

ABSTRACT
Virulence factors are often multifunctional and contribute to pathogenesis through synergistic mechanisms. For the human pathogen Streptococcus pyogenes, two factors that act synergistically are the S. pyogenes NAD+ glycohydrolase (SPN) and streptolysin O (SLO). Through distinct mechanisms, SLO forms pores in host cell membranes and translocates SPN into the host cell cytosol. Two natural variants of SPN exist, one that exhibits NADase activity and one that lacks this function, and both versions are translocated and act in concert with SLO to cause an accelerated death response in epithelial cells. While NADase+ SPN is known to trigger a metabolic form of necrosis through the depletion of NAD+, the mechanism by which NADase− SPN induces cell death was unknown. In the studies described here, we examined the pathway of NADase− cell death through analysis of activation patterns of mitogen-activated protein kinases (MAPKs). S. pyogenes infection resulted in activation of members of three MAPK subfamilies (p38, ERK, and JNK). However, only JNK was activated in an SLO-specific manner. NADase− SPN induced necrosis in HeLa epithelial cells associated with depolarization of mitochondrial membranes, activation of NF-κB, and the generation of reactive oxygen species. Remarkably, RNA interference (RNAi) silencing of JNK protected cells from NADase−-SPN-mediated necrosis, suggesting that NADase− SPN triggers a form of programmed necrosis dependent on JNK signaling. Taken together, these data demonstrate that SPN acts with SLO to elicit necrosis through two different mechanisms depending on its NADase activity, i.e., metabolic (NADase+) or programmed (NADase−), leading to distinct inflammatory profiles.
IMPORTANCE
Many bacterial pathogens produce toxins that alter how infected host cells interact with the immune system. For Streptococcus pyogenes, two toxins, a NAD+ glycohydrolase (SPN) and streptolysin O (SLO), act in combination to cause infected cells to die. However, there are two natural forms of SPN, and these variants cause dying cells to produce different types of signaling molecules. The NADase+ form of SPN kills cells by depleting reserves of NAD+ and cellular energy. The other form of SPN lacks this activity (NADase−); thus, the mechanism by which this variant induces toxicity was unknown. Here, we show that infected cells recognize NADase− SPN through a specific signaling molecule called JNK, which causes these cells to undergo a form of cellular suicide known as programmed necrosis. This helps us to understand how different forms of toxins alter host cell signaling to help S. pyogenes cause different types of diseases.
INTRODUCTION
Pore-forming toxins are perhaps the most diverse and widely distributed class of cytotoxic proteins found among bacterial pathogens. Their salient characteristic is an ability to interact with and then compromise the integrity of the host cell cytoplasmic membrane by the introduction of a transmembrane pore. This process can lead to a number of effects important for pathogenesis, ranging from alteration of host cell signaling to host cell death (for a review, see reference 1). However, despite the remarkable alterations to host cell physiology that these toxins can produce in cultured cells, determining the specific contribution that any single toxin makes to virulence can be problematic.
One complication is that pore-forming toxins typically function not in isolation but rather as members of ensembles of virulence factors that act in synergy to produce a cellular outcome. A prominent example of this phenomenon involves streptolysin O (SLO), a pore-forming toxin produced by Streptococcus pyogenes. This Gram-positive pathogen is the causative agent of superficial (e.g., pharyngitis and impetigo), invasive (e.g., cellulitis), and destructive (e.g., necrotizing fasciitis) diseases. It is also associated with postinfection sequelae (e.g., rheumatic fever and acute glomerulonephritis), including certain types of tic and obsessive-compulsive disorders in children (2–4). One difficulty in understanding the specific contribution that SLO makes to any of these diseases is the fact that it acts in synergy with a second secreted toxin, the S. pyogenes NAD+ glycohydrolase (SPN, also known as NGA). This synergy is reflected at multiple levels. First, the genes that encode SLO and SPN are located in the same operon, and when S. pyogenes is adherent to a host cell, both SPN and SLO are expressed and then exported from the bacterium by the general secretory pathway. Second, at the host cell membrane SLO facilitates the translocation of SPN across the membrane into the host cell cytosol, a process called cytolysin-mediated translocation (CMT) (5). Third, when present in an intracellular compartment, SPN acts to modify cellular responses that are initiated by SLO (6). This degree of synergy renders incomplete any conclusion regarding SLO’s contribution to pathogenesis that does not take into consideration the concomitant influence of SPN.
Although the details of the CMT mechanism and how SLO and SPN act in concert to alter host cell behaviors are not well understood, numerous details of the SLO-SPN relationship have been unraveled. For the CMT mechanism, studies have revealed that both SLO and SPN have dedicated translocation domains that are dispensable for their canonical functions (pore formation and NAD+ cleavage, respectively) (7, 8). As an archetypical member of the cholesterol-dependent cytolysin (CDC) family of hemolytic toxins, SLO forms transmembrane pores following recognition of cholesterol and the formation of a large oligomeric ring structure. However, neither cholesterol binding nor oligomerization is required for CMT (8, 9). Instead, CMT proceeds from an alternative interaction with the membrane that is codependent on SPN and can also promote subsequent oligomerization and cholesterol-dependent pore formation (9, 10). This suggests that the pathway of pore formation when S. pyogenes is adherent to host cells differs from that of the soluble toxin on bystander host cells, adding additional complexity for understanding the role of SLO in pathogenesis.
More recently, several studies that have examined the consequences of SPN-SLO synergy have found that the cotoxins interact to produce a diversity of cellular outcomes in cultured host cells, ranging from enhancement of intracellular survival of S. pyogenes in epithelial cells (11) and macrophages (12) to accelerating killing kinetics in several types of epithelial cells (13, 14). When these studies are compared, an additional complication is that diversity in SPN is rarely taken into account. Population studies have revealed that SPN exists as two distinct haplotypes, one of which has polymorphisms at three amino acid residues that act combinatorially to reduce its catalytic NADase efficiency >108-fold to essentially undetectable levels (NADase− SPN) (15). The codons for these residues have signatures of positive selection, and the NADase− haplotype is associated with a degraded version of a gene that encodes an immunity factor that is essential for expression of the NADase+ version of the protein (16). Together, these data indicate that the NADase− haplotypes are under strong selection, implying that despite their lack of NADase activity, that they remain essential for pathogenesis.
