

Abstract
Schizophrenia is a debilitating disorder that typically begins in adolescence and is characterized by perceptual abnormalities, delusions, cognitive and behavioural disturbances and functional impairments. While current treatments can be effective, they are often insufficient to alleviate the full range of symptoms. Schizophrenia is associated with structural brain abnormalities including grey and white matter volume loss and impaired connectivity. Recent findings suggest these abnormalities follow a neuroprogressive course in the earliest stages of the illness, which may be associated with episodes of acute relapse. Neuroinflammation has been proposed as a potential mechanism underlying these brain changes, with evidence of increased density and activation of microglia, immune cells resident in the brain, at various stages of the illness. We review evidence for microglial dysfunction in schizophrenia from both neuroimaging and neuropathological data, with a specific focus on studies examining microglial activation in relation to the pathology of grey and white matter. The studies available indicate that the link between microglial dysfunction and brain change in schizophrenia remains an intriguing hypothesis worthy of further examination. Future studies in schizophrenia should: (i) use multimodal imaging to clarify this association by mapping brain changes longitudinally across illness stages in relation to microglial activation; (ii) clarify the nature of microglial dysfunction with markers specific to activation states and phenotypes; (iii) examine the role of microglia and neurons with reference to their overlapping roles in neuroinflammatory pathways; and (iv) examine the impact of novel immunomodulatory treatments on brain structure in schizophrenia.
Linked Articles
This article is part of a themed section on Inflammation: maladies, models, mechanisms and molecules. To view the other articles in this section visit http://dx.doi.org/10.1111/bph.2016.173.issue-4
Abbreviations
- ACC
anterior cingulate cortex
- BP
binding potential
- DLPFC
dorsal lateral prefrontal cortex
- FC
frontal cortex
- GM
grey matter
- HLA‐DR
major histocompatibility complex class II DR alpha
- HPC
hippocampus
- Iba‐1
ionized calcium‐binding adaptor molecule 1
- MDT
mediodorsal thalamus
- PPI
pre‐pulse inhibition
- STG
superior temporal gyrus
- TC
temporal cortex
- TSPO
translocator protein
- WM
white matter
Tables of Links
| TARGETS |
|---|
| Ligand‐gated ion channels a |
| NMDA receptor |
| Catalytic receptors b |
| CD11b (Integrin, alpha M subunit) |
| Enzymes c |
| COX1 |
| LIGANDS | |||
|---|---|---|---|
| Aspirin | IL‐1ra | IL‐25 | Olanzepine |
| BDNF | IL‐4 | Interferon | PK11195 |
| Glutamate | IL‐6 | Ketamine | Quetiapine |
| GM‐CSF | IL‐8 | LPS | TNF‐α |
| IGF1 | IL‐13 | Nitric oxide (NO) | TSPO |
| IL‐1β | |||
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,2013b,2013c).
Introduction
Brain structural abnormalities are widely reported in schizophrenia across all stages of the illness. Although such abnormalities are widespread, the most consistent findings have been ventricular enlargement (Olabi et al., 2011), grey matter (GM) loss in prefrontal, temporal and subcortical structures (Ellison‐Wright and Bullmore, 2009; Fornito et al., 2009), as well as white matter (WM) tracts connecting these regions (Bora et al., 2011; Zalesky et al., 2011). Longitudinal studies have shown that grey matter loss is particularly accelerated in the early stages of schizophrenia and psychosis, which has led to suggestions that the initial phase of illness may be characterized by neuroprogression or a disturbance of neurodevelopmental processes (Pantelis et al., 2003a,2003b; 2005; 2007). However, although such structural brain studies are informative, they are also inherently limited, as the mechanisms that might underlie such changes remain unknown. A number of factors might affect the brain changes seen in schizophrenia, including medication, substance use, lifestyle, genetic and pathophysiological processes. Although the relative contributions of these factors remain controversial (see commentary by Zipursky et al., 2013), evidence for structural brain abnormalities in medication‐naïve patients (e.g. Fusar‐Poli et al., 2013) and cohorts characterized by low substance use (e.g. Jung et al., 2011) points to a role for these considerations in the primary disease processes. One such candidate pathophysiological mechanism that has received increasing attention is microglial‐mediated neuroinflammation. However, despite theoretical proposals linking neuroinflammation or microglial pathology with brain structure and function in schizophrenia, there are few reviews that have comprehensively examined evidence for this relationship. Understanding the neurobiological mechanisms for the structural brain changes in schizophrenia would open a new paradigm for treatment of psychosis and schizophrenia and would pave the way towards more targeted treatments for the disorder.
This review focuses on inflammation in schizophrenia and its relationship to structural brain changes. A comprehensive description of the structural brain changes in schizophrenia was beyond the scope of this review and has been extensively covered elsewhere (Ellison‐Wright and Bullmore, 2009; Fornito et al., 2009; Olabi et al., 2011). We provide a case for inflammation in schizophrenia, followed by a comprehensive review of studies examining the role of microglial dysfunction. Both imaging and neuropathological studies will be reviewed due to the complimentary nature of the information they provide. Subsequently, we address the link between microglial dysfunction and brain changes through a review of recent, primarily rodent, studies investigating a potential interaction between microglial activation and neuronal/white matter dysfunction. We discuss potential mechanisms involved in this interaction and propose directions for future research.
A case for inflammation in schizophrenia
There are multiple lines of evidence supporting the notion of inflammation in schizophrenia, including epidemiological, biomolecular and genetic studies, and clinical trials of adjunctive anti‐inflammatory treatments. Substantial epidemiological data implicates prenatal inflammatory disturbances in the development of schizophrenia (Yolken et al., 2009; Brown, 2011). These data are derived from population studies linking influenza epidemics to schizophrenia in adult offspring and birth cohort studies that have identified various immune‐related genetic variants and infectious agents (e.g. herpes, toxoplasmosis and rubella) that confer an elevated risk for the disorder (Yolken et al., 2009; Brown, 2011). Studies identifying an association between maternal levels of cytokines (e.g. TNF‐α and IL‐8) and risk for schizophrenia (Buka et al., 2001; Brown et al., 2004) suggest that pro‐inflammatory cytokines may also represent potential mediators between prenatal exposure and risk. Biomolecular studies have consistently reported elevated levels of pro‐inflammatory factors such as IL‐6, IL‐1β, TNF‐α, calprotectin and C‐reactive protein, and altered immune cell function, in plasma and serum of schizophrenia patients (Foster et al., 2006; Miller et al., 2011; Fineberg and Ellman, 2013). In addition, immune system‐related genes such as the major histocompatibility complex (MHC) have been associated with schizophrenia and are among the most robust hits in genome‐wide association studies in the disorder (Consortium SPG‐WAS, 2011; Consortium SWGotPG, 2014). Taken together, there is converging evidence implicating a potentially exaggerated and/or chronic immune response in the aetiology of schizophrenia. Furthermore, adjunctive treatments with anti‐inflammatory agents, such as aspirin, oestrogens, fatty acids and N‐acetyl cysteine, have been efficacious in some individuals (Amminger and McGorry, 2012; Sommer et al., 2014).
While inflammatory processes are implicated in the pathophysiology of schizophrenia, the effect of immune‐related gene mutations and peripheral cytokines on the CNS is still elusive. While it is possible for peripheral cytokines to cross the blood–brain barrier and invade the CNS (Banks, 2009), the brain itself may be subjected to an inflammatory assault via the activation of microglia, the brain's innate immune cells (Munn, 2000; Monji et al., 2009). Indeed, as microglial cells have been shown to participate in pro‐inflammatory cascades and modification of neuronal synapses (Moran and Graeber, 2004; Tremblay et al., 2010), synaptic pruning (Zhan et al., 2014) and white matter connections (Ignarro et al., 1993; Chew et al., 2013), microglial‐mediated inflammatory processes have been proposed as a potential mechanism for the structural brain changes identified in schizophrenia (Munn, 2000; Monji et al., 2009). Furthermore, treatments that modulate their activity may improve symptoms of schizophrenia. For example, oral minocycline, an antibiotic that reduces microglial activation (Kobayashi et al., 2013), has efficacy in ameliorating symptoms of schizophrenia (Miyaoka et al., 2008; Levkovitz et al., 2010; Miyaoka et al., 2012). Findings from animal studies showing that minocycline can cross the blood–brain barrier suggests that it acts directly on microglia to ameliorate symptoms (Dean et al., 2012). Antipsychotics have also been shown to inhibit microglial activation by suppressing inflammatory and oxidative reactions (Kato et al., 2011). In particular, in vitro and in vivo evidence has demonstrated that pretreatment with typical and atypical antipsychotics reduces LPS‐ and interferon‐induced secretion of cytokines such as TNF‐α, SOD and NO from activated microglia (for review, see Kato et al. (2011)).
Despite these proposals, the nature of the microglial abnormalities in schizophrenia is far from established, and few studies have directly examined the relationship between microglial pathology and brain structure in this disorder. Characterizing microglial pathology and its relationship with brain structure at various stages of schizophrenia is a necessary step in developing a framework that unites microglial dysfunction, a proposed aetiological theory of schizophrenia (Monji et al., 2013) and the well‐documented disturbances in the grey matter (Byne et al., 2007; Fornito et al., 2009; Bora et al., 2011; Mileaf and Byne, 2012) and white matter (Uranova et al., 2011; Zalesky et al., 2011; Kubicki and Shenton, 2014; Uranova et al., 2014) underlying this disorder.
Microglial cells – a brief introduction
Microglial cells, discovered by Pio Del Rio‐Hortega, represent the first line of immune defence in the CNS against insults and foreign invaders (Kettenmann et al., 2011). Microglia derive from progenitors of myeloid lineage and invade the brain from the embryonic yolk sac via the circulatory system (Ginhoux et al., 2010; Schulz et al., 2012; Nayak et al., 2014). After infiltrating the CNS parenchyma, microglial cells transition into a ramified state, which possess a small soma with long motile processes.
