

Abstract
Wnt signaling has diverse actions in cardiovascular development and disease processes. Secreted frizzled-related protein 5 (Sfrp5) has been shown to function as an extracellular inhibitor of non-canonical Wnt signaling that is expressed at relatively high levels in white adipose tissue. The aim of this study was to investigate the role of Sfrp5 in the heart under ischemic stress. Sfrp5 KO and WT mice were subjected to ischemia/reperfusion (I/R). Although Sfrp5-KO mice exhibited no detectable phenotype when compared with WT control at baseline, they displayed larger infarct sizes, enhanced cardiac myocyte apoptosis, and diminished cardiac function following I/R. The ischemic lesions of Sfrp5-KO mice had greater infiltration of Wnt5a-positive macrophages and greater inflammatory cytokine and chemokine gene expression when compared with WT mice. In bone marrow-derived macrophages, Wnt5a promoted JNK activation and increased inflammatory gene expression, whereas treatment with Sfrp5 blocked these effects. These results indicate that Sfrp5 functions to antagonize inflammatory responses after I/R in the heart, possibly through a mechanism involving non-canonical Wnt5a/JNK signaling.
Keywords: c-Jun N-terminal kinase (JNK), macrophage, myocardial infarction, obesity, Wnt signaling, Sfrp5, Wnt5a
Introduction
Inflammation is widely recognized to be involved in the pathogenesis, severity, and outcome of ischemic heart disease (1). Obesity is thought to contribute to cardiovascular disorders, at least in part, through the systemic release of pro-inflammatory adipokines by dysfunctional white adipose tissue (WAT)2 (2, 3). Inflammation has complex roles in both adaptive healing process following infarction as well as in the maladaptive processes that contribute to heart failure (4–7), and the acute healing and eventual outcome of ischemic myocardial injury are dependent upon the appropriately choreographed regulation of numerous pro- and anti-inflammatory modulators. In this regard, immune modulators produced by adipose tissue can either facilitate or impair the myocardial healing process, depending on the status of adipose tissue function and the composition of its inflammatory secretome (3, 8, 9). Thus, a better understanding of the links between obesity-mediated inflammatory processes and post-infarct remodeling of the heart is warranted.
At the cellular level, inflammatory processes are tightly orchestrated by secreted signaling molecules that bind to specific cell surface receptors and activate intracellular signaling pathways. Signaling by Wnt ligands is a major regulator of several biological processes, but its roles in modulating inflammatory responses are relatively understudied. The 19 Wnt family proteins contain cysteine-rich domains and activate signaling by binding to one or more of the 10 frizzled family receptors (10). Wnt signaling can be classified as canonical or a non-canonical (10, 11). Canonical Wnt signaling involves activation of the β-catenin signaling pathway. Non-canonical Wnt signaling involves other pathways including planar cell polarity and Ca2+ pathways (10, 12, 13). Wnt3a is the prototypical ligand that induces canonical signaling, whereas Wnt5a has mostly been associated with non-canonical signaling. Canonical and non-canonical Wnt signaling pathways are generally thought to functionally oppose each other. It is reported that both canonical and non-canonical Wnt signaling pathways play an important role in cardiac development and myocyte differentiation (12, 14), and in the adult, have been reported to be involved in maladaptive hypertrophy (15–17).
Both canonical and non-canonical Wnt signaling pathways can be regulated by several endogenous antagonists, including Wnt inhibitory factor (WIF), Cerberus (CER), and Dickkopf (Dkk) (18). Secreted frizzled-related proteins (Sfrps) represent a family of decoy receptors that negatively modulate Wnt signaling (19–22). Sfrps have cysteine-rich domain domains (22, 23), and the antagonistic effect of Sfrps on signaling is thought to be mediated by the ability to bind either Wnt ligands and/or the frizzled receptors (10, 18, 22).
Accumulating evidence implicates the participation of Sfrps in ischemic cardiovascular diseases. It is reported that Sfrp2 activates mesenchymal stem cells and promotes cardiac repair following myocardial infarction via inhibition of bone morphogenic protein (BMP) and canonical Wnt signaling (24, 25). Sfrp1 has also been shown to have a protective role in post-infarct left ventricular (LV) remodeling (26). Furthermore, the combined blockade of Wnt/frizzled signaling by Wnt3a/Wnt5a homologues suppresses development of heart failure following myocardial infarction (27). Although these studies provide compelling evidence for the importance of Sfrp/Wnt signaling in cardiac development and disease, the role of this signaling pathway in the context of inflammatory cardio-metabolic disease has not been investigated.
We and other groups have reported that Sfrp5 is highly expressed in WAT and is involved in obesity and metabolic function (28–33). Most of these reports are consistent with the interpretation that Sfrp5 has an anti-inflammatory effect and that it exerts these effects through the suppression of non-canonical Wnt5a/JNK signaling pathway. To date, nothing has been reported about the role of Sfrp5 in ischemic heart disease. Here, we investigate whether Sfrp5 affects the inflammatory response of the heart to acute ischemic injury. Specifically, we tested the effects of Sfrp5 on myocardial infarct size, apoptotic activity, and inflammatory responses following ischemia reperfusion (I/R) injury. We also tested the effects of Sfrp5 on mouse bone marrow-derived macrophage (BMDM) phenotype in vitro. Our observations indicate that Sfrp5 is cardio-protective in the context of I/R injury. This protection is associated with the inhibition of Wnt5a/JNK-dependent anti-inflammatory actions of Sfrp5.
Experimental Procedures
Materials
Antibodies against phospho-JNK (Thr-172), JNK, phospho-GSK-3β, total GSK-3β, and GAPDH were purchased from Cell Signaling Technology. Galectin3 (also known as Mac2) antibody was obtained from Santa Cruz Biotechnology. Wnt5a antibody was obtained from R&D Systems. Recombinant Sfrp5, TNFα, Wnt5a, and Wnt3a proteins (expressed in Chinese hamster ovary cells, endotoxin level is <1.0 enzyme units per 1 μg of the protein by the Limulus amebocyte lysate method) were obtained from R&D Systems. SP600125, which is a JNK inhibitor, tunicamycin, and hydrogen peroxide were purchased from Sigma-Aldrich. DMEM and RPMI 1640 medium were purchased from Life Technologies.
Mouse Lines
Sfrp5-deficient mice are viable and fertile and do not display overt abnormalities at baseline. Mice lacking Sfrp5 were backcrossed and maintained on the C57BL/6 background (Charles River Laboratories). Sfrp5−/− (Sfrp5-KO) mice were generated by replacing the first protein coding exon with the PGKneobpAloxA cassette as described previously (34). Sfrp5-KO mice and littermate WT C57BL/6 mice were used. All experiments were performed in adherence with the National Institutes of Health Guidelines on the Use of Laboratory Animals, and were approved by the Institutional Animal Care and Use Committee at Boston University.
I/R Injury Model
Ten-to-twelve-week-old male WT and Sfrp5-KO mice underwent I/R injury as described previously (9). Following anesthetization (pentobarbital 50 mg/kg intraperitoneal) and intubation, a suture was ligated around the proximal left coronary artery using a snare occluder. Ischemia followed by reperfusion was accomplished by tightening the snare occluder for 30 min and then loosening it. Myocardial reperfusion was confirmed by changes in ECG as well as by changes in the appearance of the heart from pale to bright red. The suture was left in place, and the chest was closed. During the surgical procedure, ECG and the rectal temperature were monitored, and the rectal temperature was maintained at 37 ± 0.5 °C. Twenty-four hours after reperfusion, the chest was re-opened and the suture was re-tied. Evans Blue dye was injected at the aortic root to determine the area at risk (AAR). The heart was then excised and incubated with 1.5% 2,3,5-triphenyltetrazolium chloride for 10 min at 37 °C to determine the infarction area (IA). LV area, AAR, and IA were determined by computerized planimetry using ImageJ.
