

Abstract
Patients with acquired deficiency in granulocyte macrophage colony stimulating factor (GM-CSF) are susceptible to infections with Cryptococcus (C.) neoformans and other opportunistic fungi. We previously showed that GM-CSF protects against progressive fungal disease using a murine model of cryptococcal lung infection. To better understand the cellular and molecular mechanisms through which GM-CSF enhances anti-fungal host defenses, we investigated temporal and spatial relationships between myeloid and lymphoid immune responses in GM+/+ and GM−/− mice infected with a moderately-virulent encapsulated strain of C. neoformans (strain 52D). Our data demonstrate that GM-CSF deficiency led to a reduction in: 1) total lung leukocyte recruitment; 2) Th2 and Th17 responses; 3) total numbers of CD11b+ dendritic cells (DC), and CD11b− and CD11b+ macrophages (Mϕ); 4) DC and Mϕ activation; and 5) localization of DC and Mϕ to the microanatomic sites of alveolar infection. In contrast, GM-CSF deficiency resulted in increased accumulation of DC and Mϕ precursors, namely Ly-6Chigh monocytes, in the blood and lungs of infected mice. Collectively, these results show that GM-CSF promotes the local differentiation, accumulation, activation, and alveolar localization of lung DC and macrophages in mice with cryptococcal lung infection. These findings identify GM-CSF as central to the protective immune response that prevents progressive fungal disease and thus shed new light on the increased susceptibility to these infections observed in patients with acquired GM-CSF deficiency.
Keywords: GM-CSF, dendritic cells, macrophages, lung, rodent, Cryptococcus
Introduction
The outcome of primary pulmonary infection with the encapsulated fungus Cryptococcus (C.) neoformans is profoundly shaped by the virulence of the organism and the host’s immune status (reviewed in (1)). In humans, the importance of these host-fungal interactions is underscored by the clinical burden of infection when host defenses are compromised. Worldwide, one million new cases of C. neoformans infections occur each year in HIV-infected patients resulting in 680,000 deaths (2) and cryptococcal infection is the second most common fungal infection in organ transplant patients (1). Although uncommon, clinically-significant infections have been reported in seemingly immunocompetent hosts (3–5). Recent studies by Rosen et al and Saijo et al suggest that a subset of these patients have previously-unidentified immune defects (6, 7). Specifically, these authors identified autoantibodies against granulocyte-macrophage colony stimulating factor (GM-CSF), a cytokine with pleiotropic immune effects (reviewed in references (8, 9)), in the serum and cerebrospinal fluid of a subset of patients with invasive cryptococcal infection for whom a predisposing immunocompromising condition had not previously been identified. Autoantibodies against GM-CSF or mutations in the GM-CSF receptor can lead to pulmonary alveolar proteinosis (PAP), a lung disease characterized by disrupted surfactant catabolism in alveolar macrophages ((10, 11) and reviewed in reference (12)). Thus, the studies by Rosen and Saijo confirm and extend previously published associations between patients with PAP and increased susceptibility to cryptococcal and other fungal infections (13–15). Despite this strengthened association between GM-CSF deficiency in humans and susceptibility to C. neoformans infections, the cellular and molecular mechanisms through which GM-CSF protects against progressive cryptococcal infections remain uncertain.
We previously demonstrated that lung macrophages obtained from GM-CSF deficient rats are poorly fungicidal (16). In a later study, we performed a comparative analysis of cryptococcal lung infection in C57BL/6 mice (GM+/+ mice) and GM-CSF deficient mice (GM−/− mice; C57BL/6 genetic background) and showed that progressive fungal infection occurs in the absence of GM-CSF (17). Murine models have also shown a protective role for GM-CSF in host defense against other fungal pathogens, including Pneumocystis jiroveci, Histoplasma capsulatum, and Aspergillus fumigatus (18–22). These studies implicate that lack of GM-CSF induces alterations in adaptive immunity and macrophage function, yet provide limited information about the number, immunophenotype, or microanatomic location of T and myeloid cells in response to infection.