Under healthy physiological conditions, ramified microglia actively survey the entire brain for pathogens or debris (Nimmerjahn et al., 2005). Upon detecting a threat, microglia rapidly shift phenotype, morphology and function, traditionally defined as ‘microglia activation’. Activated microglia acquire an amoeboid morphology, with swollen cell bodies and short processes to enable migration to the pathogen site (Kettenmann et al., 2011) (Figure 1). It has been suggested that microglial activation may correspond to the binary activation profile of peripheral monocytes, characterized by M1/M2 phenotypes (Mantovani et al., 2005). The M1 phenotype arises in response to inflammation and is accompanied by the release of TNF‐α, IL‐6, IL‐1β, ROS and glutamate, whereas the M2‐type microglia serve to resolve the inflammatory response and express IL‐4, IL‐13, IL‐25, IL‐1ra, insulin‐like growth factor 1 (IGF1), BDNF and COX1 (Réus et al., 2015). However, caution is warranted when applying this binary profile to microglia, as studies have shown that the gene expression profile of microglia differs from the M1/M2 profile of peripheral monocytes (Chiu et al., 2013; Yamasaki et al., 2014). In particular, microglia are highly plastic and may operate on a spectrum ranging from pro‐inflammatory to neurotrophic phenotypes depending on the local microenvironment (Biber et al., 2014).
Figure 1.
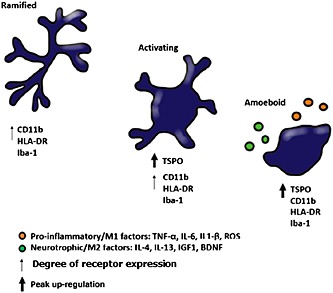
Microglial cell transitioning from ramified to amoeboid (activated state). The TSPO receptor is up‐regulated from the intermediate (‘activating’) stage of microglial activation, while CD11b, HLA‐DR and Iba‐1 reach peak up‐regulation in the amoeboid state. Activated microglia release a variety of pro‐inflammatory and neurotrophic factors that are used to identify microglia in the M1 and M2 states respectively.
The dynamic functions of microglial cells have emerged mostly through rodent studies of immune challenge and CNS damage. These dynamic cells help maintain a range of homeostatic functions in the healthy brain, including neuronal cell death, neuronal survival, synaptogenesis and synaptic pruning (Nayak et al., 2014). During activation, microglia up‐regulate a variety of molecules involved in antigen presentation, phagocytosis of cellular debris and cytokine production (Meda et al., 1995; Nakamura et al., 1999; Mack et al., 2003; Lauterbach et al., 2006; Neher et al., 2011; Brown and Neher, 2014). Microglial cells are not only activated under most pathological conditions and the drivers of repair but can also exacerbate a disease process depending on their activation profile (Napoli and Neumann, 2010). Furthermore, mutant microglia can cause behavioural symptoms of both obsessive–compulsive disorder and autism spectrum disorder in genetic mouse models, which resolved upon correction of the microglial genetic mutation (Chen et al., 2010; Derecki et al., 2012). Their neurotoxic and neuroprotective capacities suggest microglia are ‘double‐edged swords’, which has prompted interest in their potential role in neuropsychiatric disorders such as schizophrenia (Frick et al., 2013).
Microglial activation in schizophrenia – evidence from neuropathological studies
Post‐mortem studies in schizophrenia have primarily focused on assessing microglial cell number or density rather than microglial activation. The assessment of microglial number in post‐mortem samples is performed using immunohistochemical staining methods, which involve labelling microglial cells and counting these cells blinded to diagnosis utilizing stereological sampling techniques. The density of microglial cells is then obtained as counts per unit area. Overall, these studies have produced mixed results. While some studies have found increased microglial cell number in several brain regions in schizophrenia (Radewicz et al., 2000; Wierzba‐Bobrowicz et al., 2005; Fillman et al., 2013; Rao et al., 2013), others found no difference (Table 1; Steiner et al., 2006; Steiner et al., 2008; Busse et al., 2012).
Table 1.
Microglial density/number in schizophrenia from post‐mortem studies
| Study | Sample | Marker | Brain region | Density/no. | Cytokine levels | Age mean (SCZ/HC) (years) | Illness duration mean ± SD (range) (years) |
|---|---|---|---|---|---|---|---|
| Kurumaji et al. (1997) | 13 patients 10 controls | PK11195 | Putamen; VA1, superior parietal cortex | ↓ activation | N/A | 77/63 | N/A |
| Arnold et al. (1998) | 23 patients 10 controls | CD‐68 | TC; FC; OC | ≈ | N/A | 80/75 | 55 ± 9 |
| Bayer et al. (1999) | 14 patients 13 controls | HLA‐DR | FC; HPC | 4/14 SCZ positive for HLA‐DR | N/A | 64/58 | (7–43) |
| Radewicz et al. (2000) | 12 patients 11 controls | HLA‐DR | DLPFC; STG | ↑ | N/A | 80/70 | N/A |
| Wierzba‐Bobrowicz et al. (2005) | 9 patients 6 controls | HLA‐DR | FC; TC | ↑ | N/A | 56/56 | 24 (N/A SD) (10–35) |
| Steiner et al. (2006) | 16 patients 16 controls | HLA‐DR | DLPFC; HPC; ACC; MDT | ≈ | N/A | 55/58 | 23 ± 10 (3–39) |
| Steiner et al. (2008) | 16 patients 10 controls | HLA‐DR | DLPFC; HPC ACC; MDT | ≈ | N/A | 53/54 | 21 ± 12 (1–41) |
| Busse et al. (2012) | 17 patients 11 controls | HLA‐DR | HPC | ≈ except between paranoid versus residual SCZ (↑ in paranoid) | N/A | 52/56 | 19 ± 10 (2–34) |
| Kreisl et al. (2013) | 45 patients 47 controls | [3H]PBR28 | DLPFC | ↑ BP in SCZ (after controlling for binding affinity) | N/A | 55/42 | N/A |
| Fillman et al. (2013) | 37 patients 37 controls | HLA‐DR | DLPFC | ↑ density in WM ≈ in GM | ↑ IL‐6 and IL‐8 ≈ in Cox2 or IL‐1 | 51/51 | 28 ± 14 |
| Rao et al. (2013) | 10 patients 10 controls | CD11b and HLA‐DR | FC | ↑ Cd11b mRNA and protein | ↑ IL‐1b, TNF‐α, NF‐κB (but not IL‐1r) gene and protein expression | 50/59 | N/A |
| Hercher et al. (2014) | 20 patients 20 controls | Iba‐1 | DLPFC | ≈ in WM | N/A | 45/45 | 24 ± 10.9 |
No between‐group difference.
Increased in schizophrenia.
Decreased in schizophrenia.
VA1, visual area 1; CD‐68, cluster of differentiation 68; TC, temporal cortex; OC, occipital cortex; ACC, anterior cingulate cortex; MDT, mediodorsal thalamus; HPC, hippocampus; SCZ, schizophrenia.
These differences could be attributed to methodological variations including different methods of visually identifying and counting microglial cells and the use of different immunohistochemical markers. The majority of studies examining microglial density use either HLA‐DR or ionized calcium‐binding adaptor molecule 1 (Iba‐1) to label microglial cells. Both these markers, however, have a similar immunohistochemical staining profile, in that they label both activated and quiescent microglia (Steiner et al., 2006; Steiner et al., 2008) in addition to lymphocytes and peripheral monocytes (Kelemen and Autieri, 2005). It is instructive nonetheless to note that two studies examining the microglial density in white matter and conducted in the same brain region, the dorsal lateral prefrontal cortex (DLPFC), yielded conflicting results (Fillman et al., 2013; Hercher et al., 2014). The main methodological difference between these studies was the immunohistochemical marker used to label microglia, with Fillman et al. (2013) using HLA‐DR and finding increased microglial density, while Hercher et al. (2014) used Iba‐1 and found no increase. An alternative explanation for this discrepancy is that the studies differed in the visual selection process of microglial cells during the counting stage. In particular, Hercher counted only immunostained cells that were not encircling blood vessels, due to the fact that the cells encircling blood vessels are more likely to be peripheral monocytes rather than brain‐resident microglial cells. Although the marker used by Fillman et al. (2013) similarly labels both monocytes and microglia, they did not follow the strategy by Hercher et al. (2014) of eliminating potential monocytes from their cell counts, thereby, potentially overestimating microglia numbers. Of course, this would not change the results unless schizophrenia patients have increased peripheral monocytes compared with controls, an intriguing possibility that has yet to be assessed due to the lack of a suitably sensitive immunohistochemical marker.
Other technical difficulties that may account for the discrepancies between neuropathological studies of microglia in schizophrenia are differences in post‐mortem interval, brain pH (correlated in Hercher et al., 2014, with microglial density), as well as differences in age, clinical variables, illness duration and medication history in the post‐mortem samples used. Owing to the paucity of detailed ante‐mortem clinical information (Sundqvist et al., 2008), only two studies examined a potential correlation between clinical variables and microglial density, with one study finding no correlation (Arnold et al., 1998), while another found that increased microglial density was confined to the paranoid schizophrenia subgroup (Busse et al., 2012). Antipsychotic medication is another variable that may affect microglial numbers; however, the results of the studies examining this association are mixed. Two studies found no correlation between lifetime antipsychotic dosage and microglial density (Steiner et al., 2006; Rao et al., 2013), while another study found a negative correlation (Arnold et al., 1998). Finally, while the majority of post‐mortem studies include secondary analyses controlling for demographic variables, sample sizes may be insufficient for such analyses, and thus, caution should be exercised when interpreting these results (Fornito et al., 2008).
Another problem for neuropathological studies is that the immunohistochemical markers (HLA‐DR and Iba‐1) label both quiescent and amoeboid microglia; therefore, it is difficult to assess microglial activation. Attempts have been made to assess microglial activation through morphological characterization of these cells; although this is limited by the subjective nature of this classification. In their qualitative study, while no group differences were found in microglial density, Hercher et al. (2014) found that 15% of brains with schizophrenia showed an increased number of activated microglia, identified by their amoeboid morphology. Rao et al. (2013) performed a similar morphological characterization of microglial cells and found that the cell body of these cells in the frontal cortex (FC) of schizophrenia patients was more defined and their processes were less fine, indicating that microglia were potentially showing some degree of activation in patients, although not fully amoeboid. This morphological difference indicates that microglia from schizophrenia patients may be in a different state of activation when compared with controls, which may constitute a normal response to neuronal pathology or hint at an underlying, potentially genetic or epigenetic microglial dysfunction.