Studies that have conducted direct parallel comparisons of NADase+ and NADase− SPN have shown that both contribute to an accelerated cytotoxic response in several types of epithelial cells that require the pore-forming activity of SLO. A distinction is that the steps preceding death vary between the SPN haplotypes. For example, SLO pore formation results in hyperactivation of the cellular enzyme poly-ADP-ribose polymerase (PARP-1), which cleaves the same bond in NAD+ as NADase+ SPN but forms large polymers of poly-ADP-ribose (PAR). Accumulation of PAR is then differentially modified by SPN; PAR rapidly disappears in the presence of NADase+ SPN but rapidly accumulates in the presence of NADase− SPN (6). As a consequence, the proinflammatory mediator high-mobility group Box 1 protein (HMGB1) is released from the nucleus by a PARP-1-dependent mechanism in response to NADase+ SPN but is not released during an infection by NADase− SPN strains. Instead, cells infected by these latter strains release significantly higher levels of interleukin-8 (IL-8) and tumor necrosis factor alpha (TNF-α) (6). These differences in outcomes illustrate why it is important to consider SPN-SLO synergy and also SPN diversity in the context of a holistic approach to understanding how S. pyogenes interacts with host cells.
The presence or absence of SPN’s NADase activity also influences the pathway leading to cell death in epithelial cells. For NADase+ SPN, cleavage of NAD+ results in depletion of ATP and death by “metabolic catastrophe,” a type of unprogrammed necrotic cell death that occurs when demand for cellular energy outstrips its production (17). However, since NAD+ and ATP levels are not reduced in a NADase− SPN infection, it is not clear how NADase-negative SPN contributes to cell signaling and eventually to cell death (6).
In the present study, we took advantage of the fact that epithelial cells are sensitive detectors of bacterial pore-forming toxins and transduce pore-induced stimuli from the cellular membrane to nuclear and other intracellular targets via members of the highly conserved mitogen-activated protein kinase (MAPK) superfamily (18). We found that infection of epithelial cells resulted in activation of members of three prominent MAPK subfamilies (JNK, ERK, and p38). Of these, only JNK was activated specifically in response to pore formation by SLO. As with NADase+ SPN, infection by NADase− strains resulted in death by necrosis, and both haplotypes modified several SLO-dependent events downstream of JNK activation, including mitochondrial depolarization and production of reactive oxygen species (ROS). Significantly, small interfering RNA (siRNA)-based knockdown of JNK protected cells from NADase− SPN-induced necrotic cell death, implicating JNK as a key mediator of SPN-induced cellular death. These finding indicate that while both types of SPN cause necrosis, they do so by divergent mechanisms, either metabolic (NADase+) or programmed (NADase−) pathways. These data provide insight into the mechanism of NADase− cell death, how different combinations of toxic activities contribute to pathogenesis, and the selective pressures responsible for the adaptive radiation of SPN enzymatic haplotypes.
RESULTS
SPN acts with SLO to cause necrosis in epithelial cells.
Previously, we showed that SLO acts combinatorially with either of the two enzymatic haplotypes of SPN to cause accelerated cell death in HeLa cells, A549 epithelial cells, and HaCaT keratinocytes (5, 15) and that NADase+ SPN causes necrosis via the process of metabolic catastrophe (6). Furthermore, neither SPN variant induced death in these cells by pyroptosis or parthanatos, two distinct pathways of programmed necrosis (6). In order to further characterize the mechanism of NADase− cell death, intoxicated cells were examined for evidence of apoptosis, a programed noninflammatory pathway of cell death. Infection of HeLa cells revealed the characteristic accelerated cell death response dependent on both SLO and SPN, whose signature includes damage to the cellular membrane (Fig. 1A). After 5 h of infection, S. pyogenes expressing SLO in combination with either NADase+ or NADase− SPN resulted in the death of nearly all infected cells. In contrast, infection with ΔSLO or ΔSPN mutants compromised membranes in only <10% of cells at this time point, at a level similar to that observed in uninfected cells (Fig. 1A). As expected, treatment with staurosporine, a known inducer of apoptosis (19), also did not compromise membranes (Fig. 1A), although these cells did appear to have a more rounded shape (data not shown). However, similar to staurosporine treatment, cells infected with bacteria expressing SLO and SPN had depolarized mitochondrial membrane potential, as determined by staining with tetramethylrhodamine ethyl ester (TMRE) (Fig. 1A and B). Depolarization was dependent on SLO and SPN, as mitochondrial potential was unaltered for cells infected by ΔSLO or ΔSPN mutants, and these appeared similar to uninfected cells (Fig. 1A and B). Imaging nuclear morphology by staining with DAPI revealed that nuclear fragmentation, a hallmark of apoptosis (20), did not occur following infection by any strain of S. pyogenes, although it was prominent in staurosporine-treated cells (Fig. 1A and B). Similarly, infection did not result in activation of the apoptosis-associated caspases (caspases 3 and 8) activated by staurosporine (Fig. 1C). Taken together, these data show that similar to cell death caused by NADase+ SPN, cell death caused NADase− SPN is by a necrotic rather than an apoptotic mechanism.
FIG 1 .

SPN induces necrosis. HeLa cells were infected for 5 h with the indicated strains or 1 mM staurosporine (Stauro) and then analyzed as follows. (A) Cells were stained with the membrane-permeant dye Calcien AM and the impermeant dye ethidium homodimer-2 (Live/Dead) to assess membrane permeability (left). Cells with intact membranes are stained green, and cells with permeabilized membranes are stained red. Cells were stained with TMRE to assess mitochondrial membrane polarization (middle). The images are overlays of bright-field and fluorescent micrographs. Cells with mitochondria with active membrane potential appear red. Cells were stained with DAPI to visualize nuclear fragmentation (right). Membranes and mitochondria are at a magnification of ×20, and nuclei are at ×63. (B) Quantitation of images in panel A. Data are means and standard errors of the means derived from 3 independent experiments and examination of at least 1,000 total cells. (C) Cellular lysates prepared from infected monolayers were incubated with chromogenic synthetic substrates for caspases 3 and 8, as indicated. Activation is presented as the absorbance at 405 nm. Data are means and standard deviations (SD) derived from 3 independent experiments. *, P < 0.05, **, P < 0.01, and ***, P < 0.001, compared to the uninfected control.
S. pyogenes activates JNK in an SLO-dependent manner.
A variety of pore-forming toxins, including SLO have been shown to activate MAPKs (18, 21–23). Since MAPK activation can trigger several pathways of programmed necrotic cell death (24, 25), infected HeLa cells were examined for phosphorylation of members of the three most prominent MAPK subfamilies, including p38, ERK, and JNK. When cell extracts were examined using a Western blot analysis to probe for their activated phosphorylated forms, all three MAPKs were activated by S. pyogenes infection regardless of the presence or absence of SPN (NADase+ SPN, NADase− SPN, and ΔSPN) (Fig. 2A), and p38 and ERK were activated in the absence of SLO (ΔSLO) (Fig. 2A). However, activation of JNK required the presence of SLO, as JNK was not activated by infection with the SLO-deficient mutant (ΔSLO) (Fig. 2A). Pore formation enhanced JNK activation, as it was not observed following infection by a mutant that expresses an altered SLO that can bind membrane but cannot oligomerize (Fig. 2B, Mono) and was reduced upon infection with a mutant that expresses an SLO variant that can oligomerize but cannot form pores (Fig. 2B, Pre). SLO was both necessary and sufficient to activate JNK, as exposure of HeLa cells to a range of concentrations of purified SLO activated JNK in a concentration-dependent manner (Fig. 2C).
