untranslated region features revealed that GC content and the number of upstream open reading frames could play a role in selective translation occurring during germination. Gene Ontology clustering showed that the functions of polysome-associated transcripts differed between dormant and nondormant seeds and revealed actors in seed dormancy and germination. In conclusion, our results demonstrate the essential role of selective polysome loading in this biological process.
The regulation of gene expression in higher plants results from transcription but also from posttranscriptional processes, including the sequestration of mRNAs into cytosolic mRNA ribonucleoproteins, mRNA decay mechanisms, and the regulation of translation. Initiation is the key regulatory step of translation, which also includes elongation and termination. Initiation starts when a small (40S) ribosomal unit is loaded on the 5′ cap of an mRNA and forms a 43S preinitiation complex with a eukaryotic Initiation Factor2α-GTP-tRNAmet ternary complex that scans the RNA template down in a 5′-to-3′ direction until an AUG codon (Browning, 1996). Then, the coupling of the 40S subunit with a 60S ribosomal subunit gives an 80S ribosome, and the elongation phase starts (Bailey-Serres, 1999; von Arnim et al., 2014). It is worth noting that mRNAs recruit not one but multiple ribosomes to form polysomal complexes (polysomes); therefore, their number per transcript reflects the efficiency of translation and the rate of elongation (Kawaguchi et al., 2004). Initiation of translation requires the assembly of a circular mRNA protein complex and is tightly regulated by various eukaryotic Initiation Factors4 and by their phosphorylation, by RNA helicases and RNA-binding proteins, and by the availability in ATP and GTP, since translation is an energetically costly cellular process (Bailey-Serres, 1999). In addition, the characteristics of the 5′ and 3′ untranslated regions (5′UTR and 3′UTR, respectively) of mRNA also play key regulatory roles in the initiation process (Wilkie et al., 2003). For example, the length and nucleotide composition of the 5′UTR modulate the entry of the 43S preinitiation complex, probably by modifying RNA secondary structure (Kawaguchi and Bailey-Serres, 2005). The presence of upstream open reading frames (uORFs) in the 5′UTR also generally reduces translation in a length-dependent manner (von Arnim et al., 2014).
The regulation of translation in plants is mainly documented in the context of response to stresses. Indeed, translation, which requires high-energy biochemical processes, is often impaired in unfavorable environmental conditions such as hypoxia (Branco-Price et al., 2008), the presence of heavy metals (Sormani et al., 2011), or excess heat (Matsuura et al., 2010), but response to stress also involves the differential translation of specific mRNAs (Juntawong et al., 2014; von Arnim et al., 2014). The role of translational control of plant development is documented in Arabidopsis (Arabidopsis thaliana) in the context of pollen tube growth (Lin et al., 2014) and light signaling (Piques et al., 2009; Juntawong and Bailey-Serres, 2012; Liu et al., 2013) and in sunflower (Helianthus annuus) for seed germination (Layat et al., 2014). In that study, it was demonstrated that the translatome differs between germinating and nongerminating sunflower embryos, thus highlighting a timely regulated and selective recruitment of mRNAs to polysomes. This pioneering work opened fields of investigation for the understanding of germination, since until now the molecular regulators of seed germination were identified using large-scale data sets of total transcriptomes (for review, see Holdsworth et al., 2008). It is indeed widely admitted that seed germination and dormancy are regulated transcriptionally and are under the control of a balance between hormonal pathways associated with negative (abscisic acid [ABA]) and positive (ethylene and GAs) regulators of germination. Recently, system approaches have led to the design of networks of transcriptional interactions that help to define seed germination regulators (Bassel et al., 2011; Verdier et al., 2013). However, the sequence of cellular events underlying germination (i.e. occurring before radicle emergence; Bewley, 1997) and their regulation by dormancy are far from resolved, in particular when considering the putative roles of posttranscriptional mechanisms. Recent studies, such as those of Bazin et al. (2011), Layat et al. (2014), and Galland et al. (2014), led to a reconsideration of the role of mRNA metabolism in seed germination, since those authors proposed that germination would not only rely on the transcription of specific subsets of mRNAs but also on translational activity or on RNA decay.
Therefore, we propose that the modulation of seed germination by dormancy would be related to the association of specific mRNAs with polysomes, thus initiating their translation during seed imbibition, as already shown in sunflower by Layat et al. (2014). However, that work did not consider the relative parts of transcriptional versus translational regulation of germination and did not provide elements to explain the molecular bases of selective translation. Here, we have studied the changes in the transcriptome and in the translatome of dormant and nondormant Arabidopsis seeds during their imbibition at 25°C in darkness, conditions preventing the germination of dormant seeds only (Leymarie et al., 2012). We have isolated and identified the mRNAs in polysomal complexes (i.e. the translatome) and compared them with the transcripts identified at the same time points. Using bioinformatics approaches, we characterized the main features of the 5′UTR, 3′UTR, and uORF of genes that were likely to play key regulatory roles in germination. The results from this study provide insights for the understanding of seed germination regulation.
RESULTS
Polysome Profiling of Dormant and Nondormant Arabidopsis Seeds
At harvest, Arabidopsis seeds were dormant, since they only germinated to approximately 25% at 25°C in darkness (Fig. 1A). After 3 weeks of storage at 56% relative humidity and 20°C, dormant seeds germinated to 75%, and after 4 weeks of after-ripening under these conditions, seeds became nondormant, since they fully germinated at 25°C (Fig. 1A). In nondormant seeds, germination at 25°C in darkness did not begin before 2 d and was completed after 7 d (Fig. 1A). At 15°C, both dormant and nondormant seeds fully germinated after 4 d (data not shown; Leymarie et al., 2012). In this study, we chose data points at durations lower than 24 h at 25°C because they corresponded to time points during germination sensu stricto (i.e. before radicle protrusion) and allowed the comparison between dormant and nondormant seeds in a similar physiological state. Suc gradient separation (Mustroph et al., 2009) was used to purify the polysomal fraction of RNA at regular time points during seed imbibition at 25°C. The absorbance profiles of ribosome complexes showed that dry seeds contained a large amount of 80S monosomes and few or no polysomes (Fig. 1B). In dormant seeds, early imbibition, from 3 to 6 h, was first associated with a decrease in the amount of 80S monosomes but was followed by an increase of this peak after 16 and 24 h (Fig. 1B). After 24 h of imbibition, the amount of 80S was significantly higher in nondormant than in dormant seeds (Fig. 1C). However, the amount of polysomal mRNAs, which increased during seed imbibition, was similar in dormant and nondormant seeds (Fig. 1C). The amounts of polysomal components, estimated by measuring the heights of peaks relative to the baseline, confirmed the visual evaluation of absorbance profiles (Supplemental Table S1). We determined whether the 80S monosomes that accumulated in dry seeds and after 24 h of imbibition were associated with mRNA. Eukaryotic ribosomes that are not associated with mRNA dissociate into subunits in solutions of high ionic strength (0.8 m KCl; Martin, 1973), while free ribosomes are dissociated in these solutions. In dry dormant seeds, or seeds imbibed for 24 h, the height of the 80S peak was similar with KCl at 0.2 or 0.8 m (Supplemental Fig. S1, A and B), which showed that monosomes were associated with RNA. In contrast, in nondormant seeds imbibed for 24 h, treatment with 0.8 m KCl reduced the height of the 80S peak (Supplemental Fig. S1C), thus suggesting that a part of monosomes were RNA free. The increase of the 80S peak in nondormant seeds, therefore, did not correspond to active ribosomes bound to mRNA.
Figure 1.

Seed germination, polysome profiling, and changes in abundance of mRNAs in the transcriptome and translatome. A, Germination of Arabidopsis seeds on water at 25°C in darkness during 8 d: 1, freshly harvested seeds (dormant seeds); 2, dormant seeds after-ripened for 3 weeks at 20°C at 56% relative humidity; 3, dormant seeds after-ripened for 4 weeks at 20°C at 56% relative humidity (nondormant seeds). Means ± sd of triplicate experiments are shown. B and C, Suc density gradient profiles of RNA (A254) from Arabidopsis seeds. Peaks 1 and 2 correspond to 60S ribosomal subunits and to 80S monosomes, respectively. B, Absorbance profiles of mRNA obtained from dormant dry seeds and during their imbibition (3, 6, 16, and 24 h) at 25°C. C, Comparison of absorbance profiles obtained with dormant (D; black solid lines) and nondormant (ND; red dotted lines) seeds after 16 and 24 h of imbibition at 25°C. D and E, Analysis of microarray data. Venn diagrams show the distribution of mRNAs presenting abundance variations (after BH correction with P < 0.05 and log2 ratio above 0.5 or under −0.5) in total RNA (T; D) and in polysomal RNA (P; E) after 16 and 24 h of imbibition at 25°C. Numbers in parentheses correspond to the sum of all transcripts in a category.
The purified polysomal mRNAs and total mRNAs were characterized in dormant and nondormant seeds after 16 and 24 h of imbibition at 25°C using CATMA microarrays. These time points were chosen because they allowed the efficient isolation of polysomal fractions and because they corresponded to 50% and 75%, respectively, of the amount of time required for the initiation of germination of nondormant seeds (Fig. 1). MA (distribution of the log2 intensity ratio [M] plotted against the average intensity [A]) plots representing the distribution of expression ratios of all transcripts at 16 and 24 h of imbibition in either total or polysomal fractions showed that major variations were present between log2 ratio values ranging from −2 and +2 (Supplemental Fig. S2). Gene expression was considered to be different after Benjamini and Hochberg (BH) correction (P < 0.05) and for a log2 ratio above 0.5 or under −0.5, at least for one comparison. Among the 16,616 gene-specific sequence tags (GSTs) displaying signal levels above the limit of detection in at least one of the samples (representing 67% of GSTs present on the array), 4,670 and 7,028 transcripts in total or polysomal mRNA, respectively, were significantly different between dormant and nondormant seeds after both 16 and 24 h of imbibition (Fig. 1, D and E; Supplemental Data Set S1). Analysis of the transcriptome showed that 1,708 transcripts were more abundant in dormant seeds and 2,962 were more abundant in nondormant ones, when considering both durations of imbibition (Fig. 1D; Supplemental Data Set S2). Among these transcripts, 535 and 1,065 displayed the same change in abundance at 16 and 24 h of imbibition in dormant and nondormant seeds, respectively (Fig. 1D). In the translatome, 3,872 transcripts were more abundant in dormant seeds and 3,156 were more abundant in nondormant seeds at 16 and 24 h of imbibition (Fig. 1E; Supplemental Data Set S3). However, one-third of the polysome-associated transcripts (1,333) were similar at 16 and 24 h of imbibition in dormant seeds, while only 139 (among 3,156 [i.e. around 4%]) were common between 16 and 24 h of imbibition in nondormant seeds (Fig. 1E).