To overcome the subjective nature of these studies, alternative methods to quantify microglial activation include the use of markers such as [3H]‐PK11195, [3H]‐PBR28 and CD11b, which (unlike HLA‐DR and Iba‐1) are up‐regulated exclusively on the surface of activated microglial cells (Banati et al., 2000; Frick et al., 2013; Kreisl et al., 2013). The earliest post‐mortem study using the [3H]‐PK11195 ligand found decreased binding on microglial cells in patients (Kurumaji et al., 1997), while subsequent studies reported increased binding (Kreisl et al., 2013; Rao et al., 2013). A notable factor in the Kurumanji study is that it included an unusually high proportion of patients (7/13) that had been off antipsychotics for 40 days or more prior to their death. Kreisl et al. (2013) found significantly increased binding of [3H]‐PBR28 in the DLPFC of schizophrenia patients, after controlling for genetically determined binding affinity. This study illustrates the difficulty in devising immunohistochemical markers that are both specific to microglia and equally applicable across populations. However, in keeping with this study, Rao et al. (2013) found increased microglial activation in the frontal cortex using CD11b. They also found increased expression of pro‐inflammatory cytokines IL‐6, TNF‐a and NF‐κB in these patients, which may also be indicative of increased microglial activation, given that pro‐inflammatory cytokines are released by activated microglia (Kettenmann et al., 2011). Interestingly, Fillman et al. (2013) also found increased levels of IL‐6, IL‐8 and IL‐1β in the white matter of patients with schizophrenia, which provides further evidence that pro‐inflammatory cytokine gene expression is increased in post‐mortem brains of patients with schizophrenia. Therefore, evidence from post‐mortem studies suggests that there is increased microglial activation in the frontal cortex of schizophrenia patients as assessed by protein quantification (Kreisl et al., 2013; Rao et al., 2013), which is accompanied by increased expression of pro‐inflammatory cytokines in the same region (Rao et al., 2013), as well as in white matter (Fillman et al., 2013).
As we have seen however, there are conflicting results regarding the measurement of microglial density and number in schizophrenia, which may be because increased inflammation and microglial number occur only in a subgroup of patients with schizophrenia and that given the small sample sizes of earlier post‐mortem studies, it was difficult to discern the existence of such a subgroup. There is support for this hypothesis from a recent study by Fillman et al. (2013), who found that schizophrenia patients could be divided into a high and low inflammatory group on the basis of their gene expression levels of pro‐inflammatory cytokines. A subgroup of patients with a higher inflammatory profile (potentially consisting of patients in a particular stage or state of the illness) may exhibit increased microglial cell number and activation. Further evidence for the existence of a patient subgroup with a high inflammatory profile stems from the study by Busse et al. (2012), which showed that there was a significant difference in microglial number between patients with paranoid versus residual schizophrenia. This may indicate that patients experiencing active psychosis (paranoid schizophrenia group) may show a greater microglial proliferation than the patients in a non‐acute or quiescent state (residual schizophrenia group). It is also instructive to note that in the study by Busse et al. (2012) five out of the 17 patients with paranoid schizophrenia committed suicide, whereas none of the patients with residual schizophrenia took their own life, which again indicates that the paranoid schizophrenia group demonstrated both increased level of illness acuity and increased microglial number (Busse et al., 2012). Furthermore, when suicide was examined as a factor within the paranoid group, it was found to be non‐significant, suggesting that suicide was not the driving factor behind the increased microglial number. Rather, a possible driver for the increase in microglial number may have been illness acuity, medication cessation or symptom severity; although this hypothesis was not examined in their study (Busse et al., 2012). Suicide has been associated with increased microglial number in patients diagnosed with depression and schizophrenia, independently of their respective diagnosis (Steiner et al., 2008), suggesting that factors associated with suicide such as illness acuity or stress may have driven the microglial changes. Indeed, it has been proposed that microglial activation may be most apparent during acute illness relapses, which would also be consistent with the timing of structural brain changes (Cropley et al., 2013; Cropley and Pantelis, 2014).
In summary, the investigation of microglia in post‐mortem brains in schizophrenia is at a relatively early stage. The post‐mortem determination of microglial density suffers from potential confounds such as sampling bias towards violent death and variation in methodological techniques across laboratories, which may explain the mixed findings. Encouraging steps have been taken recently towards assessing microglial activation rather than density, which include the use of immunohistochemical markers specific to microglial activation (Kreisl et al., 2013; Rao et al., 2013) as well as cytokine quantification in the brain as a corollary of microglial activation (Fillman et al., 2013; Rao et al., 2013). These studies have all shown an increase in microglial activation in post‐mortem brains of patients with schizophrenia, which complement the positive findings seen in neuroimaging studies of microglial activation in this disorder discussed below (van Berckel et al., 2008; Doorduin et al., 2009).
Microglial activation in schizophrenia – evidence from PET studies
PET using specific radioligands for the 18‐kDa translocator protein (TSPO) allows quantification of activated microglia in vivo (Banati et al., 2000). TSPO is expressed primarily on the mitochondrial membrane of microglia and to a lesser extent on reactive astrocytes in the CNS (Chen and Guilarte, 2008; Lavisse et al., 2012). The relative contribution of microglia and astrocytes to the TSPO signal remains unclear but may depend on the timing and nature of the neuronal insult (Cosenza‐Nashat et al., 2009). Importantly, TSPO expression is negligible in the healthy brain but is increased in activated microglia (Ching et al., 2012) and, therefore, has been used as an indicator of microglial activation and neuroinflammation (Banati et al., 2000). However, TSPO up‐regulation does not exclusively indicate classically activated microglial cells, which are amoeboid, phagocytic and release pro‐inflammatory cytokines (Banati, 2002). Rather, its expression has also been associated with partially activated microglia, which appear less ramified with thicker and shorter processes (Marshall et al., 2013).
A number of radioligands have been developed to examine TSPO. The most commonly used is isoquinoline carboxamide, [11C]‐(R)‐PK11195, which has shown increased binding in multiple sclerosis and autoimmune encephalomyelitis (Shah et al., 1994; Vowinckel et al., 1997; Banati et al., 2000). However, this tracer has been reported to produce low penetration into the target tissue and low specificity, in part due to its lipophilic nature, which promotes binding within fatty structures of the brain (Petit‐Taboué et al., 1991; Shah et al., 1994; Chauveau et al., 2009). To overcome these limitations, over 40 second‐generation tracers for TSPO have been developed, with some yielding higher binding affinity and lower liphophilicity compared with [11C]‐(R)‐PK11195, such as [18F]‐FEPPA and [11C]‐PBR28 (Imaizumi et al., 2008; Wilson et al., 2008; Fujimura et al., 2009). Despite improved signal, inter‐subject variability in the rs6971 polymorphism can alter the binding affinity of second‐generation tracers. Therefore, PET studies using these ligands require plasma assays or genetic testing to control for differences in TSPO polymorphisms (Kreisl et al., 2010; Owen et al., 2012).
Five studies have applied PET imaging with TSPO ligands to assess microglial activation in schizophrenia (Table 2). The earliest report was by van Berckel et al. (2008), who examined [11C]‐(R)‐PK11195 ligand binding to TSPO in recent‐onset patients. They found increased ligand retention in the whole‐brain grey matter of recent‐onset patients compared with healthy controls. This suggests that an active immune pathology, characterized by increased TSPO expression, exists at the early stage of the illness. A later study by Banati and Hickie (2009) applied regions of interest to examine the spatial distribution of [11C]‐(R)‐PK11195 binding in patients with schizophrenia. This approach identified increased binding in a subset of patients within 15 of 28 regions investigated, suggesting widespread pathology. In contrast, Doorduin et al. (2009) reported a focal increase in [11C]‐(R)‐PK11195 binding potential (BP) in the hippocampus (HPC) of patients recovering from an acute psychotic episode. Regional discrepancies between studies may indicate a marked variation in TSPO expression in people with schizophrenia, possibly related to patient characteristics, such as symptom acuity.
Table 2.
PET studies examining microglial activation in schizophrenia
| Study | Sample | Mean age (years) | Mean DOI (years) | Radioligand | BP results | Correlations |
|---|---|---|---|---|---|---|
| van Berckel et al. (2008) | 10 patients | 24 | 3.1 | [11C]‐(R)‐PK11195 | ↑ BP in whole brain in patients | |
| 10 controls | 23 | |||||
| Banati and Hickie (2009) | 16 patients | 39.4 | 3 months–30 years (range) | [11C]‐(R)‐PK11195 | ↑ BP 15 of 28 regions. More marked on the right side. | |
| 8 controls | 37.6 | |||||
| Doorduin et al. (2009) | 7 recovering from psychosis | 31.3 | 5.3 | [11C]‐(R)‐PK11195 | ↑ BP in the HPC of patients. | |
| 7 controls | 26.8 | 30% ↑ BP in whole‐brain GM and WM. | ||||
| Takano et al. (2010) | 14 patients | 43.9 | 18.8 | [11C]‐DAA1106 | ≈ in BP | Relationship between BP and age, DOI, duration of drug treatment and positive symptom scores in patients. |
| 14 controls | 42.5 | |||||
| Kenk et al. (2015) | 16 patients | 42.5 | 14.8 | [18F]‐FEPPA | ≈ in BP | Positive correlation between BP and number of acute crises in the striatum. |
| 27 controls | 43.5 |
No between‐group difference.
Increased in schizophrenia.
Decreased in schizophrenia.
DOI, duration of illness.
More recently, Takano et al. (2010) used a second‐generation ligand for TSPO and did not detect a difference in [11C]‐DAA1106 BP between chronic patients and healthy controls but found a positive correlation between BP and positive symptom severity. Kenk et al. (2015) recently replicated these results in a larger sample (16 schizophrenia and 27 healthy controls). Notably, the mean illness duration was 18.8 years in Takano et al. (2010) and 14.8 years in Kenk et al. (2015), in contrast to former studies (3.1 years in van Berckel et al., 2008, and 5.3 years in Doorduin et al., 2009). These findings suggest that TSPO expression may differ as a function of illness duration or illness stage. This notion is supported by trajectories of brain loss observed in schizophrenia, which progresses faster around illness inception and plateaus later after 4–5 years (Takahashi et al., 2009a,2009b; Cropley and Pantelis, 2014).
