

Abstract
All inflammatory changes in the vessel wall are defined as vasculitis. Pediatric vasculitis may present with different clinical findings. Although Henoch-Schönlein purpura which is the most common pediatric vasculitis generally recovers spontaneously, it should be monitorized closely because of the risk of renal failure. Although Kawasaki disease is easy to diagnose with its classical findings, the diagnosis may be delayed in case of incomplete Kawasaki disease. Kawasaki disease should be considered especially in infants in case of prolonged fever even if the criteria are not fully met and intravenous immunoglobulin treatment should be administered without delay in order to prevent development of coronary artery aneurism. Reaction at the site of administration of Bacillus Calmette-Guerin (BCG) vaccine may be observed as commonly as cervical lymphadenopathy in Kawasaki disease and may be used as a valuable finding in suspicious cases. Although anti-neutrophil cytoplasmic antibody-associated vasculitides are rare in children, renal involvement is more common and progression is more severe compared to adults. Hence, efficient and aggressive treatment is required. Takayasu’s arteritis is observed commonly in young adult women and rarely in adolescent girls. Therefore, a careful physical examination and blood pressure measurement should be performed in addition to a detailed history in daily practice. In children with unexplained neurological findings, cerebral vasculitis should be considered in the absence of other systemic vasculitides and necessary radiological investigations should be performed in this regard. This review will provide an insight into the understanding of pediatric vasculitis, current diagnostic approaches and prognosis by the aid of new studies.
Keywords: ANCA-associated vasculitis, Behcet’s disease in children, Henoch-Schonlein purpura/IgA vasculitis, Kawasaki disease, pediatric vasculitis, primary cerebral vasculitis
Introduction
There are some vasculitides which do not occur or which are observed very rarely in adults, but are encountered frequently in the childhood in clinical practice in addition to vasculitides observed very rarely in children compared to adults. Blood vessel inflammation is called vasculitis. Stenosis, obstruction, aneurysm or rupture which may occur as a result of blood vessel inflammation may lead to transient or persistent tissue damage. Inflammation may develop primarily or secondary to any underlying disease. Inflammatory changes which occur only in the outer most layer of the blood vessels are called periarteritis. The clinical picture observed in vasculitides varies depending on the size of the involved vessel and the disease severity. The complaints and clinical symptoms in vasculitides in children show marked variability and differences. In presence of clinical findings including fever of unknown origin, weight loss and fatigue, cutaneous lesions (urticaria, livedo reticularis, palpable purpura, nudules, ulcer necrosis), unexplained myalgia, arthritis or arhtralgia, hypertension and soft tissue edema, vasculitis should be considered. Henoch-Schonlein purpura (HSP) and Kawasaki Disease (KD) are the most common vasculitedes observed in the childhood. Other vasculitides are observed rarely in the childhood (1). In this review, the vasculitides which we encounter in pediatric daily practice will be classified by vessel size and the current changes related with treatment and prognosis of vasculitis will be reviewed and summarized.
Classification
Classification criteria of vasculitides are established based only on clinical and laboratory findings. The classification criteria have been developed in order to arrange the clinical definitions of different vasculitides in the most comrehensible way, but not to make the diagnosis (2, 3). Initially, the “American College of Rheumatology” (ACR) criteria were developed in 1990 mainly for adulthood vasculitides and also included the criteria used for childhood vasculitides. Subsequently, vasculitis terminology was developed in the Chapel Hill Consensus Conference (CHCC) in 1994 and the same classification was updated in 2012 with its new form (Table 1) (4). The terminology developed in the Chapel Hill Consensus Conference is the definitions which can also be used for childhood vasculitides. The EULAR/PRINTO/PRES criteria which were started to be developed by way of internet questionnaires in the mid 2000s to classify childhood vasculitides were established in 2008 in Ankara Conference (5). These classification criteria were developed for HSP, childhood polyarteritis nodosa (PAN), Takayasu arteritis (TA) and granulomatous polyarteritis (GPA). In this study, KD which is one of the most common vasculitides observed in the childhood is not evaluated. The diagnostic criteria developed by the ‘American Heart Association’ are used for these patients (Table 2).
Table 1.
Classification of vasculitis accepted in the 2012 International Chapel Hill Consensus Conference
| Large vessel vasculitides |
| Takayasu arteritis |
| Giant cell arteritis |
| Medium vessel vasculitides |
| Polyarteritis nodosa |
| Kawasaki disease |
| Small vessel vasculitides |
|
| Vasculitides involving vessels with variable size |
| Behçet’s disease |
| Cogan syndrome |
| Single organ vasculitides |
| Cutaneous leukocytoclastic vasculitis |
| Cutaneous arteritis |
| Primary central nervous system vasculitides |
| Isolated aortitis |
| Other |
| Vasculitides related with systemic diseases |
| Lupus vasculitis |
| Rheumatoid vasculitis |
| Sarcoid vasculitis |
| Other |
| Vasculitides with known etiology |
| Hepatitis C virus-related cryoglobulinemic vasculitis |
| Hepatitis B virus-related vasculitis |
| Syphilis-related aortitis |
| Drug-induced immune complex vasculitis |
| Drug-induced ANCA-associated vasculitis |
| Cancer-related vasculitis |
| Other |
ANCA: anti-neutrophilic cytoplasmic antibody
Table 2.
Classification criteria in childhood vasculitides
| Henoch-Schonlein purpura/IgA vasculitis classification criteria |
| Purpura or petechiae (mandatory) more extensive in the lower extremities and one of the following four criteria. |
| Abdominal pain |
| Histopathology (Iga accumulation on biopsy) |
| Arthritis or arthralgia |
| Renal involvement |
| Kawasaki disease diagnostic criteria |
| Fever (mandatory) in addition to four of the following five criteria |
| Non- purulent conjunctivitst |
| Mucosal changes |
| Cervical lymphadenopathy |
| Polymorphic rash on the trunk |
| Extremity changes |
| Takayasu arteritis classification criteria |
| Aneurysm or dilatation in the aorta or its main branches and pulmonary artery shown by angiography (mandatory) plus one of the following five criteria. |
| Pulsenessness or claudication |
| Blood pressure difference in four extremities |
| Hearing of murmur |
| Hypertension |
| Increased acute phase response |
| Polyarteritis nodosa classification criteria |
| Histopathological or angiographic abnormalities (mandatory) plus one of the following five criteria. |
| Cutaneous involvement |
| Myalgia/muscle tenderness |
| Hypertensiyon |
| Peripheral neuropathy |
| Renal involvement |
| Granulomatous polyangiitis classification criteria (at least three of six criteria) |
| Histopathology (granulomatous inflammation) |
| Upper respiratory tract involvement |
| Laryngotracheobronchial obstruction |
| Lung involvement |
| ANCA positivity |
| Renal involvement |
| Classification criteria for primary vasculitis of the central nervous system |
| Acquired neurological or psychiatric finding which can not be explained by another cause |
| Histopathological or angiographic changes indicating vasculitis in CNS. |
| Absence of systemic vasculitis or any disease which could cause to these patological changes in CNS |
The sequencing used in vasculitis classifications and the clinical pictures and current therapeutic options in childhood vasculitides will be reviewed below.
1. Vasculitides which involve large vessels
Giant cell (temporal) arteritis and Takayasu arteritis are included in this class. Since giant cell arteritis is not observed in the childhood, only information about Takayasu arteritis will be reviewed.
Takayasu arteritis
Takayasu arteritis is a chronic vasculitis with an unknown cause which frequently involves the aorta and its main branches. It is most commonly observed in young women and especially in Japanese people. Although it occurs rarely below the age of 16 years, it is the third most common vasculitis after KD and HSP in the childhood. In children, the clinical picture is not specific and may usually involve fever, malaise, arthralgia, myalgia, abdominal pain, hypertension, hypertensive retinopathy, heart failure, headache and convulsions. Murmurs are heard in the ischemic phase of the disease and reduction in pulses is noted. The most common clinical finding is hypertension. Reduction in pulses and murmurs are the second most common clinical finding. Therefore, detailed physical examination including auscultation in terms of murmurs is very important in children with hypertension and non-specific findings (weight loss, headache, abdominal pain, dyspnea) (6–9). The initial findings in Takayasu arteritis including fever, malaise, myalgia and weight loss are related with interleukin 6 (IL-6) which is produced excessively in the area of inflammation. Observation of granulomas and Langerhan’s giant cells noted in tuberculosis also in TA patients suggests that possible tuberculosis infection may be involved in the etiology. A positive PPD test is observed frequently in patients with Takayasu arteritis (10).