Regulation of the Transcriptome and Translatome by Dormancy
This work was highly appropriate to determine whether the molecular regulation of seed germination is mainly transcriptional or translational. Therefore, we compared the abundance of individual transcripts in the transcriptome and in the translatome by plotting the polysomal RNA ratios (nondormant to dormant) against total RNA ratios (nondormant to dormant) for all GSTs (Fig. 2, A and B). In this representation, the diagonal line represents the behavior of transcripts that are regulated in the same way in the translatome and in the transcriptome. There was no correlation between the transcriptome versus the translatome after 16 h (r2 = 0.045; Fig. 2A) or even 24 h (r2 = 0.019; Fig. 2B) of imbibition. We constructed Venn diagrams to assess the dynamics of polysome loading with regard to dormancy release (Fig. 2, C and D). At both points of imbibition, we compared the differentially abundant transcripts in the transcriptome and translatome for dormant and nondormant seeds. At 16 h of imbibition, 529 transcripts (approximately 26%) more abundant in the transcriptome of dormant seeds were found in the translatome of nondormant seeds, suggesting that after-ripening allowed this subset of mRNAs to become associated with polysomes (Fig. 2C). Among the 2,022 transcripts specifically more abundant in the transcriptome of nondormant seeds, 699 (approximately 34%) were associated with polysomes in nondormant seeds, whereas 496 (approximately 25%) were found in the translatome of dormant seeds (Fig. 2C). Conversely, only 317 transcripts (approximately 28%) among 1,113 found in the transcriptome of dormant seeds were addressed to polysomes in these seeds (Fig. 2C). At 24 h of imbibition, 450 (approximately 39%) transcripts that were more abundant in the transcriptome of dormant seeds became addressed to polysomes when seeds became nondormant (Fig. 2D). Fifteen percent (305) of the transcripts identified as being specific to the transcriptome of nondormant seeds were in the translatome of dormant seeds, and 40% (415) were in the translatome of nondormant seeds (Fig. 2D). Finally, only 15% (175) of the transcripts from the dormant transcriptome were addressed to polysomes in dormant seeds (Fig. 2D). The transcriptome and translatome of dormant and nondormant seeds were compared using SeedNet, a model designed from transcriptomic data of seed samples in relation to germination and dormancy. SeedNet topology allows the identification of three regions: region 1, specific to dormancy, and regions 2 and 3, associated with germination (Bassel et al., 2011). Figure 2, E and F, show localization within the SeedNet model of transcripts identified in the transcriptome and translatome of dormant and nondormant seeds, combining data of both durations of imbibition. For the transcriptome, the transcripts more abundant in dormant seeds were mainly found in region 1, while those more abundant in nondormant seeds were plotted in regions 2 and 3 (Fig. 2E). Transcripts associated with polysomes in nondormant seeds were mainly localized in region 3, but transcripts associated with polysomes in dormant seeds were localized randomly in regions 1, 2, and 3 (Fig. 2F).
Figure 2.

Comparison of changes in mRNA abundance between the transcriptome and translatome. A and B, Scatterplots show the change (abundance in nondormant seeds/abundance in dormant seeds [ND/D]) in total mRNA abundance (x axis; transcriptome) versus change in polysomal mRNA (y axis; translatome) seeds after 16 h (A) or 24 h (B) of imbibition at 25°C. All transcripts present on the microarray are shown as dots on the graphs, which represent their ratio of log2 (abundance in nondormant seeds) to log2 (abundance in dormant seeds). The transcripts above or below the solid diagonal line are positively or negatively regulated at the level of translation, respectively. C and D, Venn diagrams show the number of transcripts displaying significant changes in abundance (after BH correction with P < 0.05 and log2 ratio above 0.5 or under −0.5) in transcriptome (T) and translatome (P) at 16 h (C) or 24 h (D) of imbibition at 25°C. E and F, Localization of transcripts associated with the transcriptome and translatome in the SeedNet transcript coexpression network (www.vseed.nottingham.ac.uk). The regions outlined in orange correspond to clusters associated with dormancy (region 1) or germination (regions 2 and 3; Bassel et al., 2011). The red dots represent the transcripts identified in this study as more abundant in dormant seeds, and the blue dots represent the transcripts more abundant in nondormant seeds in the transcriptome (E) or translatome (F) after 16 and 24 h of imbibition at 25°C.
Regulation of Polysome Loading by mRNA Sequence Features and by Translational Efficiency
The polysomal ratio (the proportion of individual mRNA species in polysomes; Fig. 3A) was determined for transcripts detected in polysomal fractions. While the distribution of polysomal ratios was similar for both durations of imbibition, it appeared significantly (P < 0.0001) different between dormant and nondormant seeds (Fig. 3A). This distribution shows that the transcripts that were more abundant in dormant seeds presented a lower polysomal ratio (i.e. a higher number of transcripts with a polysomal ratio lower than 80%) than the transcripts more abundant in nondormant seeds (a higher number of transcripts with a polysomal ratio of 92%), whatever the duration of imbibition (Fig. 3A). The major characteristics of 5′UTR and 3′UTR (length and GC content) of differentially abundant transcripts in the translatome with a described 5′UTR described in The Arabidopsis Information Resource (TAIR; Supplemental Table S2) have been analyzed in relation to their polysomal ratio. At both durations of imbibition, higher polysome ratios were observed when 5′UTR length was between 50 and 80 nucleotides, and polysomal ratio was lower for mRNAs with short (less than 25 nucleotides) or long (greater than 100 nucleotides) 5′UTR lengths, whether seeds were dormant or not (Fig. 3B). However, the relationship between polysomal ratio and 5′UTR length was not affected by dormancy, since the slopes of the curves shown in Figure 3B were similar. The relationship between polysome ratio and GC content in the 5′UTR revealed differences between dormant and nondormant seeds (Fig. 3C). Low GC contents (20%–30%) in the 5′UTR were associated with high polysomal ratio, and higher GC content significantly decreased ribosome loading (Fig. 3C). The effect of GC content on polysomal ratio was considerably more pronounced in dormant than in nondormant seeds, since polysomal ratio decreased to 25% in dormant seeds but only to 55% in nondormant ones when GC content increased to 60% (Fig. 3C). The effect of 3′UTR features on polysomal ratio was also investigated but did not indicate any relationship between length or GC content and the ability of seeds to germinate (Supplemental Fig. S3).
Figure 3.

Regulation of polysome loading by 5′UTR. For all graphs, black symbols and black lines correspond to nondormant (ND) seeds, while white symbols and gray lines correspond to dormant seeds (D). Triangles and circles correspond to 16 and 24 h of imbibition, respectively. A, Distribution of the transcripts according to their polysomal ratio (P/T) in dormant seeds (n = 2,582 at 16 h and n = 2,581 at 24 h of imbibition) and nondormant seeds (n = 1,629 at 16 h and n = 1,601 at 24 h of imbibition). Distributions were statistically different between dormant and nondormant seeds at both durations of imbibition, as determined by Student’s t test (P < 0.0001). B, Relationship between the length of the 5′UTR and the polysomal ratio calculated for transcripts that were found in polysomal fractions of dormant and nondormant seeds. Transcripts were grouped based on 45 nucleotide (nt) classes. C, Effect of GC content in the 5′UTR on polysomal ratio. mRNAs were grouped based on 5% GC content windows, and the polysomal ratio was calculated for each subset. D, Consensus sequences of motifs overrepresented in the 5′UTR for the first 100 mRNAs identified as differentially abundant in the polysomal fraction of dormant and nondormant seeds at 16 and 24 h of imbibition. The motif enrichment (ME) represents the frequency of the motifs overrepresented in the 100 first differentially abundant transcripts relative to their frequency in the 5′UTR from the whole genome of Arabidopsis. The E value calculated by MEME reveals the statistical significance of the motif. E, Relationship between the number of uORFs per gene and the polysomal ratio in the polysomal fraction of dormant and nondormant seeds at 16 and 24 h of imbibition. F, Relationship between the length of uORFs and the polysomal ratio in the polysomal fraction of dormant and nondormant seeds at 16 and 24 h of imbibition.
Overrepresented motifs in the 5′UTR of polysomal transcripts more abundant (analysis of 100 transcripts) in dormant and nondormant seeds were determined with the unsupervised Multiple Expectation Maximization for Motif Elicitation (MEME) algorithm (Bailey et al., 2009; Fig. 3D). We identified U-rich motifs in the 5′UTR of polysomal RNAs from dormant seeds and the same consensus motif, AGAGAGAAGA, at both durations of imbibition in polysomal transcripts from nondormant seeds (Fig. 3D). In all cases, the motif enrichment (which corresponds to the factor of enrichment of this motif in comparison with its occurrence in the whole Arabidopsis genome) and the E value (which refers to its statistical significance) demonstrated the high specificity of the motifs (Fig. 3D). Putative uORFs were searched in the 5′UTRs of all the transcripts associated with polysomes (listed in Supplemental Data Set S4). More than 75% of the transcripts did not present any uORF (Table I), but transcripts from nondormant seeds displayed few uORFs (one to three), whereas transcripts with more than three uORFs were more frequent in dormant seeds (Table I). Figure 3E shows that the polysomal ratio decreased with the increasing number of uORFs per transcript, but this effect was more pronounced in dormant seeds. The longer the length of the uORF, the lower the polysomal ratio (Fig. 3F); however, the inhibition of polysome loading by the length of uORF appeared to be stronger for transcripts more abundant in nondormant seeds (higher slope values on the graph).
Table I. Number of uORFs per transcript found in the translatome of dormant and nondormant seeds at 16 and 24 h of imbibition at 25°C.
Letters a and b indicate homogenous groups in a corresponding class (Fisher’s test, P = 0.05).
| No. of uORFs per Transcript | Percentage of Transcripts Having the Corresponding No. of uORFs in the Translatome of |
|||
|---|---|---|---|---|
| Dormant Seeds Imbibed for |
Nondormant Seeds Imbibed for |
|||
| 16 h | 24 h | 16 h | 24 h | |
| 0 | 77.90 a | 77.63 a | 76.23 a | 76.10 a |
| 1 | 1.8 a | 1.96 a | 7.55 b | 7.41 b |
| 2 | 2.66 a | 2.75 a | 6.14 b | 6.16 b |
| 3 | 3.42 a | 3.33 a | 4.63 b | 4.70 b |
| 4 | 3.98 a | 3.83 a | 2.42 b | 2.50 b |
| 5 | 4.59 a | 4.41 a | 1.82 b | 1.88 b |
| 6 | 6.07 a | 6.09 a | 1.21 b | 1.25 b |
In addition, we compared the translation efficiency in dormant and nondormant seeds. We adapted the method from David et al. (2012), where the amount of puromycin-labeled proteins detected on the gel is directly related to the translational activity. Puromycylation is based on the incorporation of puromycin into the C terminus of elongation nascent chains, and the ribosome-bound nascent chains are detected with a puromycin-specific antibody, as shown in Figure 4. Incubation of seeds with sodium arsenite (an irreversible inhibitor of translation initiation [Kedersha and Anderson, 2002] and puromycylation [David et al., 2012]) and absence of puromycin treatment suppressed the signal, thus demonstrating the efficiency of this technique for assessing translation in seeds (Fig. 4). Densitometric quantification of the labeled bands showed that translation remained unchanged between 16 and 24 h of imbibition and was similar in dormant and in nondormant seeds (Fig. 4). Nevertheless, a longer duration of imbibition (i.e. 72 h) resulted in a higher translational activity in nondormant seeds, but at this time point germination had occurred in nondormant seeds (Fig. 4).