These studies implicate microglial activation in some individuals with schizophrenia; in particular, those in the early stages of schizophrenia and with increased symptom acuity. However, positive findings were produced by studies using [11C]‐(R)‐PK11195 and tended to target individuals with shorter illness durations. Therefore, it is difficult to determine whether discrepant findings across studies were due to illness duration or the specific radioligand utilized. This could be addressed in a future study that directly compared microglial activation between recent‐onset and chronic patients with schizophrenia, using the same radioligand. Furthermore, differences in signal detection across studies may be due to a variation in the region‐of‐interest selection during image processing. In particular, four of the five studies either examined whole‐brain BP or applied coarse regions of interest to detect patterns of activation, which may lack specificity. Voxel‐based methods may be more suited to capturing the spatial distribution of microglial activation. Lastly, TSPO expression may be attributed to microglial or astrocytic cell expression, as well as microglia across various activation states and phenotypes; therefore, in vivo PET studies would benefit from markers specific to microglia phenotype and activation state to identify precise therapeutic targets.
Linking microglia to brain changes: the search for mechanisms
Microglia dysfunction may involve both M1/inflammatory and M2/neuroprotective profiles, which would have wide‐ranging implications in the protection of the CNS and its development and maintenance. In particular, a balance between these two profiles has been shown to be crucial for the recovery of the brain from acute injury (Bodhankar et al., 2015; Han et al., 2015). However, little is known regarding impaired M2 polarization and its effect on the CNS, particularly with respect to schizophrenia. Microglia in a pro‐inflammatory state are recognized to contribute to neuronal and axonal degeneration in human neurodegenerative diseases (Schmitz and Chew, 2008; Bernstein et al., 2009; Ilieva et al., 2009). The ‘neuroinflammatory hypothesis’ states that the cytotoxic effects of persistent microglial activation might cause secondary neuronal degeneration, decreased neurogenesis and synaptic dysfunction, and thus, disease progression (Munn, 2000). This hypothesis has been used to explain the progression of not only schizophrenia (Munn, 2000; Monji et al., 2009), but other neuropsychiatric syndromes including bipolar disorder (Stertz et al., 2013), delirium (Cerejeira et al., 2010), Alzheimer's disease (Rojo et al., 2008) and normal ageing (Bilbo et al., 2012). However, there are few empirical studies that address this issue. In vivo, increased [11C]‐(R)‐PK11195 binding in dementia was reported to overlap with regions showing progressive atrophy on MRI scans (Cagnin et al., 2001) and, further, tissue loss indexed by volumetric brain changes in relapsing multiple sclerosis was associated with periods of active inflammation preceded by an initial volume increase (Cheriyan et al., 2012). Together, these findings implicate microglial activation and inflammatory processes in structural brain changes.
In schizophrenia, both neuropathological and MRI studies suggest that grey matter alterations are most pronounced in frontal and temporal regions (Glantz and Lewis, 2000; Fornito et al., 2009; Olabi et al., 2011; Konopaske et al., 2014). This overlap extends to studies of microglial dysfunction, with increases reported in the DLPFC and superior temporal gyrus (STG) (Radewicz et al., 2000; Wierzba‐Bobrowicz et al., 2005; Banati and Hickie, 2009; Rao et al., 2013). Similarly, grey matter alterations have been detected in frontal and subcortical structures, such as the HPC (Uranova et al., 2007; Whitford et al., 2007; Andreasen et al., 2011; Uranova et al., 2011; Fung et al., 2014), which also show microglial activation (Doorduin et al., 2009; Fillman et al., 2013). These studies support the notion that structural brain changes co‐occur with microglial dysfunction in both white and grey matter. However, findings from studies examining microglial cells (Arnold et al., 1998; Steiner et al., 2008; Takano et al., 2010; Busse et al., 2012; Hercher et al., 2014; Kenk et al., 2015) and structural brain change (Ikeda et al., 2004; Eastwood and Harrison, 2005; Connor et al., 2009; Mitelman et al., 2009) are mixed, involve multiple brain regions and have not been examined in the same sample; therefore, the spatial overlap remains tentative. Crucially, anatomical co‐occurrence of microglial alterations and brain pathology does not imply that the two are necessarily functionally related. This assessment is critical and should be examined in future studies in order to elucidate whether microglial dysfunction contributes to the morphological changes seen in schizophrenia.
Very few studies in schizophrenia have addressed a possible association between inflammatory processes and structural brain changes in the same sample. Most of these studies examined a potential interaction between peripheral cytokine levels and the changes in white and/or grey matter in schizophrenia. Specifically, decreased anti‐inflammatory cytokine levels have been associated with smaller hippocampal volume (Bossu et al., 2015), while increased pro‐inflammatory cytokines were associated with decreased prefrontal cortical thickness (Cannon et al., 2015), reduced grey matter volume (Fillman et al., 2015), as well as decreased white matter integrity (Prasad et al., 2014). The finding that decreased anti‐inflammatory cytokines are associated with reduced brain volume may indicate a deficiency in the M2/neurotrophic role of microglia in schizophrenia. Conversely, the finding that increased pro‐inflammatory cytokines are associated with reduced cortical thickness and white matter integrity implicates the M1 pro‐inflammatory phenotype. These findings are supported by associations between genetic polymorphisms of these cytokines and alterations in the structure (Meisenzahl et al., 2001; Papiol et al., 2005; Kalmady et al., 2014) and function (Papiol et al., 2007; Fatjo‐Vilas et al., 2012) of the brain in schizophrenia. While these studies are informative, they did not examine microglial function. To our knowledge, the only study that examined microglial activation (TSPO) and regional grey matter volume (MRI) did not find any association between the two (Kenk et al., 2015). However, the association between microglial activation and brain changes may exist in an ‘inflammatory’ subset of patients (e.g. Fillman et al., 2013) and vary as a function of illness severity or duration. Therefore, further clinical studies are required to clarify the existence of a link between various functions of microglia and structural brain changes in subgroups of schizophrenia patients.
Despite the paucity of clinical studies directly examining microglial activation in association with structural brain changes, recent animal models have addressed this link. Below, we focus on a range of animal models of schizophrenia that: (i) investigate the association between microglial activation and behavioural impairments of schizophrenia and (ii) explore the impact of microglial dysfunction on the brain. While rodent models cannot capture the full scope of the disorder, they can be used to model certain features or endophenotypes and, unlike clinical studies, allow for the investigation of mechanisms.
A number of animal studies indicate an association between microglial activation and a range of schizophrenia endophenotypes such as sensorimotor gating measured by pre‐pulse inhibition (PPI) and working memory deficits. These studies employ a pre‐ or postnatal infection paradigm that involves the exposure of rodents to pro‐inflammatory agents such as human immunodeficiency virus (HIV; Paris et al., 2015), polyinosinic‐polycytidylic acid (poly I:C; Ribeiro et al., 2013; Van den Eynde et al., 2014) and GM‐CSF (Zhu et al., 2014). Notably, the rodents in all these studies exhibited at least one behavioural correlate of schizophrenia, specifically impaired PPI (Ribeiro et al., 2013; Liaury et al., 2014; Zhu et al., 2014; Paris et al., 2015) and cognitive deficits (Ribeiro et al., 2013; Liaury et al., 2014; Van den Eynde et al., 2014), in conjunction with increased microglial density or activation. Both the behavioural impairments and microglial activation were then reversed upon treatment with atypical antipsychotics (Ribeiro et al., 2013) or treatments targeting microglia and inflammation (e.g. minocycline (Zhu et al., 2014). In one study, the rodent strain itself demonstrated spontaneous symptoms of schizophrenia that were then alleviated by similar treatments (Liaury et al., 2014). Taken together, these preclinical studies: (i) support the epidemiological evidence for an association between prenatal/perinatal infection and an increased risk of schizophrenia and (ii) imply a causative role for microglia in mediating this process, as treatments targeting microglia (e.g. minocycline) are successful in alleviating both microglial activation and behavioural impairments.
It is noteworthy that some of these animal studies show that microglial activation does not arise instantaneously in response to the infectious agent but rather grows steadily throughout the lifespan, reaching a peak in late adolescence and early adulthood, which coincides with the emergence of behavioural impairments (Ribeiro et al., 2013; Van den Eynde et al., 2014). These studies indicate that a pre‐ or perinatal infection may serve to prime rather than explosively activate microglia, which then interact with cells in the developing nervous system leading to a subtle rearrangement of synaptic circuitry that may over time result in behavioural impairments in adolescence. These studies point to a subtle role for microglia and provide a compelling argument for schizophrenia emerging within an overarching neurodevelopmental context. Future studies are required to elucidate whether the symptoms of schizophrenia arise from an acute inflammatory reaction, a chronic low‐grade microglial response or an interplay between the two states played out against the background of the developing nervous system.
While the above studies address a potential interaction between microglial activation and behavioural impairment, they do not address the mechanism by which microglial activation can mediate brain pathology. This question is addressed by the following set of preclinical studies that illustrate a wide variety of potential mechanisms by which microglial dysfunction may contribute to brain changes and behavioural deficits in schizophrenia. Three main findings emerge from animal studies investigating the effect of microglial activation on grey matter. The most replicated finding is decreased hippocampal neurogenesis in response to microglial activation (Furuya et al., 2013; Mattei et al., 2014). Pro‐inflammatory cytokines (TNF‐α and IL‐1β) were shown to mediate the relationship between microglial activation (M1 phenotype) and hippocampal neurogenesis (Mattei et al., 2014), which implicates a disruption of this inflammatory cascade in schizophrenia. Importantly, both studies linking microglial activation to neurogenesis, resulted in behavioural impairments analogous to schizophrenia, including PPI (Mattei et al., 2014) and working‐memory deficits (Furuya et al., 2013).