FIG 2 .
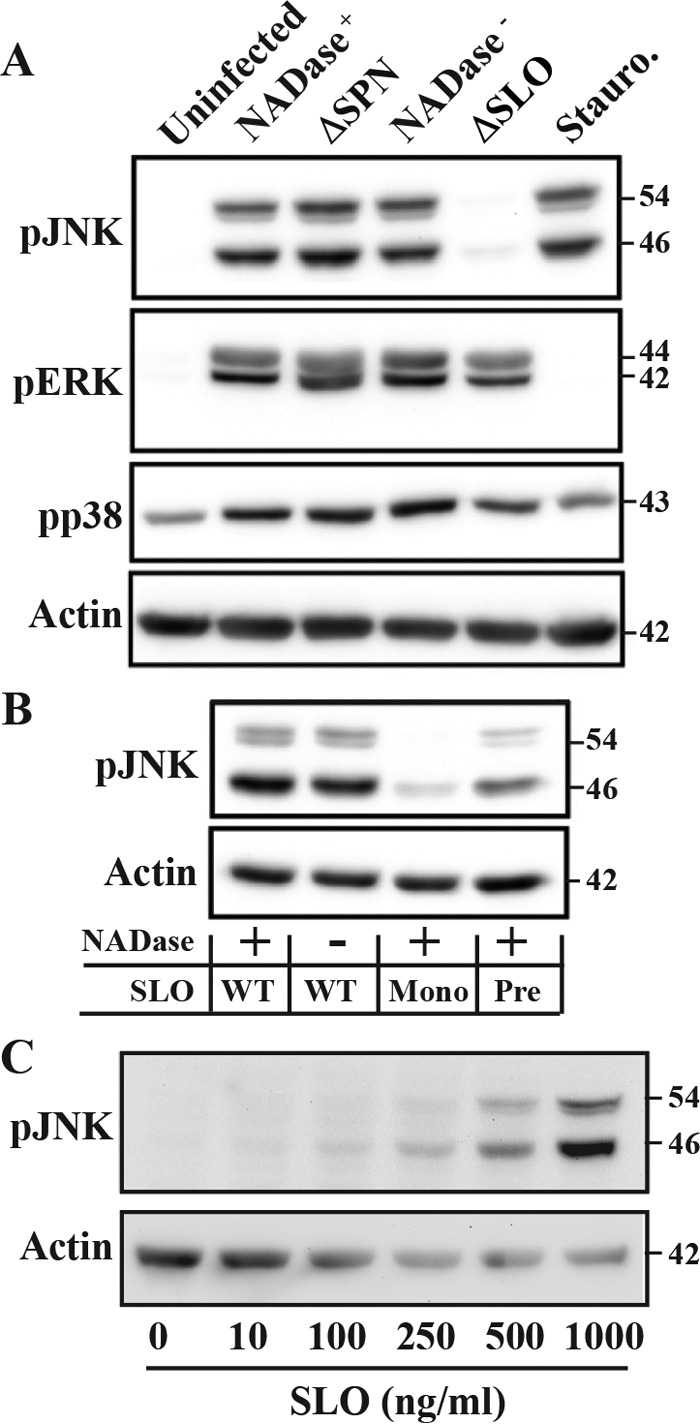
Activation of JNK requires SLO and is enhanced by pore formation. (A) HeLa cells were infected with the indicated strains for 5 h, and activation of several MAP kinases was assessed by Western blotting analyses of cellular lysates to detect their phosphorylated forms. Two isoforms exist for JNK and ERK. Lysates were compared to uninfected cells and to cells treated with staurosporine (Stauro; 1 mM) and to actin. (B) Activation of JNK was assessed by Western blotting following infection by various S. pyogenes strains expressing NADase+ or NADase− SPN in combination with wild-type (WT), monomer-locked (Mono), or prepore-locked (8) SLO mutants, as indicated below the gels. (C) Western blot analysis of JNK activation in HeLa cells following a 30-min exposure to the indicated concentrations of purified SLO. Blots presented are representative of at least 3 independent experiments. The molecular masses of the various proteins detected are on the right, in kilodaltons.
JNK activation is time and concentration dependent.
Transient activation of JNK can protect cells from pore-forming toxins (26), while sustained activation of JNK leads to deadly cellular outcomes (27–29). To assess the character of JNK activation in response to S. pyogenes, HeLa cells were infected over a range of multiplicities of infection to determine whether there is a minimum amount of bacteria needed to activate this pathway and whether this threshold was influenced by NADase activity. Cell lysates were prepared following 5 h of infection and analyzed as described above, revealing that activation was still apparent when the inoculum was reduced by approximately 50% from the standard concentration (optical density [OD] = 0.5). Additionally, cells appeared slightly more sensitive to infection by the NADase+ strain (Fig. 3). For both SPN haplotypes, JNK activation was evident at 3 h and increased until the final time point (5 h) (Fig. 3), indicating that JNK activation is sustained in HeLa cells in response to S. pyogenes infection.
FIG 3 .
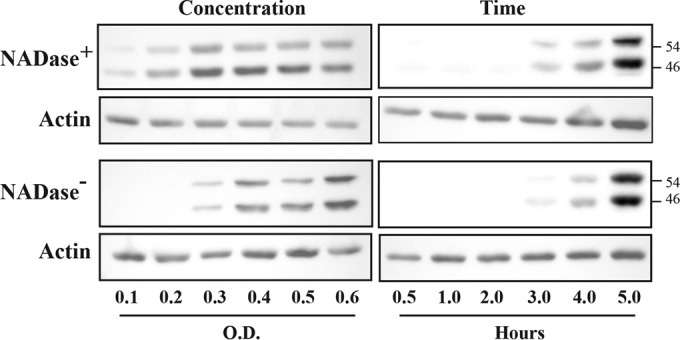
JNK activation is concentration and time dependent. Activation of JNK was assessed by Western blotting to detect its two phosphorylated forms in HeLa cell lysates following infection. The top and bottom panels contain data from cells infected with NADase+ and NADase− strains, respectively, as indicated and are compared to actin. Concentration is represented by the optical density of the infecting suspension of bacteria determined at 600 nm, and lysates for all concentration experiments were prepared at 5 h postinfection. The times shown are the hours postinfection at which cellular lysates were prepared from cells infected at an OD of 0.5. The molecular masses of proteins detected are on the right, in kilodaltons. Data are representative of at least two independent experiments.
JNK and PARP contribute to mitochondrial depolarization.