In laboratory tests, acute phase response is usually found to be increased. Magnetic resonance angiography and traditional angiography are substantially valuable in the diagnosis (Figure 1). It is difficult to evaluate the disease activity. Clinical reliability of acute phase response is not sufficient especially in the childhood. Since the clinical findings are mostly non-specific, vascular damage progresses insidiously. Active vasculitis may be observed also in cases where acute phase response is absent and scintigraphic data are normal (10, 11).
Figure 1.
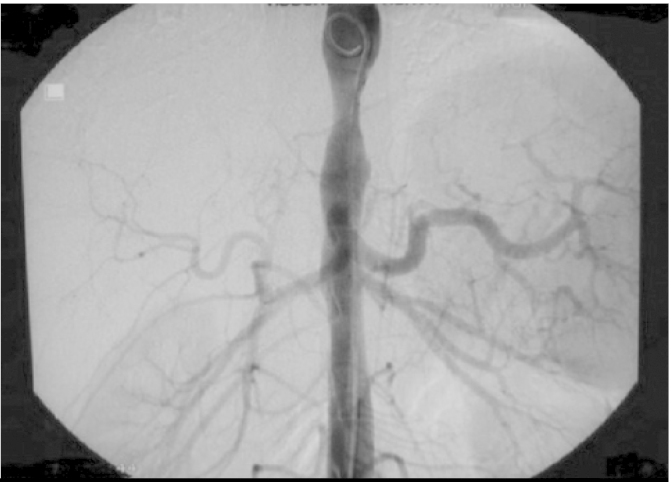
Angiographic findings in Takayasu arteritis
There is no controlled study related with treatment of Takayasu arteritis. Ozen et al. (12) showed that initiation treatment with cyclophosphamid and steroid and maintenence treatment with methotrexate were efficient in children. The clinical prognosis of the disease is variable; use of corticosteroid decreases progression of the lesion by 50%. Immunosupressive treatment provides better disease control and prevents recurrence of stenosis (13). Angioplasty is efficient in treatment of TA and increases 5-year survivial up to 80–95%. Recurrence of stenosis occurs with a rate of 31,7% in the first year and use of corticosteroid and immunosupressive agents decreases the risk by 50% (14, 15). Stenosis is irreversible and may result in severe complications including renovascular hypertension, severe claudication (limping), myocardial infarction and stroke. Early angioplasty is needed in patients with stenosis in renal arteries, aortic valve failure and stenosis in more than three sites (6–9). Tosilizumab which was shown to be efficient in adult studies has been reported to be also efficient in pediatric TA cases in case reports (15,16). Recurrence is frequent in Takayasu arteritis. Increase in the extension of vascular involvement, aortic failure and pulmonary hypertension in the follow-up are related with increased mortality rates.
2. Vasculitides which involve medium vessels
Polyarteritis nodosa
Polyarteritis nodosa is a necrotising vasculitis which involves medium vessles. It may occur at any age. Since organ involvement is extremely variable in polyarteritis nodosa, very different complaints and findings may occur. While the skin, musculo-skeletal system, kidney, gastrointestinal system and neurological system are involved frequently, the respiratory system is involved less frequently. Patients may show involvement of all these organs or only cutaneous involvement or single organ involvement may be present. The frequency of PAN decreased worldwide with the reduction in the frequency of Hepatitis B virus infection. It is thought that infectious triggers may be involved in pathogenesis (1, 17–19).
Both PAN and anti-neutrophilic cytoplasmic antibody (ANCA)-related vasculitis are necrotizing vasculitides which involve medium- and small vessels and which are difficult to differentiate both clinically and pathologically. Ludici et al. (17) found similar recurrence rates in 35 patients with a diagnosis of childhood-onset ANCA-related vasculitis and 21 patients with a diagnosis of PAN, but reported that the patients with a diagnosis of ANCA-related vasculitis had more frequent attacks, the Birmingham Vasculitis Activity Score (BVAS) rates were higher during these attacks and as a result the total amount of damage was higher in these patients. Although the majority of the cases of polyarteritis nodosa are in the form of minor attacks, recurrence is observed in ¾ and the mortality rate has been found to be 10%. Childhood PAN is classified according to the EULAR/PRINTO/PRES criteria (Table 2) (5).
Eleftheriou et al. (18) retrospectively examined a large number of patients (n=69) who were followed up in their own center for 32 years and were diagnosed with PAN in a paper related with childhood PAN published in 2013. The most common initial complaints and findings included cutaneous involvement (88%), fever (87%), myalgia (83%), arthritis/arthralgia (75%), weight loss (64%), fatigue (62%), hypertension (16%), peripheral neuropathy (4%) and the systems involved included the urinary system (19%), gastrointestinal system (10%) and nervous system (10%). Forty of 50 skin biopsies were compatible with necrotizing vasculitis and the results of 96% of the patients who underwent angiography were found to be compatible with PAN.
In laboratory evaluation, leukocytosis, thrombocytosis and increased acute phase reactants are noted. Positive hepatitis serology may be present in children with polyarteritis nodosa, though rarely. Antineutrophilic cytoplasmic antibodies are expected to be negative. If positive ANCA is associated with typical renal involvement and possible lung involvement, this supports microscopic polyangiitis (MPA). Traditional angiogram is the gold standart in the diagnosis of PAN. Magnetic resonance angiography may be inadequate to identify microaneursims. Biopsy of the organs involved (kidney, intestines, nerve, muscle, skin) shows necrotizing arteritis and is important for a definite diagnosis (20).
In the study conducted by Eleftheriou et al. (18), it was reported that cyclophosphamide and corticosteroid were found to be the most commonly used drugs (83%) and the other therapeutical options included plasma exchange (9%) and biological drugs (after 2002; 13%). The recurrence rate was found to be 35% and the mortality rate was found to be 4%.
High dose corticosteroid and cyclophosphamide (generally given as monthly intravenous doses) are used until remission is achieved in treatment of polyarteritis nodosa. When remission is achieved, azathioprine which is less toxic is given as maintenance treatment instead of cyclophosphamide (20).
Adenosine deaminase -2 deficiency
Adenosine deaminase 2 (ADA2) deficiency which is a severe disease is manifested in a wide spectrum ranging from fatal vasculopathy to a latent disease limited to the skin. In adenosine deaminase-2 deficiency, the most common types of clinical presentation include early onset stroke, childhood PAN, fever, livedo racemosa, lacunary infarctions, hepatosplenomegaly accompanied by portal hypertension, Sneddon syndrome and humoral immune deficiency (21, 22). When Elkan et al. (21) found PAN in 19 members of five different georgian jewish families, they proposed the hypothesis that this disease may be related with an autosomal recessive single gene mutation and performed exome sequencing in 16 patients. They found CECR1 gene mutation which leads to ADA2 deficiency in all these 16 patients. A patient followed up by our group presented with early onset stroke and fatal vasculopathy accompanied with signs mimicking autoinflammatory disease (23). It has been reported that dermatological lesions ranging from livedo reticularis to nodules may occur and non-granulomatous necrotizing arteritis may be observed histopathologically (24). No consensus has been reached on any method for treatment of adenosine deaminase-2 deficiency. Plasma infusion and use of etanercept are recommended (21, 22).
Kawasaki disease
Kawasaki disease (KD) is an acute, febrile childhood vasculitis which mainly involves the coronary arteries and is frequently observed in children aged between 6 months and 5 years. It may result in severe complications, if it is not diagnosed in the acute phase or if treatment is delayed. The risk of development of coronary artery aneurysm is increased five-fold in untreated patients. It is the second most common acquired heart disease in children. Although the cause is not known exactly, acute onset, seasonal and epidemic characteristics suggest that infectious agents are involved in the etiology. The fact that the frequency is variable in different ethnic groups and tendency to development of aneurysm have been attempted to be explained by various HLA types and polymorphisms (25). Salo et al. (26) investigated the incidence of KD in North European countries and found the annual incidence to be 11.4 per 100 000 children in Finland, 5.4 in Norway and 7.4 in Sweden. It is more frequent in Asia countries and especially in Japan. In 2010, the annual incidence in children below the age of five years was found to be 239.6/100 000 in Japan (27).