In addition to neurogenesis, microglial activation has been shown to decrease reelin expression in the HPC, which was accompanied by impairments in working‐memory and motor skills (Ratnayake et al., 2012). Interestingly, decreased reelin expression has been reported in post‐mortem studies of schizophrenia (Eastwood and Harrison, 2003) and may underlie impaired neuronal migration and connectivity during development (Folsom and Fatemi, 2013). Finally, and in support of impaired connectivity in schizophrenia, aberrant synapse formation was found in response to microglial activation (Sargin et al., 2009). With regard to white matter, several studies report that elevated microglial cells are accompanied by myelin and oligodendrocyte loss particularly in the frontal cortex (Zhang et al., 2008; Morita et al., 2014; Zhang et al., 2014), which were reversed by the administration of the antipsychotics quetiapine (Zhang et al., 2008) and olanzapine (Zhang et al., 2014). The association between microglial activation, myelin and oligodendrocyte loss in these animal models may underlie connectivity impairments observed in schizophrenia (Zalesky et al., 2011). These findings are in contrast to results from post‐mortem studies predominantly showing morphological deficits rather than reduced oligodendrocyte number (Uranova et al., 2011). However, such studies did not examine oligodendrocyte number in conjunction with microglial activation. Nonetheless, these rodent models demonstrate a wide variety of mechanisms by which microglial activation may induce grey and white matter deficits in schizophrenia.
Given the diverse mechanisms through which microglial activation affects the brain, it is plausible that the changes in the brain in response to microglial dysfunction will be similarly complex. The role of synaptic pruning in neuronal circuitry is of particular interest in a neurodevelopmental context (Carr, 2015; Riccomagno and Kolodkin, 2015). However, few studies have addressed the link between microglial dysfunction, synaptic impairment and whole‐brain connectivity. One notable exception is Zhan et al. (2014), who examined synaptic plasticity in CX3CR1 (fractalkine receptor)‐knockout mice. These mice exhibited a transient reduction in microglial number, which resulted in impaired synaptic pruning and concomitant functional dysconnectivity as shown through MRI in the adult knockout mice. This indicates that microglia abnormalities are sufficient to cause circuit‐level functional dysconnectivity, mediated through abnormal synaptic maturation. Although this study does make significant inroads in associating microglial dysfunction with synaptic plasticity, it is noteworthy that this rodent model produced social interaction deficits, which are seen in a variety of psychiatric disorders including schizophrenia, suggesting that the process may be relevant to a number of psychiatric and neurological conditions and depend on specific circuits that are disrupted (Ratnayake et al., 2012).
Furthermore, it is possible that microglial cells are themselves responding to a pre‐existing brain pathology and, therefore, the direction of this relationship remains unclear. For example, myelin damage in a mouse was shown to induce microglial activation, while both myelin damage and microglial activation were alleviated by administration of the antipsychotic quetiapine (Zhang et al., 2008). This indicates that the pathology of white matter may be the primary factor triggering an inflammatory‐degenerative process. This ‘chicken or egg’ question is further complicated by studies demonstrating neuronal damage in the absence of microglial activation. For example, Hou et al. (2013) demonstrated that chronic ketamine exposure in mice was associated with increased oxidative stress, decreased hippocampal neurogenesis and cognitive and sensorimotor symptoms analogous to schizophrenia, notably without microglial increase. As ketamine is an NMDA receptor antagonist and the hypofunctioning of the NMDA receptor is a key aetiological theory of schizophrenia (Moghaddam and Javitt, 2012), this study indicates that microglial activation may represent one of many pathogenetic mechanisms underlying neuronal damage.
Summary and road map
Neuroimaging and neuropathological studies have revealed microglial activation or dysfunction in some schizophrenia patients. The existence of a neuroinflammatory subset could contribute to the mixed findings across studies, as sample sizes may have been insufficient to expose such a subset. The ‘neuroinflammatory’ hypothesis proposes that this group of patients would undergo progressive brain changes as a result of microglial dysfunction. However, the relationship between microglial activation and brain abnormalities remains unclear, as there are limited clinical studies addressing this link. Nonetheless, animal models of schizophrenia have addressed potential mechanisms that may underlie this relationship and implicate decreased neurogenesis and reelin expression, as well as reduced oligodendrocyte number and myelination in response to microglial activation.
What emerges from these preliminary investigations is that microglia are highly diverse cells, serving both pro‐inflammatory and neuroprotective functions. Therefore, disrupted microglia in schizophrenia would likewise have diverse and wide‐ranging consequences for brain function. In light of this complexity, future studies should focus on elucidating the functions of microglia during healthy neurodevelopment, which would provide a foundation for understanding microglial dysfunction in relation to key developmental windows of relevance to schizophrenia and other neurodevelopmental disorders. Furthermore, microglial markers could be developed that identify microglia across different activation states and phenotypes. This could determine the relative contribution of the M1/pro‐inflammatory and M2/neurotrophic phenotypes to the pathophysiology of schizophrenia. In particular, the effect of the M1 and M2 microglial activation states on neurons could be examined in relation to microglial functions such as synaptic pruning and strengthening. Such molecular investigations may shed light on the manner in which microglial activation is associated with the macroscopic structural brain changes in schizophrenia. Furthermore, given that MHC class proteins are expressed on neurons, as well as microglia (Boulanger and Shatz, 2004), immune system markers capable of identifying neurons could determine potentially shared mechanisms between CNS and immune system pathways, which may have implications for the proposed inflammatory hypothesis of schizophrenia. In particular, schizophrenia may not be primarily a disorder of inflammation but arise from disturbances of overlapping downstream molecular pathways, common to both the immune system and CNS.
Future PET and post‐mortem studies could address unresolved questions pertaining to microglia in schizophrenia. Firstly, increased sample sizes in patients with carefully documented clinical characteristics could clarify whether a subset of patients exhibit microglial dysfunction. These studies could address the characteristics of a ‘neuroinflammatory’ subset; in particular, the relative contribution of genotype, illness severity and duration, medication history and age. Secondly, more work is required to establish a link between microglial dysfunction and brain changes in schizophrenia. This could be achieved through longitudinal and multimodal imaging studies concurrently measuring microglial activation and structural brain changes. In doing so, these studies can determine whether microglial activation precedes or follows structural brain change, which may point to potential novel therapeutic targets. Furthermore, given microglia modify neuronal synapses; diffusion imaging could be used to identify connectivity impairments in conjunction with PET in order to elucidate a potential association between microglial activation and connectivity. Finally, treatment intervention studies could address whether microglia play a casual role in brain changes by utilizing a variety of novel drugs targeting microglial cells and assessing their effect on microglia‐specific markers and brain structure in schizophrenia.
Conflict of interest
Authors report no competing interests.
Acknowledgements
This study was supported by an Australian National Health and Medical Research Council (NHMRC) project grant (1065742), a NARSAD Distinguished Investigator grant awarded to C. P. (ID: 18722), and a University of Melbourne Early Career Researcher grant awarded to V. C. (601253). V. C. was supported by an NHMRC Early Career Fellowship (628880). M. A. D. was supported by the Rotary Health Ian Scott PhD Scholarship in Mental Health. L. L. was supported by the Australian Post‐Graduate Award PhD Scholarship. C. P. was supported by an NHMRC Senior Principal Research Fellowship (ID: 628386) and A. C. was supported by an NHMRC Principal Research Fellowship (APP1041875). E. S. was supported by the Clifford Chair in Neural Engineering. Thanks to Benjamin Rossi for creating the microglial activation figure.
Laskaris, L. E. , Di Biase, M. A. , Everall, I. , Chana, G. , Christopoulos, A. , Skafidas, E. , Cropley, V. L. , and Pantelis, C. (2016) Microglial activation and progressive brain changes in schizophrenia. British Journal of Pharmacology, 173: 666–680. doi: 10.1111/bph.13364.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013a). The concise guide to PHARMACOLOGY 2013/14: ligand‐gated ion channels. Br J Pharmacol 170: 1582–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013b). The concise guide to PHARMACOLOGY 2013/14: catalytic receptors. Br J Pharmacol 170: 1676–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M et al. (2013c). The concise guide to PHARMACOLOGY 2013/14: enzymes. Br J Pharmacol 170: 1797–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amminger GP, McGorry PD (2012). Update on omega‐3 polyunsaturated fatty acids in early‐stage psychotic disorders. Neuropsychopharmacology 37: 309–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC, Nopoulos P, Magnotta V, Pierson R, Ziebell S, Ho B‐C (2011). Progressive brain change in schizophrenia: a prospective longitudinal study of first‐episode schizophrenia. Biol Psychiatry 70: 672–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Trojanowski JQ, Gur RE, Blackwell P, Han LY, Choi C (1998). Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch Gen Psychiatry 55: 225–232. [DOI] [PubMed] [Google Scholar]
- Banati R, Hickie IB (2009). Therapeutic signposts: using biomarkers to guide better treatment of schizophrenia and other psychotic disorders. Med J Aust 190: S26. [DOI] [PubMed] [Google Scholar]