Figure 4.

Quantification of translational activity by incorporation of puromycin and western-blot detection (anti-puromycin antibodies) after 16, 24, and 72 h of imbibition at 25°C (with 4 h with puromycin) with dormant (D) and nondormant (ND) seeds. Dormant seeds were incubated for 4 h with 500 µm sodium arsenite and puromycin (SA) or without puromycin (control [C]). Means ± sd of three biological replicates are shown.
Identification of Transcripts in the Transcriptome and the Translatome
The microarrays data were validated by performing quantitative real-time (qRT)-PCR with 10 selected transcripts. This showed a high correlation between qRT-PCR and microarray transcript abundance, with r2 coefficients ranging from 0.71 to 0.88 (Supplemental Fig. S4).
A full list of differentially abundant transcripts in the transcriptome and translatome is given in Supplemental Data Sets S2 and S3; however, we focused our functional analysis of gene identity on the transcripts that were identified in the translatome only, since we previously demonstrated the discrepancy between the transcriptome and translatome (Fig. 2). Putative functions were assigned to transcripts from Supplemental Data Set S3 using the Gene Ontology (GO) TAIR categorization and were visualized as functional clusters after redundancy reduction via REVIGO (Fig. 5). In this representation, colors indicate the user-provided P values (see legend) and the sizes of the circles are relative to the frequency of GO terms in the underlying GO database. Polysomal RNAs associated with germination fell essentially into the categories cell wall, hormone metabolism, and redox pathway, whatever the duration of imbibition (Fig. 5, B and D). Furthermore, the categories response to stress and hormone metabolism pathways were associated with polysomal RNAs related to dormancy (Fig. 5, A and C). The more relevant GO categories are discussed below, as are the sequential modifications of the transcripts belonging to the different GO categories in the translatome after 16 and 24 h of imbibition (Supplemental Table S3). Finally, the abundance of some transcripts involved in the translational regulation of dormancy and germination was analyzed by qRT-PCR in dormant and nondormant seeds after 16 and 24 h of imbibition (Fig. 6). Figure 6 shows that the abundance of these transcripts was generally much higher in the translatome than in the transcriptome in both dormant (Fig. 6A) and nondormant (Fig. 6B) seeds.
Figure 5.

GO classification of mRNAs specifically found in the translatome of dormant (D) and nondormant (ND) seeds after 16 h (A and B) and 24 h (C and D) of imbibition at 25°C. TAIR accession numbers were used to perform the classification according to GO categories with the Classification SuperViewer Tool in the Bio-Analytic Resource for Plant Biology, and plots were obtained using REVIGO (http://revigo.irb.hr; Supek et al., 2011), which allows a two-dimensional space representation according to the semantic similarity of GO terms (semantically similar GO terms remain close together in the plot). The size of the circles indicates the frequency of the GO term in the GO database (more general terms are represented by larger circles), and the color of the circles indicates the P value (scale at right; blue and green circles are GO terms with more significant P values than orange and red circles).
Figure 6.

Pattern of expression in the transcriptome and translatome of key transcripts more abundant in dormant seeds (log2 ratio ND:D < 0; A) and more abundant in nondormant seeds (log2 ratio ND:D > 0; B) after 16 and 24 h of imbibition. Expression data were obtained by qRT-PCR on three biological replicates. RGL2, At3g03450; ABI2, At5g57050; PIF6-PIL2, At3g62090; MPK4, At4g01370; HSP70, At5g02500; ABA1, At5g67030; LEA, At2g21490; SLP2, At4g34980; LTP5, At3g51600; EXPA4, At2g39700; EXPA1, At1g69530; AC1, At2g19590; GASA6, At1g74670. Green corresponds to changes measured in total RNA (T; transcriptome), and blue corresponds to changes in polysomal RNA (P; translatome) at 16 h (solid bars) and 24 h (striped bars) of imbibition.
DISCUSSION
The control of translational activity in plants has emerged these last years to increasing experimental evidence demonstrating low coupling between mRNAs and proteins (Bailey-Serres et al., 2009; von Arnim et al., 2014). Translation is mainly regulated at its initiation step, when 60S and 40S ribosomal subunits join together at the initiating AUG codon of an mRNA, before the formation of a polysome on the translated mRNA due to the recruitment of additional ribosomes (Bailey-Serres et al., 2009). The role of this process in seed germination has been documented recently by Layat et al. (2014), who were the first to show that translatomes of dormant and nondormant sunflower seeds differed markedly. Here, we demonstrate the role of the translatome in the germination of Arabidopsis seeds and its regulation by dormancy status.
In Arabidopsis, the Columbia ecotype is dormant at harvest, since it germinates poorly at 25°C in darkness (Fig. 1A; Leymarie et al., 2012). Dormancy is alleviated by germinating seeds under continuous light, stratification at 5°C, or 4 to 5 weeks of after-ripening (Leymarie et al., 2012). Polysomal profiles of dry dormant and nondormant seeds showed that monosomes were abundant at the end of seed maturation but that RNA-associated polysomes were scarce or absent (Fig. 1B), in agreement with results obtained with endosperm of castor bean (Ricinus communis) seeds (Marrè et al., 1965). Increase of the salt concentration in Suc gradients revealed that monosomes in dry seeds were already associated with mRNA (Supplemental Fig. S1A). They probably contained the so-called long-lived mRNAs that are stored at the end of seed development and thought to be translated after the onset of imbibition (Kimura and Nambara, 2010). This would mean that translation in the early steps of seed imbibition is determined during late seed development, and it would be worth identifying the mRNA associated with these monosomes. Seed imbibition was associated with a transient decrease of monosomes followed by their progressive recovery at 24 h of imbibition (Fig. 1B) and with an increase of polysomal mRNAs, thus revealing translational activity. This is in accordance with polysome profiles obtained from seeds of other plant species, such as castor bean (Marrè et al., 1965), wheat (Triticum aestivum; Spiegel et al., 1975), and sunflower (Layat et al., 2014). The similarity of polysome profiles in dormant and nondormant seeds suggests that the translational activity was similar in both kinds of seeds, even if nondormant seeds contained more free monosomes at 24 h of imbibition (Fig. 1C). These were not all bound to mRNA (inactive ribosome; Supplemental Fig. S1C), but they may accumulate to sustain the increased protein synthesis that is associated with the subsequent radicle protrusion (Beltrán-Peña et al., 1995). In yeast (Saccharomyces cerevisiae), it has been proposed that inactive ribosomes that are accumulated during stress dissociate upon stress release, promoting recovery (van den Elzen et al., 2014). Moreover, this mechanism was also identified in nonstressed cells and would be a new level of translation regulation (van den Elzen et al., 2014). We suggest that the storage of ribosomes in an unproductive state in nondormant seeds might serve as a reservoir of active ribosomes for sustaining the active translation that occurs at the onset of radicle protrusion.
Microarray analysis allowed the identification of transcripts in the transcriptome and translatome of dormant and nondormant seeds at the two time points of imbibition. After BH correction, 4,670 and 7,028 transcripts were found to be differentially abundant in total and polysomal RNA, respectively, for dormant and nondormant seeds after 16 and 24 h of imbibition (Fig. 1, D and E). By comparison, a time-course analysis of transcriptomic changes during Arabidopsis seed germination also revealed that approximately 8,000 transcripts were similarly up- or down-regulated at each time point studied (i.e. 1, 6, 12, and 24 h of imbibition; Narsai et al., 2011). In sunflower, approximately 7,300 transcripts were continuously associated with polysomes from 3 to 24 h in both dormant and nondormant embryos (Layat et al., 2014), a number that is very similar to the one determined here. It is also worth noting that germination was associated with a marked change in the nature of the transcripts associated with polysomes, since they differed noticeably between 16 and 24 h (Fig. 1E). This clearly demonstrates that completion of the germination program relies on dynamic and qualitative changes in translation. Recent articles (Galland et al., 2014; Layat et al., 2014) also revealed the sequential regulation of protein synthesis during seed germination. In addition, the microarray data demonstrate that dormancy was associated with an active association of transcripts with polysomes, but contrary to nondormant seeds, many polysome-associated transcripts were similar at 16 and 24 h of imbibition (Fig. 1E). This is interesting because it demonstrates that dormancy cannot be explained by the absence of translation of some transcripts but by an active association of selected transcripts with polysomes, as already pointed out by Layat et al. (2014).
The divergence between the number of transcripts associated with dormancy and germination in the transcriptome (4,670) and the translatome (7,028) clearly raises the question of transcriptional versus translational regulation of germination. Figure 2, A and B, shows that there was little or no correlation between the translatome and transcriptome at both durations of seed imbibition. The discrepancy between transcript abundance and the translation of specific mRNAs was also shown in yeast (Lackneret al., 2007), mouse fibroblasts (Schwanhäusser et al., 2011), transformed cancerous cells (Spence et al., 2006), primary T cells (Grolleau et al., 2002), and Arabidopsis (Sormani et al., 2011; Juntawong and Bailey-Serres, 2012). This suggests that RNA in the transcriptome was not necessarily recruited to polysomes and that dormancy can regulate this process. The Venn diagrams (Fig. 2, C and D) are of highest interest, since they bring unexpected conclusions about the dynamics of transcript association with polysomes. First, they demonstrate that the regulation of germination is both transcriptional and translational and that the relative part of each process evolves during germination. At 16 h of imbibition, 25% (418 among 1,646) of the polysomal transcripts more abundant in nondormant seeds were not found as up-regulated in the transcriptome of either dormant or nondormant seeds, suggesting a translational regulation. This percentage increased to 47% (784 among 1,649) at 24 h, which indicates that the regulation at the translation level is more important in the latter phases of germination. Second, this analysis demonstrates that numerous transcripts found in the transcriptome of either dormant or nondormant seeds are not addressed to polysomes or, even more surprisingly, can be found in the polysomes of their counterparts. For example, many transcripts (529 and 450 at 16 and 24 h, respectively) of dormant seeds were also found in the polysomes of nondormant seeds (Fig. 2C). Their association with polysomes occurred after dormancy alleviation, which suggests that after-ripening would modify the ability of preexisting transcripts to become polysomal. Similarly, 496 and 305 transcripts found as being specific to nondormant seeds at 16 and 24 h, respectively, were in fact associated with polysomes when seeds were dormant (i.e. before after-ripening; Fig. 2, C and D). This suggests that dormancy alleviation may also trigger the dissociation of polysomal complexes, leading the released transcripts to be present in the nondormant transcriptome. These are major findings, since current knowledge of the molecular regulation of dormancy is based on transcriptomic studies, and we show here that they are not fully relevant for studying this process. The use of SeedNet provided additional insights into the relationship between the transcriptome and translatome in germinating seeds. While our transcriptome data clearly fitted with the model, the translatome did not match with the regions designed for transcriptomic data (Fig. 2, E and F). Particularly, the transcripts belonging to the translatome of dormant seeds were not preferentially found in region 1 of the model, which is defined as the dormancy region. Using sunflower seeds, Layat et al. (2014) also pointed out that SeedNet failed to predict functional areas of translated transcripts.