Echocardiography
To measure LV systolic function and chamber dimensions, echocardiography was performed with a Vevo 770 machine using a probe 707B (30 MHz). Mice were anesthetized with 1–2% isoflurane and placed on the heating pad (37 °C) during observation. The heart was first visualized in long/short axis views followed by M-mode analysis of the short axis. The symmetrical short axis images were obtained at the level of papillary muscles. Diastolic intraventricular septum, diastolic posterior wall thickness (PWd), LV end diastolic diameter (LVEDD), and LV end systolic diameter (LVESD) were assessed from M-mode images. %LV fractional shortening (%FS) was calculated by the following equation: %FS = LVESD/(LVEDD − LVESD) × 100. At least three measurements were obtained and averaged for every data point from each mouse.
Immunohistochemistry and Immunofluorescence
Myocardial tissues from WT or Sfrp5-KO mice were fixed by 4% paraformaldehyde overnight. Fixed tissues were replaced sucrose solution before embedding by OCT compound. Cryostat sections were cut at a thickness of 5 μm. Endogenous peroxidase activity was blocked with 0.3% H2O2 in PBS for 10 min at room temperature, and nonspecific binding sites were blocked with 3% horse serum in PBS for 1 h at room temperature. Samples were incubated with goat polyclonal Wnt5a antibody (R&D Systems; 1:100) overnight at 4 °C, followed by incubation with biotin-conjugated secondary antibody and horseradish peroxidase-avidin. After incubation, a VECTASTAIN Elite ABC kit (Vector Laboratories) was used according to the manufacturer's instructions. For immunofluorescence, tissues were blocked with BSA and then incubated with Wnt5a (R&D Systems) antibody and Mac2 (Santa Cruz Biotechnology) antibody, followed by incubation with anti-goat IgG conjugated with Alexa Fluor 488 (Life Technologies) for detection of Wnt5a and anti-rabbit IgG conjugated with Alexa Fluor 594 (Life Technologies) for detection of Mac2. DAPI was used for nuclear staining. Images were recorded using a Zeiss LSM 710-Live Duo scan confocal microscope.
Western Blot Analysis
Tissue samples were homogenized in radioimmunoprecipitation assay buffer (Thermo Scientific) with protease and phosphatase inhibitor mixture (Thermo Scientific). Protein concentration was determined by the BCA method (Thermo Scientific). Equal amounts of protein (10–50 μg) were separated in denaturing SDS 10% polyacrylamide gels and transferred to PVDF membranes. After blocking with 5% nonfat dried skim milk or BSA in TBS containing 0.1% Tween-20 at room temperature for 1 h, the membranes were incubated with the primary antibodies(1:100–1:1000) at 4 °C overnight, followed by incubation with the secondary antibody conjugated with HRP (1:2500–1:10000) at room temperature for 1 h. Proteins were visualized using the ECL Plus Western blotting detection kit (Amersham Biosciences).
RNA Isolation and Quantitative Real-time PCR
Total RNA was prepared from mouse tissues and cultured cells by using an RNeasy mini or micro kit (Qiagen), respectively. cDNA was synthesized from 500 ng of total RNA using the QuantiTect reverse transcription kit (Qiagen). Quantitative Real-Time PCR (qRT-PCR) was carried out on a ViiA7 (Applied Biosystems) using SYBR Green real-time PCR master mix (Applied Biosystems). The Δ-Δ Ct method using 36B4 or GAPDH RNA and sham/control as the reference and calibrator, respectively, was employed for relative quantification of gene expression.
Cell Culture
Primary cultures of neonatal rat ventricular myocytes (NRVMs) were prepared as described previously (9). NRVMs were incubated in DMEM supplemented with 10% FBS for 18 h after preparation. Assays were performed after 24–48 h of serum deprivation. A GasPak system (BD Biosciences) was used to create hypoxic conditions. BMDMs were obtained by in vitro differentiation of mouse bone marrow progenitors. Bone marrow cells were aseptically flushed from femurs and tibias of mice through a 27-gauge needle. Cells were then plated and cultured in RPMI 1640 containing 2 mm glutamine, 25 mm HEPES, 10% heat-inactivated FBS (low endotoxin level), and 10 ng/ml M-CSF (R&D Systems). Assays were performed at day 5 after plating and induction of BMDM differentiation by M-CSF treatment. Mouse 3T3-L1 cells (ATCC) were maintained in DMEM with 10% FBS and differentiated into adipocytes by treatment with DMEM supplemented with 5 μg/ml insulin, 0.5 mm 1-methyl-3-isobutyl-xanthine, and 1 μm dexamethasone (35). At day 7 after differentiation, 3T3-L1 adipocytes were treated with tunicamycin, H2O2, TNFα, or vehicle for 24 h.
TUNEL Staining
TUNEL staining for both hearts after I/R and cultured NRVMs after hypoxia/reoxygenation (H/R) with or without pretreatment (Wnt5a, Sfrp5, or SP600125), was performed using an in situ cell death detection kit (Roche Applied Science). For heart tissues, specimens were fixed by 4% paraformaldehyde in PBS overnight at 4 °C, permeabilized with 0.1% Triton X-100, and then incubated with anti-sarcomeric actinin antibody (Sigma; clone EA53, 1:100) for 60 min followed by incubation with Alexa Fluor 594-conjugated anti-mouse IgG antibody (Life Technologies). Specimens were then incubated with TUNEL staining solution for 1 h according to the manufacturer's protocol. For NRVMs, equal numbers of cells were exposed to 12 h of hypoxia followed by 24 h of reoxygenation, and then treated as described above with the 4% paraformaldehyde fixation for 15 min at room temperature. DAPI was used for nuclear staining. TUNEL-positive cardiomyocytes were counted in randomly selected three fields of the slide.
Statistical Analysis
Data are presented as mean ± S.E. Analyses were performed using StatView software. To compare three or more groups, a one- or two-way analysis of variance was performed with post hoc Student's t tests. For echocardiographic analysis, a two-way repeated measured analysis of variance was performed with Bonferroni post hoc tests. A value of p < 0.05 was considered statistically significant.
Results
Characterization of Sfrp5 Gene Expression
Consistent with prior studies (28, 36), Sfrp5 transcript expression is enriched in adipose tissue, particularly in epididymal and pericardial depots, whereas expression in subcutaneous WAT and brown adipose tissue was much lower (Fig. 1A). Sfrp5 mRNA expression in the myocardium is low when compared with epididymal WAT (Fig. 1B). In contrast to Sfrp5, levels of Sfrp1, Sfrp3, and Sfrp4 were low or undetectable in epididymal WAT, whereas Sfrp2 showed modest expression (Fig. 1C). Deficiency of Sfrp5 did not detectably affect the expression of other Sfrp family members (Fig. 1C).
FIGURE 1.