- Banati R, Newcombe J, Gunn RN, Cagnin A, Turkheimer F, Heppner F et al. (2000). The peripheral benzodiazepine binding site in the brain in multiple sclerosis: quantitative in vivo imaging of microglia as a measure of disease activity. Brain 123: 2321–2337. [DOI] [PubMed] [Google Scholar]
- Banati RB (2002). Visualising microglial activation in vivo . Glia 40: 206–217. [DOI] [PubMed] [Google Scholar]
- Banks WA (2009). The blood–brain barrier in psychoneuroimmunology. Immunol Allergy Clin North Am 29: 223–228. [DOI] [PubMed] [Google Scholar]
- Bayer TA, Buslei R, Havas L, Falkai P (1999). Evidence for activation of microglia in patients with psychiatric illnesses. Neurosci Lett 271: 126–128. [DOI] [PubMed] [Google Scholar]
- Bernstein H‐G, Steiner J, Bogerts B (2009). Glial cells in schizophrenia: pathophysiological significance and possible consequences for therapy. Expert Review of Neurotherapeutics 7: 1059–1071. [DOI] [PubMed] [Google Scholar]
- Biber K, Owens T, Boddeke E (2014). What is microglia neurotoxicity (not)? Glia 62: 841–854. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Smith SH, Schwarz JM (2012). A lifespan approach to neuroinflammatory and cognitive disorders: a critical role for glia. J Neuroimmune Pharmacol 7: 24–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodhankar S, Lapato A, Chen Y, Vandenbark AA, Saugstad JA, Offner H (2015). Role for microglia in sex differences after ischemic stroke: importance of M2. Metab Brain Dis 30: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora E, Fornito A, Radua J, Walterfang M, Seal M, Wood SJ et al. (2011). Neuroanatomical abnormalities in schizophrenia: a multimodal voxelwise meta‐analysis and meta‐regression analysis. Schizophr Res 127: 46–57. [DOI] [PubMed] [Google Scholar]
- Bossu P, Piras F, Palladino I, Iorio M, Salani F, Ciaramella A et al. (2015). Hippocampal volume and depressive symptoms are linked to serum IL‐18 in schizophrenia. Neurol Neuroimmunol Neuroinflammation 2 e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger LM, Shatz CJ (2004). Immune signalling in neural development, synaptic plasticity and disease. Nat Rev Neurosci 5: 521–531. [DOI] [PubMed] [Google Scholar]
- Brown AS (2011). The environment and susceptibility to schizophrenia. Prog Neurobiol 93: 23–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Hooton J, Schaefer CA, Zhang H, Petkova E, Babulas V et al. (2004). Elevated maternal interleukin‐8 levels and risk of schizophrenia in adult offspring. Am J Psychiat 161: 889–895. [DOI] [PubMed] [Google Scholar]
- Brown GC, Neher JJ (2014). Microglial phagocytosis of live neurons. Nat Rev Neurosci 15: 209–216. [DOI] [PubMed] [Google Scholar]
- Buka SL, Tsuang MT, Torrey EF, Klebanoff MA, Wagner RL, Yolken RH (2001). Maternal cytokine levels during pregnancy and adult psychosis. Brain Behav Immun 15: 411–420. [DOI] [PubMed] [Google Scholar]
- Busse S, Busse M, Schiltz K, Bielau H, Gos T, Brisch R et al. (2012). Different distribution patterns of lymphocytes and microglia in the hippocampus of patients with residual versus paranoid schizophrenia: further evidence for disease course‐related immune alterations? Brain Behav Immun 26: 1273–1279. [DOI] [PubMed] [Google Scholar]
- Byne W, Fernandes J, Haroutunian V, Huacon D, Kidkardnee S, Kim J et al. (2007). Reduction of right medial pulvinar volume and neuron number in schizophrenia. Schizophr Res 90: 71–75. [DOI] [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE et al. (2001). In‐vivo measurement of activated microglia in dementia. Lancet 358: 461–467. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Chung Y, He G, Sun D, Jacobson A, van Erp TG et al. (2015). Progressive reduction in cortical thickness as psychosis develops: a multisite longitudinal neuroimaging study of youth at elevated clinical risk. Biol Psychiatry 77: 147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr F (2015). Neural circuits: pruning the projections. Nat Rev Neurosci 16: 375–375. [DOI] [PubMed] [Google Scholar]
- Cerejeira J, Firmino H, Vaz‐Serra A, Mukaetova‐Ladinska EB (2010). The neuroinflammatory hypothesis of delirium. Acta Neuropathol 119: 737–754. [DOI] [PubMed] [Google Scholar]
- Chauveau F, Van Camp N, Dollé F, Kuhnast B, Hinnen F, Damont A et al. (2009). Comparative evaluation of the translocator protein radioligands 11C‐DPA‐713, 18 F‐DPA‐714, and 11C‐PK11195 in a rat model of acute neuroinflammation. J Nucl Med 50: 468–476. [DOI] [PubMed] [Google Scholar]
- Chen M‐K, Guilarte TR (2008). Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther 118: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S‐K, Tvrdik P, Peden E, Cho S, Wu S, Spangrude G et al. (2010). Hematopoietic origin of pathological grooming in Hoxb8 mutant mice. Cell 141: 775–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheriyan J, Kim S, Wolansky LJ, Cook SD, Cadavid D (2012). Impact of inflammation on brain volume in multiple sclerosis. Arch Neurol 69: 82–88. [DOI] [PubMed] [Google Scholar]
- Chew LJ, Fusar‐Poli P, Schmitz T (2013). Oligodendroglial alterations and the role of microglia in white matter injury: relevance to schizophrenia. Dev Neurosci 35: 102–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching ASC, Kuhnast B, Damont A, Roeda D, Tavitian B, Dollé F (2012). Current paradigm of the 18‐kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging 3: 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP et al. (2013). A neurodegeneration‐specific gene‐expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 4: 385–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor CM, Guo Y, Akbarian S (2009). Cingulate white matter neurons in schizophrenia and bipolar disorder. Biol Psychiatry 66: 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium SPG‐WAS (2011). Genome‐wide association study identifies five new schizophrenia loci. Nat Genet 43: 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium SWGotPG (2014). Biological insights from 108 schizophrenia‐associated genetic loci. Nature 511: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosenza‐Nashat M, Zhao M‐L, Suh H‐S, Morgan J, Natividad R, Morgello S et al. (2009). Expression of the translocator protein of 18 kDa by microglia, macrophages and astrocytes based on immunohistochemical localization in abnormal human brain. Neuropathol Appl Neurobiol 35: 306–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cropley V, Wood SJ, Pantelis C (2013). Brain structural, neurochemical and neuroinflammatory markers of psychosis onset and relapse: is there evidence for a psychosis relapse signature? Int Clin Psychopharmacol 28: 0268–1315. [Google Scholar]
- Cropley VL, Pantelis C (2014). Using longitudinal imaging to map the ‘relapse signature’ of schizophrenia and other psychoses. Epidemiol Psychiatr Sci 23: 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean OM, Data‐Franco J, Giorlando F, Berk M (2012). Minocycline. CNS Drugs 26: 391–401. [DOI] [PubMed] [Google Scholar]
- Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG et al. (2012). Wild‐type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484: 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doorduin J, de Vries EFJ, Willemsen ATM, de Groot JC, Dierckx RA, Klein HC (2009). Neuroinflammation in schizophrenia‐related psychosis: a PET study. J Nucl Med 50: 1801–1807. [DOI] [PubMed] [Google Scholar]
- Eastwood S, Harrison P (2003). Interstitial white matter neurons express less reelin and are abnormally distributed in schizophrenia: towards an integration of molecular and morphologic aspects of the neurodevelopmental hypothesis. Mol Psychiatry 8: 821–831. [DOI] [PubMed] [Google Scholar]
- Eastwood SL, Harrison PJ (2005). Interstitial white matter neuron density in the dorsolateral prefrontal cortex and parahippocampal gyrus in schizophrenia. Schizophr Res 79: 181–188. [DOI] [PubMed] [Google Scholar]
- Ellison‐Wright I, Bullmore E (2009). Meta‐analysis of diffusion tensor imaging studies in schizophrenia. Schizophr Res 108: 3–10. [DOI] [PubMed] [Google Scholar]
- Fatjo‐Vilas M, Pomarol‐Clotet E, Salvador R, Monte GC, Gomar JJ, Sarro S et al. (2012). Effect of the interleukin‐1beta gene on dorsolateral prefrontal cortex function in schizophrenia: a genetic neuroimaging study. Biol Psychiatry 72: 758–765. [DOI] [PubMed] [Google Scholar]
- Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T et al. (2013). Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 18: 206–214. [DOI] [PubMed] [Google Scholar]
- Fillman SG, Weickert TW, Lenroot RK, Catts SV, Bruggemann JM, Catts VS et al. (2015). Elevated peripheral cytokines characterize a subgroup of people with schizophrenia displaying poor verbal fluency and reduced Broca's area volume. Mol Psychiatry 18: 1359–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineberg A, Ellman L (2013). Inflammatory cytokines and neurological and neurocognitive alterations in the course of schizophrenia. Biol Psychiatry 73: 951–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folsom TD, Fatemi SH (2013). The involvement of reelin in neurodevelopmental disorders. Neuropharmacology 68: 122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornito A, Yücel M, Patti J, Wood S, Pantelis C (2009). Mapping grey matter reductions in schizophrenia: an anatomical likelihood estimation analysis of voxel‐based morphometry studies. Schizophr Res 108: 104–113. [DOI] [PubMed] [Google Scholar]
- Fornito A, Yung AR, Wood SJ, Phillips LJ, Nelson B, Cotton S et al. (2008). Anatomic abnormalities of the anterior cingulate cortex before psychosis onset: an MRI study of ultra‐high‐risk individuals. Biol Psychiatry 64: 758–765. [DOI] [PubMed] [Google Scholar]
- Foster R, Kandanearatchi A, Beasley C, Williams B, Khan N, Fagerhol MK et al. (2006). Calprotectin in microglia from frontal cortex is up‐regulated in schizophrenia: evidence for an inflammatory process? Eur J Neurosci 24: 3561–3566. [DOI] [PubMed] [Google Scholar]
- Frick LR, Williams K, Pittenger C (2013). Microglial dysregulation in psychiatric disease. Clin Dev Immunol 2013: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura Y, Zoghbi SS, Simèon FG, Taku A, Pike VW, Innis RB et al. (2009). Quantification of translocator protein (18 kDa) in the human brain with PET and a novel radioligand, 18 F‐PBR06. J Nucl Med 50: 1047–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung SJ, Joshi D, Fillman SG, Weickert CS (2014). High white matter neuron density with elevated cortical cytokine expression in schizophrenia. Biol Psychiatry 75: e5–e7. [DOI] [PubMed] [Google Scholar]