Epidemiology
Three large KD epidemics were examined in Japan and a relation was established between Kawasaki patients in Japan, Hawai and SanDiego and strong winds of Asian origin. This study showed that environmental triggers might have spread by way of winds (28). Jaggi et al. (29) showed adenoviral infections with a rate of 10% in patients with KD, though with a low titer. It is thought that similar viral agents may be involved, because seasonal condensation occurs especially in winter and spring months (30).
Pathogenesis
Following inflammation in the coronary arteries, vascular endothelial growth factor (VEGF), matrix metalloproteinase 9, tumor necrosis factor alpha (TNF- α) and other proinflammatory cytokines are released. This immune reaction causes to damage in the intima layer of vessels, the internal and external elastic lamina is fragmented and ballooning occurs. It is thought that coronary artery aneurysm (CAA) occurs as a result of this (31).
Genetics
Although a tendency to infectious diseases occurs in children with genetic sensitivity, our information about genetic sensitivity and infectious agents in KD is insufficient. Although many genetic factors have been predicted hypothetically in KD, none of these is predicted as a definite triggering factor. It is thought that infection may play a triggering role for KD in patients with a genetic predisposition, but maternal antibodies transferred from the mother in the first months of life and antibodies developed by the immune system as a response to infective agents play a preventive role in terms of KD which is obvserved rarely in the adulthood (25, 31).
Clinical findings
The disease is manifested with prolonged fever despite all antibiotic therapies. The first 10 days from the time of onset of fever is called “acute phase”. The following second 10 day-period is called “subacute phase”. Many permanent sequelae of the disease occur in this phase. The following period is the time period during which permanent sequealae occur, if the disease is not treated. The main finding is prolonged fever. The body temperature ranges between 39 and 40 C°. Fever does not respond to the antibiotics and antipyretic drugs used. Therefore, KD should be absolutely considered in any child below the age of four years with high fever of unknown origin irresponsive to antibiotic treatment. In the acute phase, other clinical findings included in the diagnostic cirteria accompany fever. Unilateral or bilateral conjunctival hemorrhage, intraoral and lip changes are found in children with Kawasaki disease. This finding is manifested as fissuring of lips and intraoral changes (Figure 2a). Extensive aphtous stomatitis and erythema are found inside the mouth. The most specific sign of the disease is fissures of lips which may be hemorrhagic. In addition, extensive maculopapular rash is observed in the trunk suggesting exanthematous measles. Changes in hands and feet accompany the clinical picture. This picture which initially shows itself as soft tissue edema is subsequently manifested by peeling of the skin beginning at the fingertips and toenails (Figure 2b). Bilateral swollen palms of the hands and soles of the feet is typical for vasculitic pictures in the childhood. In patients with Kawasaki disease, cervical lymphadenopathy (mostly bilateral and rarely unilateral) is found in the acute phase (25).
Figure 2. a, b.
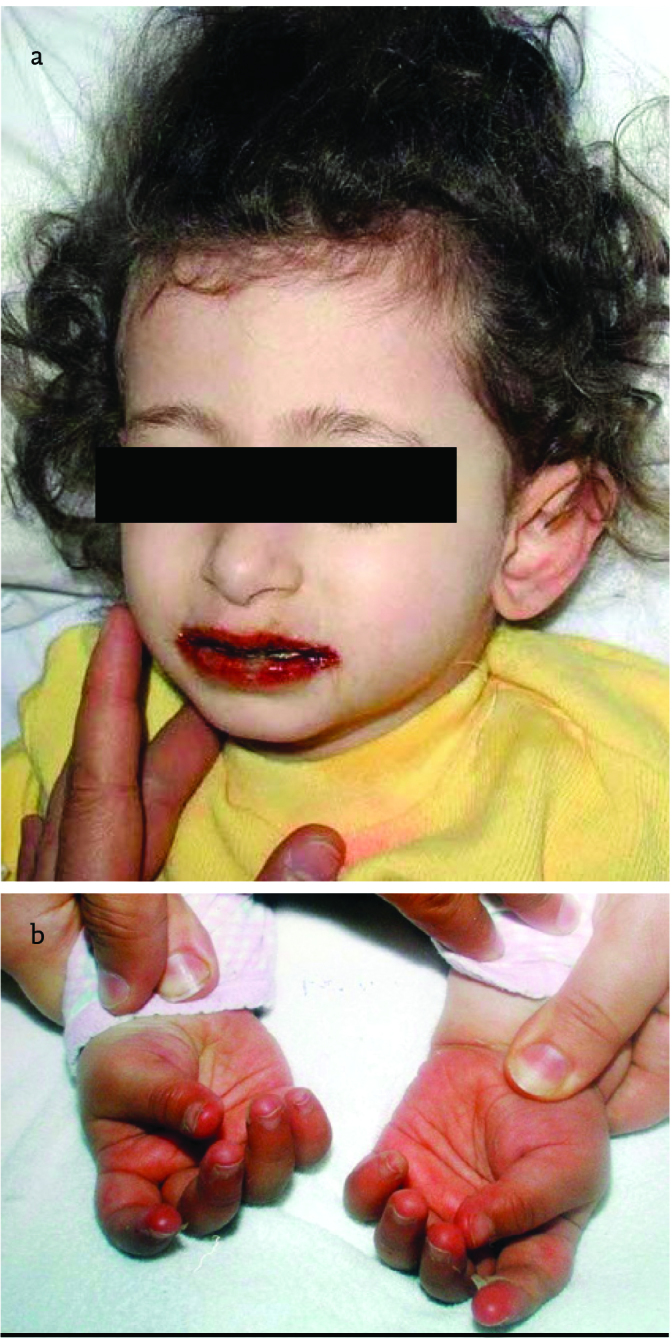
Appearance of lips in Kawasaki disease and peeling of fingertips occuring in the subacute period
In addition to the common classical signs, there are also signs which may be determinative. In the study conducted by Rezai et al. (32), 15 articles related with the BCG vaccine injection site reaction were examined. BCG vaccine injection site reaction was found with a rate of 49.8% in patients with KD. It was reported that this reaction was observed more frequently compared to cervical lymphadenopathy and rash and it was emphasized that it may be included as a diagnostic cirterion especially in incomplete Kawasaki disease. The other clinical findings which are observed rarely in Kawasaki disease include restlessness, diarrhea, bile tract inflammation, hepatitis, gallbladder hydrops, jaundice, pancreatitis, aseptic menigitis, anterior uveitis, arthralgia and arthritis, respiratory findings, otitis media or tympanitis, urethritis, meatitis and facial nerve palsy (33).
The most significant clinical finding which occurs in the subacute phase of the disease and may lead to severe morbidity and mortality in the acute phase and long-term is cardiovascular involvement. Although coronary artery involvement is observed primarily, complication including myocarditis and pericarditis may also occur. Although classical coronary artery involvement is in the form of coronary aneurysm, we observe coronary artery dilatation in clinical practice. The risk of aneurysm can be substantially reduced by recognition of the disease and administration of intravenous immunoglobulin (IVIG) in the acute phase. The most common risk factors for coronary artery involvement include male gender, young age, prolonged fever, delayed diagnosis, persistence of fever after treatment, low hemoglobin level, increased leukocyte number, increased acute phase level, low platelet count and low albumin level (25, 33).
Giacchi et al. (34) compared 32 patients with KD and 26 healthy controls and found significiant intimal thickenning in the coronary artery walls in the patients with KD. In addition, they found higher wall thickness in the acute phase even in the patients without coronary involvement. It has been reported that this thickenning would carry a higher risk for cardiovascular disease.