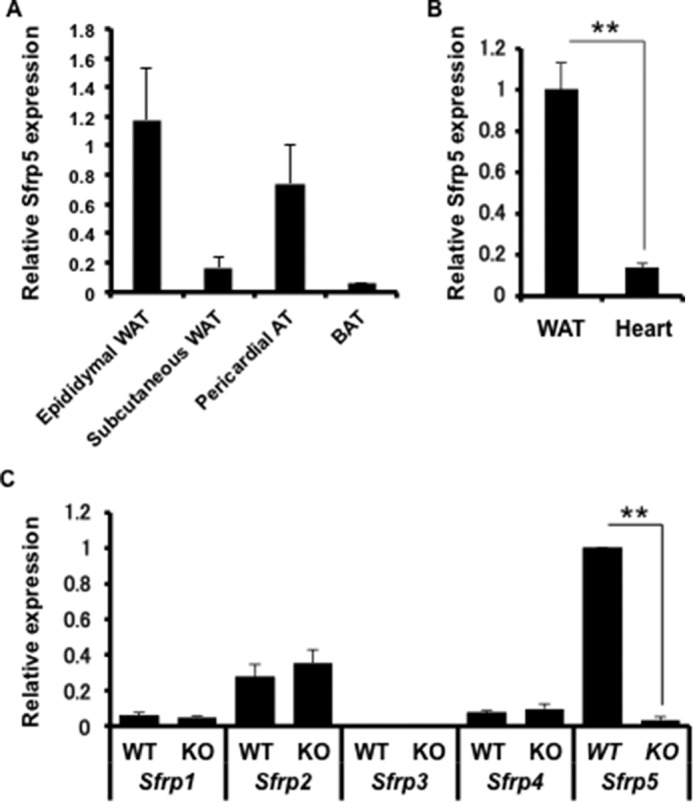
Tissue-specific expression of Sfrp5. Total RNA was extracted from adipose tissue, and cDNA was obtained by reverse transcription. Quantification was carried out by qRT-PCR. A, Sfrp5 mRNA in adipose tissue depots from WT mice (n = 4 each). BAT, brown adipose tissue. BAT, brown adipose tissue. B, expression of Sfrp5 mRNA in epididymal adipose tissue and heart of WT mice. (n = 4 each). **, p < 0.05. C, basal expression of Sfrp1, Sfrp2, Sfrp3, Sfrp4, and Sfrp5 mRNA in Sfrp5-KO mice and WT controls. (n = 4 each). **, p < 0.05.
WT and Sfrp5-KO mice were subjected to 30 min of left anterior descending artery ligation followed by 24 h of reperfusion. Myocardial I/R in WT mice led to a marked reduction in Sfrp5 transcript expression in epididymal and pericardial fat (Fig. 2). The expression of other adipokines, adiponectin and C1q/TNF-related protein 9 (CTRP9), which have cardiovascular- and metabolic-protective activities (8, 9, 37–41), was also examined in the myocardial I/R model. Like Sfrp5, the CTRP9 transcript was down-regulated following ischemic myocardial injury in both pericardial and epididymal fat depots (Fig. 2). Ischemic myocardial injury also led to the down-regulation of adiponectin transcript in the pericardial adipose tissue, but this down-regulation was not detected in epididymal WAT. Sfrp5 deficiency did not have any detectable effect on the transcript levels of adiponectin or CTRP9 at baseline or in response to myocardial injury in either adipose tissue depot (Fig. 2). Similarly, Sfrp deficiency did not have any detectable effect on the expression of TNF-α, IL-1β, or IL-6 transcripts at baseline or in response to myocardial injury in either pericardial or epididymal adipose tissue (data not shown).
FIGURE 2.

Modulation of adipokine expression following myocardial I/R injury. Total RNA was extracted from epididymal WAT and pericardial adipose tissue (AT) following sham and I/R injury, and obtained cDNA by reverse transcription reaction. Quantification was carried out by qRT-PCR. Expression of Sfrp5, adiponectin, and CTRP9 mRNA in the indicated adipose tissue depot following sham operation and I/R injury was determined (n = 4 each). *, p < 0.05.
Studies were performed in cultured adipocytes to better define the regulation of Sfrp5. The time course of Sfrp5 transcript induction was similar to that of adiponectin during the differentiation of 3T3-L1 adipocytes (Fig. 3). Sfrp5 expression was down-regulated under conditions of endoplasmic reticulum stress, conferred by treatment with tunicamycin, reactive oxygen species stress, conferred by treatment of H2O2, and by incubation with TNFα in differentiated 3T3-L1 adipocytes. These conditions also led to the down-regulation of adiponectin in cultured adipocytes.
FIGURE 3.
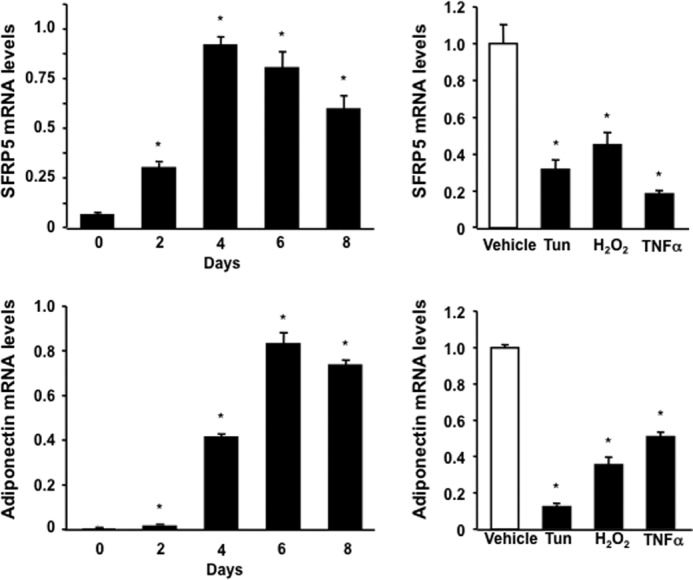
Regulation of Sfrp5 expression in cultured 3T3-L1 adipocytes. Left panel, mRNA expression of Sfrp5 and adiponectin at the different time points during differentiation of 3T3-L1 cells into adipocytes and expressed relative to 18S levels (n = 3). *, p < 0.01 versus day 0. Right panel, expression of Sfrp5 and adiponectin in response to various stimuli in adipocytes. Differentiated 3T3-L1 adipocytes were treated with tunicamycin (Tun, 5 μg/ml), H2O2 (0.2 mm), TNFα (10 ng/ml), or vehicle for 24 h. Transcript levels of Sfrp5 and adiponectin were determined by qRT-PCR and expressed relative to 18S levels (mean ± S.E., n = 3). *, p < 0.01 versus vehicle.
Increased Myocardial Infarct Size and Myocyte Apoptosis in Sfrp5-KO Mice after Ischemia/Reperfusion Injury
Sfrp5-KO mice are viable and fertile and do not display obvious morphological abnormalities when they are fed a normal diet (28, 34, 42). Body weight and echocardiographic data at baseline in 8–10-week-old male mice did not differ between WT and Sfrp5-KO mice (Table 1). To evaluate the functional role of Sfrp5 in the heart under conditions of transient ischemia, we induced myocardial I/R injury in both WT and Sfrp5-KO mice. All mice survived the surgical induction of I/R injury. Representative photographs of myocardial tissues stained with Evans blue dye, to delineate AAR, and 2,3,5-triphenyltetrazolium chloride, to delineate IA, in WT and Sfrp5-KO mice are shown in Fig. 4A. The ratio of AAR to left ventricular area (AAR/LV) was statistically equivalent between WT and Sfrp5-KO mice (52.2 ± 2.5% versus 52.1 ± 2.1%) (Fig. 4B). In contrast, the ratios of IA to AAR (IA/AAR) and IA to left ventricular area (IA/LV) in Sfrp5-KO mice were statistically higher when compared with those of WT mice (IA/AAR; 28.6 ± 2.3% versus 39.1 ± 2.9%, IA/LV; 15.3 ± 1.7% versus 20.6 ± 1.9%). Thus, Sfrp5 deficiency leads to greater infarct size following I/R injury.