Our more recent studies demonstrated that GM-CSF promotes the in vitro differentiation of Ly-6Chigh “inflammatory” monocytes into CD11b+ DC and exudate macrophages (ExM), a subset of primarily recruited lung macrophages expressing CD11b which distinguishes them from resident CD11b− alveolar macrophages (23–25). Additional data demonstrated that CD11b+ DC interact with newly-recruited lung T cells to influence adaptive immune responses and that ExM are phagocytic and can become fungicidal. The objective of the current study was to further investigate the in vivo effects of GM-CSF on pulmonary DC and macrophages in mice infected by the intratracheal route with a moderately-virulent strain of C. neoformans (strain 52D). Our studies were performed using GM+/+ mice (C57BL/6J mice) which develop persistent cryptococcal lung infection (17, 26, 27) and GM−/− mice (C57BL/6J genetic background) which develop progressive lung infection (17). Our results show that GM-CSF promotes the differentiation, accumulation, activation, and alveolar localization of lung DC and macrophages. These findings help define the critical role of GM-CSF and myeloid cells in pulmonary host defenses against fungal pathogens and enhance our understanding of cryptococcal infections in susceptible patient populations.
Materials and Methods
Mice
Wild-type (C57BL/6J) mice were obtained from Charles River Laboratory Inc. (Wilmington, MA). GM-CSF deficient mice (GM−/− mice), were originally developed (28) and provided by Dr. J Whitsett (Children’s Hospital, Cincinnati) and extensively backcrossed against C57BL/6J mice as previously described (17). For our studies, GM−/− mice were bred on site. All mice were housed under specific pathogen-free conditions in the Animal Care Facility at the Ann Arbor Veterans Affairs Health System. All studies were conducted according to a protocol approved by the VA Institutional Animal Care and Use Committee. Mice were 6–8 weeks of age at the time of infection.
C. neoformans
C. neoformans strain 52D was obtained from the American Type Culture Collection (24067; Manassas, VA); this strain displayed smooth colony morphology when grown on Sabouraud dextrose agar. For intratracheal (i.t.) inoculation, C. neoformans was grown to a late logarithmic phase (48–72 h) at 37°C in Sabouraud dextrose broth (1% neopeptone and 2% dextrose: DIFCO, Detroit, MI) on a shaker. Cultured yeasts were then washed in non-pyrogenic saline, counted in the presence of Trypan Blue using a hemocytometer, and diluted to 3.3 × 105 CFU/ml in sterile non-pyrogenic saline immediately prior to i.t. inoculation.
Surgical intratracheal inoculation
Mice were anesthetized by i.p. injection of ketamine (100 mg/kg; Fort Dodge Laboratories, Fort Dodge, IA) and xylazine (6.8 mg/kg; Lloyd Laboratories, Shenandoah, IA). Through a small midline neck incision, the strap muscles were divided and retracted laterally to expose the trachea. Intratracheal inoculation was performed under direct vision using a 30 gauge needle attached to a 1 ml syringe mounted on a repetitive pipette (stepper, Tridak, Brookfield, CT). An inoculum of 104 CFU (30 μL) was injected into the trachea. Skin was closed using cyanoacrylate adhesive.
Tissue Collection
Lungs were perfused in situ via the right heart using PBS containing 0.5 mM EDTA until pulmonary vessels became grossly clear (~5 ml). Lungs were then excised, minced, and enzymatically digested to a single cell suspension as previously described (25). After erythrocyte lysis, cells were washed, filtered over 70 μm mesh, and resuspended in complete medium. Dead cells were removed by centrifugation over a percoll gradient. Total numbers of viable lung leukocytes were assessed in the presence of Trypan Blue using a hemocytometer. PBMC were isolated from peripheral blood obtained from the retro-orbital vein of deeply anesthetized mice and processed as previously described (23). All cell preparations were washed twice in sterile PBS before use for culture or antibody staining.