As shown above, SPN is required for mitochondrial depolarization (Fig. 1B). Since both activated JNK and PAR can be released from the nucleus to interact with the mitochondrial membrane and cause membrane depolarization (25, 30), we evaluated the possibility that these molecules contribute to depolarization. Cells were first treated with siRNAs to silence JNK1 and PARP-1, resulting in transfection efficiencies approaching 100% (data not shown) and a >98% reduction of JNK1 and formation of PAR following infection by the NADase− SPN strain compared to cells treated with control siRNA (see Fig. S1A in the supplemental material). In addition, these treatments did not alter the efficiency of CMT (see Fig. S1B). Activated JNK can activate PARP-1 (27); however, this is not the case for S. pyogenes infection, as PAR was formed in cells treated with siRNAs targeting JNK (see Fig. S1A). When mitochondrial membrane potential was evaluated by staining with TMRE, cells treated with control siRNAs lost potential in an SPN-dependent manner (Fig. 4) similar to untreated cells (Fig. 1B). Silencing PARP-1 did not protect loss of mitochondrial potential from infection by the NADase+ strain but did protect from the NADase− strain (Fig. 4), whereas silencing JNK protected mitochondrial potential from infection by both the NADase+ and NADase− strains, and in the latter case, protection was observed in >90% of cells (Fig. 4). These data indicate that pathways leading to depolarization are not identical between the SPN variants, as JNK is required for NADase+ SPN, while NADase− SPN requires both the production of PAR polymers and activation of JNK.
FIG 4 .

Silencing JNK and PARP inhibits NADase− SPN-induced mitochondrial depolarization. (A) HeLa cells were treated with siRNA targeting JNK1 and PARP-1 72 h prior to infection with the indicated strains. At 5 h postinfection, cells were stained with TMRE to assess mitochondrial polarization. Data are means and standard errors of the means derived from 2 independent experiments and examination of at least 1,000 total cells and are compared to siRNA-treated but uninfected cells and cells treated with siControl, consisting of a nonspecific siRNA and siGLO, an indicator of transfection efficiency (Dharmacon). **, P < 0.01; ***, P < 0.001.
SPN and SLO affect events downstream of JNK and PAR.
Activation of JNK and production of PAR can result in multiple outcomes, including the production of reactive oxygen species (ROS) and the translocation of the inflammatory-associated signaling molecule NF-κB from the cytoplasm into the nucleus (24, 27, 30). Immunofluorescence was used to detect nuclear translocation of the NF-κB p65 subunit during infection with various S. pyogenes strains. A nuclear location could be visualized in the majority of infected cells (~50 to 75%), irrespective of SPN or SPN activity phenotype (Fig. 5A). The exception was infection by the ΔSLO mutant, in which only ~10% of cells demonstrated nuclear translocation (Fig. 5). Similarly, staining with the fluorophore CM-H2DCFDA to detect ROS revealed that ROS could not be detected in cells infected with the ΔSLO mutant but was abundant in cells infected by strains expressing either variant of SPN (Fig. 5A). However, unlike NF-κB, the production of ROS required SPN, as infection by the ΔSPN strain did not result the production of ROS (Fig. 5B). These data indicate that activation of NF-κB requires SLO, while production of ROS requires both SLO and intracellular SPN of either haplotype.
FIG 5 .

ROS production, but not NF-κB activation, requires SPN. HeLa cells were infected with the strains indicated for 5 h and then analyzed as follows. (A) NF-κB activation was assessed by immunofluorescence to enumerate the nuclear translocation of NF-κB p65 subunit compared to the number of cells where p65 is located only in the cytoplasm. (B) Staining with the fluorescent probe CM-H2DCFDA to detect the presence of reactive oxygen species (ROS). Infected cells were compared to cell treated for 3.5 h with 1 mM H2O2. Data are means and SD derived from 3 independent experiments and the examination of at least 1,000 cells. **, P < 0.01, and ***, P < 0.001, compared to the uninfected control.
NADase− cell death requires JNK activation.
Since the data presented above shows that SLO and SPN are associated with activation of JNK and PARP-1 along with modulation of various cellular responses downstream of activation, it was of interest to determine the contribution of JNK and PARP-1 to NADase− SPN cell death. HeLa cells were treated with siRNAs targeting either JNK1 or PARP-1 and infected with NADase+ or NADase− S. pyogenes strains for 5 h, and then cell viability was assessed as described above. This analysis revealed that silencing PARP-1 had no protective effect, with nearly all cells demonstrating compromised membranes indistinguishable from those in cells treated with control siRNAs or cells that were untreated (Fig. 6). In contrast, silencing JNK resulted in a significantly higher percentage of viable cells following infection by the NADase+ strain than in control treated and untreated cells and, remarkably, protected ~70% of the cells infected by the NADase− strain from cytotoxicity (Fig. 6). These data indicate that the necrotic cell death promoted by the NADase− variant of SPN is dependent on activation of JNK1.
FIG 6 .
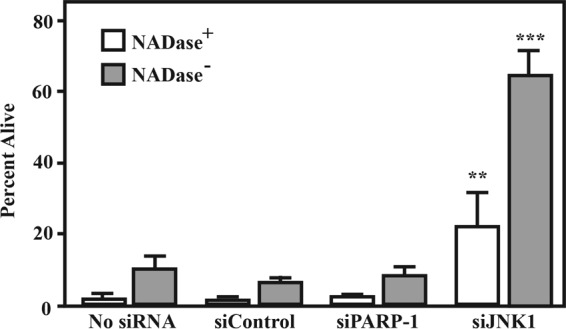
Silencing of JNK, but not PARP, increases cell survival. HeLa cells were treated with siRNAs targeting PARP-1 and JNK and after 72 h infected by a NADase+ or NADase− strain, as indicated, and compared to untreated cells (no siRNA) and siControl-treated cells. Following 5 h of infection, the number of cells that were viable was determined by fluorescence microscopy using a vital probe (Live/Dead) as described for Fig. 1. Cells with compromised membrane permeability were considered nonviable. Data are means and SD derived from 3 independent experiments and the examination of at least 1,000 cells. **, P < 0.01; ***, P < 0.001.
DISCUSSION
In this study, we have expanded our understanding of how SPN and SLO interact to produce cellular responses that neither produces in isolation and how their synergy is influenced by SPN diversity. While both SPN variants cooperate with SLO to induce an accelerated death response in HeLa cells, only the NADase+ SPN, with its ability to cleave available NAD+, has an obvious function. Here, we have shown that cell death in response to the NADase− SPN involves events downstream of the SLO-mediated activation of the c-Jun NH-terminal (JNK) MAPK. The fact that this cell death is dependent on cellular signaling suggests that unlike the unprogrammed metabolic catastrophe induced by NADase+ SPN, JNK-dependent NADase− cell death represents a cell-directed pathway of programmed necrosis.