TABLE 1.
Baseline characteristics of Sfrp5-KO and WT mice
n, sample size for parameters listed below; HW/BW, heart weight/body weight; HW/TL, heart weight/tibia length; HR, bpm, heart rate in beats per minute; IVSd, interventricular septal thickness at diastole; PWd, posterior wall thickness at diastole.
| WT | KO | |
|---|---|---|
| n | 8 | 7 |
| BW, g | 27.9 ± 1.03 | 28.5 ± 0.98 |
| HW/BW | 6.36 ± 0.25 | 6.23 ± 0.15 |
| HW/TL | 9.89 ± 0.49 | 9.86 ± 0.24 |
| n | 8 | 12 |
| HR, bpm | 492.5 ± 7.1 | 484.9 ± 6.81 |
| LVEDD, mm | 3.64 ± 0.07 | 3.75 ± 0.06 |
| LVESD, mm | 2.04 ± 0.07 | 2.20 ± 0.05 |
| FS, % | 43.91 ± 0.98 | 41.48 ± 0.81 |
| IVSd, mm | 0.683 ± 0.06 | 0.682 ± 0.007 |
| PWd, mm | 0.696 ± 0.01 | 0.685 ± 0.008 |
FIGURE 4.

Sfrp5 deficiency promotes functional deterioration following I/R injury. A and B, AAR and IA were determined by Evans blue staining and 2,3,5-triphenyltetrazolium chloride staining. Ratios of area at risk to left ventricle (AAR/LV), infarct area to area at risk (IA/AAR), and infarct area to left ventricle (IA/LV), respectively, were determined in WT (n = 10) and Sfrp5-KO (n = 14) mice at 24 h after I/R. *, p < 0.05 when compared with WT mice. NS, not significant. C and D, echocardiographic analyses were performed at pre-surgery (baseline), and at 2 weeks (day 14) and 4 weeks (day 28) after I/R injury. Shown are measurements of LVEDD, LVESD, and %FS for WT (n = 6–8) and Sfrp5-KO (n = 7–9) mice. *, p < 0.05 when compared with WT mice. A two-way repeated measured analysis of variance was performed with Bonferroni post hoc tests.
Hearts were also assessed echocardiographically at baseline and at 1, 14, and 28 days after I/R injury (Fig. 4C). At 1 day after ischemic injury, the reperfused hearts showed a trend toward functional deterioration, but this was not statistically significant. At 14 and 28 days after I/R, the deterioration in the percentage of fractional shortening (%FS) was greater in the Sfrp5-KO mice than WT mice, consistent with the greater infarct size.
To investigate the extent of apoptotic cell death in the heart following I/R injury, TUNEL staining was performed on the excised hearts of the WT and Sfrp5-KO experimental groups at the 24-h reperfusion time point. Representative photographs of TUNEL-positive, α-sarcomeric actin-positive cardiac myocyte nuclei in the ischemic portions of the LV are shown in Fig. 5A. Quantitative analysis showed abundant TUNEL-positive cells in the ischemic LV of both Sfrp5-KO and WT mice, with a significantly higher proportion of positive cells in the hearts of the Sfrp5-KO strain (Fig. 5B). In contrast, TUNEL-positive cells were not detected in the hearts of sham-treated mice.
FIGURE 5.

Sfrp5 deficiency leads to greater myocyte death in the infarct zone. A, TUNEL staining of WT (n = 3, 5) and Sfrp5-KO (n = 3, 7) mice in the sham-operated heart and I/R-treated heart (respectively) at 24 h after I/R. Apoptotic nuclei were identified by TUNEL staining (green). Total nuclei were identified by DAPI (blue). Myocytes were counterstained by α-sarcomeric actinin (red). B, TUNEL-positive nuclei are expressed as a percentage of the total nuclei. *, p < 0.05, **, p < 0.01. C, myocyte apoptosis was assessed by TUNEL staining. *, p < 0.05. D, Wnt5a-induced increase in myocyte apoptosis was abrogated by pretreatment with the JNK inhibitor SP600125. *, p < 0.05.
To analyze the involvement of Sfrp5 in greater detail, cultured NRVMs were exposed to hypoxia/reoxygenation, to simulate I/R, in the presence and absence of Sfrp5 and Wnt5a, one of its cognate Wnt ligands (28, 43). Myocyte apoptosis was assessed by TUNEL staining (Fig. 5C). Hypoxia/reoxygenation led to a modest increase in TUNEL-positive NRVMs, but treatment with recombinant Wnt5a protein led to a marked increase in the frequency of TUNEL-positive nuclei. The inclusion of recombinant Sfrp5 completely reversed the Wnt5a-induced increase in NRVM apoptosis. Sfrp5 alone had no effect on the frequency of TUNEL-positive NRVMs, ostensibly because cardiac myocytes do not express Wnt5a.
JNK, a component of planar cell polarity, is activated by non-canonical Wnt signaling in multiple systems (28, 44, 45) including cardiac myocytes (Ref. 46 and data not shown). To evaluate the role of JNK in mediating Wnt5a-induced myocyte apoptosis, cultured NRVMs were treated with recombinant Wnt5a under conditions of hypoxia/reoxygenation. The Wnt5a-induced increase in myocyte apoptosis was abrogated by pretreatment with the JNK inhibitor SP600125 (Fig. 5D). SP600125 was as effective as recombinant Sfrp5 in inhibiting apoptosis, returning it to baseline levels.
Enhanced Accumulation of Wnt5a-positive Macrophages in the Ischemic Myocardium of Sfrp5-KO Mice
Because it has been reported that Sfrp5 antagonizes Wnt5a-mediated signaling in other systems (28, 43), we analyzed the presence of Wnt5a-positive cells in the infarcted heart. Immunohistochemical analysis showed that Wnt5a-positive cells are present in the damaged myocardium, and Wnt5a-positive cells largely co-localized with a subset of cells that were positive for Mac2, a marker for macrophages (Fig. 6A). Quantitative analyses revealed a marked increase in Wnt5a-positive as well as Mac2-positive cells in the ischemic myocardium (Fig. 6B). Notably, the number of Wnt5a-positive and Mac2-positive cells present in the ischemic myocardium was greater in the Sfrp5-KO mouse than in WT mice. The Wnt5a-positive cells did not co-localize with CD31, a vascular endothelial cell marker, nor with Fsp1 (fibroblast-specific protein 1; also known as S100A), a fibroblast marker (data not shown). No signal for Wnt5a or Mac2 could be detected in sham-treated hearts (Fig. 6B).
FIGURE 6.