- Furuya M, Miyaoka T, Tsumori T, Liaury K, Hashioka S, Wake R et al. (2013). Yokukansan promotes hippocampal neurogenesis associated with the suppression of activated microglia in Gunn rat. J Neuroinflammation 10: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar‐Poli P, Smieskova R, Kempton MJ, Ho BC, Andreasen NC, Borgwardt S (2013). Progressive brain changes in schizophrenia related to antipsychotic treatment? A meta‐analysis of longitudinal MRI studies. Neurosci Biobehav Rev 37: 1680–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65–73. [DOI] [PubMed] [Google Scholar]
- Han L, Cai W, Mao L, Liu J, Li P, Leak RK et al. (2015). Rosiglitazone promotes white matter integrity and long‐term functional recovery after focal cerebral ischemia. Stroke 46: 2628–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hercher C, Chopra V, Beasley CL (2014). Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J Psychiat Neurosci 39: 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Zhang H, Xie G, Cao X, Zhao Y, Liu Y et al. (2013). Neuronal injury, but not microglia activation, is associated with ketamine‐induced experimental schizophrenia model in mice. Prog Neuropsychopharmacol Biol Psychiatry 45: 107–116. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Fukuto JM, Griscavage JM, Rogers NE, Byrns RE (1993). Oxidation of nitric oxide in aqueous solution to nitrite but not nitrate: comparison with enzymatically formed nitric oxide from l‐arginine. Proc Natl Acad Sci 90: 8103–8107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Ikeda K, Iritani S, Ueno H, Niizato K (2004). Distribution of neuropeptide Y interneurons in the dorsal prefrontal cortex of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 28: 379–383. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW (2009). Non‐cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol 187: 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaizumi M, Briard E, Zoghbi SS, Gourley JP, Hong J, Fujimura Y et al. (2008). Brain and whole‐body imaging in nonhuman primates of [11 C] PBR28, a promising PET radioligand for peripheral benzodiazepine receptors. Neuroimage 39: 1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung WH, Kim JS, Jang JH, Choi J‐S, Jung MH, Park J‐Y et al. (2011). Cortical thickness reduction in individuals at ultra‐high‐risk for psychosis. Schizophr Bull 37: 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmady SV, Venkatasubramanian G, Shivakumar V, Gautham S, Subramaniam A, Jose DA et al. (2014). Relationship between interleukin‐6 gene polymorphism and hippocampal volume in antipsychotic‐naive schizophrenia: evidence for differential susceptibility? PLoS One 9 e96021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato TA, Monji A, Mizoguchi Y, Hashioka S, Horikawa H, Seki Y et al. (2011). Anti‐Inflammatory properties of antipsychotics via microglia modulations: are antipsychotics a ‘fire extinguisher’ in the brain of schizophrenia? Mini Rev Med Chem 11: 565–574. [DOI] [PubMed] [Google Scholar]
- Kelemen SE, Autieri MV (2005). Expression of allograft inflammatory factor‐1 in T lymphocytes: a role in T‐lymphocyte activation and proliferative arteriopathies. Am J Pathol 167: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G et al. (2015). Imaging neuroinflammation in gray and white matter in schizophrenia: an in‐vivo PET study with [18 F]‐FEPPA. Schizophr Bull 41: 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011). Physiology of microglia. Physiol Rev 91: 461–553. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K et al. (2013). Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis 4 e525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopaske GT, Lange N, Coyle JT, Benes FM (2014). Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA psychiat 71: 1323–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl WC, Fujita M, Fujimura Y, Kimura N, Jenko KJ, Kannan P et al. (2010). Comparison of [(11)C]‐(R)‐PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: implications for positron emission tomographic imaging of this inflammation biomarker. Neuroimage 49: 2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL et al. (2013). A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab 33: 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicki M, Shenton ME (2014). Diffusion tensor imaging findings and their implications in schizophrenia. Curr Opin Psychiatry 27: 179–184. [DOI] [PubMed] [Google Scholar]
- Kurumaji A, Wakai T, Toru M (1997). Decreases in peripheral‐type benzodiazepine receptors in postmortem brains of chronic schizophrenias. J Neural Transm 104: 1361–1370. [DOI] [PubMed] [Google Scholar]
- Lauterbach H, Zuniga EI, Truong P, Oldstone MBA, McGavern DB (2006). Adoptive immunotherapy induces CNS dendritic cell recruitment and antigen presentation during clearance of a persistent viral infection. J Exp Med 203: 1963–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavisse S, Guillermier M, Hérard A‐S, Petit F, Delahaye M, Van Camp N et al. (2012). Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci 32: 10809–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkovitz Y, Mendlovich S, Riwkes S, Braw Y, Levkovitch‐Verbin H, Gal G et al. (2010). A double‐blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early‐phase schizophrenia. J Clin Psychiat 71: 138. [DOI] [PubMed] [Google Scholar]
- Liaury K, Miyaoka T, Tsumori T, Furuya M, Hashioka S, Wake R et al. (2014). Minocycline improves recognition memory and attenuates microglial activation in Gunn rat: a possible hyperbilirubinemia‐induced animal model of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 50: 184–190. [DOI] [PubMed] [Google Scholar]
- Mack CL, Vanderlugt‐Castaneda CL, Neville KL, Miller SD (2003). Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler's virus model of multiple sclerosis. J Neuroimmunol 144: 68–79. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Sica A, Locati M (2005). Macrophage polarization comes of age. Immunity 23: 344–346. [DOI] [PubMed] [Google Scholar]
- Marshall SA, McClain JA, Kelso ML, Hopkins DM, Pauly JR, Nixon K (2013). Microglial activation is not equivalent to neuroinflammation in alcohol‐induced neurodegeneration: the importance of microglia phenotype. Neurobiol Dis 54: 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattei D, Djodari‐Irani A, Hadar R, Pelz A, de Cossio LF, Goetz T et al. (2014). Minocycline rescues decrease in neurogenesis, increase in microglia cytokines and deficits in sensorimotor gating in an animal model of schizophrenia. Brain Behav Immun 38: 175–184. [DOI] [PubMed] [Google Scholar]
- Meda L, Cassatella MA, Szendrei GI, Otvos L, Baron P, Villalba M et al. (1995). Activation of microglial cells by [beta]‐amyloid protein and interferon‐[gamma]. Nature 374: 647–650. [DOI] [PubMed] [Google Scholar]
- Meisenzahl EM, Rujescu D, Kirner A, Giegling I, Kathmann N, Leinsinger G et al. (2001). Association of an interleukin‐1beta genetic polymorphism with altered brain structure in patients with schizophrenia. Am J Psychiatry 158: 1316–1319. [DOI] [PubMed] [Google Scholar]
- Mileaf MI, Byne W (2012). Neuronal deficit in medial pulvinar from right but not left hemisphere in schizophrenia. Schizophr Res 134: 291–292. [DOI] [PubMed] [Google Scholar]
- Miller B, Buckley P, Seabolt W, Mellor A, Kirkpatrick B (2011). Meta‐analysis of cytokine alterations in schizophrenia: clinical status and antipsychotic effects. Biol Psychiatry 70: 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitelman M, Canfield EL, Newmark R, Brickman AM, Torosjan Y, Chu K et al. (2009). Longitudinal assessment of gray and white matter in chronic schizophrenia: a combined diffusion‐tensor and structural magnetic resonance imaging study. Open Neuroimag J 3: 31–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaoka T, Wake R, Furuya M, Liaury K, Ieda M, Kawakami K et al. (2012). Minocycline as adjunctive therapy for patients with unipolar psychotic depression: an open‐label study. Prog Neuropsychopharmacol Biol Psychiatry 37: 222–226. [DOI] [PubMed] [Google Scholar]
- Miyaoka T, Yasukawa R, Yasuda H, Hayashida M, Inagaki T, Horiguchi J (2008). Minocycline as adjunctive therapy for schizophrenia: an open‐label study. Clin Neuropharmacol 31: 287–292. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Javitt D (2012). From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology 37: 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monji A, Kato T, Kanba S (2009). Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci 63: 257–265. [DOI] [PubMed] [Google Scholar]
- Monji A, Kato TA, Mizoguchi Y, Horikawa H, Seki Y, Kasai M et al. (2013). Neuroinflammation in schizophrenia especially focused on the role of microglia. Prog Neuropsychopharmacol Biol Psychiatry 42: 115–121. [DOI] [PubMed] [Google Scholar]
- Moran LB, Graeber MB (2004). The facial nerve axotomy model. Brain Res Rev 44: 154–178. [DOI] [PubMed] [Google Scholar]
- Morita S, Tatsumi K, Makinodan M, Okuda H, Kishimoto T, Wanaka A (2014). Geissoschizine methyl ether, an alkaloid from the Uncaria hook, improves remyelination after cuprizone‐induced demyelination in medial prefrontal cortex of adult mice. Neurochem Res 39: 59–67. [DOI] [PubMed] [Google Scholar]
- Munn NA (2000). Microglia dysfunction in schizophrenia: an integrative theory. Med Hypotheses 54: 198–202. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Si QS, Kataoka K (1999). Lipopolysaccharide‐induced microglial activation in culture: temporal profiles of morphological change and release of cytokines and nitric oxide. Neurosci Res 35: 95–100. [DOI] [PubMed] [Google Scholar]
- Napoli I, Neumann H (2010). Protective effects of microglia in multiple sclerosis. Exp Neurol 225: 24–28. [DOI] [PubMed] [Google Scholar]
- Nayak D, Roth TL, McGavern DB (2014). Microglia development and function. Annu Rev Immunol 32: 367–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Neniskyte U, Zhao J‐W, Bal‐Price A, Tolkovsky AM, Brown GC (2011). Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol 186: 4973–4983. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo . Science 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- Olabi B, Ellison‐Wright I, McIntosh AM, Wood SJ, Bullmore E, Lawrie SM (2011). Are there progressive brain changes in schizophrenia? A meta‐analysis of structural magnetic resonance imaging studies. Biol Psychiatry 70: 88–96. [DOI] [PubMed] [Google Scholar]
- Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A et al. (2012). An 18‐kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab 32: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelis C, Pantelis C, Yücel M, Wood SJ, McGorry PD, Velakoulis D (2003a). Early and late neurodevelopmental disturbances in schizophrenia and their functional consequences. Aust N Z J Psychiatry 37: 399–406. [DOI] [PubMed] [Google Scholar]