The involvement of JNK in cell death offers a striking example of SLO-SPN synergy. While purified SLO and other pore-forming toxins have been shown to activate several classes of MAPK (18, 21, 22), only JNK was specifically activated by SLO in the context of S. pyogenes infection. The fact that purified SLO could also activate JNK suggests that SLO is both necessary and sufficient to trigger activation. However, this sustained activation of JNK was not sufficient to promote accelerated cellular death, as this response also required the presence of intracellular NADase− SPN. Interestingly, silencing JNK also provided some protection against NADase+ SPN. Since the two SPN alleles differ at just three catalytic residues and only nine residues overall (16), this suggests that NADase+ SPN has dual activities, JNK-associated cell death activity and a NAD+-glycohydrolase-promoted death activity. However, in the context of these infections, the data demonstrate that the NAD+-glycohydrolase-promoted cell death pathway is dominant.
In contrast, the mechanism by which NADase− SPN promotes JNK-dependent death is less clear. Examination of several known cell death-related processes revealed that mitochondrial depolarization and production of ROS directly correlated with death by SPN, suggesting that these may represent the convergence of JNK1 and PARP-1. Both abundant PAR and activated JNK1 can interact with mitochondria, resulting in depolarization and the stimulation of ROS production to the point of cell death (27, 31, 32). Depending on context, activated JNK can stimulate production of PAR (27), or conversely, accumulation of PAR can lead to activation of JNK (32). The order of these events can dictate whether apoptosis or necrosis occurs (24, 33, 34). In the present study, silencing either PARP-1 or JNK1 protected mitochondrial potential when cells were challenged with NADase− SPN, suggesting that the production of PAR and the activation of JNK1 are related events in a common pathway leading to cell death. However, other data argue against this mechanism, including the observation that while silencing PARP-1 protected mitochondrial membrane potential, it did not protect against cell death. This indicates that depolarization does not always lead to cell death and that PARP-1 is not an essential component of the JNK1-mediated pathway of programmed necrosis. Consistent with this, activation of PARP-1 and that of JNK are not identical. While SLO is required to activate both during infection, purified SLO activates JNK but not PARP-1 (6). Thus, while PAR production can act to modulate cell signaling by S. pyogenes-infected cells, PARP-1 is activated independently from JNK (6).
The finding that signaling through JNK can trigger necrosis is not without precedent. However, the role of JNK in cellular health is influenced by multiple interactions in ways that are not well understood. JNK has been implicated in protection of cells from damage by pore-forming toxins (35) and can both inhibit and promote apoptosis or necrosis (24). In the RIP1 kinase-dependent pathway of programmed necrosis known as necroptosis, JNK functions as the cellular executioner by directing the production of toxic levels of ROS (36), possibly by the direct interaction of activated JNK with the mitochondria (31) or by inhibition of the cellular protective response to ROS (24). Whether JNK activation leads to survival, apoptosis, or necrosis depends on events that happen downstream of its activation. This is illustrated in the present study by the fact that the SLO-directed sustained activation of JNK was not sufficient to trigger the accelerated death response, as this also required the presence of SPN. How SPN links JNK to necrotic cell death remains to be determined; however, the outcome of JNK activation can be dictated by the activation of NF-κB, which was also activated in response to SLO. NF-κB can suppress JNK-activated death-promoting processes through the activation of genes that act as countermeasures, including genes whose products can protect against ROS toxicity (24). This suggests that SPN may function to antagonize an NF-κB-activated cellular defense that would normally support viability during sustained activation of JNK. It is possible that the interaction of SPN and SLO at the membrane contributes to perturbations leading to inhibition of pore healing that may occur in the absence of SPN.
Production of ROS plays a central role in many JNK-dependent pathways of cellular death. In the present study, ROS was a specific outcome of SPN intoxication, although the disparate effects of silencing JNK and PARP-1 on cell death and mitochondrial depolarization cast doubt on the role of mitochondrial membrane damage as the primary source of ROS. It should be noted that this is not the only JNK-related pathway for ROS production. In the version of necroptosis triggered by the cytokine TNF-α, ROS plays a central role in cell death and is produced by the membrane-associated NADPH oxidase (NOX), which becomes activated by the TNF-α receptor and the RIP1 kinase (37). In this regard, it is interesting that inhibition of JNK can prevent NOX activation (38). Also interesting is that the TcdB cytotoxin of Clostridium difficile produces necrosis by activation of NOX (39), which, reminiscent of SPN, occurs independently of its canonical glucosyltransferase enzymatic activity (40). It will be of interest to determine if there is a common mechanism underlying TcdB-induced necrosis, NOX activation, ROS toxicity, and NADase−-SPN-directed necrotic cell death.
In this study, we have shown that despite a >108-fold decrease in activity, NADase− SPN is not inert but actively contributes to mitochondrial depolarization, production of ROS, and a necrotic cell death dependent on JNK. These data provide insight into why NADase-negative SPN is still under positive selection and why the NADase− SPN allele has remained intact through the evolution of this pathogen. The different death pathways induced by NADase+ versus NADase− SPN dictate the inflammatory profile of these dying epithelial cells (6), which may play a role in the differential distribution of the two variants between lineages of S. pyogenes that differ in niche selection (16). The recent findings that Mycobacterium tuberculosis uses a NAD+ glycohydrolase to induce necrotic death in infected macrophages versus apoptosis driven by host immunity and that this results in differential modulation of adaptive immunity (41) suggest that the use of NAD+ glycohydrolases to induce differential pathways of host cell death to manipulate signaling may be an emerging theme in pathogenesis. How the differential signaling induced by SPN variants contributes to virulence will provide important insights into S. pyogenes population biology and pathogenesis of its large array of different diseases.
MATERIALS AND METHODS
Bacterial strains and media.
All experiments involving S. pyogenes utilized isogenic strains derived from the M serotype 6 strain JRS4 (42) (wild type [WT]) that were engineered to express different SPN variants or to lack expression of SPN or SLO due to in-frame deletions in the relevant genes. These included SPNJ4 (NADase+), SPNH5 (NADase−), ΔSPN (SPN deficient), and ΔSLO (SLO deficient). The construction of these strains has been described in detail (15). The various SPN proteins expressed by these strains all contain a carboxy-terminal HA epitope tag (15). Routine culture of S. pyogenes for all assays was conducted using Todd-Hewitt medium supplemented with 0.2% yeast extract (THY medium). The strains of Escherichia coli and media used for protein purification are described below.
Infection of HeLa cells.
HeLa cells (a gift from Dong Yu) were infected using a standard method (15). Briefly, prior to infection, the various S. pyogenes strains were inoculated into THY medium and grown overnight at 37°C. Strains were then subcultured by inoculating 10 ml of fresh THY medium at a dilution of 1:10 with incubation at 37°C to allow two rounds of doubling as determined by the OD at 600 nm (OD600). Streptococci were harvested by centrifugation (6,000 × g for 5 min) at room temperature, washed once with 10 ml of phosphate-buffered saline (PBS), and resuspended in tissue culture medium consisting of Dulbecco’s modified Eagle medium (DMEM), 10% fetal bovine serum (FBS), 12.5 mM glutamine, and 50 mM HEPES to an OD600 of 0.5. HeLa cells were cultured in 6-well plates (Techno Plastic Products; Midsci) under the conditions described previously (5) and were infected with 125 µl of the streptococcal suspensions, as described in detail elsewhere (15). Infections were continued for 5 h, unless otherwise specified, and were analyzed as described below.