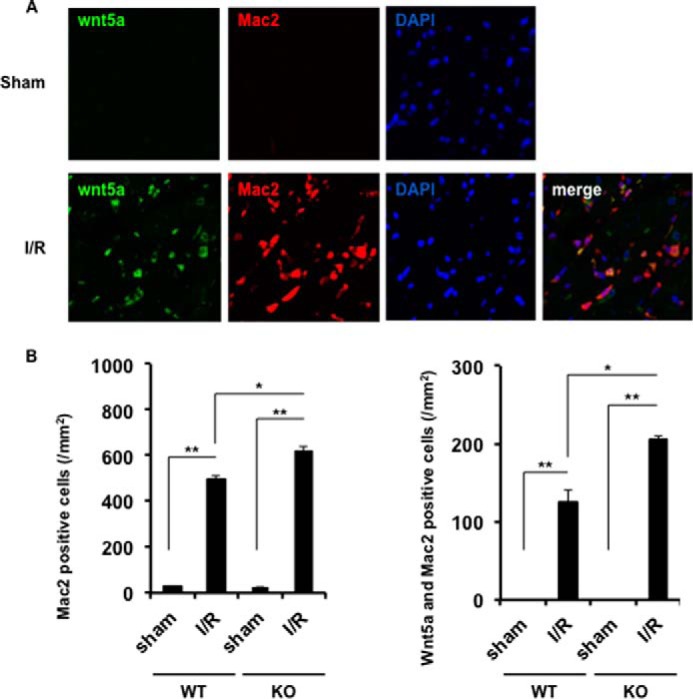
Sfrp5 deficiency leads to a greater influx of Wnt5a-positive macrophages after I/R injury. A, representative immunofluorescence image of co-localization with Wnt5a and Mac2. Wnt5a was expressed by Alexa Fluor 488 (green), and Mac2 was expressed by Alexa Fluor 594 (red). Nuclei were expressed by DAPI (blue). B, quantitative analysis of the number of Mac2 single-positive cells and the number of Mac2 and Wnt5a double-positive cells. Sham, n = 3 per group; I/R, n = 5 per group; *, p < 0.05, **, p < 0.01.
Sfrp5/Wnt5a-mediated Modulation of Macrophage Signaling
Accumulating evidence suggests that Wnt5a can function as an immune system modulator that acts in an autocrine manner (45, 47–50). Thus, we evaluated the effects of Wnt5a and Sfrp5 on murine BMDMs in vitro. Stimulation with LPS led to a 5-fold increase in Wnt5a transcript expression (Fig. 7A). Moreover, Wnt5a mRNA was elevated more than 2-fold after stimulation with exogenous Wnt5a (Fig. 7A). LPS treatment also led to an up-regulation of Wnt5a protein levels (Fig. 7B). These results suggest that Wnt5a is up-regulated upon macrophage activation, consistent with a recent study (47). Next, we evaluated effects of Wnt5a stimulation on cellular signaling in BMDMs. JNK phosphorylation was activated after stimulation with exogenous Wnt5a in a time-dependent manner (Fig. 7C). Notably, treatment with Sfrp5 led to the inhibition of Wnt5a-enhanced JNK phosphorylation in a dose-dependent manner (Fig. 7D). In contrast, stimulation with Wnt3a, a canonical Wnt, activated GSK-3β, which is downstream of canonical Wnt signaling, but did not activate JNK (Fig. 7E).
FIGURE 7.

Wnt5a activates macrophage JNK signaling. A, Wnt5a expression in activated macrophages. qRT-PCR was performed to evaluate Wnt5a transcript level in BMDMs under both LPS and Wnt5a stimulation for 24 h. *, p < 0.05, **, p < 0.01. B, Wnt5a protein expression in BMDMs following LPS stimulation for 24 h (n = 4). C, Wnt5a activation of JNK in BMDMs in a time-dependent manner. BMDMs were depleted of serum overnight, and then incubated with Wnt5a (200 ng/ml) for the indicated times. p-JNK, phosphorylated JNK. *, p < 0.05. D, Sfrp5 inhibits Wnt5a induced phosphorylation of JNK. BMDMs were pretreated with increasing concentrations of recombinant Sfrp5 (50, 100, 200, and 500 ng/ml) for 30 min., and then stimulated with Wnt5a (200 ng/ml) for 30 min. *, p < 0.05. E, Wnt3a activates canonical Wnt signaling, but not JNK. BMDMs were incubated with increasing concentrations of recombinant Wnt3a. p-GSK-3β, phosphorylated GSK-3β; p-JNK, phosphorylated JNK.
Sfrp5 Inhibits JNK Activation and Inflammatory Cytokine/Chemokine Expression in Macrophages and Ischemic Myocardium
Treatment with Wnt5a enhanced the expression of the inflammatory cytokines TNFα and IL-1β in BMDMs (Fig. 8A). The activation of cytokines by Wnt5a was completely inhibited by treatment with recombinant Sfrp5. Furthermore, Wnt5a-stimulated cytokine induction is abrogated by the JNK inhibitor SP600125 (Refs. 28 and 45 and data not shown). Collectively, these findings document the activation of inflammatory signaling pathways downstream of Wnt5a in macrophages.
FIGURE 8.

Enhanced activation of JNK signaling and increased expression of inflammatory mediators in Sfrp5-KO mice after I/R injury. A, Sfrp5 inhibits Wnt5a-induced expression of pro-inflammatory cytokines. BMDMS were stimulated with Wnt5a in the presence and absence of recombinant Sfrp5, and TNFα and IL-1β expression was analyzed by qRT-PCR (n = 3). *, p < 0.05. B, Western immunoblot of JNK phosphorylation (p-JNK) in sham and ischemic hearts of WT and Sfrp5-KO mice. C, results of quantitative RT-PCR analysis of pro-inflammatory myocardial expression levels of IL1-β, TNF-α, and MCP-1 in the different experimental groups. Transcript levels were normalized to GAPDH in sham WT mice. n = 4 samples in each group. *, p < 0.05, **, p < 0.01.
To validate the in vitro findings in BMDMs in vivo, JNK signaling and the expression of pro-inflammatory mediators were evaluated in the hearts of WT and Sfrp5-KO mice. As expected, JNK signaling was elevated following I/R in WT mice, but the level of JNK phosphorylation was much greater in the Sfrp5-KO mice (Fig. 8B). Consistent with observations of enhanced macrophage infiltration, transcript levels of the inflammatory cytokines IL-1β and TNFα and the chemokine MCP-1 (also known as CCL2) were up-regulated in the ischemic area from Sfrp5-KO mice to a greater extent than the ischemic LV of WT mice (Fig. 8C). There were little or no differences in the levels of JNK phosphorylation or inflammatory mediator expression between WT and Sfrp5-KO in sham-treated mice (Fig. 8), consistent with the lack of macrophage infiltration under these conditions (Fig. 6).
Discussion
The data from this study show, for the first time, that Sfrp5 confers resistance to damage caused by acute myocardial ischemia. The genetic deficiency of Sfrp5 in mice led to a greater myocardial infarct size following I/R injury, and this was accompanied by a greater amount of apoptotic cell death of cardiac myocytes and a greater degree of inflammation in the infarct zone. These data indicate a new mechanism by which obesity contributes to heart disease. A growing body of evidence suggests that obesity contributes to cardiovascular disease through unbalanced adipokine secretion that contributes to a chronic low-grade inflammatory state (2). In this regard, we identified Sfrp5 as an anti-inflammatory adipokine that antagonizes the pro-inflammatory activity of Wnt5a, a regulator of non-canonical Wnt signaling (28). Utilizing genetic models, it was shown that Sfrp5/Wnt5a signaling is a regulator of adipose tissue inflammation and systemic metabolic health (28, 45). Here we show that genetic ablation of Sfrp5, a condition that is mimicked by severe metabolic dysfunction, is sufficient to promote adverse effects on the ischemic heart that are associated with elevated inflammation in the injured myocardium. Notably, the effects of Sfrp5 deficiency can be observed in mice fed a normal chow diet, which have normal systemic metabolic function (28), suggesting a direct effect on the injured myocardium. Furthermore, these data in metabolically normal mice document that the modulation of Sfrp5 expression is sufficient to confer changes in cardiovascular function independent of any potential confounding metabolic actions.