In some studies, it has been shown that typical mucosal and lymph node changes are absent in babies younger than six months. In addition, it has been reported that strawberry tongue and swollen palms of the hands and soles of the feet are observed with a lower rate compared to older children, echocardiography should be performed without delay considering incomplete KD in presence of systemic inflammation findings in infants, infants have a higher risk in terms of severe coronary abnormalities compared to older patients with KD and treatment should be initiated immediately to prevent CAA considering KD in case of prolonged fever even if the cirteria are not met fully (35).
Laboratory
There is no diagnostic laboratory test. Typically, acute phase reactants are increased. Leukocytosis, neutrophil left shift and thrombocytosis after the first week may be observed. Normochormic normocytic anemia, pyuria, increased ALT and GGT and hyponatremia may also be found (36).
Treatment
Immunoglobulin treatment should be administered as soon as the diagnosis is made. Intravenous immunoglobulin is administered at a dose of 2 gr/kg/dose (maximum dose 40 g/dose). During administration of intravenous immunoglobulin, the patient’s increased temperature drops rapidly and all systemic findings begin to disappear rapidly. In addition to intravenous immunoglobulin treatment acetylsalicylic acid (ASA) treatment is initiated at a dose of 60–80 mg/kg/day. ASA treatment is continued for a mean time of 21 days or for a shorter time depending on the patient’s clinical status. Subsequently, the ASA dose should be reduced to the antiaggregant dose (3–5 mg/kg/day, maximum dose 80–100 mg/day). ASA at antiaggregant dose should be continued for a life time, if coronary involvement is present and for one year, if coronary involvement is absent. About 10–20% of the patients are resistant to IVIG treatment. A second IVIG dose may be given, if clinical findings and fever do not improve until up to the 36th hour following IVIG infusion. Despite a second dose, irresponsiveness is present in 3–4% of the patients. In such cases, it is recommended to add intensive high dose steroid at a dose of 30 mg/kg/day in combination with IVIG. The indicators of resistance to IVIG in Kawasaki disease include male gender, being younger than one year old, administration of IVIG after the 10th day, thrombocytopenia, low albumin level, low hemoglobin level, low sodium level, persistent increased CRP after IVIG infusion and persistent low serum albumin level after IVIG infusion. In resistant cases, other treatment options include TNF alpha inhibitors, calsineurin inhibitors, plasma exhange, rituksimab and anakinra (25, 37).
3. Vasculitides which involve small vessels
a. Anti neutrophilic cytoplasmic antibody (ANCA)-related small vessel vasculitides
Systemic necrotizing vasculitides constitute 3% of the cases referred to pediatric rheumatology centers (17). PAN and ANCA-related vasculitides (ARV) are included in this group. ANCA-related vasculitides are examined under three main titles including granulomatous polyangiitis (GPA), eosinophilic granulomatous polyangiitis (EGPA) and microscopic polyangiitis (MPA). The annual incidence of granulomatous polyangiitis ranges between 0.2/100 000 and 1.2/100 000. It most commonly occurs between the midfourties and mid-sixties. It occurs in the second decade of life in pediatric patients and is more common in girls (38).
Upper and lower respiratory tract inflammation and renal involvement are the three involvement sites which are very significant for the diagnosis. Characteristic findings related with involvement of these organs have been identified in studies conducted with children (39, 40). The most common findings at the time of onset of the disease include fatigue, wieght loss and fever and otorhinolaryngological, respiratory system and renal findings are the most commonly observed findings. Less commonly, findings of musculoskeletal, gastrointestinal, opthalmic, cutaneous and neurological involvement are observed. Cardiac involvement has not been found in any patient (39). In the study conducted by Bohm et al. (40), the otorhinolaryngological findings were reported to include septum perforation, saddle nose, auricular chondritis, subglottic stenosis, coarse voice and stridor. The respiratory tract findings include wheezing, expiratory dyspnea, nodule on lung graphy, patchy or diffuse opacity, hilar lymphadenopathy and transient pulmonary infiltrations (Figure 3). Ophtalmic findings have been reported to include conjonctivitis, keratitis and blepharitis.
Figure 3.
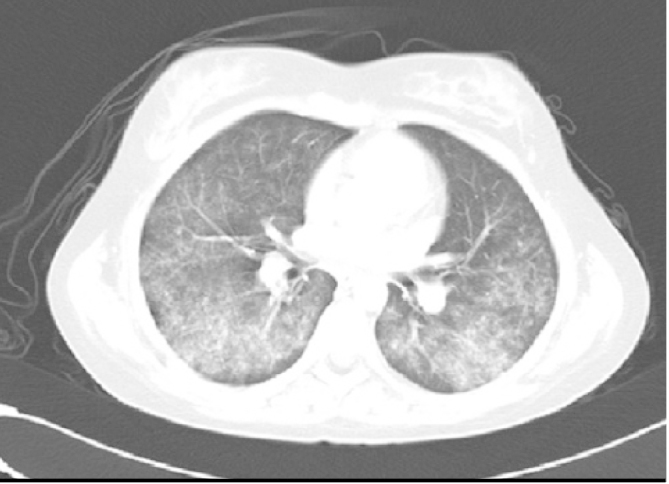
Tomographic appearance of the lungs in ANCA-related vasculitis
ANCA: anti-neutrophilic cytoplasmic antibody
When patients were evaluated in terms of ANCA positivity at the time of diagnosis in the study of Bohm et al. (40), anti proteinase 3 (anti-PR3) positivity was found with a rate of 67%, anti myeloperoxidase (anti-MPO) positivity was found with a rate of 26% and ANCA positivity was found with a rate of 82.7%. Positivity of antineutrophilic cytoplasmic antibodies is substantially significant in the pathogenesis and diagnosis of ARV. However, presence of ANCA alone is not sufficient for development of vasculitis. Primarily, circulating neutrophils should be activated and be exposed to cytokine effect. Tumor necrosis factor α causes to proteinase 3 (PR3) and myeloperoxidase (MPO) migration from the granule to the neutrophil cell surface. Vasculitis is initiated when antineutrophilic cytoplasmic antibodies bind to these structures reaching the cell surface. Antineutrophilic cytoplasmic antibodies stimulate neutrophils and factors which provide complement activation. Other inflammatory cells also accumulate in the area of inflammation. The characteristics which differentiate antineutrophilic cytoplasmic antibody-related vasculitis from small vessel vasculitis include being necrotizing vasculitis, absence of immune complexes (or presence of very few immune complexes) and being related with PR3 and MPO-ANCA.
The criteria used for vasculitis activity and damage in adults are not sufficiently appropriate for childhood ARV. There is a limited number of pediatric studies in this area. In the study conducted by Morishita et al. (41), BVAS was administered in 152 patients and a weak corrleation was found with physician global assessment scale and a moderate correlation was found with treatment decision and erythrocyte sedimentation rate (ESR). The classification criteria used for childhood GPA are the EULAR/PRINTO/PRES criteria published in 2010 (Table 2) (5).
There is no treatment guide related with ANCA-related vasculitides in children. Currently, there is no validated study related with maintenence treatment for ANCA-related childhood vasculitis. Therefore, treatment approches are based on adult protocols and the results of the largest evaluation in this area have been published recently (42). Granulomatous polyangiitis is a disease which may be fatal both in children and adults. Mortality is related with renal involvement, pulmonary hemorrhage or failure and infection. Previously, steroid and cyclophosphamide (CYC) were being used intensively (42, 43). Although CYC is accepted to be an efficient drug in providing remission, it should be considered that recurrence in 5 years occurs with a rate of 50% and toxic risks in the short- and long-term (e.g. increased risk of life threatening infection) and malignancy and infertility risks should also be considered (38). Therefore, monthly intravenous regimes have been adopted rather than daily oral regime for CYC treatment (43). The mortality rate is 90% in the first year if it is not treated. Although steroid and cyclophosphamide are generally used in combination for treatment, currently rituksimab is also used in resistant cases to achieve remission. In the follow-up, azathioprine (AZT) or other disease-modifying drugs are used in combination with steroid (40, 42). Plasma exchange binds circulating ANCAs and may be used as an assistive method in the acute period (42).