Purification of SLO.
Expression and purification of SLO with a C-terminal His6 tag from E. coli followed an established method (43), as described in detail elsewhere (44). Protein concentrations were determined using a bicinchoninic acid (BCA) assay (Pierce) with a bovine serum albumin (BSA) standard, and purity was assessed by SDS-PAGE and staining with Coomassie brilliant blue. Activity was verified by an erythrocyte hemolysis assay, as described elsewhere (8). Yields were typically 4 mg protein, 95% pure, via Coomassie brilliant blue stain starting from 300 ml of culture.
Western blot analysis.
After infection, cells were washed with 10 ml Dulbecco’s medium–phosphate-buffered saline (D-PBS), scraped, and subjected to centrifugation (700 × g, 10 min) to pellet intact cells. Supernatants were discarded, and cells were lysed with M-PER (Thermo Scientific) by incubation in the appropriate volume of reagent according to the manufacturer’s recommendations. After 10 min at room temperature, the mixture was clarified by centrifugation (20,000 × g, 10 min), and aliquots of the supernatant were mixed with SDS loading buffer (4×). The samples were boiled for 10 min and stored at −20°C until analysis by Western blotting as previously described (15) using the antibodies listed below.
Antibodies.
Antibodies for P-JNK, P-ERK, P-p38, JNK1, and NF-κB p65 were purchased from Cell Signaling Technologies (Danvers, MA). PAR antibody was purchased from BD Biosciences (San Jose, CA). Actin AC-40 antibody (Sigma, St. Louis, MO) was used as a loading control. For Western blot analyses, primary antibodies were used at a dilution of 1:1,000, and secondary horseradish peroxidase (HRP)-conjugated goat anti-rabbit antibodies (Sigma, St. Louis, MO) were used at 1:10,000.
Caspase activation, cell permeability and cytotoxicity.
Caspase activation was assessed using a colorimetric substrate assay (catalog no. ALX-850-228; Enzo Life Sciences, Farmingdale, NY). HeLa cells were infected for 5 h and lysed according to the manufacturer’s instructions. Briefly, 50 µl of 2× reaction buffer with 10 mM dithiothreitol (DTT) was added to 50 µl lysed cells followed by the addition of p-Nitroanilide-conjugated substrates for caspases 3 and 8 to a final concentration of 200 µM. Reactions were developed for 1 h at 37°C, and then absorbance (405 nm) was determined using a 96-well plate reader (M200 Infinite Pro; Tecan, Mannedorf, Switzerland). Cell membrane integrity was assessed by visualization of the ability of infected HeLa cells to exclude the membrane-impermeant fluorescent dye ethidium homodimer-1 (Live/Dead; Invitrogen) as described previously (15). Infected cells were compared to uninfected cells and cells treated with 1 µM staurosporine (Sigma, St. Louis, MO) for an equivalent period of time. Cells with permeable membranes after 5 h of infection or treatment were considered nonviable.
Nuclear integrity, mitochondrial permeability, and reactive oxygen species.
For determination of nuclear integrity, cells were stained with DAPI (Prolong Gold with DAPI; Invitrogen) according to the manufacturer’s instructions. Mitochondrial membrane potential was assessed by staining with tetramethylrhodamine ethyl ester (TMRE; Invitrogen) diluted in D-PBS for a final concentration of 150 nM, as follows. Following infection, cells were washed once with D-PBS; TMRE was then added and incubated with the cells for 20 min at 37°C. After this time, the stain was removed and replaced with D-PBS. For a positive depolarization of the mitochondrial membrane, carbonyl cyanide 3-chlorophenylhydrazone (CCCP) was used at 50 µM for 1 h prior to staining. For identification of reactive oxygen species, infected cells were washed with D-PBS and then stained with CM-H2DCFDA (Molecular Probes) at a concentration of 1 mM in D-PBS for 15 min. Cells were washed with D-PBS and then incubated in phenol red-free complete tissue culture medium prior to visualization. Cells treated with all stains were imaged using a Leica DMRE2 fluorescence microscope, and images were obtained with a Q Imaging Retiga cooled charge-coupled device (CCD) digital camera using Volocity software (Perkin Elmer; Waltham, MA). Infected cells were compared to uninfected cells and to cells treated with H2O2 or staurosporine, as described above.
Nuclear translocation of NF-κB.
Two days prior to infection, HeLa cells were seeded in 8-well chamber slides (Lab-Tek), cultured until confluent, and then infected as described above. Following infection, monolayers were washed with PBS and fixed with 4% paraformaldehyde for 15 min at room temperature. Fixed cells were washed twice with PBS and then permeabilized with saponin (0.5% [vol/vol] in PBS). After three PBS washes, cells were blocked with PBS–T-BSA (0.1% [vol/vol] Tween 20 and 1% [vol/vol] BSA in PBS), and anti-NF-κB antibody (see above) diluted 1:100 in PBS–T-BSA was added to monolayers and incubated overnight at 4°C. Fluorescent secondary anti-rabbit antibodies, Alexa Fluor 594 or 488 (Invitrogen) at a dilution of 1:300 was added and incubated for 2 h at room temperature in the dark. Cells were washed in PBS, the chamber was removed, and 10 µl of ProLong Gold with DAPI was placed on each chamber square, which was overlaid with a glass slide and allowed to set overnight. Cells were imaged by fluorescence microscopy as described above and scored as the percentage of cells with nuclear versus cytoplasmic staining.
siRNA silencing of JNK and PARP.
Knockdown of JNK and PARP was performed with 100 nM of targeting siRNA duplexes (catalog no. 6232 and 6304, respectively; Cell Signaling, Inc., Danvers, MA) delivered as lipid-siRNA complexes using Lipofectamine 2000 and Opti-MEM I (Invitrogen) according to the manufacturer’s instructions. In parallel, siControl and siGLO (catalog no. D-001810-10 and D-001630-02; Dharmacon) at a concentration of 100 nM were similarly complexed. By using a reverse transfection method, complexes were added to trypsinized cells within each well in complete tissue culture medium for 24 h, at which point medium was replaced with fresh medium. Western blot analyses verified knockdown compared to actin using the antibodies described above and untreated cells with typical efficiencies of decrease of >95%. Cells were infected 72 h after treatment.
Statistical analyses and image processing.