Mechanistically, the infarcted hearts of the Sfrp5-deficient mice displayed enhanced inflammation and greater levels of myocyte death. A loss of Sfrp5 led to greater cytokine and chemokine production in the infarct area, and this was accompanied by the greater influx of Wnt5a-positive macrophages. In cell culture studies, Wnt5a promotes the expression of pro-inflammatory mediators, and this activity is completely blocked by co-incubation with Sfrp5. Although Wnt signaling has been extensively studied in the context of development and differentiation, it has more recently been appreciated that Wnt5a can function as a modulator of innate immunity in adult organisms (45, 50–52). Wnt5a is recognized as the prototypical regulator of non-canonical Wnt signaling, partly through its ability to activate the planar cell polarity pathway. JNK is a key component of the planar cell polarity pathway, and this study found that JNK is activated to a greater extent in the infarcted myocardium of the Sfrp5-deficient mice and that Wnt5a activates JNK in cultured macrophages in an Sfrp5-repressible manner. JNK signaling participates in inflammatory responses and is central in the process of macrophage-driven metabolic dysfunction in obesity (53). Consistent with these data, we recently provided evidence that macrophage Wnt5a expression is associated with greater inflammation and impaired revascularization in a murine model of peripheral artery disease (44).
There is a high degree of complexity in the Wnt signaling system (54, 55). There are 19 Wnt ligands, and it is possible that other non-canonical Wnts, in addition to Wnt5a, participate in the suppression of JNK and the anti-apoptotic and anti-inflammatory actions of Sfrp5. Other Wnts that activate non-canonical signaling include Wnt5b, Wnt11, and possibly Wnt4. However, transcripts that encode at least two of these candidates are expressed at low or undetectable levels in the heart (Wnt5b and Wnt4). Although additional experiments will be required to document a causal link between Sfrp5 and Wnt5a with mouse genetic models, multiple lines of evidence support the notion that Sfrp5 acts via Wnt5a in the ischemic heart. These data include the observations that Sfrp5 deficiency leads to the greater influx of Wnt5a-positive macrophages into the infarct zone and cell culture studies showing that Sfrp5 can suppress the pro-inflammatory and pro-apoptotic actions of Wnt5a on macrophages and cardiac myocytes, respectively.
It has also been reported that Wnt5a stimulates hypertrophy in cultured myocytes (46). Cardiac hypertrophy was not examined in our study because it is not a significant end point in the acute I/R model. However, the pro-hypertrophic effect of Wnt5a would be expected to be greater under conditions where Sfrp5 levels are limiting, such as severe metabolic dysfunction, suggesting that an imbalance in Sfrp5/Wnt5a expression could also contribute to worsened post-myocardial infarction remodeling at later time points or in the development of left ventricle hypertrophy, which is often associated with obesity-related metabolic dysfunction.
Sfrp5 expression is highly enriched in adipocytes, and it is now appreciated to be a marker of adipose tissue (30, 31, 36, 56, 57) and to function as an adipokine (28). Sfrp5 shows a biphasic pattern of regulation in adipose tissue upon diet-induced obesity. Sfrp5 is transiently up-regulated in the early stages of murine obesity, but its expression declines at later time points (28). A similar pattern of Sfrp5 expression has been reported where it was noted that the decrease in Sfrp5 expression coincides with the plateau in adipose tissue expansion, suggesting that Sfrp5 is a marker of “healthy” fat (30). Here, we document another facet of Sfrp5 regulation, that ischemic injury in a remote tissue will lead to a reduction in Sfrp5 expression in pericardial and epididymal adipose tissue depots. Similarly, levels of other anti-inflammatory adipokines, adiponectin and CTRP9, were down-regulated in pericardial adipose tissue following myocardial I/R injury, and CTRP9 was also down-regulated in epididymal adipose tissue under these conditions. Relatively little is known about the mechanisms of Sfrp5 regulation. However, it has features in common with adiponectin, including marked up-regulation during adipocyte differentiation and suppression by agents that promote adipocyte dysfunction including endoplasmic reticulum stress, oxidant stress, and inflammation. With regard to the regulation of Sfrp5 expression, the acute production of pro-inflammatory cytokines following myocardial I/R may contribute to its down-regulation, as has been proposed for the down-regulation of adiponectin under these conditions (58). Furthermore, because adiponectin, Sfrp5, and potentially CTRP9 represent markers of functional adipose tissue, their down-regulation following myocardial I/R is suggestive of a mechanism of heart-to-fat crosstalk that could contribute to metabolic dysfunction under conditions of cardiovascular disease.
In summary, we show that Sfrp5 functions to limit infarct size in the heart following ischemia-reperfusion injury. Because excessive inflammation contributes to infarct expansion, we propose that the myocardium-sparing properties of Sfrp5 are mediated, at least in part, by its abilities to suppress pro-inflammatory Wnt5a/JNK signaling within the macrophages that infiltrate the infarct and pro-apoptotic Wnt5a/JNK signaling within myocytes. Because Sfrp5 is a secreted factor that is highly expressed by adipose tissue, we proposed further that it functions as a cardio-protective adipokine, and that its reduced expression could contribute to the increased prevalence of myocardial infarction in obese individuals.
Author Contributions
K. W. and K. N. designed the study. K. N., S. S., and J. J. F. performed the experiments, analyzed the data, and wrote the paper. R. K., I. S., K. O., Y. K., and N. O. provided technical assistance and contributed to the preparation of the figures. All authors analyzed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grants HL081587, HL116591, HL126141, and HL120160 (to K. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as a Paper of the Week.
- WAT
- white adipose tissue
- I/R
- ischemia/reperfusion
- Sfrp
- secreted frizzled-related protein
- BMDM
- bone marrow-derived macrophage
- NRVM
- neonatal rat ventricular myocyte
- AAR
- area at risk
- IA
- infarction area
- LV
- left ventricular
- LVEDD
- LV end diastolic diameter
- LVESD
- LV end systolic diameter
- %FS
- percentage of fractional shortening
- qRT-PCR
- quantitative real-time PCR
- GSK-3β
- glycogen synthase kinase 3β.