Measurement of cytokine production
Lung leukocytes obtained from mice infected for 14 days were cultured at 5×106 cells/ml in 24 well plates in 2 ml of complete RPMI1640 media at 37° and 5% CO2 for 24 hours without additional stimulation. At the end of culture, supernatants were harvested and cytokine concentration was assessed in triplicates by Luminex assay (Luminex Corporation, Austin, TX) per the manufacturer’s instructions.
Histology and Fluorescent Immunohistochemistry
Lungs were perfused with PBS via the right ventricle of the heart and inflated with 50% optimal cutting temperature (OCT) compound (Tissue Tek, Sakura Finetek, Torrance, CA) in PBS via the trachea. Lung lobes were then harvested and frozen in OCT. Twenty μm sections were stained using hematoxylin-eosin (H&E) and viewed by light microscopy. Microphotographs were taken using the Digital Microphotography system DFX1200 with ACT-1 software (Nikon Co, Tokyo, Japan). For immunohistochemistry, sections were fixed in cold acetone, blocked using purified anti-mouse CD16/32 antibody (clone 93, BioLegend, San Diego, CA, 10 μg/ml), and stained with Alexa Fluor 488 anti-mouse CD11c antibody (clone N418, Biolegend, diluted 1:100). Negative control sections were stained with an isotype-matched antibody recommended by the manufacturer (Alexa Fluor 488 Armenian Hamster IgG, clone HTK888, BioLegend, at the same dilution). Sections were mounted using ProLong Gold Antifade Mountant with DAPI (Life Technologies, Grand Island, NY). Confocal Microscopy was performed using an Olympus BX51WI microscope (Olympus, Center Valley, PA). Images were collected using a Hamamatsu EM-CCD color digital camera C9100 and the Stereo Investigator software (mbf BioScience, MicroBright Field, Williston, VT).
Monoclonal Abs used for flow cytometric analysis
The following mAbs purchased from BioLegend (San Diego, CA) were used: N418 (anti-murine CD11c); 2.4G2 (“Fc block”, anti-murine CD16/CD32); 30-F11 (anti-murine CD45); C/23 (anti-murine CD40); 16-10A1 (anti-murine CD80); GL1 (anti-murine CD86); AF6-120.1 (anti-murine I-Ab); 10F.9G2 (anti-murine PD-L1); TY25 (anti-murine PD-L2); 6d5 (anti-murine CD19); 1A8 (anti-murine Ly-6G); 145-2C11 (anti-murine CD3ε); BM8 (anti-murine F4/80); NLDC-145 (anti-murine CD205); C068C2 (anti-murine CD206); XMG1.2 (anti-IFNγ); TC11-18H10.1 (anti-IL-17A); and TRFK4 (anti-IL-5). The mAb, AL-21 (anti-murine Ly-6C); M1/70 (anti-murine CD11b) and ebio13A (anti-IL-13) were purchased from BD Biosciences (San Diego, CA). The mAb 2f8 (anti-murine CD204) was purchased from AbD Serotec. Monoclonal Abs were primarily conjugated with Alexa Fluor 700, FITC, PE, PE-Cy7, PerCP-Cy5.5, allophycocyanin (APC), APC-Cy7, Brilliant Violet 421, or Pacific blue. Isotype-matched control mAbs (BioLegend) were tested simultaneously in all experiments.
Cell staining and flow cytometric analysis
Cell staining, including blockade of Fc receptors, and sample analysis by flow cytometry were performed as described previously (25). A minimum of 10,000 events were acquired per sample on an LSRII flow cytometer (BD, San Jose, CA) and analyzed using FlowJo (Treestar, Ashland, Oregon).