There is no specific finding which could support a less agressive treatment regime in milder disease states in children. Although some approaches for less aggressive treatment have been published in a few adult studies, they have not been applied in children, since these studies have been performed in recent years. Many pediatric rheumatologists recommend efficient and agressive treatment in childhood ARV, because it is thought that the long-term toxic findings of steroid and inflammation lead to potential negative effects in growing children with a higher rate compared to adults. Since clinic trials and consensus guidelines are absent in childhood ARV, treatment selection depends on the pysician’s decision and the severity of the disease is very significant in this decision (41, 42).
b. Immune complex-related small vessel vasculitis
The vasculitides in this class include anti-glomerular basal membrane disease, cryoglobulinemic vasculitis, IgA vasculitis (Henoch-Schonlein) and hypocomplementemic urticarial vasculitis (anti-C1q vasculitis) (2). In this review, Henoch-Schonlein purpura/IgA vasculitis which is encountered commonly in clinical practice will be explained.
Henoch-Schonlein purpura/IgA vasculitis
Henoch-Schonlein purpura is the most common vasculitis in the childhhod and usually has a benign prognosis. It occurs mainly in the childhood and is observed rarely in adults. It occurs most commonly between the ages of 3 and 15 years and more frequently in boys compared to girls (1.5/1). The disease is associated with leucocytoclastic vasculitis involving small vessels. Renal involvement is the main cause of chronic disease in HSP. In this case, renal functions should be monitored closely and adequate and efficient treatment should be given in case of severe involvement (44). The factors which lead to occurence of the disease are not known clearly. The incidence is generally increased in autumn and winter months. Although the eitology is not known clearly, there is a history of prior upper respiratory tract infection. However, a clear microbiological factor or correlation has not been demonstrated. It may be triggered by antigenic stimuli including infective agents, vaccination, drugs and insect bites (45, 46).
Clinical findings
The classical quadruple of the disease includes palpable purpura, arthritis or artralgia which does not cause to sequela, gastrointestinal involvement characterized with abdominal pain and renal disease. Recurrence of purpura may be associated with severe renal disease and may be observed in 25% of HSP patients (47). Especially in countries where familial Mediteranean fever (FMF) is prevalent, FMF should be considered in the differential diagnosis in children with recurrent HSP episode. Soft tissue edema which is mostly prominent in the scalp is observed in almost all patients. Purpuric rash may sometimes be necrotic (Figure 4). Joint involvement is in the form of acute polyarthritis. Involvement in all joints may be observed in association with soft tissue edema. Scrotal and penile edema may also be observed. The severity of renal involvement determines long-term prognosis in Henoch-Schonlein purpura. Different forms of renal involment may be observed. Nephritis episode may develop years after improvement of HSP findings. The most common finding in renal involvement is hematuria. It generally develops in the first four weeks. Varying degrees of proteinuria may occur. Severe proteinuria including nephrotic syndrome may also be observed. Hypertension may be observed in the beginning or in the recovery period. Renal function tests are generally normal, but sometimes severe renal involvement and progressive glomerulonephritis may be observed (47). Neurological involvement may rarely be observed in HSP. Central nervous system (CNS) damage occurs with the direct effect of vasculitis or indirect effect of systemic inflammation. Severe neurological involvement is observed rarely in this disease. Mild involvement has been reported more frequently. In one series, convulsion and confusion were screened retrospectively in 244 patients and found in 17 (6.9%) patients. In recent studies, lower incidences have been reported (48). Henoch Schonlein purpura has a substantially benign prognosis below the age of 8 years. The disease may lead to more severe clinical findings in the adolescence.
Figure 4.
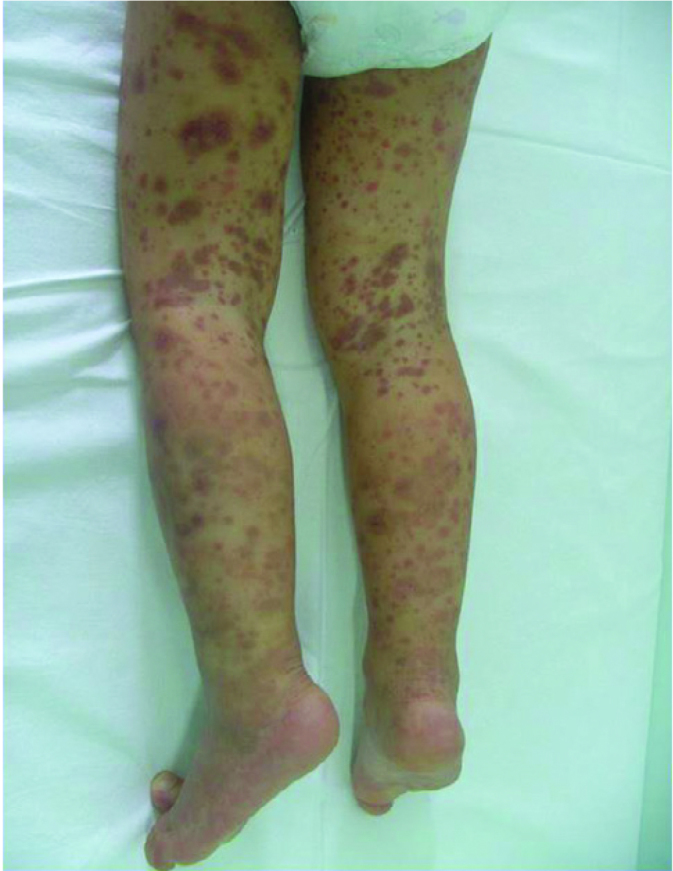
Appearance of purpuric rash in Henoch-Schonlein purpura
Treatment
Generally, supportive treatment is sufficient in the majority of the patients. In addition, symptomatic treatment and immunosuppresive treatment (in some patients) is applied. In presence of arthritis and arthralgia, non-steroid anti-inflammatory drugs (NSAID) are sufficient. Steroid may be administered in patients with severe gastrointestinal involvement, proteinuria at nephrotic level and crescentic glomerulonephritis. Steroid rapidly improves abdominal pain and decreases the risk of invagination and surgical intervention. Although early use of glucocorticoid has been reported to reduce the risk of development of renal disease in some studies, this has not been confirmed with randomized, controlled studies (47).
Since hemoperfusion could eliminate immune mediators in many diseases, its effect in HSP nephritis was investigated and it was thought that steroid in combination with hemoperfusion was more efficient in treating HSP nephritis and this effect was related with decreased circulating cytokines (49).
Prognosis
Signs and symptoms in Henoch-Schonlein purpura spontanously resolve. Recurrence may be observed in 1/3 patients in the first months of the disease. In recurent HSP, the findings are similar to the previous episode, but the clinical picture is milder. In the study conducted by Cohen et ve al. (50), the determinants which affected hospitalization time were reported to include abdominal pain, systemic fever, a CRP value above 45 mg/dL and being older than six years.
4. Vasculitides involving vessels with different size
This group includes Behçet’s disease and Cogan’s syndrome. Behçet’s disease which is commonly observed in clinical practice will be explained below. Cogan’s syndrome may be manifested with non-syphilitic interstitial keratitis, neurosensory hearing defect and aortic failure in the childhood.
Behçet’s disease
This vasculitis which may involve arteries and veins with any size is characterized with cutaneous, opthalmic, gastrointestinal and central nervous system lesions which may accompany recurrent oral and/or genital ulcers (4). The diagnostic criteria most commonly used at the present time were established in 1990 by the ‘International Study Group’ (ISG). According to these criteria, at least two of the following criteria should accompany oral ulcers recurred for at least 3 times in a 12-month period: recurrent genital ulcers, anterior uveitis, posterior uveitis, observation of cells in the vitreus, opthalmic findings including retinal vasculitis, erythema nodosum, pseudofolliculitis, papulopustular lesions, cutaneous findings including acneiform nodules, positive pathergy test.
The incidence has been reported to be 1/250 in Turkey which is located in the Middle East where the disease is prevalent (52). The onset of disease occurs before 16 years in 4–26% of the patients with Behçet’s disease. Although it is observed with a two-fold higher frequency in male patients in adults, gender difference is not observed in children (53, 54). However, neurological involvement in Behçet’s disease is observed with a 5.5-fold higher frequency in male patients (53). In an international pediatric study including 110 patients, it was shown that 19% of the children with a diagnosis of Behçet’s disease had a familial history of Behçet’s disease (54). Behçet’s disease which has an unclear etiology is mostly related with HLA-B51 molecule genetically. In recent years, all genome studies [genome-wide association (GWAS)] have shown that IL-23/IL-17 axis is activated in inflammation in Behçet’s disease and TH17 is a conducter here (55, 56). Again, a recent study found that the IL-17 level was markedly increased in the patients with Behçet’s disease and PAN compared to the control group (57).