References
- 1.Ridker P. M., and Lüscher T. F. (2014) Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 35, 1782–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ouchi N., Parker J. L., Lugus J. J., and Walsh K. (2011) Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 11, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura K., Fuster J. J., and Walsh K. (2014) Adipokines: a link between obesity and cardiovascular disease. J. Cardiol. 63, 250–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mortensen R. M. (2012) Immune cell modulation of cardiac remodeling. Circulation 125, 1597–1600 [DOI] [PubMed] [Google Scholar]
- 5.Frangogiannis N. G. (2012) Regulation of the inflammatory response in cardiac repair. Circ. Res. 110, 159–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nahrendorf M., Pittet M. J., and Swirski F. K. (2010) Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121, 2437–2445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawaguchi M., Takahashi M., Hata T., Kashima Y., Usui F., Morimoto H., Izawa A., Takahashi Y., Masumoto J., Koyama J., Hongo M., Noda T., Nakayama J., Sagara J., Taniguchi S., and Ikeda U. (2011) Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123, 594–604 [DOI] [PubMed] [Google Scholar]
- 8.Shibata R., Izumiya Y., Sato K., Papanicolaou K., Kihara S., Colucci W. S., Sam F., Ouchi N., and Walsh K. (2007) Adiponectin protects against the development of systolic dysfunction following myocardial infarction. J. Mol. Cell. Cardiol. 42, 1065–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shibata R., Sato K., Pimentel D. R., Takemura Y., Kihara S., Ohashi K., Funahashi T., Ouchi N., and Walsh K. (2005) Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat. Med. 11, 1096–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kikuchi A., Yamamoto H., and Kishida S. (2007) Multiplicity of the interactions of Wnt proteins and their receptors. Cell. Signal. 19, 659–671 [DOI] [PubMed] [Google Scholar]
- 11.Logan C. Y., and Nusse R. (2004) The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 [DOI] [PubMed] [Google Scholar]
- 12.Rao T. P., and Kühl M. (2010) An updated overview on Wnt signaling pathways: a prelude for more. Circ. Res. 106, 1798–1806 [DOI] [PubMed] [Google Scholar]
- 13.van Amerongen R. (2012) Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 4, a007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gessert S., and Kühl M. (2010) The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ. Res. 107, 186–199 [DOI] [PubMed] [Google Scholar]
- 15.Malekar P., Hagenmueller M., Anyanwu A., Buss S., Streit M. R., Weiss C. S., Wolf D., Riffel J., Bauer A., Katus H. A., and Hardt S. E. (2010) Wnt signaling is critical for maladaptive cardiac hypertrophy and accelerates myocardial remodeling. Hypertension 55, 939–945 [DOI] [PubMed] [Google Scholar]
- 16.ter Horst P., Smits J. F., and Blankesteijn W. M. (2012) The Wnt/Frizzled pathway as a therapeutic target for cardiac hypertrophy: where do we stand? Acta Physiol. (Oxf.) 204, 110–117 [DOI] [PubMed] [Google Scholar]
- 17.Bergmann M. W. (2010) WNT signaling in adult cardiac hypertrophy and remodeling: lessons learned from cardiac development. Circ. Res. 107, 1198–1208 [DOI] [PubMed] [Google Scholar]
- 18.Kawano Y., and Kypta R. (2003) Secreted antagonists of the Wnt signalling pathway. J. Cell Sci. 116, 2627–2634 [DOI] [PubMed] [Google Scholar]
- 19.Finch P. W., He X., Kelley M. J., Uren A., Schaudies R. P., Popescu N. C., Rudikoff S., Aaronson S. A., Varmus H. E., and Rubin J. S. (1997) Purification and molecular cloning of a secreted, Frizzled-related antagonist of Wnt action. Proc. Natl. Acad. Sci. U.S.A. 94, 6770–6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Melkonyan H. S., Chang W. C., Shapiro J. P., Mahadevappa M., Fitzpatrick P. A., Kiefer M. C., Tomei L. D., and Umansky S. R. (1997) SARPs: a family of secreted apoptosis-related proteins. Proc. Natl. Acad. Sci. U.S.A. 94, 13636–13641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayr T., Deutsch U., Kühl M., Drexler H. C., Lottspeich F., Deutzmann R., Wedlich D., and Risau W. (1997) Fritz: a secreted frizzled-related protein that inhibits Wnt activity. Mech. Dev. 63, 109–125 [DOI] [PubMed] [Google Scholar]
- 22.Bovolenta P., Esteve P., Ruiz J. M., Cisneros E., and Lopez-Rios J. (2008) Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J. Cell Sci. 121, 737–746 [DOI] [PubMed] [Google Scholar]
- 23.Rattner A., Hsieh J. C., Smallwood P. M., Gilbert D. J., Copeland N. G., Jenkins N. A., and Nathans J. (1997) A family of secreted proteins contains homology to the cysteine-rich ligand-binding domain of frizzled receptors. Proc. Natl. Acad. Sci. U.S.A. 94, 2859–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alfaro M. P., Vincent A., Saraswati S., Thorne C. A., Hong C. C., Lee E., and Young P. P. (2010) sFRP2 suppression of bone morphogenic protein (BMP) and Wnt signaling mediates mesenchymal stem cell (MSC) self-renewal promoting engraftment and myocardial repair. J. Biol. Chem. 285, 35645–35653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He W., Zhang L., Ni A., Zhang Z., Mirotsou M., Mao L., Pratt R. E., and Dzau V. J. (2010) Exogenously administered secreted frizzled related protein 2 (Sfrp2) reduces fibrosis and improves cardiac function in a rat model of myocardial infarction. Proc. Natl. Acad. Sci. U.S.A. 107, 21110–21115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barandon L., Casassus F., Leroux L., Moreau C., Allières C., Lamazière J. M., Dufourcq P., Couffinhal T., and Duplàa C. (2011) Secreted frizzled-related protein-1 improves postinfarction scar formation through a modulation of inflammatory response. Arterioscler. Thromb. Vasc. Biol. 31, e80–87 [DOI] [PubMed] [Google Scholar]
- 27.Laeremans H., Hackeng T. M., van Zandvoort M. A., Thijssen V. L., Janssen B. J., Ottenheijm H. C., Smits J. F., and Blankesteijn W. M. (2011) Blocking of frizzled signaling with a homologous peptide fragment of Wnt3a/Wnt5a reduces infarct expansion and prevents the development of heart failure after myocardial infarction. Circulation 124, 1626–1635 [DOI] [PubMed] [Google Scholar]
- 28.Ouchi N., Higuchi A., Ohashi K., Oshima Y., Gokce N., Shibata R., Akasaki Y., Shimono A., and Walsh K. (2010) Sfrp5 is an anti-inflammatory adipokine that modulates metabolic dysfunction in obesity. Science 329, 454–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schulte D. M., Müller N., Neumann K., Oberhäuser F., Faust M., Güdelhöfer H., Brandt B., Krone W., and Laudes M. (2012) Pro-inflammatory wnt5a and anti-inflammatory sFRP5 are differentially regulated by nutritional factors in obese human subjects. PLoS One 7, e32437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jura M., Jaroslawska J., Chu D. T., and Kozak L. P. (2015) Mest and Sfrp5 are biomarkers for healthy adipose tissue. Biochimie 10.1016/j.biochi.2015.05.006 [DOI] [PubMed] [Google Scholar]