Lung leukocyte subsets of DC, CD4+ T cells, macrophages, and Ly-6Chigh monocytes were identified using established gating strategies (23, 24) and as described in additional detail in figure legends 3, 4, and 5 and the Results section. Briefly, initial gates eliminated debris and doublets, identified CD45+ leukocytes, and excluded CD3+ T cells, CD19+ B cells, and Ly-6G+ granulocytes as described previously (23, 24). Thereafter, additional gating identified CD11c+ DC and macrophages (Mϕ). Autofluorescence, assessed in the PerCP-Cy5.5 channel using a 488 nm laser, was used to distinguish non-autofluorescent DC from autofluorescent Mϕ. Within the non-autofluorescent population of DC, a CD11c versus CD11b plot was used to identify total CD11c+ CD11b+ DC (described as “CD11b+ DC” in the text). Within the autofluorescent population of Mϕ, a CD11c versus CD11b plot was used to identify CD11b− Mϕ and CD11b+ Mϕ. The phenotype of DC and macrophages is summarized as follows: CD11b+ DC (CD45+ CD3− CD19− FSCmoderate Ly-6G− non-autofluorescent CD11c+ CD11b+); CD11b− Mϕ (CD45+ CD3− CD19−FSCmoderate/high Ly-6G− autofluorescent CD11c+ CD11b−); and CD11b+ Mϕ (CD45+ CD3− CD19− FSCmoderate/high Ly-6G− autofluorescent CD11c+ CD11b+). In some studies, CD11b− and CD11b+ Mϕ were further subdivided into CD11chigh and CD11clow subsets. Ly-6Chigh monocytes were identified within the CD11c− FSClow population of myeloid cells as having the following phenotype: (CD45+ CD3− CD19− FSClow Ly-6G− CD11c− F4/80+ CD11b+ Ly-6Chigh). Myeloid cell differentiation and activation was further assessed by measuring cell surface expression of MHC class II (I-Ab), CD40, CD80, CD86, PD-L2, CD204, CD205, CD206, and Ly-6C relative to an isotype-matched control antibody.
Within the CD45+ population, CD4+ T cells were identified as CD45+ FSClow SSClow CD4+ CD8−. To detect intracellular cytokine production, lung leukocytes were stimulated with PMA (50ng/ml) and ionomycin (1μg/ml) (Sigma, St. Louis, MO) in the presence of brefeldin A and monensin (Biolegend, San Diego, CA) for 6 hours, and then stained with Aqua Live/Dead fixable viability dye and antibodies targeting CD45, CD3, CD4, and CD8, and then fixed with 2% buffered formaldehyde. Intracellular cytokines staining was performed with antibodies against IFNγ, IL-17A, IL-5 and IL-13 in permeabilization buffer (eBioscience).
To ensure consistency in data analysis, gate positions were held constant for all samples. To calculate the total number of cells in each population of interest in each sample, the corresponding percentage was multiplied by the total number of CD45+ cells in that sample. The latter value was calculated for each sample as the product of the percentage of CD45+ cells and the original hemocytometer count of total cells identified within that sample.
Real-time quantitative RT-PCR analysis
Lung Mϕ were obtained by plating lung leukocytes (isolated as described above) in 6-well plates (5×106/ml of 5% complete media, 2 ml/well), and incubating them at 5% CO2, 37°C for 90 minutes. At the end of incubation, RNA was extracted from adherent cells using TRIzol (Invitrogen), and DNase treated using the DNA-free Kit (Ambion by Life Technologies). CD11b− and CD11b+ lung Mϕ were obtained by flow sorting of lung leukocytes stained using fluorochrome-conjugated antibodies as detailed above. One-step qRT-PCR was performed using the QuantiTect Sybr Green Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol with GAPDH serving as an endogenous reference. Reactions were run in triplicates using a StepOne Plus real-time PCR system (Applied Biosystems). Primers were synthesized by Sigma based on the following 5′-3′ sequences: Arg-1 sense, CAGAAGAATGGAAGAGTCAG; Arg-1 antisense, CAGATATGCAGGGAGTCACC; Retnla (Relmα) sense, TTCTTGCCAATCCAGCTAAC; Retnla (Relmα) antisense, GGGTTCTCCACCTCTTCATT; Mrc1 (CD206) sense, CTCTGTTCAGCTATTGGACGC; Mrc1 (CD206) antisense, CGGAATTTCTGGGATTCAGCTTC; iNOS sense, GTTCTCAGCCCAACAATACAAGA; iNOS antisense, GTGGACGGGTCGATGTCAC; GAPDH sense, TATGTCGTGGAGTCTACTGGT, GAPDH antisense, GAGTTGTCATATTTCTCGTGG. Data were analyzed using the 2−ΔΔCT method.