These data clearly demonstrate that germination, as modulated by dormancy, relies on the specific and timely regulated association of transcripts with polysomes. This is confirmed by the distribution of transcripts in relation to their polysomal ratio, which was affected by dormancy (Fig. 3A) and which indicates that differences exist in selective translation between dormant and nondormant seeds, even though they displayed similar polysomal profiles and translational activities (Figs. 1C and 4). The use of the puromycin method (Fig. 4) nicely demonstrated that the efficiency of the translation machinery was similar during imbibition of either dormant or nondormant seeds. This is in accordance with measurements of translational activity obtained by radiolabeled Met incorporation by dormant and nondormant seeds after 1 d of imbibition (Chibani et al., 2006). Since global translation was not repressed by dormancy but polysomal ratio was (Figs. 3A and 4), we propose that dormancy regulates the selective translation of individual transcripts. In other words, dormancy-associated translational control (which can be seen in 16 and 24 h) is selective to particular sets of genes and is not a general translational repression. This means that transcripts in nondormant seeds must be translated with a higher efficiency, as shown by their higher polysome-loading values, and this can be explained by the regulatory role of 5′UTR.
Selective translation in eukaryotes is known to be driven by 5′UTR or 3′UTR (Wilkie et al., 2003; Kawaguchi and Bailey-Serres, 2005), but here, the 5′UTR sequences only appeared to play a role in seed dormancy regulation. In 5′UTR, secondary structure, length, GC content, 7-methylguanylate cap, and initiation sequence context are likely to play roles in translational efficiency (Kawaguchi and Bailey-Serres, 2005). Here, the relationship between 5′UTR length and polysomal ratio (Fig. 3B) corresponds to classical observations in eukaryotes (Pestova and Kolupaeva, 2002; Kawaguchi and Bailey-Serres, 2005; Jackson et al., 2010). Besides the control of protein levels, elements in 5′UTR can also affect transcription or mRNA degradation (Dvir et al., 2013). Short 5′UTRs (20 nucleotides) are known to inhibit the entry of the 40S preinitiation complex or recognition of the AUG initiation codon, whereas moderately long 5′UTRs (60–100 nucleotides) promoted initiation (Kozak, 1991) but longer lengths decreased polysomal ratio, as shown in Figure 3B. However, the effect of 5′UTR on translation cannot be directly related to germination, since dormant and nondormant seeds displayed the same response to this parameter (Fig. 3B). Besides, the GC content of 5′UTR appeared to be more important for regulating the polysomal ratio of transcripts in relation to dormancy. Indeed, the increase of GC content dramatically altered polysomal loading in dormant seeds, whereas it had only a slight effect in nondormant seeds (Fig. 3C). Polysomal ratio has been shown to be negatively correlated with 5′UTR GC content in Arabidopsis plants subjected to dehydration stress, which can be related to a higher requirement for ATP during the initiation step (Kawaguchi and Bailey-Serres, 2005). Our results also show that the 5′UTRs of transcripts more abundant in the translatome of dormant and nondormant seeds contain specific overrepresented motifs (Fig. 3D), thus suggesting that translated mRNA can be selected according to these motifs. The UUUC motif appeared to be specific for transcripts addressed to polysomes in dormant seeds (Fig. 3D). To our knowledge, no similar motif was found in eukaryotes, but in viruses, pyrimidine-rich motifs are involved in translation initiation enhancement (Silveira Carneiro et al., 1995).
The AG-rich motif found in 5′UTR of nondormant seed transcripts has already been identified as a regulator of exon recognition in the splicing process in plants (McCullough and Schuler, 1997). In our data set, around 75% of the polysomal transcripts had no uORF (Table I), which is quite similar to the proportion found in the whole Arabidopsis genome (Kim et al., 2007). However, there were more transcripts containing a high number of uORFs in the translatome of dormant seeds than in nondormant seeds, which might explain the lower polysome loading evidenced in these seeds (Fig. 3E). In addition, our results clearly show the major influence of length of the uORF on polysome loading, particularly in nondormant seeds (Fig. 3F). Although uORFs are known to repress in vitro translation in a length-dependent manner (Kozak, 2002), only a moderate correlation between the inhibition of gene expression and uORF length had been shown in plants until now (von Arnim et al., 2014). Therefore, we suggest that the presence and the length of uORFs, in addition to specific motifs in 5′UTR and GC content, also contribute to the selective translation of mRNA in dormant and nondormant seeds. However, it will be necessary to determine whether the features of 5′UTR are key determinants in this process. This can be achieved by carrying out a series of mutations in 5′UTR (Dvir et al., 2013; Kim et al., 2014) in order to identify important regions for differential mRNA translation during seed germination or dormancy maintenance. Such an approach has already proved to be efficient for demonstrating the role of 5′UTR in the regulation of mRNA translation during heat stress in Arabidopsis (Khurana et al., 2013; Matsuura et al., 2013).
The clusters representative of transcripts found in dormant and nondormant polysomal fractions have been visualized as scatterplots (Fig. 5). Some GO categories appear to be largely represented in both dormant and nondormant seeds (Fig. 5). Similar to what was shown by Layat et al. (2014), translatomes of dormant and nondormant seeds contain many up-regulated transcripts (196 and 153, respectively) belonging to the GO category transport, but dormant seeds were markedly enriched into transporters from the subcategories metabolite transporters at the mitochondrial membrane and sugars. The role of transporters in seed germination is poorly described, but among the transcripts identified here, one can point out the ATP-binding cassette transporters, which are a large family transporting different molecules, including hormones. For example, ABCG25 (At1g71960), found in the translatome of dormant seeds at 16 and 24 h, is an efflux transporter that exports ABA out of the vessels, thus mediating ABA import into cells (Kuromori et al., 2010). Interestingly, COMATOSE (At4g39850) is also found in the translatome of dormant seeds, whereas it has been proposed that its expression was associated with the late phases of germination (Footitt et al., 2002; Carrera et al., 2007). The GO categories transcription, DNA dependent and transcription, RNA binding were also largely represented in the translatome of dormant and nondormant seeds. They include many transcription factors, which demonstrates that germination, but also dormancy maintenance, both require an active transcription at some time points of the whole process, as suggested by Layat et al. (2014), but the nature of the transcripts drives the cellular functioning toward germination or not. For example, 13 WRKY transcription factors were identified in this work (four in nondormant seeds and nine in dormant seeds), and they are known to act as negative or positive regulators of ABA signaling (Tripathi et al., 2014). WRKY33 (At2g38470), which is found in the translatome of dormant seeds, has already been shown to be involved in the salt stress response and ABA signaling (Jiang and Deyholos, 2009). Among the transcription factors, MYC2 (At1g01260), specifically present in the translatome of dormant seeds, is known as an activator of jasmonic acid responses (Fernández-Calvo et al., 2011), a phytohormone related to germination inhibition (Oh et al., 2004). Our data set also underlines that many chromatin-remodeling factors are present in the translatome and are likely to play a role in both germination and dormancy (Supplemental Data Sets S2 and S3). Chromatin structure regulates gene transcription but is still poorly characterized in the context of seed germination, since only a few studies have addressed this point (Molitor et al., 2014). The prominence of this GO category suggests that this mechanism should deserve more attention, as proposed by Nonogaki (2014).
The microarray analysis of translatomes also allowed the identification of functional GO categories that were specific of either dormant or nondormant states (Fig. 5; Supplemental Data Set S3). Clustering analysis shows that polysome-associated transcripts specific to nondormant embryos fell mainly into the categories hormone metabolism, cell wall, or redox (Supplemental Data Set S3). In the GO category hormone metabolism, we found regulators of the hormonal balance that drives the germination process (Finch-Savage and Leubner-Metzger, 2006). This category contains, for example, the inhibitors of ABA signaling HIGHLY ABSCISIC ACID-INDUCED PP2C GENE1 (At5g59220; Bhaskara et al., 2012) and ABSCISIC ACID-HYPERSENSITIVE GERMINATION1 (At5G51760; Nishimura et al., 2007), positive regulators of ethylene synthesis 1-AMINOCYCLOPROPANE-1-CARBOXYLIC ACID OXIDASE1 (ACO1; At2g19590, Fig. 6) or of GA synthesis GIBBERELLIN20-OXIDASE3 (At5g07200; Yamaguchi, 2008) and GA signaling SUBTILISIN-LIKE SERINE PROTEASE2 (At4g34980; Ogawa et al., 2003; Fig. 6) and GIBBERELLIN-STIMULATED ARABIDOPSIS6 (GASA6; At1g74670; Roxrud et al., 2007; Fig. 6). Reactive oxygen species have been proposed to play a signaling role in Arabidopsis seed germination (Leymarie et al., 2012). This assumption is confirmed here by the overrepresentation of the GO category redox in the polysomal fraction of nondormant seeds, which contained transcripts related to enzymatic reactive oxygen species scavenging, such as CATALASE2 (At4g35090), MONODEHYDROASCORBATE REDUCTASE4 (At3g27820), or GLUTATHIONE PEROXIDASE8 (At1g63460; Bailly et al., 2004) but also GLUTAREDOXIN13 (At1g03850) and GLUTAREDOXIN C1 (At5g63030), which regulate the cellular redox state through the formation of reversible disulfide bonds from thiol groups (Buchanan and Balmer, 2005), thus playing a role in seed germination (Montrichard et al., 2003; El-Maarouf-Bouteau et al., 2013). As proposed by Bailly et al. (2008) and Leymarie et al. (2012), regulation of enzymatic and nonenzymatic detoxifying systems would result from an increased reactive oxygen species production in germinating seeds. Transcripts involved in cell wall modifications were found to be more abundant in the translatome of nondormant seeds than dormant seeds. Overrepresentation of cell wall-related transcripts in the transcriptome was shown in Lepidium sativum (Morris et al., 2011) and Arabidopsis (Endo et al., 2012) germinating seeds and in the translatome of sunflower seeds (Layat et al., 2014). Transcripts for expansins such as EXPA1, EXPA10, EXPA4, and EXPA2 (At1g69530, At1g26770, At2g39700, and At5g05290; Fig. 6), mannanase such as MAN7 (At5g66460), and pectin lyases (At4g33440 and At3g15720) were highly abundant in the translatome of nondormant seeds. Enzymes coded by these transcripts participate in cell wall extensibility modification, which subsequently allows cell elongation, a process often mentioned as being critical for radicle elongation in Arabidopsis seeds (Bewley, 1997; Nonogaki et al., 2007). For example, EXPA2 (At5g05290) was recently shown to be involved in GA-induced seed germination in Arabidopsis (Yan et al., 2014). The GO category lipid metabolism also appeared to be more important in the translatome of nondormant seeds at 24 h of imbibition (Fig. 5; Supplemental Table S3). For example, the transcripts for LTP6 and LTP5 (At3g51600 and At3g08770, respectively; Fig. 6), which ranged within this category, correspond to lipid transfer proteins, and they have been shown to participate in the intracellular transfer of lipids during seed germination (Pagnussat et al., 2012).