In Behçet’s disease, occurence of the major clinical finding may take years after the first clinical finding. Hence, the mean time of occurence of the second main clinical finding was 7 years in 40 Korean children. In another international study, the mean time between the first sign and the diagnosis of Behçet’s disease was found to be 3.7 years (54, 58). However, it was observed that neurological involvement occured about 6 months after the onset of the disease (very early), when 26 patients with a diagnosis of neuro-Behçet’s disease were examined retrospectively (53).
Uveitis is mostly in the form of panuveitis and is observed more frequently and has a more severe prognosis in male patients. In addition, it has been reported that genital ulcers are observed more frequently in girls (54).
The frequency of neuro-Behçet’s disease has been reported to be 5%–15% in all pediatric patients with Behçet’s disease (53). The frequency of neurological involvement is 2.9–5.5-fold higher in male patients compared to female patients (53, 59). The disease may be manifested in three forms: parencymal, vascular and mixed types. While parenchymatous involvement which occurs as meningitis or encephalomyelitis occurs more commonly in adults, vascular type is in the form of benign intracranial hypertension caused mostly by an underlying dural sinus thrombosis and accompanied by papilledema (53, 59). It has been noted that 36–42% of the children with neuro-Behçet’s disease present primarily with neurological involvement without meeting the ISG criteria(before the other main findings occur) (53, 59). Mora et al. (59) examined 53 patients aged 16 years and below who were diagnosed with neuro-Behçet’s disease between 1971 and 2011 and whose imaging findings related with Behçet’s disease were clearly described by way of web screening. They observed that most of these patients showed non-parenchymal involvement rather than parenchymal involvement (vascular type) and thus the most common complaint was headache related with intracranial hypertension in these patients.
In treatment, colchicin is used in uncomplicated Behçet’s disease with skin involvement, while steroid, azatioprine, cyclophosphamide, methotrexate, interferon- α and anti-TNF drugs are used in vascular and neurological system involvement (52).
5. Single organ vasculitis
Central nervous system vasculitides
This is the vasculitic picture which includes only CNS findings without accompaniment of systemic findings. On imaging, primarily infarct or ischemia findings are found. The diagnosis is possible with angiography or brain biopsy. The prognosis is variable. Presence or absence of systemic findings determines the picture. Childhood CNS vasculitis (CPANS) which is also known as primary CNS angiitis is one of the most important causes of acquired neurological deficits in children (60). Calabrese CNS vasculitis classification criteria may also be used in children for making a diagnosis. The disease has been defined as three different subtypes:
Non progressive (NP) medium-large vessel CNS vasculitides (cPACNS) (angiography positive),
Progressive (P) medium-large vessel CNS vasculitides (cPACNS) (angiography positive)
Small vessel CNS (cPACNS) vasculitis (angiography negative, biopsy positive).
In the study conducted by Celucci et al. (61), it was found that von willebrand factor (vWF) antigene level indicated activity of the disease in children with primary CNS vasculitis. A reduction in vWF antigene level indicates improvement in the activity of primary CNS vasculitis in children. Treatment mathods for different subtypes are different. In non-progressive CPACNS, corticosteroid treatment reduces progression of inflammation in the vascular wall, corrects vascular stenosis, reduces the risk of development of thrombosis and prevents recurrent ischemic episodes. In treatment of progressive angiography-positive cPACNS and small vessel CPACNS, combined immunosupressive agents are used for long-term (60).
In the childhood, vasculitic diseases are menifested with very different signs and symptoms and it may be difficult to make a diagnosis, because the findings including un-healing rash, malaise and fatigue are also freqently observed in other diseases. In suspected cases, the findings are usually permanent and elevated acute phase reactants accompany. Vasculitis specific investigations, radiological imaging methods and skin and other organ biopsies should be performed to make a diagnosis. HSP which is the most common vasculitis in the childhood usually has a benign prognosis, but it should be kept in mind that renal involvement may be present in a significant portion and some of these may progress to end stage renal disease. Therefore, patients diagnosed with HSP should be examined and necessary tests should be performed with certain intervals according to the routine follow-up schedule. Development of CAA is observed very rarely in patients diagnosed with Kawasaki disease who have received treatment in the first 10 days, while it is observed more frequently in atypical KD and in patients with a delayed diagnosis, because the process of inflammation is prolonged. One should be careful in this respect and rare findings which may be present in KD should be searched for with a careful physical examination. Takayasu arteritis is observed very rarely in the childhood. Recovery is achievable, though with difficulty. Meticulous follow-up is mandatory and blood presüsure measurements should be done carefully. Use of tosilizumab should be considered in treatment of resistant TA patients. Since there is a limited number of studies related with the prognosis of vasculitides in children, our information in this area is limited. On the other hand, an early and agressive treatment method has been adopted in many pediatric rheumatology centers, because especially ARVs show a severe prognosis. Since childhood vasculitides are observed rarely, further studies should be designed as multi-center studies with long-term follow periods.
Footnotes
Author Contributions: Concept - Ö.K.; Design - K.B., A.A., S.Ş., Ö.K.; Supervision - Ö.K.; Data Collection and/or Processing - K.B., A.A., S.Ş.; Analysis and/or Interpretation - K.B., Ö.K.; Literature Review - K.B., A.A., S.Ş.; Writing - K.B., A.A., S.Ş., Ö.K.; Critical Review - Ö.K.
Conflict of Interest: No conflict of interest was declared by the authors.