Statistical analysis
All data were expressed as mean ± SEM. Continuous ratio scale data were evaluated by unpaired Student t test (for comparison between two samples). Statistical calculations were performed on a Dell 270 computer using GraphPad Prism version 6.00 for Windows (GraphPad Software, San Diego California USA). Statistical difference was accepted at p < 0.05.
Results
GM-CSF promotes Th2 and Th17 responses during cryptococcal lung infection
We previously compared fungal lung burden in GM-CSF deficient (GM−/−) and wild type C57BL/6 (GM+/+) mice infected by the intratracheal route with C. neoformans strain 52D and showed that CFUs in GM−/− mice increased 2, 3, and 6 fold relative to GM+/+ mice at 14, 21, and 35 days post infection (dpi), respectively (17). In the current study, similar increases in pulmonary CFU were confirmed in GM−/− mice at 14 and 28 dpi (Supplementary Figure 1). As GM-CSF improves control of cryptococcal lung infection, we sought to identify cellular and molecular mechanisms through which GM-CSF mediates this beneficial effect.
To determine whether GM-CSF impacted lung leukocyte accumulation, the number of CD45+ lung leukocytes was assessed in GM+/+ and GM−/− mice using flow cytometric analysis (FCA) at day 0 (uninfected) and 7, 14, 21, and 28 dpi. In GM+/+ mice, the number of CD45+ lung leukocytes increased by 7 dpi (7 fold relative to uninfected GM+/+ mice), peaked at 14 dpi (14 fold), and remained elevated at 21 and 28 dpi (Figure 1A). In contrast, significantly fewer leukocytes accumulated in the lungs of GM−/− mice at 7, 14, and 21 dpi, whereas CD45+ numbers were equivalent to GM+/+ mice at 28 dpi.
Figure 1.

Accumulation of CD45+ leukocytes and Th2 cytokine response are impaired in GM−/− mice with cryptococcal lung infection. (A) Flow cytometric analysis (FCA) was performed on lung leukocytes isolated at day 0 (uninfected) and 7, 14, 21, and 28 dpi from GM+/+ and GM−/− mice. Initial gates (not shown) eliminated debris and doublets, and identified CD45+ leukocytes. Total numbers of CD45+ leukocytes were calculated as the product of the percentage of CD45+ cells and the original hemocytometer count of total cells identified within that sample. Data represent mean ± SEM of 6 mice per experimental cohort assayed individually in two separate experiments (GM+/+ mice, solid black lines; GM−/− mice, dashed gray lines). (B) Total lung leukocytes isolated from GM+/+ mice and GM−/− mice at 14 dpi were cultured for 24 hours at 5 × 106 cells/ml in the absence of additional stimulation. Supernatants were harvested and cytokine levels were detected by Luminex assay. Data represent mean cytokine concentration ± SEM (pg/ml) of 5 mice per experimental cohort assayed individually (GM+/+ mice, black bars; GM−/− mice, gray bars). * p < 0.05 by unpaired Student t test for comparisons between GM+/+ and GM−/− mice.
To broadly determine whether GM-CSF impacted T cell polarization profiles, we next assessed lung leukocytes obtained from GM+/+ and GM−/− mice at 14 dpi for their production of cytokines associated with Th1 (IFNγ and TNFα), Th2 (IL-4, IL-5, IL-13, IL-10), and Th17 (IL-17) responses. In GM+/+ mice, we found robust production of IL-4, IL-5, IL-10, and IL-13 as well as modest expression of IFNγ, IL-17, and TNFα (Figure 1B). In contrast, secretion of IL-4, IL-5, IL-10, and IL-13 were significantly decreased in GM−/− relative to GM+/+ mice; non-significant trends towards reduced IL-17 and TNFα were also noted. Interestingly, leukocyte production of IFNγ was increased in GM−/− versus GM+/+ mice.