Attention also has to be paid to the transcripts that were specifically present in the translatome of dormant seeds, since their active translation was associated with the inhibition of germination. They mainly ranged in the categories response to stress and signaling. For example, REPRESSOR OF GIBBERELLIN-LIKE2 (At3g03450; Fig. 6), which encodes a DELLA protein known to repress germination (Lee et al., 2002), was highly abundant in the translatome of dormant seeds at 24 h of imbibition, suggesting that GA signaling might be impaired in those seeds. In addition, signaling components of ABA are overrepresented in the translatome of these seeds, such as ABSCISIC ACID INSENSITIVE3 (ABI3; At3g24650), ABI2 (At5g57050; Fig. 6), and ABSCISIC ACID DEFICIENT1 (At5g67030), major negative regulators of germination (Finkelstein et al., 2002). Interestingly, PHYTOCHROME INTERACTING FACTOR3-LIKE2 (At3g62090; Fig. 6), a phytochrome-interacting factor that was suggested to play a role in Arabidopsis primary dormancy (Penfield et al., 2010), was more abundant in the translatome of dormant seeds. Translatome profiling of dormant seeds also revealed that many mitogen-activated protein kinases (MPKs) were involved in the inhibition of germination, such as MPK1 (At1g10210) and MPK4 (At4g01370; Fig. 6) or upstream kinases such as MITOGEN-ACTIVATED PROTEIN KINASE KINASE2 (MKK2; At4g29810) and MKK9 (At1g73500). MPKs are involved in numerous signaling pathways that control plant development via the phosphorylation of target molecules and are involved in hormone signaling pathways (Colcombet and Hirt, 2008). MPK1 has been shown to be activated by ABA (Umezawa et al., 2013) and MPK4 is activated by jasmonic acid (Gomi et al., 2005), but their roles in the regulation of germination are poorly described. Inhibition of germination was also related to the association of many transcripts of heat shock proteins (HSPs) with polysomes such as Hsp70 (At3g09440, At5g02500, and At1g11660; Fig. 6), as shown in sunflower (Layat et al., 2014), thus suggesting that chaperone activity can play a role in the maintenance of dormancy. Interestingly it has been shown recently in mammals that chaperones, and particularly HSP70, would regulate translation elongation by affecting ribosome dynamics (Shalgi et al., 2013). LATE EMBRYOGENESIS ABUNDANT (At2g21490; Fig. 6), also involved in the response to stress and activated by ABA (Chen et al., 2008), was also more abundant in dormant seeds (Supplemental Table S4).
Further insights about the dynamic changes of GO categories between 16 and 24 h of seed imbibition are given Supplemental Table S3. This table shows the dynamics of association of the various categories of transcripts with polysomes, taking into account changes in both P values and frequency within a kind of seed. Even if it is difficult to determine the more relevant parameter at the biological level (i.e. frequency or P value), this table allows the identification of GO categories whose abundance and number of individual transcripts increased or decreased during seed imbibition. For example, in nondormant seeds, photosystem and cell organization and biogenesis increased, while dormancy maintenance was associated with a decrease of the GO categories redox and lipid metabolism (Supplemental Table S3). This demonstrates that the selective association of transcripts with polysomes led to the regulation of major cellular events, preparing subsequent seedling growth after radicle protrusion or dormancy maintenance. For example, the GO category photosystem includes cellular components preparing autotrophy acquisition (Supplemental Data Set S3), which will take place in the growing seedling.
Finally, the relevance of our findings was confirmed when studying by PCR the abundance of key transcripts in the transcriptome and translatome (Fig. 6). For some key genes identified as being important for regulating germination and dormancy (see above), we confirmed by qRT-PCR that they are all highly abundant in the polysomal fraction of either dormant or nondormant seeds. As demonstrated previously, in some cases there was a correlation between the amount of transcripts and their abundance in the translatome (ACO1, GASA6, and HSP70), but in most cases they were not related, demonstrating again the translational component of the regulation of germination and dormancy.
In conclusion, this comparison between the transcriptome and translatome of dormant and nondormant Arabidopsis seeds clearly emphasizes the key fundamental role of the polysomal fraction in dormancy, which should lead us to consider in a new light the dogma of a transcriptional regulation of seed germination. We demonstrate that germination results for a timely and selective recruitment of mRNA to polysomes and bring comprehensive insights related to the regulation of this process. GO analyses of translatome data allowed the identification of key players in the active regulation of either germination or dormancy. Figure 7 summarizes the major findings of this study and provides a comprehensive model showing how the dynamics of the association of transcripts with polysomes regulates Arabidopsis seed germination. The robustness of omics analysis using the plant model Arabidopsis permitted us to demonstrate that the 5′UTR sequence regulates the translation efficiency at multiple levels and underlined in particular the major contribution of its GC content and uORF. We are convinced that future studies dealing with the molecular regulation of dormancy will have to focus on this novel scale of posttranscriptional mechanisms in order to provide cutting-edge theories to explain this complex phenomenon.
Figure 7.

Simplified model showing the dynamics of transcript association with polysomes and their related GO categories during germination or dormancy maintenance at 25°C. In the dry state, dormancy release occurs during after-ripening, but the stored mRNAs are roughly similar in both categories of seeds (Meimoun et al., 2014). Imbibition results in the transcription of genes specifically associated with dormant (D) and nondormant (ND) seeds, while a pool of mRNA is similar in dormant and nondormant seeds (nondifferentially expressed mRNA and stored mRNA). Horizontal colored bars indicate the percentage of transcripts recruited by polysomes among the different RNA pools in dormant and nondormant seeds at 16 and 24 h of imbibition (calculated from the transcriptome and translatome analysis presented in Fig. 2). This demonstrates that up-regulated transcripts in dormant seeds participate only in a very minor way in dormancy maintenance (12% and 7% addressed to polysomes at 16 and 24 h, respectively) and that up-regulated transcripts in nondormant seeds are more likely to become polysome associated (45% and 25% addressed to polysomes at 16 and 24 h, respectively). This suggests that the regulation of germination and dormancy is mainly translational. Three major GO categories specifically translated at each time point in dormant and nondormant seeds are indicated. Vertical bars on the right show that the transcripts translated at 16 and 24 h have 36% similarity in dormant seeds but only 4% in nondormant seeds, demonstrating the dynamics of translational activity in germinating seeds.
MATERIALS AND METHODS
Plant Material and Germination Assays
Arabidopsis (Arabidopsis thaliana ecotype Columbia-0) plants were grown in a greenhouse at 20°C and 22°C under long day (16 h of light/8 h of dark) conditions. Seeds were first sown in a mixture of soil and vermiculite (9:1), and plantlets were transferred after 12 to 15 d on soil:perlite:vermiculite (2:1:1). Floral stems were cut at silique maturity (about 3 months after sowing), dried at room temperature for 4 d in a paper envelope, and seeds were collected. Freshly harvested seeds were either stored at −20°C to preserve their dormancy or after-ripened at 20°C and 56% relative humidity in the dark for 4 weeks to alleviate dormancy. Nondormant seeds were then also stored at −20°C. Germination assays were performed by placing seeds in darkness in 9-cm petri dishes (100 seeds per dish, three replicates) on a filter paper on the top of a layer of cotton wool moistened with deionized water. Germination was evaluated daily, a seed being considered as germinated when the radicle protruded through the testa. Dormant and nondormant seeds used for other experiments were imbibed at 25°C in darkness in petri dishes on a filter paper placed on a layer of cotton wool imbibed with water for the durations mentioned in each experiment description. After imbibition, seeds were collected, frozen in liquid nitrogen, and stored at −80°C. For each experiment, a germination test was always performed to check the germination rate for dormant and nondormant seeds during storage in the freezer.
Polysome Preparation and RNA Extraction
All chemicals used in this study without specific annotation were furnished by Sigma. Polysomes were prepared according to Mustroph et al. (2009). Frozen imbibed dormant or nondormant seeds were ground in a mortar into a fine powder in liquid nitrogen and then solubilized (1 mL per 100 mg of seeds) in polysome extraction buffer containing 200 mm Tris-HCl, pH 9, 200 mm KCl, 25 mm EGTA, 36 mm MgCl2, 1% (v/v) octylphenyl-polyethylene glycol (Igepal CA-630), 1% (v/v) polyoxyethylene(23) laurylether (Brij 35), 1% (v/v) Triton X-100, 1% (v/v) Tween 20, 1% (v/v) polyoxyethylene 10 tridecyl ether, 1% (v/v) sodium deoxycholate, 1 mm dithiothreitol, 0.5 mm phenylmethylsulfonyl fluoride, 50 μg mL−1 cycloheximide, 50 μg mL−1 chloramphenicol, and 10 µL mL−1 protease inhibitor cocktail. Cells debris were eliminated by centrifugation at 14,000g for 15 min at 4°C, and the supernatant was filtrated with Miracloth. An aliquot of 800 μL of the supernatant was reserved for the isolation of total RNA. Four-milliliter extracts were loaded on an 11-mL 15% to 60% Suc gradient (2 m Suc, 0.4 m Tris-HCl, pH 8.4, 0.2 m KCl, 0.1 m MgCl2, 100 mg mL−1 chloramphenicol, and 100 mg mL−1 cycloheximide). After ultracentrifugation at 16,000g for 2.5 h in a Beckman SW41 rotor, the Suc gradient was fractionated in 10 fractions with the Density Gradient Fractionator system (Teledyne ISCO) with a 254-nm detector and 2 m Suc as displacement fluid. The concentration of KCl used in Suc salt solution of the Suc gradient was 0.2 m for the polysome preparation and extraction of mRNA, and 0.8 m of KCl was used to separate free monosomes from monosomes associated with mRNA (Martin, 1973; Fennoy and Bailey-Serres, 1995). The four fractions corresponding to the polysomes (fractions 7–10) were pooled. The total and polysomal RNA were extracted by the addition of 500 µL of 5% SDS-0.2 m EDTA and 1 volume of phenol/chloroform/isoamyl alcohol (Barkan, 1993). After 5 min of centrifugation at 2,200g (room temperature), 1 mL of 100% ethanol was added to the aqueous phase, and the mix was centrifuged at 2,200g for 15 min. After resuspension of the pellet in water, the RNA was purified with Macherey-Nagel NucleoSpin XS columns according to the manufacturer’s instruction. The RNA quantity was measured with Nanovue (GE Healthcare), and RNA quality was checked with the Experion electrophoresis system (Bio-Rad).