- Pantelis C, Velakoulis D, McGorry PD, Wood SJ, Suckling J, Phillips LJ et al. (2003b). Neuroanatomical abnormalities before and after onset of psychosis: a cross‐sectional and longitudinal MRI comparison. Lancet 361: 281–288. [DOI] [PubMed] [Google Scholar]
- Pantelis C, Velakoulis D, Wood S, Yücel M, Yung A, Phillips L et al. (2007). Neuroimaging and emerging psychotic disorders: the Melbourne ultra‐high risk studies. Int Rev Psychiatry 19: 371–379. [DOI] [PubMed] [Google Scholar]
- Pantelis C, Yücel M, Wood SJ, Velakoulis D, Sun D, Berger G et al. (2005). Structural brain imaging evidence for multiple pathological processes at different stages of brain development in schizophrenia. Schizophr Bull 31: 672–696. [DOI] [PubMed] [Google Scholar]
- Papiol S, Molina V, Desco M, Rosa A, Reig S, Gispert JD et al. (2005). Ventricular enlargement in schizophrenia is associated with a genetic polymorphism at the interleukin‐1 receptor antagonist gene. Neuroimage 27: 1002–1006. [DOI] [PubMed] [Google Scholar]
- Papiol S, Molina V, Rosa A, Sanz J, Palomo T, Fananas L (2007). Effect of interleukin‐1beta gene functional polymorphism on dorsolateral prefrontal cortex activity in schizophrenia patients. Am J Med Genet B Neuropsychiatric Genet Off Publ Int Soc Psychiatric Genet 144b: 1090–1093. [DOI] [PubMed] [Google Scholar]
- Paris JJ, Singh HD, Carey AN, McLaughlin JP (2015). Exposure to HIV‐1 Tat in brain impairs sensorimotor gating and activates microglia in limbic and extralimbic brain regions of male mice. Behav Brain Res 291: 209–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS Guide to PHARMACOLOGY: an expert‐driven knowledgebase of drug targets and their ligands. Nucl Acids Res 42 (Database Issue): D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit‐Taboué M‐C, Baron J‐C, Barré L, Travère J‐M, Speckel D, Camsonne R et al. (1991). Brain kinetics and specific binding of [11 C] PK 11195 to ω 3 sites in baboons: positron emission tomography study. Eur J Pharmacol 200: 347–351. [DOI] [PubMed] [Google Scholar]
- Prasad KM, Upton CH, Nimgaonkar VL, Keshavan MS (2014). Differential susceptibility of white matter tracts to inflammatory mediators in schizophrenia: an integrated DTI study. Schizophr Res 161: 119–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radewicz K, Garey LJ, Gentleman SM, Reynolds R (2000). Increase in HLA‐DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenias. J Neuropath Exp Neur 59: 137–150. [DOI] [PubMed] [Google Scholar]
- Rao JS, Kim H‐W, Harry GJ, Rapoport SI, Reese EA (2013). Increased neuroinflammatory and arachidonic acid cascade markers, and reduced synaptic proteins, in the postmortem frontal cortex from schizophrenia patients. Schizophr Res 147: 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratnayake U, Quinn TA, Castillo‐Melendez M, Dickinson H, Walker DW (2012). Behaviour and hippocampus‐specific changes in spiny mouse neonates after treatment of the mother with the viral‐mimetic poly I:C at mid‐pregnancy. Brain Behav Immun 26: 1288–1299. [DOI] [PubMed] [Google Scholar]
- Réus G, Fries G, Stertz L, Badawy M, Passos I, Barichello T et al. (2015). The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience 300: 141–154. [DOI] [PubMed] [Google Scholar]
- Ribeiro BMM, do Carmo MRS, Freire RS, Rocha NFM, Borella VCM, de Menezes AT et al. (2013). Evidences for a progressive microglial activation and increase in iNOS expression in rats submitted to a neurodevelopmental model of schizophrenia: reversal by clozapine. Schizophr Res 151: 12–19. [DOI] [PubMed] [Google Scholar]
- Riccomagno MM, Kolodkin AL (2015). Sculpting neural circuits by axon and dendrite pruning. Annu Rev Cell Dev Biol 31: 779–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo LE, Fernández JA, Maccioni AA, Jimenez JM, Maccioni RB (2008). Neuroinflammation: implications for the pathogenesis and molecular diagnosis of Alzheimer's disease. Arch Med Res 39: 1–16. [DOI] [PubMed] [Google Scholar]
- Sargin D, Hassouna I, Sperling S, Sirén A‐L, Ehrenreich H (2009). Uncoupling of neurodegeneration and gliosis in a murine model of juvenile cortical lesion. Glia 57: 693–702. [DOI] [PubMed] [Google Scholar]
- Schmitz T, Chew L‐J (2008). Cytokines and myelination in the central nervous system. Scientific World Journal 8: 1119–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz C, Perdiguero EG, Chorro L, Szabo‐Rogers H, Cagnard N, Kierdorf K et al. (2012). A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336: 86–90. [DOI] [PubMed] [Google Scholar]
- Shah F, Hume SP, Pike VW, Ashworth S, McDermott J (1994). Synthesis of the enantiomers of [N‐methyl‐11 C] PK 11195 and comparison of their behaviours as radioligands for PK binding sites in rats. Nucl Med Biol 21: 573–581. [DOI] [PubMed] [Google Scholar]
- Sommer IE, van Westrhenen R, Begemann MJH, de Witte LD, Leucht S, Kahn RS (2014). Efficacy of anti‐inflammatory agents to improve symptoms in patients with schizophrenia: an update. Schizophr Bull 40: 181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C et al. (2008). Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res 42: 151–157. [DOI] [PubMed] [Google Scholar]
- Steiner J, Mawrin C, Ziegeler A, Bielau H, Ullrich O, Bernstein HG et al. (2006). Distribution of HLA‐DR‐positive microglia in schizophrenia reflects impaired cerebral lateralization. Acta Neuropathol 112: 305–316. [DOI] [PubMed] [Google Scholar]
- Stertz L, Magalhães PV, Kapczinski F (2013). Is bipolar disorder an inflammatory condition? The relevance of microglial activation. Curr Opin Psychiatry 26: 19–26. [DOI] [PubMed] [Google Scholar]
- Sundqvist N, Garrick T, Bishop I, Harper C (2008). Reliability of post‐mortem psychiatric diagnosis for neuroscience research. Aust N Z J Psychiatry 42: 221–227. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Wood SJ, Soulsby B, McGorry PD, Tanino R, Suzuki M et al. (2009a). Follow‐up MRI study of the insular cortex in first‐episode psychosis and chronic schizophrenia. Schizophr Res 108: 49–56. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Wood SJ, Yung AR, Soulsby B, McGorry PD, Suzuki M et al. (2009b). Progressive gray matter reduction of the superior temporal gyrus during transition to psychosis. Arch Gen Psychiatry 66: 366–376. [DOI] [PubMed] [Google Scholar]
- Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R et al. (2010). Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsychopharmacol 13: 943–950. [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL, Majewska AK (2010). Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8 e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD (2011). Ultrastructural alterations of myelinated fibers and oligodendrocytes in the prefrontal cortex in schizophrenia: a postmortem morphometric study. Schizophr Res Treatment 2011: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uranova NA, Vikhreva OV, Rachmanova VI, Orlovskaya DD (2014). Microglial activation in white matter in schizophrenia: findings from a postmortem electron microscopic morphometric study. Neurol Psychiat Brain Res 20: 25. [Google Scholar]
- Uranova NA, Vostrikov VM, Vikhreva OV, Zimina IS, Kolomeets NS, Orlovskaya DD (2007). The role of oligodendrocyte pathology in schizophrenia. Int J Neuropsychopharmacol 10: 537–545. [DOI] [PubMed] [Google Scholar]
- van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E et al. (2008). Microglia activation in recent‐onset schizophrenia: a quantitative (R)‐[11C]PK11195 positron emission tomography study. Biol Psychiatry 64: 820–822. [DOI] [PubMed] [Google Scholar]
- Van den Eynde K, Missault S, Fransen E, Raeymaekers L, Willems R, Drinkenburg W et al. (2014). Hypolocomotive behaviour associated with increased microglia in a prenatal immune activation model with relevance to schizophrenia. Behav Brain Res 258: 179–186. [DOI] [PubMed] [Google Scholar]
- Vowinckel E, Reutens D, Becher B, Verge G, Evans A, Owens T et al. (1997). PK11195 binding to the peripheral benzodiazepine receptor as a marker of microglia activation in multiple sclerosis and experimental autoimmune encephalomyelitis. J Neurosci Res 50: 345–353. [DOI] [PubMed] [Google Scholar]
- Whitford TJ, Grieve SM, Farrow TFD, Gomes L, Brennan J, Harris AWF et al. (2007). Volumetric white matter abnormalities in first‐episode schizophrenia: a longitudinal, tensor‐based morphometry study. Am J Psychiatry 164: 1082–1089. [DOI] [PubMed] [Google Scholar]
- Wierzba‐Bobrowicz T, Lewandowska E, Lechowicz W, Stepień T, Pasennik E (2005). Quantitative analysis of activated microglia, ramified and damage of processes in the frontal and temporal lobes of chronic schizophrenias. Folia Neuropathol Assoc Pol Neuropathol Med Res Centre Pol Acad Sci 43: 81–89. [PubMed] [Google Scholar]
- Wilson AA, Garcia A, Parkes J, McCormick P, Stephenson KA, Houle S et al. (2008). Radiosynthesis and initial evaluation of [18 F]‐FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol 35: 305–314. [DOI] [PubMed] [Google Scholar]
- Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R et al. (2014). Differential roles of microg1lia and monocytes in the inflamed central nervous system. J Exp Med 211: 1533–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yolken RH, Dickerson FB, Fuller Torrey E (2009). Toxoplasma and schizophrenia. Parasite Immunol 31: 706–715. [DOI] [PubMed] [Google Scholar]
- Zalesky A, Fornito A, Seal ML, Cocchi L, Westin CF, Bullmore ET et al. (2011). Disrupted axonal fiber connectivity in schizophrenia. Biol Psychiatry 69: 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F et al. (2014). Deficient neuron‐microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17: 400–406. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhang Y, Xu H, Wang L, Adilijiang A, Wang J et al. (2014). Olanzapine ameliorates neuropathological changes and increases IGF‐1 expression in frontal cortex of C57BL/6 mice exposed to cuprizone. Psychiatry Res 216: 438–445. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu H, Jiang W, Xiao L, Yan B, He J et al. (2008). Quetiapine alleviates the cuprizone‐induced white matter pathology in the brain of C57BL/6 mouse. Schizophr Res 106: 182–191. [DOI] [PubMed] [Google Scholar]
- Zhu F, Liu Y, Zhao J, Zheng Y (2014). Minocycline alleviates behavioral deficits and inhibits microglial activation induced by intrahippocampal administration of granulocyte–macrophage colony‐stimulating factor in adult rats. Neuroscience 266: 275–281. [DOI] [PubMed] [Google Scholar]
- Zipursky RB, Reilly TJ, Murray RM (2013). The myth of schizophrenia as a progressive brain disease. Schizophr Bull 39: 1363–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]