To establish whether the observed differences in total lung leukocyte cytokine production reflected specific differences in T cell polarization profiles, we enumerated CD4+ T cells and assessed their cytokine production using intracellular FCA of leukocytes obtained from the lungs of GM+/+ and GM−/− mice at 14 and 28 dpi. Total numbers of CD4+ T cells were increased in GM−/− mice at both time points (Figure 2A). As expected based on our lung leukocyte cytokine data (Figure 1B), the percent and total numbers of CD4+ T cells expressing IL-5 and IL-13 (at 14 and 28 dpi) was high in GM+/+ mice and significantly decreased in GM−/− mice (Figure 2B and C; upper panels and graphs). In contrast, the percentage (at 14 dpi) and total numbers (at 14 and 28 dpi) of CD4+ T cells expressing IFNγ was increased in GM−/− mice whereas the percentage (at 14 and 28 dpi) and total numbers (at 28 dpi) of CD4+ T cells expressing IL-17 was significantly decreased (in GM−/− mice; Figure 2B and C; lower panels and graphs). No difference in the total numbers of CD8+ T cells, or the subset of CD8+ T cells expressing IFNγ was observed between GM+/+ and GM−/− mice (data not shown). Taken together, these findings show that GM-CSF enhances Th2 and Th17 responses and diminishes IFNγ responses in mice that develop persistent cryptococcal lung infection.
Figure 2.

Th2 and Th17 cells are diminished in GM−/− mice with cryptococcal lung infection. (A–C) Lung leukocytes obtained from GM+/+ and GM−/− mice at 14 dpi were analyzed by flow cytometry to identify CD4+ T cells and to evaluate their cytokine production using intracellular staining (as detailed in the Materials and Methods section). Shown are total numbers of CD4+ T cells (A), and the percentage (B) and total number (C) of CD4+ T cells positive for IL-5, IL-13, IFNγ, and IL-17. Bar graph data (A and C) represent mean ± SEM of 5 mice per experimental cohort assayed individually (GM+/+ mice, black bars; GM−/− mice, gray bars). * p < 0.05 by unpaired Student t test for comparisons between GM+/+ and GM−/− mice at the same time point. Contour plots (B) show representative data from one mouse per experimental cohort, yet the numbers in the plots represent mean percentage out of CD4+ cells ± SEM of 5 mice per group.
GM-CSF promotes accumulation and activation of CD11b+ DC during cryptococcal lung infection
The differential effects of GM-CSF on T helper cell polarization focused our attention on CD11b+ DC, as prior studies have shown that they play a crucial role in orchestrating the adaptive immune response to C. neoformans (23, 25). We used established gating strategies (references (23, 27), the Materials and Methods section, and Figure 3A) to identify and enumerate CD11b+ lung DC by FCA at baseline and throughout the infection (Figures 3A and B). We found similar numbers of CD11b+ DC in the lungs of uninfected GM+/+ and GM−/− mice (day 0). Following infection in GM+/+ mice, the numbers of CD11b+ lung DC increased rapidly and substantially (21 fold), peaking at 7 dpi, and then slowly declining. In contrast, the numbers of CD11b+ lung DC in GM−/− mice were significantly reduced (relative to their numbers in GM+/+ mice) at 7, 14, and 21 dpi. Furthermore, their total expansion was low (4 fold) and delayed, peaking at 28 dpi.
Figure 3.