DNA Microarray Hybridization Studies and Analysis of Polysome Loading
Microarray analysis was carried out at the Unité de Recherche en Génomique Végétale in Evry, France, using the CATMA version 6.2 array based on Roche-NimbleGen technology. A single high-density CATMA version 6.2 microarray slide contains 12 chambers, each containing 219,684 primers representing all the Arabidopsis genes: 30,834 probes corresponding to the Coding DNA Sequence TAIR version 8 annotation (including 476 probes of mitochondrial and chloroplast genes) + 1,289 probes corresponding to EUGENE software predictions. Moreover, it included 5,352 probes corresponding to repeat elements, 658 probes for microRNA, 342 probes for other RNAs (ribosomal RNA, transfer RNA, small nuclear RNA, and small nucleolar RNA), and finally 36 controls. Each long primer was triplicate in each chamber for robust analysis and in both strands. For each comparison, one technical replicate with fluorochrome reversal (dye swap) was performed for each of the three biological replicates (i.e. four hybridizations per comparison). Starting from 500 ng of total RNA, labeling of complementary RNAs with Cy3-dUTP or Cy5-dUTP (Perkin-Elmer-NEN Life Science Products) was performed as described by Lurin et al. (2004). The hybridization and washing were performed according to NimbleGen Arrays User’s Guide version 5.1 instructions. Two-micrometer scanning was performed with the InnoScan900 scanner (Innopsys), and raw data were extracted using Mapix software (Innopsys). For each array, the raw data comprised the logarithm of median feature pixel intensity at wavelengths of 635 nm (red) and 532 nm (green). For each array, a global intensity-dependent normalization using the Loess procedure (Yang et al., 2002) was performed to correct the dye bias. The differential analysis is based on log ratio averaging over the duplicate probes and over the technical replicates. Hence, the numbers of available data for each gene equals the number of biological replicates and are used to calculate the moderated Student’s t test (Smyth, 2004). Under the null hypothesis, no evidence that the specific variances vary between probes is highlighted by Limma, and consequently, the moderated Student’s t statistic is assumed to follow a standard normal distribution. To control the false discovery rate, adjusted P values found using the optimized BH approach of Storey and Tibshirani (2003) were calculated. We considered as differentially abundant probes with an adjusted P ≤ 0.05. Analysis was done with R software. The function SqueezeVar of the Limma library was used to smooth the specific variances by computing empirical Bayes posterior means. The library kerfdr was used to calculate the adjusted P values.
Microarray data from this article were deposited at the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/; accession no. GSE61809) and at CATdb (http://urgv.evry.inra.fr/CATdb/; project AU13-12_Polysome) according to Minimum Information about a Microarray Experiment standards.
In silico comparison allowed us to compare the transcriptome and translatome for each point and type of seeds and to compare the time points (16 versus 24 h) for each type of sample. Relative RNA abundance in polysomal (P) and total (T) fractions was quantified with the polysomal ratio (P/T), which was calculated as follows:
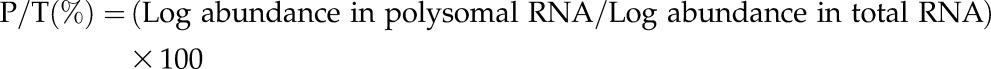 |
Student’s t tests were used to determine whether polysomal ratios of dormant and nondormant seeds for both durations of imbibition were significantly different from each other at P = 0.0001. Fisher’s test was used to compare the distribution of transcripts in each category (according to uORF occurrences) at P = 0.05.
Distribution of Differentially Abundant mRNAs in the SeedNet Network
The network was constructed using the SeedNet online tool (www.vseed.nottingham.ac.uk; Bassel et al., 2011), with the list of transcripts identified as up-regulated in dormant and nondormant seeds for both duration of imbibition in the transcriptome or translatome.
qRT-PCR
RNA (100 ng) was reverse transcribed and amplified, and the relative expression was calculated as described by Leymarie et al. (2012). Primer sequences are presented in Supplemental Table S4.
Translation Quantification
Quantification of translation was adapted from David et al. (2012). Seeds (30 mg) were first imbibed for 12, 20, or 68 h in water at 25°C, before being dried 1 h at 20°C, and then incubated for 4 h with 200 µg mL−1 puromycin at 25°C, in order to increase penetration of the product, thus giving total durations of imbibition of 16, 24, and 72 h. Seeds were then frozen and ground in liquid nitrogen in a mortar and solubilized in 300 µL of extraction buffer (50 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 25 mm KCl, 1× protease inhibitor cocktail, 0.2 units L−1 Ribolock [Thermo], and 10% [v/v] Nonidet P-40). Cells debris were eliminated by centrifugation at 14,000g for 15 min at 4°C, and the supernatant was filtrated with Miracloth. Proteins were precipitated by adding 2 volumes of ethanol and incubated at 4°C for 6 h (Merret et al., 2013). After 15 min of centrifugation at 14,000g at 4°C, the pellet was solubilized with 2× Laemmli buffer. The protein concentration was determined using the 2-D Quant Kit, and 30 µg was separated by SDS-PAGE (10%–20% [v/v] precast polyacrylamide gel; BioRad) and electroblotted onto a polyvinylidene difluoride membrane, which was blocked with 1% (w/v) casein in phosphate-buffered saline-Tween for 1 h, and incubated with the primary antibody (rabbit anti-puromycin antibody from Merck Millipore used at 1:25,000 dilution) for 1 h at room temperature. The secondary antiserum, anti-rabbit IgG-Alkaline Phosphatase (Sigma), was used at 1:30,000 dilution and incubated for 1 h at room temperature. Blots were revealed with nitroblue tetrazolium/5-bromo-4-chloro-3-indolyl phosphate, and all the bands were quantified with ImageJ software.
GO Analysis and Clustering
Subsets of transcripts were classified according to GO categories with the Classification SuperViewer Tool in the Bio-Analytic Resource for Plant Biology (http://bar.utoronto.ca). GO analysis results were presented using the software REVIGO (http://revigo.irb.hr; Supek et al., 2011). REVIGO summarizes long, unintelligible lists of GO terms by finding a representative subset of the terms using a simple clustering algorithm that relies on semantic similarity measures. REVIGO allows one to visualize the clusters in scatterplots in two-dimensional space, assigning x and y coordinates to each term so that more semantically similar GO terms are also closer on the plot. The disc color gradient (blue to red) indicates the degree of GO enrichment corresponding to P values, while the disc size is proportional to the frequency of the GO term in the underlying database. The spatial arrangement of discs approximately reflects a grouping of GO categories by semantic similarity. The allowed similarity used was small (0.5), and the semantic similarity measure was SimRel.
Characterization of mRNA Sequence Features
Information about 5′UTR and 3′UTR was searched in TAIR (http://www.arabidopsis.org) for transcripts being differentially abundant in the translatome of dormant and nondormant seeds, and analyses on the untranslated region sequences were performed on these genes (Supplemental Table S2). The nucleotide composition of the untranslated regions was determined using MFOLD (http://mfold.rna.albany.edu/). Overrepresented motifs in 5′UTR and 3′UTR were identified on untranslated regions of the 100 first differentially abundant transcripts (according to BH test) by use of the MEME suite (http://meme.nbcr.net/meme/; Bailey et al., 2009) using the two-component mixture with set minimum and maximum widths of six and 50 bases, respectively. The motif consensus obtained by MEME was represented using WebLogo application (http://weblogo.berkeley.edu/; Crooks et al., 2004). The motif enrichment represents the ratio of the frequency of the motif overrepresented in the coexpressed transcripts (calculated with MEME) to the frequency of this motif in 5′UTR (or 3′UTR) in the whole genome of Arabidopsis (calculated with TAIR). The uORFs were determined only for transcripts having a described 5′UTR (Supplemental Table S2). In the 5′UTR sequence, we considered having one uORF when an initiation codon (ATG) was followed by a stop codon (TAA, TGA, and TAG). We determined the number of uORFs using Microsoft Excel, and the lengths of uORFs were determined using MFOLD. Transcripts presenting one or several uORFs are listed in Supplemental Data Set S4.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Absorbance profiles of RNA complexes from Arabidopsis seeds obtained in the presence of KCl.
Supplemental Figure S2. MA (distribution of the log2 intensity ratio [M] plotted against the average intensity [A]) plots of total and polysomal mRNA in dormant and nondormant seeds.
Supplemental Figure S3. Characterization of 3′UTR sequences of mRNA associated with polysomes in dormant and nondormant seeds.
Supplemental Figure S4. Validation of microarray results by qRT-PCR.
Supplemental Table S1. Change in 80S and 60S abundance during the time course of seed imbibition.
Supplemental Table S2. Number of transcripts differentially abundant in the translatome of dormant and nondormant seeds having described 5′UTR and 3′UTR in TAIR.
Supplemental Table S3. Changes in polysomal transcripts, clustered by GO categories, during imbibition at 25°C of dormant and nondormant seeds.
Supplemental Table S4. Sequences of primers used for qRT-PCR experiments.
Supplemental Data Set S1. mRNA abundance in dormant and nondormant seeds in total and polysomal fractions.
Supplemental Data Set S2. GO function of transcripts displaying changes in abundance in dormant and nondormant seeds in the transcriptome.
Supplemental Data Set S3. GO clustering of transcripts differentially abundant in dormant and nondormant seeds in the translatome.
Supplemental Data Set S4. List of transcripts differentially abundant in the translatome of dormant and nondormant seeds having one or several uORFs.