Unless otherwise indicated, data presented are means and standard errors derived from a minimum of three independent experiments. Differences between means were tested for significance using the Tukey-Kramer multiple-comparison test with the null hypothesis rejected for P values of >0.05. For image-based data, cells were directly enumerated from captured images, and the data presented are the means and standard errors of the means derived from at least three independent experiments and the examination of a minimum of 1,000 total cells. For publication, images were processed using Adobe Photoshop CS6 and prepared using Adobe Illustrator CS6.
SUPPLEMENTAL MATERIAL
Silencing PARP and JNK is efficient and does not inhibit CMT. (A) HeLa cells were transfected with 100 nM of siRNA duplexes targeting JNK1 and PARP-1 (Cell Signaling Technology) using Lipofectamine 2000 and Opti-MEM I (Invitrogen) by a reverse transfection method per the manufacturers’ protocol with complexes added to trypsinized cells in complete medium. Cells and complexes were incubated for at least 24 h before medium was removed and fresh complete medium was added. Cells were then infected with indicated strains at 72 h after treatment with siRNAs. Protein level comparisons were to levels of actin in lysates and to treated but infected cells and to cells transfected with siControl, consisting of a nonspecific siRNA and siGLO, an indicator of transfection efficiency (Dharmacon). Analyses using Western blotting of cellular lysates assessed the presence of JNK1 and activation of PARP-1 compared to actin. (B) CMT efficiencies in treated cells were assessed by Western blotting of cellular lysates from monolayers infected by strains expressing NADase+ or NADase− SPN tagged with an influenza hemagglutinin epitope (HA) tag using an antiserum specific for the HA epitope (Sigma). Data are representative of at least 2 independent experiments. Download
ACKNOWLEDGMENTS
We thank Dong Yu (Washington University) for providing HeLa cells.
Footnotes
Citation Chandrasekaran S, Caparon MG. 2016. The NADase-negative variant of the Streptococcus pyogenes toxin NAD+ glycohydrolase induces JNK1-mediated programmed cellular necrosis. mBio 7(1):e02215-15. doi:10.1128/mBio.02215-15.
REFERENCES
- 1.Los FC, Randis TM, Aroian RV, Ratner AJ. 2013. Role of pore-forming toxins in bacterial infectious diseases. Microbiol Mol Biol Rev 77:173–207. doi: 10.1128/MMBR.00052-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cunningham MW. 2000. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walker MJ, Barnett TC, McArthur JD, Cole JN, Gillen CM, Henningham A, Sriprakash KS, Sanderson-Smith ML, Nizet V. 2014. Disease manifestations and pathogenic mechanisms of group A Streptococcus. Clin Microbiol Rev 27:264–301. doi: 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leckman JF, King RA, Gilbert DL, Coffey BJ, Singer HS, Dure LS, Grantz H, Katsovich L, Lin H, Lombroso PJ, Kawikova I, Johnson DR, Kurlan RM, Kaplan EL. 2011. Streptococcal upper respiratory tract infections and exacerbations of tic and obsessive-compulsive symptoms: a prospective longitudinal study. J Am Acad Child Adolesc Psychiatry 50:108–118.e3. doi: 10.1016/j.jaac.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Madden JC, Ruiz N, Caparon M. 2001. Cytolysin-mediated translocation (CMT): a functional equivalent of type III secretion in gram-positive bacteria. Cell 104:143–152. doi: 10.1016/S0092-8674(01)00198-2. [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekaran S, Caparon MG. 2015. The Streptococcus pyogenes NAD(+) glycohydrolase modulates epithelial cell PARylation and HMGB1 release. Cell Microbiol 17:1376–1390. doi: 10.1111/cmi.12442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghosh J, Caparon MG. 2006. Specificity of Streptococcus pyogenes NAD(+) glycohydrolase in cytolysin-mediated translocation. Mol Microbiol 62:1203–1214. doi: 10.1111/j.1365-2958.2006.05430.x. [DOI] [PubMed] [Google Scholar]
- 8.Magassa N, Chandrasekaran S, Caparon MG. 2010. Streptococcus pyogenes cytolysin-mediated translocation does not require pore formation by streptolysin O. EMBO Rep 11:400–405. doi: 10.1038/embor.2010.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mozola CC, Magassa N, Caparon MG. 2014. A novel cholesterol-insensitive mode of membrane binding promotes cytolysin-mediated translocation by streptolysin O. Mol Microbiol 94:675–687. doi: 10.1111/mmi.12786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mozola CC, Caparon MG. 2015. Dual modes of membrane binding direct pore formation by streptolysin O. Mol Microbiol 97:1036–1050. doi: 10.1111/mmi.13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Seaghdha M, Wessels MR. 2013. Streptolysin O and its co-toxin NAD-glycohydrolase protect group A streptococcus from Xenophagic killing. PLoS Pathog 9:e1003394. doi: 10.1371/journal.ppat.1003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bastiat-Sempe B, Love JF, Lomayesva N, Wessels MR. 2014. Streptolysin O and NAD-glycohydrolase prevent phagolysosome acidification and promote group A streptococcus survival in macrophages. mBio 5:e01690-14. doi: 10.1128/mBio.01690-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cywes C, Hakansson A, Christianson J, Wessels MR. 2005. Extracellular group A streptococcus induces keratinocyte apoptosis by dysregulating calcium signalling. Cell Microbiol 7:945–955. doi: 10.1111/j.1462-5822.2005.00525.x. [DOI] [PubMed] [Google Scholar]
- 14.Michos A, Gryllos I, Håkansson A, Srivastava A, Kokkotou E, Wessels MR. 2006. Enhancement of streptolysin O activity and intrinsic cytotoxic effects of the group A streptococcal toxin, NAD-glycohydrolase. J Biol Chem 281:8216–8223. doi: 10.1074/jbc.M511674200. [DOI] [PubMed] [Google Scholar]
- 15.Chandrasekaran S, Ghosh J, Port GC, Koh EI, Caparon MG. 2013. Analysis of polymorphic residues reveals distinct enzymatic and cytotoxic activities of the Streptococcus pyogenes NAD+ glycohydrolase. J Biol Chem 288:20064–20075. doi: 10.1074/jbc.M113.481556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riddle DJ, Bessen DE, Caparon MG. 2010. Variation in Streptococcus pyogenes NAD+ glycohydrolase is associated with tissue tropism. J Bacteriol 192:3735–3746. doi: 10.1128/JB.00234-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin S, DiPaola RS, Mathew R, White E. 2007. Metabolic catastrophe as a means to cancer cell death. J Cell Sci 120:379–383. doi: 10.1242/jcs.03349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ratner AJ, Hippe KR, Aguilar JL, Bender MH, Nelson AL, Weiser JN. 2006. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem 281:12994–12998. doi: 10.1074/jbc.M511431200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belmokhtar CA, Hillion J, Ségal-Bendirdjian E. 2001. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene 20:3354–3362. doi: 10.1038/sj.onc.1204436. [DOI] [PubMed] [Google Scholar]