Financial Disclosure: The authors declared that this study has received no financial support
References
- 1.Weiss PF. Pediatric vasculitis. Pediatr Clin North Am. 2012;59:407–23. doi: 10.1016/j.pcl.2012.03.013. http://dx.doi.org/10.1016/j.pcl.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mahr A, de Menthon M. Classification and classification criteria for vasculitis: achievements, limitations and prospects. Curr Opin Rheumatol. 2015;27:1–9. doi: 10.1097/BOR.0000000000000134. http://dx.doi.org/10.1097/BOR.0000000000000134. [DOI] [PubMed] [Google Scholar]
- 3.Batu ED, Ozen S. Vasculitis: do we know more to classify better? Pediatr Nephrol. 2015;30:1425–30. doi: 10.1007/s00467-014-3015-0. http://dx.doi.org/10.1007/s00467-014-3015-0. [DOI] [PubMed] [Google Scholar]
- 4.Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised international Chapell Hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1–11. doi: 10.1002/art.37715. http://dx.doi.org/10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 5.Ozen S, Pistorio A, Iusan SM, et al. EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara 2008. Part II: Final classification criteria. Ann Rheum Dis. 2010;69:798–806. doi: 10.1136/ard.2009.116657. http://dx.doi.org/10.1136/ard.2009.116657. [DOI] [PubMed] [Google Scholar]
- 6.Goel R, Kumar TS, Danda D, et al. Childhood onset Takayasu arteritis experience from a tertiary care center in South India. J Rheumatol. 2014;41:1183–9. doi: 10.3899/jrheum.131117. http://dx.doi.org/10.3899/jrheum.131117. [DOI] [PubMed] [Google Scholar]
- 7.Szugye HS, Zeft AS, Spalding SJ. Takayasu arteritis in the pediatric population: a contemporary United States-based single center cohort. Pediatr Rheumatol Online J. 2014;12:21. doi: 10.1186/1546-0096-12-21. http://dx.doi.org/10.1186/1546-0096-12-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mendiola Ramírez K, Portillo Rivera AC, Galicia Reyes A, García Montes JA, Maldonado Velázquez Mdel R, Faugier Fuentes E. Type III Takayasu’s arteritis in a pediatric patient. Case report and review of the literature. Reumatol Clin. 2012;8:216–9. doi: 10.1016/j.reuma.2011.11.008. http://dx.doi.org/10.1016/j.reuma.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Eleftheriou D, Varnier G, Dolezalova P, McMahon AM, Al-Obaidi M, Brogan PA. Takayasu arteritis in childhood: retrospective experience from a tertiary referral centre in the United Kingdom. Arthritis Res Ther. 2015;17:36. doi: 10.1186/s13075-015-0545-1. http://dx.doi.org/10.1186/s13075-015-0545-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watson L, Brogan P, Peart I, Landes C, Barnes N, Cleary G. Diagnosis and assessment of disease activity in Takayasu arteritis: a childhood case illustrating the challenge. Case Rep Rheumatol. 2014;2014:603171. doi: 10.1155/2014/603171. http://dx.doi.org/10.1155/2014/603171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tombetti E, Di Chio M, Sartorelli S, et al. Systemic pentraxin-3 levels reflect vascular enhancement and progression in Takayasu arteritis. Arthritis Res Ther. 2014;16:479. doi: 10.1186/s13075-014-0479-z. http://dx.doi.org/10.1186/s13075-014-0479-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozen S, Duzova A, Bakkaloglu A, et al. Takayasu arteritis in children: preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate. J Pediatr. 2007;150:72–6. doi: 10.1016/j.jpeds.2006.10.059. http://dx.doi.org/10.1016/j.jpeds.2006.10.059. [DOI] [PubMed] [Google Scholar]
- 13.Osman M, Aaron S, Noga M, Yacyshy E. Takayasu’s arteritis progression on anti-TNF biologics: a case series. Clin Rheumatol. 2011;30:703–6. doi: 10.1007/s10067-010-1658-1. http://dx.doi.org/10.1007/s10067-010-1658-1. [DOI] [PubMed] [Google Scholar]
- 14.Reddy E, Robbs VJ. Surgical management of Takayasu’s arteritis in children and adolescents. Cardiovasc J Africa. 2007;18:393–7. [PubMed] [Google Scholar]
- 15.Keser G, Direskeneli H, Aksu K. Management of Takayasu arteritis: a systematic review. Rheumatology. 2014;53:793–801. doi: 10.1093/rheumatology/ket320. http://dx.doi.org/10.1093/rheumatology/ket320. [DOI] [PubMed] [Google Scholar]
- 16.Bravo Mancheno B, Perin F, Guez Vázquez Del Rey Mdel M, García Sánchez A, Alcazar Romero PP. Successful tocilizumab treatment in a child with refractory Takayasu arteritis. Pediatrics. 2012;130:e1720–4. doi: 10.1542/peds.2012-1384. [DOI] [PubMed] [Google Scholar]
- 17.Iudici M, Puechal X, Pagnoux C, et al. Brief report: childhood-onset systemic necrotizing vasculitides: long-term data from the french vasculitis study group registry. Arthritis Rheumatol. 2015;67:1959–65. doi: 10.1002/art.39122. http://dx.doi.org/10.1002/art.39122. [DOI] [PubMed] [Google Scholar]
- 18.Eleftheriou D, Dillon MJ, Tullus K, et al. Systemic polyarteritis nodosa in the young: a single-center experience over thirty-two years. Arthritis Rheum. 2013;65:2476–85. doi: 10.1002/art.38024. http://dx.doi.org/10.1002/art.38024. [DOI] [PubMed] [Google Scholar]
- 19.Merlin E, Mouy R, Pereira B, et al. Long-term outcome of children with pediatric-onset cutaneous and visceral polyarteritis nodosa. Joint Bone Spine. 2015;82:251–7. doi: 10.1016/j.jbspin.2015.01.007. http://dx.doi.org/10.1016/j.jbspin.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 20.Eleftheriou D, Batu ED, Ozen S, Brogan PA. Vasculitis in children. Nephrol Dial Transplant. 2015;30:i94–103. doi: 10.1093/ndt/gfu393. [DOI] [PubMed] [Google Scholar]
- 21.Navon Elkan P, Pierce SB, Segel R, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370:921–31. doi: 10.1056/NEJMoa1307362. http://dx.doi.org/10.1056/NEJMoa1307362. [DOI] [PubMed] [Google Scholar]
- 22.Zhou Q, Yang D, Ombrello AK, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911–20. doi: 10.1056/NEJMoa1307361. http://dx.doi.org/10.1056/NEJMoa1307361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garg N, Kasapcopur O, Foster J, 2nd, et al. Novel adenosine deaminase 2 mutations in a child with a fatal vasculopathy. Eur J Pediatr. 2014;173:827–30. doi: 10.1007/s00431-014-2320-8. http://dx.doi.org/10.1007/s00431-014-2320-8. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Santiago TM, Zavialov A, Saarela J, et al. Dermatologic features of ADA2 deficiency in cutaneous polyarteritis nodosa. JAMA Dermatol. 2015:1. doi: 10.1001/jamadermatol.2015.1635. (Epub ahead of print). http://dx.doi.org/10.1001/jamadermatol.2015.1635. [DOI] [PubMed] [Google Scholar]
- 25.Greco A, De Virgilio A, Rizzo M. Kawasaki disease: an evolving paradigm. Autoimmun Rev. 2015;14:703–9. doi: 10.1016/j.autrev.2015.04.002. http://dx.doi.org/10.1016/j.autrev.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Salo E, Griffiths EP, Farstad T, et al. Incidence of Kawasaki disease in Northern European Countries. Pediatr Int. 2012;54:770–2. doi: 10.1111/j.1442-200X.2012.03692.x. http://dx.doi.org/10.1111/j.1442-200X.2012.03692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura Y, Yashiro M, Uehara R, et al. Epidemiologic features of Kawasaki disease in Japan: results of the 2009–2010 nationwide survey. J Epidemiol. 2012;22:216–21. doi: 10.2188/jea.JE20110126. http://dx.doi.org/10.2188/jea.JE20110126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodo X, Ballester J, Cayan D, et al. Association of Kawasaki disease with tropospheric wind patterns. Sci Rep. 2011;1:152. doi: 10.1038/srep00152. http://dx.doi.org/10.1038/srep00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jaggi P, Kajon A, Mejias A, et al. Human adenovirus infection in Kawasaki disease: a confounding bystander? Clin Inf Dis. 2012;56:57–63. doi: 10.1093/cid/cis807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burns JC, Herzog L, Fabri O, et al. Seasonality of Kawasaki disease: a global perspective. PLoS One. 2013;8:e74529. doi: 10.1371/journal.pone.0074529. http://dx.doi.org/10.1371/journal.pone.0074529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanchez-Manubens J, Bou R, Anton J. Diagnosis and classification of Kawasaki disease. J Autoimmun. 