- 31.Flehmig G., Scholz M., Klöting N., Fasshauer M., Tönjes A., Stumvoll M., Youn B. S., and Blüher M. (2014) Identification of adipokine clusters related to parameters of fat mass, insulin sensitivity and inflammation. PLoS One 9, e99785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu W., Li L., Yang M., Luo X., Ran W., Liu D., Xiong Z., Liu H., and Yang G. (2013) Circulating Sfrp5 is a signature of obesity-related metabolic disorders and is regulated by glucose and liraglutide in humans. J. Clin. Endocrinol. Metab. 98, 290–298 [DOI] [PubMed] [Google Scholar]
- 33.Prats-Puig A., Soriano-Rodríguez P., Carreras-Badosa G., Riera-Pérez E., Ros-Miquel M., Gomila-Borja A., de Zegher F., Ibáñez L., Bassols J., and López-Bermejo A. (2014) Balanced duo of anti-inflammatory SFRP5 and proinflammatory WNT5A in children. Pediatr. Res. 75, 793–797 [DOI] [PubMed] [Google Scholar]
- 34.Satoh W., Matsuyama M., Takemura H., Aizawa S., and Shimono A. (2008) Sfrp1, Sfrp2, and Sfrp5 regulate the Wnt/β-catenin and the planar cell polarity pathways during early trunk formation in mouse. Genesis 46, 92–103 [DOI] [PubMed] [Google Scholar]
- 35.Maeda N., Takahashi M., Funahashi T., Kihara S., Nishizawa H., Kishida K., Nagaretani H., Matsuda M., Komuro R., Ouchi N., Kuriyama H., Hotta K., Nakamura T., Shimomura I., and Matsuzawa Y. (2001) PPARγ ligands increase expression and plasma concentrations of adiponectin, an adipose-derived protein. Diabetes 50, 2094–2099 [DOI] [PubMed] [Google Scholar]
- 36.Koza R. A., Nikonova L., Hogan J., Rim J. S., Mendoza T., Faulk C., Skaf J., and Kozak L. P. (2006) Changes in gene expression foreshadow diet-induced obesity in genetically identical mice. PLoS Genet. 2, e81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kambara T., Ohashi K., Shibata R., Ogura Y., Maruyama S., Enomoto T., Uemura Y., Shimizu Y., Yuasa D., Matsuo K., Miyabe M., Kataoka Y., Murohara T., and Ouchi N. (2012) CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J. Biol. Chem. 287, 18965–18973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ouchi N., and Walsh K. (2012) Cardiovascular and metabolic regulation by the adiponectin/C1q/tumor necrosis factor-related protein family of proteins. Circulation 125, 3066–3068 [DOI] [PubMed] [Google Scholar]
- 39.Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Ge G., Spooner E., Hug C., Gimeno R., and Lodish H. F. (2009) Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J. 23, 241–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shibata R., Ouchi N., Ito M., Kihara S., Shiojima I., Pimentel D. R., Kumada M., Sato K., Schiekofer S., Ohashi K., Funahashi T., Colucci W. S., and Walsh K. (2004) Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat. Med. 10, 1384–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y., Yi W., Yuan Y., Lau W. B., Yi D., Wang X., Wang Y., Su H., Wang X., Gao E., Koch W. J., and Ma X. L. (2013) C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation 128, S113–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leaf I., Tennessen J., Mukhopadhyay M., Westphal H., and Shawlot W. (2006) Sfrp5 is not essential for axis formation in the mouse. Genesis 44, 573–578 [DOI] [PubMed] [Google Scholar]
- 43.Li Y., Rankin S. A., Sinner D., Kenny A. P., Krieg P. A., and Zorn A. M. (2008) Sfrp5 coordinates foregut specification and morphogenesis by antagonizing both canonical and noncanonical Wnt11 signaling. Genes Dev. 22, 3050–3063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kikuchi R., Nakamura K., MacLauchlan S., Ngo D. T., Shimizu I., Fuster J. J., Katanasaka Y., Yoshida S., Qiu Y., Yamaguchi T. P., Matsushita T., Murohara T., Gokce N., Bates D. O., Hamburg N. M., and Walsh K. (2014) An antiangiogenic isoform of VEGF-A contributes to impaired vascularization in peripheral artery disease. Nat. Med. 20, 1464–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fuster J. J., Zuriaga M. A., Ngo D. T., Farb M. G., Aprahamian T., Yamaguchi T. P., Gokce N., and Walsh K. (2015) Noncanonical Wnt signaling promotes obesity-induced adipose tissue inflammation and metabolic dysfunction independent of adipose tissue expansion. Diabetes 64, 1235–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hagenmueller M., Riffel J. H., Bernhold E., Fan J., Katus H. A., and Hardt S. E. (2014) Dapper-1 is essential for Wnt5a induced cardiomyocyte hypertrophy by regulating the Wnt/PCP pathway. FEBS Lett. 588, 2230–2237 [DOI] [PubMed] [Google Scholar]
- 47.Rauner M., Stein N., Winzer M., Goettsch C., Zwerina J., Schett G., Distler J. H., Albers J., Schulze J., Schinke T., Bornhäuser M., Platzbecker U., and Hofbauer L. C. (2012) WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Miner. Res. 27, 575–585 [DOI] [PubMed] [Google Scholar]
- 48.Kim J., Chang W., Jung Y., Song K., and Lee I. (2012) Wnt5a activates THP-1 monocytic cells via a β-catenin-independent pathway involving JNK and NF-κB activation. Cytokine 60, 242–248 [DOI] [PubMed] [Google Scholar]
- 49.Blumenthal A., Ehlers S., Lauber J., Buer J., Lange C., Goldmann T., Heine H., Brandt E., and Reiling N. (2006) The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 108, 965–973 [DOI] [PubMed] [Google Scholar]
- 50.Pereira C., Schaer D. J., Bachli E. B., Kurrer M. O., and Schoedon G. (2008) Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler. Thromb. Vasc. Biol. 28, 504–510 [DOI] [PubMed] [Google Scholar]
- 51.Naskar D., Maiti G., Chakraborty A., Roy A., Chattopadhyay D., and Sen M. (2014) Wnt5a-Rac1-NF-κB homeostatic circuitry sustains innate immune functions in macrophages. J. Immunol. 192, 4386–4397 [DOI] [PubMed] [Google Scholar]
- 52.Pereira C. P., Bachli E. B., and Schoedon G. (2009) The wnt pathway: a macrophage effector molecule that triggers inflammation. Curr. Atheroscler. Rep. 11, 236–242 [DOI] [PubMed] [Google Scholar]
- 53.Han M. S., Jung D. Y., Morel C., Lakhani S. A., Kim J. K., Flavell R. A., and Davis R. J. (2013) JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 339, 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dawson K., Aflaki M., and Nattel S. (2013) Role of the Wnt-Frizzled system in cardiac pathophysiology: a rapidly developing, poorly understood area with enormous potential. J. Physiol. 591, 1409–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deb A. (2014) Cell-cell interaction in the heart via Wnt/β-catenin pathway after cardiac injury. Cardiovasc. Res. 102, 214–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anunciado-Koza R. P., Higgins D. C., and Koza R. A. (2015) Adipose tissue Mest and Sfrp5 are concomitant with variations of adiposity among inbred mouse strains fed a non-obesogenic diet. Biochimie 10.1016/j.biochi.2015.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang R., Hong J., Liu R., Chen M., Xu M., Gu W., Zhang Y., Ma Q., Wang F., Shi J., Wang J., Wang W., and Ning G. (2014) SFRP5 acts as a mature adipocyte marker but not as a regulator in adipogenesis. J. Mol. Endocrinol. 53, 405–415 [DOI] [PubMed] [Google Scholar]
- 58.Shibata R., Sato K., Kumada M., Izumiya Y., Sonoda M., Kihara S., Ouchi N., and Walsh K. (2007) Adiponectin accumulates in myocardial tissue that has been damaged by ischemia-reperfusion injury via leakage from the vascular compartment. Cardiovasc. Res. 74, 471–479 [DOI] [PubMed] [Google Scholar]