- 20.Fink SL, Cookson BT. 2005. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun 73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Husmann M, Dersch K, Bobkiewicz W, Beckmann E, Veerachato G, Bhakdi S. 2006. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem Biophys Res Commun 344:1128–1134. doi: 10.1016/j.bbrc.2006.03.241. [DOI] [PubMed] [Google Scholar]
- 22.Kloft N, Busch T, Neukirch C, Weis S, Boukhallouk F, Bobkiewicz W, Cibis I, Bhakdi S, Husmann M. 2009. Pore-forming toxins activate MAPK p38 by causing loss of cellular potassium. Biochem Biophys Res Commun 385:503–506. doi: 10.1016/j.bbrc.2009.05.121. [DOI] [PubMed] [Google Scholar]
- 23.Stassen M, Müller C, Richter C, Neudörfl C, Hültner L, Bhakdi S, Walev I, Schmitt E. 2003. The streptococcal exotoxin streptolysin O activates mast cells to produce tumor necrosis factor alpha by p38 mitogen-activated protein kinase- and protein kinase C-dependent pathways. Infect Immun 71:6171–6177. doi: 10.1128/IAI.71.11.6171-6177.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papa S, Bubici C, Zazzeroni F, Pham CG, Kuntzen C, Knabb JR, Dean K, Franzoso G. 2006. The NF-kappaB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ 13:712–729. doi: 10.1038/sj.cdd.4401865. [DOI] [PubMed] [Google Scholar]
- 25.Virág L, Robaszkiewicz A, Vargas JM, Oliver FJ. 2013. Poly(ADP-ribose) signaling in cell death. Mol Aspects Med 34:1153–1167. doi: 10.1016/j.mam.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 26.Huffman DL, Abrami L, Sasik R, Corbeil J, van der Goot FG, Aroian RV. 2004. Mitogen-activated protein kinase pathways defend against bacterial pore-forming toxins. Proc Natl Acad Sci U S A 101:10995–11000. doi: 10.1073/pnas.0404073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, Shen HM. 2007. c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ 14:1001–1010. doi: 10.1038/sj.cdd.4402088. [DOI] [PubMed] [Google Scholar]
- 28.Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. 2001. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. 2005. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 30.Chambers JW, LoGrasso PV. 2011. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J Biol Chem 286:16052–16062. doi: 10.1074/jbc.M111.223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chambers JW, Cherry L, Laughlin JD, Figuera-Losada M, Lograsso PV. 2011. Selective inhibition of mitochondrial JNK signaling achieved using peptide mimicry of the sab kinase interacting motif-1 (KIM1). ACS Chem Biol 6:808–818. doi: 10.1021/cb200062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Y, Huang S, Liu ZG, Han J. 2006. Poly(ADP-ribose) polymerase-1 signaling to mitochondria in necrotic cell death requires RIP1/TRAF2-mediated JNK1 activation. J Biol Chem 281:8788–8795. doi: 10.1074/jbc.M508135200. [DOI] [PubMed] [Google Scholar]
- 33.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV, Franzoso G. 2004. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell 119:529–542. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 34.Ventura JJ, Cogswell P, Flavell RA, Baldwin AS Jr., Davis RJ. 2004. JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev 18:2905–2915. doi: 10.1101/gad.1223004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kao CY, Los FC, Huffman DL, Wachi S, Kloft N, Husmann M, Karabrahimi V, Schwartz JL, Bellier A, Ha C, Sagong Y, Fan H, Ghosh P, Hsieh M, Hsu CS, Chen L, Aroian RV. 2011. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog 7:e1001314. doi: 10.1371/journal.ppat.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chambers JW, Pachori A, Howard S, Iqbal S, LoGrasso PV. 2013. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J Biol Chem 288:4000–4011. doi: 10.1074/jbc.M112.406777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 38.Tobar N, Toyos M, Urra C, Méndez N, Arancibia R, Smith PC, Martínez J. 2014. c-Jun N terminal kinase modulates NOX-4 derived ROS production and myofibroblasts differentiation in human breast stromal cells. BMC Cancer 14:640. doi: 10.1186/1471-2407-14-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrow MA, Chumbler NM, Lapierre LA, Franklin JL, Rutherford SA, Goldenring JR, Lacy DB. 2013. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc Natl Acad Sci U S A 110:18674–18679. doi: 10.1073/pnas.1313658110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wohlan K, Goy S, Olling A, Srivaratharajan S, Tatge H, Genth H, Gerhard R. 2014. Pyknotic cell death induced by Clostridium difficile TcdB: chromatin condensation and nuclear blister are induced independently of the glucosyltransferase activity. Cell Microbiol 16:1678–1692. doi: 10.1111/cmi.12317. [DOI] [PubMed] [Google Scholar]
- 41.Sun J, Siroy A, Lokareddy RK, Speer A, Doornbos KS, Cingolani G, Niederweis M. 2015. The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat Struct Mol Biol 22:672–678. doi: 10.1038/nsmb.3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Port GC, Paluscio E, Caparon MG. 2015. Complete genome sequences of emm6 Streptococcus pyogenes JRS4 and parental strain D471. Genome Announc 3:e00275-15. doi: 10.1128/genomeA.00725-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, Johnson AE, Tweten RK. 1998. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an alpha-helical to beta-sheet transition identified by fluorescence spectroscopy. Biochemistry 37:14563–14574. doi: 10.1021/bi981452f. [DOI] [PubMed] [Google Scholar]
- 44.Ghosh J, Anderson PJ, Chandrasekaran S, Caparon MG. 2010. Characterization of Streptococcus pyogenes beta-NAD+ glycohydrolase: re-evaluation of enzymatic properties associated with pathogenesis. J Biol Chem 285:5683–5694. doi: 10.1074/jbc.M109.070300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Silencing PARP and JNK is efficient and does not inhibit CMT. (A) HeLa cells were transfected with 100 nM of siRNA duplexes targeting JNK1 and PARP-1 (Cell Signaling Technology) using Lipofectamine 2000 and Opti-MEM I (Invitrogen) by a reverse transfection method per the manufacturers’ protocol with complexes added to trypsinized cells in complete medium. Cells and complexes were incubated for at least 24 h before medium was removed and fresh complete medium was added. Cells were then infected with indicated strains at 72 h after treatment with siRNAs. Protein level comparisons were to levels of actin in lysates and to treated but infected cells and to cells transfected with siControl, consisting of a nonspecific siRNA and siGLO, an indicator of transfection efficiency (Dharmacon). Analyses using Western blotting of cellular lysates assessed the presence of JNK1 and activation of PARP-1 compared to actin. (B) CMT efficiencies in treated cells were assessed by Western blotting of cellular lysates from monolayers infected by strains expressing NADase+ or NADase− SPN tagged with an influenza hemagglutinin epitope (HA) tag using an antiserum specific for the HA epitope (Sigma). Data are representative of at least 2 independent experiments. Download