Acknowledgments
We thank Dr. Dominique Weil (Centre National de la Recherche Scientifique, Unité Mixte de Recherche 7622, Compartmentation and Intracellular Traffic of mRNPs) for the use of the Suc gradient fractionator and for helpful discussions, George Bassel (University of Birmingham) for help with the use of the SeedNet tool, and Dr. Alexandre David (Institut de Génomique Fonctionnelle) for explanations about the translation quantification method with puromycin.
Glossary
- 5′UTR
5′ untranslated region
- 3′UTR
3′ untranslated region
- uORF
upstream open reading frame
- BH
Benjamini and Hochberg
- GSTs
gene-specific sequence tags
- TAIR
The Arabidopsis Information Resource
- MEME
Multiple Expectation Maximization for Motif Elicitation
- qRT
quantitative real-time
- GO
Gene Ontology
- ABA
abscisic acid
References
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey-Serres J. (1999) Selective translation of cytoplasmic mRNAs in plants. Trends Plant Sci 4: 142–148 [DOI] [PubMed] [Google Scholar]
- Bailey-Serres J, Sorenson R, Juntawong P (2009) Getting the message across: cytoplasmic ribonucleoprotein complexes. Trends Plant Sci 14: 443–453 [DOI] [PubMed] [Google Scholar]
- Bailly C, El-Maarouf-Bouteau H, Corbineau F (2008) From intracellular signaling networks to cell death: the dual role of reactive oxygen species in seed physiology. C R Biol 331: 806–814 [DOI] [PubMed] [Google Scholar]
- Bailly C, Leymarie J, Lehner A, Rousseau S, Côme D, Corbineau F (2004) Catalase activity and expression in developing sunflower seeds as related to drying. J Exp Bot 55: 475–483 [DOI] [PubMed] [Google Scholar]
- Barkan A. (1993) Nuclear mutants of maize with defects in chloroplast polysome assembly have altered chloroplast RNA metabolism. Plant Cell 5: 389–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassel GW, Lan H, Glaab E, Gibbs DJ, Gerjets T, Krasnogor N, Bonner AJ, Holdsworth MJ, Provart NJ (2011) Genome-wide network model capturing seed germination reveals coordinated regulation of plant cellular phase transitions. Proc Natl Acad Sci USA 108: 9709–9714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazin J, Langlade N, Vincourt P, Arribat S, Balzergue S, El-Maarouf-Bouteau H, Bailly C (2011) Targeted mRNA oxidation regulates sunflower seed dormancy alleviation during dry after-ripening. Plant Cell 23: 2196–2208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán-Peña E, Ortíz-López A, Sánchez de Jiménez E (1995) Synthesis of ribosomal proteins from stored mRNAs early in seed germination. Plant Mol Biol 28: 327–336 [DOI] [PubMed] [Google Scholar]
- Bewley JD. (1997) Seed germination and dormancy. Plant Cell 9: 1055–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara GB, Nguyen TT, Verslues PE (2012) Unique drought resistance functions of the Highly ABA-Induced clade A protein phosphatase 2Cs. Plant Physiol 160: 379–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco-Price C, Kaiser KA, Jang CJ, Larive CK, Bailey-Serres J (2008) Selective mRNA translation coordinates energetic and metabolic adjustments to cellular oxygen deprivation and reoxygenation in Arabidopsis thaliana. Plant J 56: 743–755 [DOI] [PubMed] [Google Scholar]
- Browning KS. (1996) The plant translational apparatus. Plant Mol Biol 32: 107–144 [DOI] [PubMed] [Google Scholar]
- Buchanan BB, Balmer Y (2005) Redox regulation: a broadening horizon. Annu Rev Plant Biol 56: 187–220 [DOI] [PubMed] [Google Scholar]
- Carrera E, Holman T, Medhurst A, Peer W, Schmuths H, Footitt S, Theodoulou FL, Holdsworth MJ (2007) Gene expression profiling reveals defined functions of the ATP-binding cassette transporter COMATOSE late in phase II of germination. Plant Physiol 143: 1669–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Zhang J, Neff MM, Hong SW, Zhang H, Deng XW, Xiong L (2008) Integration of light and abscisic acid signaling during seed germination and early seedling development. Proc Natl Acad Sci USA 105: 4495–4500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibani K, Ali-Rachedi S, Job C, Job D, Jullien M, Grappin P (2006) Proteomic analysis of seed dormancy in Arabidopsis. Plant Physiol 142: 1493–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colcombet J, Hirt H (2008) Arabidopsis MAPKs: a complex signalling network involved in multiple biological processes. Biochem J 413: 217–226 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE (2004) WebLogo: a sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David A, Dolan BP, Hickman HD, Knowlton JJ, Clavarino G, Pierre P, Bennink JR, Yewdell JW (2012) Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J Cell Biol 197: 45–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir S, Velten L, Sharon E, Zeevi D, Carey LB, Weinberger A, Segal E (2013) Deciphering the rules by which 5′-UTR sequences affect protein expression in yeast. Proc Natl Acad Sci USA 110: E2792–E2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Maarouf-Bouteau H, Meimoun P, Job C, Job D, Bailly C (2013) Role of protein and mRNA oxidation in seed dormancy and germination. Front Plant Sci 4: 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo A, Tatematsu K, Hanada K, Duermeyer L, Okamoto M, Yonekura-Sakakibara K, Saito K, Toyoda T, Kawakami N, Kamiya Y, et al. (2012) Tissue-specific transcriptome analysis reveals cell wall metabolism, flavonol biosynthesis and defense responses are activated in the endosperm of germinating Arabidopsis thaliana seeds. Plant Cell Physiol 53: 16–27 [DOI] [PubMed] [Google Scholar]
- Fennoy SL, Bailey-Serres J (1995) Post-transcriptional regulation of gene expression in oxygen-deprived roots of maize. Plant J 7: 287–295 [DOI] [PubMed] [Google Scholar]
- Fernández-Calvo P, Chini A, Fernández-Barbero G, Chico JM, Gimenez-Ibanez S, Geerinck J, Eeckhout D, Schweizer F, Godoy M, Franco-Zorrilla JM, et al. (2011) The Arabidopsis bHLH transcription factors MYC3 and MYC4 are targets of JAZ repressors and act additively with MYC2 in the activation of jasmonate responses. Plant Cell 23: 701–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finch-Savage WE, Leubner-Metzger G (2006) Seed dormancy and the control of germination. New Phytol 171: 501–523 [DOI] [PubMed] [Google Scholar]
- Finkelstein RR, Gampala SSL, Rock CD (2002) Abscisic acid signaling in seeds and seedlings. Plant Cell (Suppl) 14: S15–S45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Footitt S, Slocombe SP, Larner V, Kurup S, Wu Y, Larson T, Graham I, Baker A, Holdsworth M (2002) Control of germination and lipid mobilization by COMATOSE, the Arabidopsis homologue of human ALDP. EMBO J 21: 2912–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galland M, Huguet R, Arc E, Cueff G, Job D, Rajjou L (2014) Dynamic proteomics emphasizes the importance of selective mRNA translation and protein turnover during Arabidopsis seed germination. Mol Cell Proteomics 13: 252–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomi K, Ogawa D, Katou S, Kamada H, Nakajima N, Saji H, Soyano T, Sasabe M, Machida Y, Mitsuhara I, et al. (2005) A mitogen-activated protein kinase NtMPK4 activated by SIPKK is required for jasmonic acid signaling and involved in ozone tolerance via stomatal movement in tobacco. Plant Cell Physiol 46: 1902–1914 [DOI] [PubMed] [Google Scholar]
- Grolleau A, Bowman J, Pradet-Balade B, Puravs E, Hanash S, Garcia-Sanz JA, Beretta L (2002) Global and specific translational control by rapamycin in T cells uncovered by microarrays and proteomics. J Biol Chem 277: 22175–22184 [DOI] [PubMed] [Google Scholar]
- Holdsworth MJ, Bentsink L, Soppe WJJ (2008) Molecular networks regulating Arabidopsis seed maturation, after-ripening, dormancy and germination. New Phytol 179: 33–54 [DOI] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Deyholos MK (2009) Functional characterization of Arabidopsis NaCl-inducible WRKY25 and WRKY33 transcription factors in abiotic stresses. Plant Mol Biol 69: 91–105 [DOI] [PubMed] [Google Scholar]
- Juntawong P, Bailey-Serres J (2012) Dynamic light regulation of translation status in Arabidopsis thaliana. Front Plant Sci 3: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juntawong P, Girke T, Bazin J, Bailey-Serres J (2014) Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc Natl Acad Sci USA 111: E203–E212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R, Bailey-Serres J (2005) mRNA sequence features that contribute to translational regulation in Arabidopsis. Nucleic Acids Res 33: 955–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi R, Girke T, Bray EA, Bailey-Serres J (2004) Differential mRNA translation contributes to gene regulation under non-stress and dehydration stress conditions in Arabidopsis thaliana. Plant J 38: 823–839 [DOI] [PubMed] [Google Scholar]
- Kedersha N, Anderson P (2002) Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans 30: 963–969 [DOI] [PubMed] [Google Scholar]
- Khurana R, Kathuria H, Mukhopadhyay A, Kapoor S, Tyagi AK (2013) A 286 bp upstream regulatory region of a rice anther-specific gene, OSIPP3, confers pollen-specific expression in Arabidopsis. Biotechnol Lett 35: 455–462 [DOI] [PubMed] [Google Scholar]
- Kim BH, Cai X, Vaughn JN, von Arnim AG (2007) On the functions of the h subunit of eukaryotic initiation factor 3 in late stages of translation initiation. Genome Biol 8: R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lee G, Jeon E, Sohn EJ, Lee Y, Kang H, Lee DW, Kim DH, Hwang I (2014) The immediate upstream region of the 5′-UTR from the AUG start codon has a pronounced effect on the translational efficiency in Arabidopsis thaliana. Nucleic Acids Res 42: 485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Nambara E (2010) Stored and neosynthesized mRNA in Arabidopsis seeds: effects of cycloheximide and controlled deterioration treatment on the resumption of transcription during imbibition. Plant Mol Biol 73: 119–129 [DOI] [PubMed] [Google Scholar]
- Kozak M. (1991) A short leader sequence impairs the fidelity of initiation by eukaryotic ribosomes. Gene Expr 1: 111–115 [PMC free article] [PubMed] [Google Scholar]