Accumulation and activation of CD11b+ DC is impaired in GM−/− mice with cryptococcal lung infection. (A) Gating strategy to identify CD11b+ DC in mice infected with C. neoformans (representative plots, uninfected GM+/+ mice and GM−/− mice). After exclusion of cell debris and doublets, consecutive gates (not shown, detailed in the Materials and Methods section) identified CD45+ leukocytes and eliminated CD3+ T cells, CD19+ B cells, and Ly-6G+ granulocytes. Next, CD11c+ cells were identified (gate R1; depicts DC and MΦ) and non-autofluorescent DC were distinguished from large autofluorescent macrophages (gate R2). Thereafter, a plot of CD11c versus CD11b identified CD11b+ DC (gate R3). (B) Total numbers of CD11b+ DC in lungs of GM+/+ and GM−/− mice at 0 (uninfected), 7, 14, 21, and 28 dpi. Data represent mean ± SEM of 6 mice per experimental cohort assayed individually in two separate experiments (GM+/+ mice, solid black; GM−/− mice, dashed gray). (C) CD11b+ DC identified at 14 dpi from GM+/+ or GM−/− mice were evaluated for expression of I-Ab (MHC Class II), CD40, CD80, CD86, and programmed cell death ligand-1 (PD-L1). Displayed are representative histogram overlays (shaded histogram, isotype control; open histogram, specific Ab staining) and bar graphs of delta mean fluorescent intensity ± SEM of 5 mice per group (GM+/+ mice, black bars; GM−/− mice, gray bars). (B–D) * p < 0.05 by unpaired Student t test for comparisons between GM+/+ and GM−/− mice at the same time point.
We also evaluated the expression of MHC Class II and costimulatory molecules CD40, CD80, and CD86 by CD11b+ DC in GM+/+ and GM−/− mice as prior studies have demonstrated a positive association between their expression and clearance or containment of cryptococcal lung infection (27, 29–32). No differences in the expression of these molecules were observed on CD11b+ DC in the lungs of uninfected GM+/+ and GM−/− mice (data not shown). At 14 dpi, expression of CD80 was reduced in GM−/− mice; a non-significant trend towards decreased expression of MHC Class II was also observed (relative to GM+/+ mice; Figure 3C). Expression of CD40 and CD86 did not differ. We also analyzed the expression of the immunomodulatory receptor, Programmed Cell Death Ligand 2 (PD-L2) by CD11b+ lung DC. PD-L2 was not expressed by CD11b+ DC in the lungs of uninfected GM+/+ or GM−/− mice (data not shown). We observed modest PD-L2 expression by CD11b+ DC in the infected GM+/+ mice and minimal expression on CD11b+ lung DC obtained from GM−/− mice (Figure 3C). In addition to assessing DC activation markers, we also examined the expression level of Ly-6C on CD11b+ DC since we have previously shown that expression of this molecule decreases as DC differentiate from Ly-6Chigh monocytes (23). Ly-6C was significantly higher on CD11b+ DC in the lungs of GM−/− relative to GM+/+ mice (data not shown) suggesting they were less differentiated. Collectively, these studies indicate that GM-CSF promotes the accumulation and activation of CD11b+ DC in mice with cryptococcal lung infection.
GM-CSF deficiency does not impair the accumulation of Ly-6Chigh lung monocytes in response to cryptococcal lung infection
Our next objective was to determine whether the reduction in CD11b+ DC observed in GM−/− mice was attributable to impaired accumulation of their Ly-6Chigh monocyte precursors. Established gating schemes and FCA ((23, 24) and Figure 4A) were used to identify and enumerate Ly-6Chigh monocytes in GM+/+ and GM−/− mice at baseline and following infection. In uninfected mice, no differences were identified in the frequency or total numbers of Ly-6Chigh monocytes (Figure 4B). The numbers of Ly-6Chigh monocytes in the lungs of GM+/+ mice increased markedly (23 fold) and rapidly, peaking at 7 dpi (Figure 4B). Interestingly, the percentage of Ly-6Chigh monocytes in the lungs of GM−/− mice significantly exceeded that observed in GM+/+ mice at 7, 14, and 21 dpi (Figure 4B, left graph). Despite the reduction in total lung leukocytes in infected GM−/− mice relative to GM+/+ mice (refer to Figure 1A), total numbers of Ly-6Chigh monocytes in GM−/− and GM+/+ mice were similar at 7, 14, and 28 dpi and were increased in GM−/− mice at 21 dpi (Figure 4B, right graph). Lastly, the percentage of Ly-6Chigh monocytes in peripheral blood of GM+/+ and GM−/− mice was determined. Results show that the percentage of Ly-6Chigh monocytes in peripheral blood of GM+/+ and GM−/− mice was similar at baseline and increased with infection. Furthermore, similar to what was observed in the lung, the percentage of Ly-6Chigh monocytes in GM−/− mice was comparable or significantly elevated relative to GM+/+ mice (Figure 4C). Collectively, these data demonstrate that GM-CSF deficiency does not impair the accumulation of Ly-6Chigh monocytes in the blood or lungs of mice with cryptococcal lung infection.