2014;49:113–7. doi: 10.1016/j.jaut.2014.01.010. http://dx.doi.org/10.1016/j.jaut.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Rezai MS, Shahmohammadi S. Erythema at BCG inoculation site in Kawasaki disease patients. Mater Sociomed. 2014;26:256–60. doi: 10.5455/msm.2014.26.256-260. http://dx.doi.org/10.5455/msm.2014.26.256-260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bayers S, Shulman ST, Paller AS. Kawasaki disease Part I. Diagnosis, clinical features, and pathogenesis. J Am Acad Dermatol. 2013;69:501.e1–11. doi: 10.1016/j.jaad.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 34.Giacchi V, Sciacca P, Stella I, et al. Assessment of coronary artery intimal thickening in patients with a previous diagnosis of Kawasaki disease by using high resolution transthoracic echocardiography: our experience. BMC Cardiovasc Disord. 2014;14:106. doi: 10.1186/1471-2261-14-106. http://dx.doi.org/10.1186/1471-2261-14-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeom JS, Woo HO, Park JS, Park ES, Seo JH, Youn HS. Kawasaki disease in infants. Korean J Pediatr. 2013;56:377–82. doi: 10.3345/kjp.2013.56.9.377. http://dx.doi.org/10.3345/kjp.2013.56.9.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sundel RP. Kawasaki disease. Rheum Dis Clin North Am. 2015;41:63–73. doi: 10.1016/j.rdc.2014.09.010. http://dx.doi.org/10.1016/j.rdc.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 37.Bayers S, Shulman ST, Paller AS. Kawasaki disease Part II. Complications and treatment. J Am Acad Dermatol. 2013;69:513.e1–8. doi: 10.1016/j.jaad.2013.06.040. [DOI] [PubMed] [Google Scholar]
- 38.Twilt M, Benseler S, Cabral D. Granulomatosis with polyangiitis in childhood. Curr Rheumatol Rep. 2012;14:107–15. doi: 10.1007/s11926-012-0238-6. http://dx.doi.org/10.1007/s11926-012-0238-6. [DOI] [PubMed] [Google Scholar]
- 39.Cabral DA, Uribe AG, Benseler S, et al. Classification, presentation, and initial treatment of Wegener’s granulomatosis in childhood. Arthritis Rheum. 2009;60:3413–24. doi: 10.1002/art.24876. http://dx.doi.org/10.1002/art.24876. [DOI] [PubMed] [Google Scholar]
- 40.Bohm M, Gonzalez Fernandez MI, Ozen S, et al. Clinical features of childhood granulomatosis with polyangiitis (Wegener’s granulomatosis) Pediatr Rheumatol Online J. 2014;12:18. doi: 10.1186/1546-0096-12-18. http://dx.doi.org/10.1186/1546-0096-12-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morishita K, Li SC, Muscal E, et al. Assessing the performance of the Birmingham Vasculitis Activity Score at diagnosis for children with antineutrophil cytoplasmic antibody-associated vasculitis in a registry for childhood vasculitis (ARChiVe) J Rheumatol. 2012;39:1088–94. doi: 10.3899/jrheum.111030. http://dx.doi.org/10.3899/jrheum.111030. [DOI] [PubMed] [Google Scholar]
- 42.Morishita K, Brown K, Cabral D. Pediatric vasculitis: advances in treatment. Curr Opin Rheumatol. 2015;27:493–9. doi: 10.1097/BOR.0000000000000203. http://dx.doi.org/10.1097/BOR.0000000000000203. [DOI] [PubMed] [Google Scholar]
- 43.Langford CA. Cyclophosphamide as induction therapy for Wegener’s granulomatosis and microscopic polyangiitis. Clin Exp Immunol. 2011;164:31–4. doi: 10.1111/j.1365-2249.2011.04364.x. http://dx.doi.org/10.1111/j.1365-2249.2011.04364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pohl M. Henoch–Schönlein purpura nephritis. Pediatr Nephrol. 2015;30:245–52. doi: 10.1007/s00467-014-2815-6. http://dx.doi.org/10.1007/s00467-014-2815-6. [DOI] [PubMed] [Google Scholar]
- 45.Ercan G, Kasapçopur O, Akdenizli E, Arisoy N. The role of streptococcal infection in Henoch-Schönlein purpura. J Trop Pediatr. 2004;50:187–8. doi: 10.1093/tropej/50.3.187. http://dx.doi.org/10.1093/tropej/50.3.187. [DOI] [PubMed] [Google Scholar]
- 46.Rigante D, Castellazzi L, Bosco A, Esposito S. Is there a crossroad between infections, genetics, and Henoch-Schönlein purpura? Autoimmun Rev. 2013;12:1016–21. doi: 10.1016/j.autrev.2013.04.003. http://dx.doi.org/10.1016/j.autrev.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 47.Trnka P. Henoch-Schönlein purpura in children. J Paediatr Child Health. 2013;49:995–1003. doi: 10.1111/jpc.12403. http://dx.doi.org/10.1111/jpc.12403. [DOI] [PubMed] [Google Scholar]
- 48.Berube MD, Blais N, Lanthier S. Neurologic manifestations of Henoch-Schönlein purpura. Handb Clin Neurol. 2014;120:1101–11. doi: 10.1016/B978-0-7020-4087-0.00074-7. http://dx.doi.org/10.1016/B978-0-7020-4087-0.00074-7. [DOI] [PubMed] [Google Scholar]
- 49.Chen L, Wang Z, Zhai S, Zhang H, Lu J, Chen X. Effects of hemoperfusion in the treatment of childhood Henoch-Schönlein purpura nephritis. Int J Artif Organs. 2013;36:489–97. doi: 10.5301/ijao.5000223. http://dx.doi.org/10.5301/ijao.5000223. [DOI] [PubMed] [Google Scholar]
- 50.Cohen N, Mimouni FB, Friedel N, Amarilyo G. Predictors of hospital length of stay in pediatric Henoch-Schonlein purpura. Rheumatol Int. 2015;35:1561–4. doi: 10.1007/s00296-015-3257-6. http://dx.doi.org/10.1007/s00296-015-3257-6. [DOI] [PubMed] [Google Scholar]
- 51.International Study Group for Behçet’s Disease, criteria for diagnosis of Behçet’s disease. Lancet. 1990;335:1070–80. [PubMed] [Google Scholar]
- 52.Hatemi G, Yazici Y, Yazici H. Behcet’s syndrome. Rheum Dis Clin North Am. 2013;89:245–61. doi: 10.1016/j.rdc.2013.02.010. http://dx.doi.org/10.1016/j.rdc.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 53.Uluduz D, Kurtuncu M, Yapici Z, et al. Clinical characteristics of pediatric-onset neuro-Behçet disease. Neurology. 2011;77:1900–5. doi: 10.1212/WNL.0b013e318238edeb. http://dx.doi.org/10.1212/WNL.0b013e318238edeb. [DOI] [PubMed] [Google Scholar]
- 54.Koné-Paut, Darce-Bello M, Shahram F, et al. Registries in rheumatological and musculoskeletal conditions. Paediatric Behcet’s disease: an international cohort study of 110 patients. One-year follow-up data. Rheumatology. 2010;50:184–8. doi: 10.1093/rheumatology/keq324. [DOI] [PubMed] [Google Scholar]
- 55.Hamzaoui K. Th17 cells in Behçet’s disease: a new immunoregulatory axis. Clin Exp Rheumatol. 2011;29:S71–6. [PubMed] [Google Scholar]
- 56.Liu X, Yang P, Wang C, et al. IFN-alpha blocks IL-17 production by peripheral blood mononuclear cells in Behçet’s disease. Rheumatology (Oxford) 2010;50:293–8. doi: 10.1093/rheumatology/keq330. http://dx.doi.org/10.1093/rheumatology/keq330. [DOI] [PubMed] [Google Scholar]
- 57.Taskiran EZ, Batu ED, Kilic L, et al. New Insights to the pathogenesis of two orphan vasculitides: childhood polyarteritis nodosa and Behçet disease. Ann Paediatr Rheumatol. 2015;4:7–11. http://dx.doi.org/10.5455/apr.030320151114. [Google Scholar]
- 58.Kim DK, Chang SN, Bang D, et al. Clinical analysis of 40 cases of childhood-onset Behçet’s disease. Pediatr Dermatol. 1994;11:95–101. doi: 10.1111/j.1525-1470.1994.tb00559.x. http://dx.doi.org/10.1111/j.1525-1470.1994.tb00559.x. [DOI] [PubMed] [Google Scholar]
- 59.Mora P, Menozzi C, Orsoni JG, et al. Neuro-Behçet’s disease in childhood: a focus on the neuro-ophthalmological features. Orphanet J Rare Dis. 2013;8:18. doi: 10.1186/1750-1172-8-18. http://dx.doi.org/10.1186/1750-1172-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Twilt M, Benseler SM. Childhood inflammatory brain diseases: pathogenesis, diagnosis and therapy. Rheumatology (Oxford) 2014;53:1359–68. doi: 10.1093/rheumatology/ket398. http://dx.doi.org/10.1093/rheumatology/ket398. [DOI] [PubMed] [Google Scholar]
- 61.Cellucci T, Tyrrell PN, Pullenayegum E, Benseler SM. von Willebrand factor antigen--a possible biomarker of disease activity in childhood central nervous system vasculitis? Rheumatology (Oxford) 2012;51:1838–45. doi: 10.1093/rheumatology/kes156. http://dx.doi.org/10.1093/rheumatology/kes156. [DOI] [PubMed] [Google Scholar]
- 62.Barut K, Sezgin S, Kasapcopur O. Pediatric Vasculitis. Curr Opin Rheumatol. 2016;28:29–38. doi: 10.1097/BOR.0000000000000236. [DOI] [PubMed] [Google Scholar]