- Kozak M. (2002) Pushing the limits of the scanning mechanism for initiation of translation. Gene 299: 1–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuromori T, Miyaji T, Yabuuchi H, Shimizu H, Sugimoto E, Kamiya A, Moriyama Y, Shinozaki K (2010) ABC transporter AtABCG25 is involved in abscisic acid transport and responses. Proc Natl Acad Sci USA 107: 2361–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner DH, Beilharz TH, Marguerat S, Mata J, Watt S, Schubert F, Preiss T, Bähler J (2007) A network of multiple regulatory layers shapes gene expression in fission yeast. Mol Cell 26: 145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layat E, Leymarie J, El-Maarouf-Bouteau H, Caius J, Langlade N, Bailly C (2014) Translatome profiling in dormant and nondormant sunflower (Helianthus annuus) seeds highlights post-transcriptional regulation of germination. New Phytol 204: 864–872 [DOI] [PubMed] [Google Scholar]
- Lee S, Cheng H, King KE, Wang W, He Y, Hussain A, Lo J, Harberd NP, Peng J (2002) Gibberellin regulates Arabidopsis seed germination via RGL2, a GAI/RGA-like gene whose expression is up-regulated following imbibition. Genes Dev 16: 646–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leymarie J, Vitkauskaité G, Hoang HH, Gendreau E, Chazoule V, Meimoun P, Corbineau F, El-Maarouf-Bouteau H, Bailly C (2012) Role of reactive oxygen species in the regulation of Arabidopsis seed dormancy. Plant Cell Physiol 53: 96–106 [DOI] [PubMed] [Google Scholar]
- Lin SY, Chen PW, Chuang MH, Juntawong P, Bailey-Serres J, Jauh GY (2014) Profiling of translatomes of in vivo-grown pollen tubes reveals genes with roles in micropylar guidance during pollination in Arabidopsis. Plant Cell 26: 602–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MJ, Wu SH, Wu JF, Lin WD, Wu YC, Tsai TY, Tsai HL, Wu SH (2013) Translational landscape of photomorphogenic Arabidopsis. Plant Cell 25: 3699–3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lurin C, Andrés C, Aubourg S, Bellaoui M, Bitton F, Bruyère C, Caboche M, Debast C, Gualberto J, Hoffmann B, et al. (2004) Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16: 2089–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrè E, Cocucci S, Sturani E (1965) On the development of the ribosomal system in endosperm of germinating castor bean seeds. Plant Physiol 40: 1162–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TE. (1973) A simple general method to determine the proportion of active ribosomes in eukaryotic cells. Exp Cell Res 80: 496–498 [DOI] [PubMed] [Google Scholar]
- Matsuura H, Ishibashi Y, Shinmyo A, Kanaya S, Kato K (2010) Genome-wide analyses of early translational responses to elevated temperature and high salinity in Arabidopsis thaliana. Plant Cell Physiol 51: 448–462 [DOI] [PubMed] [Google Scholar]
- Matsuura H, Takenami S, Kubo Y, Ueda K, Ueda A, Yamaguchi M, Hirata K, Demura T, Kanaya S, Kato K (2013) A computational and experimental approach reveals that the 5′-proximal region of the 5′-UTR has a cis-regulatory signature responsible for heat stress-regulated mRNA translation in Arabidopsis. Plant Cell Physiol 54: 474–483 [DOI] [PubMed] [Google Scholar]
- McCullough AJ, Schuler MA (1997) Intronic and exonic sequences modulate 5′ splice site selection in plant nuclei. Nucleic Acids Res 25: 1071–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meimoun P, Mordret E, Langlade NB, Balzergue S, Arribat S, Bailly C, El-Maarouf-Bouteau H (2014) Is gene transcription involved in seed dry after-ripening? PLoS ONE 9: e86442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merret R, Descombin J, Juan YT, Favory JJ, Carpentier MC, Chaparro C, Charng YY, Deragon JM, Bousquet-Antonelli C (2013) XRN4 and LARP1 are required for a heat-triggered mRNA decay pathway involved in plant acclimation and survival during thermal stress. Cell Rep 5: 1279–1293 [DOI] [PubMed] [Google Scholar]
- Molitor AM, Bu Z, Yu Y, Shen WH (2014) Arabidopsis AL PHD-PRC1 complexes promote seed germination through H3K4me3-to-H3K27me3 chromatin state switch in repression of seed developmental genes. PLoS Genet 10: e1004091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrichard F, Renard M, Alkhalfioui F, Duval FD, Macherel D (2003) Identification and differential expression of two thioredoxin h isoforms in germinating seeds from pea. Plant Physiol 132: 1707–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris K, Linkies A, Müller K, Oracz K, Wang X, Lynn JR, Leubner-Metzger G, Finch-Savage WE (2011) Regulation of seed germination in the close Arabidopsis relative Lepidium sativum: a global tissue-specific transcript analysis. Plant Physiol 155: 1851–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustroph A, Juntawong P, Bailey-Serres J (2009) Isolation of plant polysomal mRNA by differential centrifugation and ribosome immunopurification methods. Methods Mol Biol 553: 109–126 [DOI] [PubMed] [Google Scholar]
- Narsai R, Rocha M, Geigenberger P, Whelan J, van Dongen JT (2011) Comparative analysis between plant species of transcriptional and metabolic responses to hypoxia. New Phytol 190: 472–487 [DOI] [PubMed] [Google Scholar]
- Nishimura N, Yoshida T, Kitahata N, Asami T, Shinozaki K, Hirayama T (2007) ABA-Hypersensitive Germination1 encodes a protein phosphatase 2C, an essential component of abscisic acid signaling in Arabidopsis seed. Plant J 50: 935–949 [DOI] [PubMed] [Google Scholar]
- Nonogaki H. (2014) Seed dormancy and germination: emerging mechanisms and new hypotheses. Front Plant Sci 5: 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonogaki H, Chen F, Bradford K (2007) Mechanisms and genes involved in germination sensu stricto. In Bradford K, Nonogaki H, eds, Seed Development, Dormancy and Germination. Blackwell Publishing, Oxford, pp 264–304 [Google Scholar]
- Ogawa M, Hanada A, Yamauchi Y, Kuwahara A, Kamiya Y, Yamaguchi S (2003) Gibberellin biosynthesis and response during Arabidopsis seed germination. Plant Cell 15: 1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh E, Kim J, Park E, Kim JI, Kang C, Choi G (2004) PIL5, a phytochrome-interacting basic helix-loop-helix protein, is a key negative regulator of seed germination in Arabidopsis thaliana. Plant Cell 16: 3045–3058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagnussat L, Burbach C, Baluška F, de la Canal L (2012) An extracellular lipid transfer protein is relocalized intracellularly during seed germination. J Exp Bot 63: 6555–6563 [DOI] [PubMed] [Google Scholar]
- Penfield S, Josse EM, Halliday KJ (2010) A role for an alternative splice variant of PIF6 in the control of Arabidopsis primary seed dormancy. Plant Mol Biol 73: 89–95 [DOI] [PubMed] [Google Scholar]
- Piques M, Schulze WX, Höhne M, Usadel B, Gibon Y, Rohwer J, Stitt M (2009) Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Mol Syst Biol 5: 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roxrud I, Lid SE, Fletcher JC, Schmidt ED, Opsahl-Sorteberg HG (2007) GASA4, one of the 14-member Arabidopsis GASA family of small polypeptides, regulates flowering and seed development. Plant Cell Physiol 48: 471–483 [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Shalgi R, Hurt JA, Krykbaeva I, Taipale M, Lindquist S, Burge CB (2013) Widespread regulation of translation by elongation pausing in heat shock. Mol Cell 49: 439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silveira Carneiro J, Equestre M, Pagnotti P, Gradi A, Sonenberg N, Perez Bercoff R (1995) 5′ UTR of hepatitis A virus RNA: mutations in the 5′-most pyrimidine-rich tract reduce its ability to direct internal initiation of translation. J Gen Virol 76: 1189–1196 [DOI] [PubMed] [Google Scholar]
- Smyth GK. (2004) Linear models and empirical Bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: e3. [DOI] [PubMed] [Google Scholar]
- Sormani R, Delannoy E, Lageix S, Bitton F, Lanet E, Saez-Vasquez J, Deragon JM, Renou JP, Robaglia C (2011) Sublethal cadmium intoxication in Arabidopsis thaliana impacts translation at multiple levels. Plant Cell Physiol 52: 436–447 [DOI] [PubMed] [Google Scholar]
- Spence J, Duggan BM, Eckhardt C, McClelland M, Mercola D (2006) Messenger RNAs under differential translational control in Ki-ras-transformed cells. Mol Cancer Res 4: 47–60 [DOI] [PubMed] [Google Scholar]
- Spiegel S, Obendorf RL, Marcus A (1975) Transcription of ribosomal and messenger RNAs in early wheat embryo germination. Plant Physiol 56: 502–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R (2003) Statistical methods for identifying differentially expressed genes in DNA microarrays. Methods Mol Biol 224: 149–157 [DOI] [PubMed] [Google Scholar]
- Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6: e21800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi P, Rabara RC, Rushton PJ (2014) A systems biology perspective on the role of WRKY transcription factors in drought responses in plants. Planta 239: 255–266 [DOI] [PubMed] [Google Scholar]
- Umezawa T, Sugiyama N, Takahashi F, Anderson JC, Ishihama Y, Peck SC, Shinozaki K (2013) Genetics and phosphoproteomics reveal a protein phosphorylation network in the abscisic acid signaling pathway in Arabidopsis thaliana. Sci Signal 6: rs8. [DOI] [PubMed] [Google Scholar]
- van den Elzen AM, Schuller A, Green R, Séraphin B (2014) Dom34-Hbs1 mediated dissociation of inactive 80S ribosomes promotes restart of translation after stress. EMBO J 33: 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdier J, Lalanne D, Pelletier S, Torres-Jerez I, Righetti K, Bandyopadhyay K, Leprince O, Chatelain E, Vu BL, Gouzy J, et al. (2013) A regulatory network-based approach dissects late maturation processes related to the acquisition of desiccation tolerance and longevity of Medicago truncatula seeds. Plant Physiol 163: 757–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Arnim AG, Jia Q, Vaughn JN (2014) Regulation of plant translation by upstream open reading frames. Plant Sci 214: 1–12 [DOI] [PubMed] [Google Scholar]
- Wilkie GS, Dickson KS, Gray NK (2003) Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem Sci 28: 182–188 [DOI] [PubMed] [Google Scholar]
- Yamaguchi S. (2008) Gibberellin metabolism and its regulation. Annu Rev Plant Biol 59: 225–251 [DOI] [PubMed] [Google Scholar]
- Yan A, Wu M, Yan L, Hu R, Ali I, Gan Y (2014) AtEXP2 is involved in seed germination and abiotic stress response in Arabidopsis. PLoS ONE 9: e85208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YH, Dudoit S, Luu P, Lin DM, Peng V, Ngai J, Speed TP (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30: e15. [DOI] [PMC free article] [PubMed] [Google Scholar]