Figure 4.

Ly-6Chigh monocytes are increased in the lungs and peripheral blood of GM−/− mice with cryptococcal lung infection. (A) Gating strategy to identify Ly-6Chigh monocytes in the lungs of mice infected with C. neoformans (representative plots, GM+/+ mice and GM−/− mice at 7 dpi). Initial gates eliminated cell debris and doublets, selected CD45+ leukocytes, and excluded lymphocytes (not shown). Sequential gates identified SSClow CD11c− cells (gate R7) that expressed F4/80 (gate R8), CD11b, and Ly-6C (gates R9). (B–C) The percent (left panel of B and C) and total number (right panel of B) of Ly-6Chigh monocytes in the lungs (B) and peripheral blood (C) of GM+/+ and GM−/− mice in uninfected mice (D0) and at 7, 14, 21, and 28 dpi. Data represent mean ± SEM of 6 mice per experimental cohort assayed individually in two separate experiments (GM+/+ mice, solid black lines or black bars; GM−/− mice, dashed gray lines or gray bars); * p < 0.05 by unpaired Student t test for comparisons between GM+/+ and GM−/− mice at the same time point.
GM-CSF promotes the accumulation and alternative activation of CD11b− and CD11b+ lung macrophages in mice with cryptococcal lung infection
Increased fungal burdens in GM−/− mice suggested a macrophage defect as these are the critical effector cells responsible for cryptococcal killing. To investigate this possibility, we utilized established gating strategies and FCA (references (24, 27), Materials and Methods section and Figure 5A) to identify and enumerate two sub-populations of lung macrophages (Mϕ) based on their expression of CD11b. In uninfected GM+/+ mice, we identified a large population of autofluorescent CD11c+ CD11b− Mϕ and a smaller population of CD11c+ CD11b+ Mϕ (Figure 5A). Note that in prior studies we have identified CD11b− and CD11b+ Mϕ as alveolar and exudate macrophages, respectively (24, 27, 33). In uninfected GM−/− mice, the sub-population of CD11b+ Mϕ predominated (Figure 4A) consistent with other reports demonstrating that few, if any, CD11b− Mϕ are present in the lung of uninfected GM-CSF-deficient mice; those that are present are felt to represent an immature subset (34). In GM+/+ and GM−/− mice, both CD11b− and CD11b+ Mϕ expressed the F4/80 cell surface marker, further confirming their myeloid origin (Figure 5B).
Figure 5.

Accumulation of CD11chigh subsets of CD11b− and CD11b+ macrophages are impaired in GM−/− mice with cryptococcal lung infection. (A) Gating strategy to identify CD11b− and CD11b+ macrophages (Mϕ) in mice infected with C. neoformans (representative plots, uninfected GM+/+ mice and GM−/− mice). Initial gates (not shown and as per Figure 4 gates R1–R3) excluded cell debris and doublets, identified CD45+ cells, and excluded lymphocytes and granulocytes. Subsequent gates identified large CD11c+ autofluorescent Mϕ (gate R4) and distinguished CD11b− Mϕ (gate R5) from CD11b+ Mϕ (gate R6). (B) Representative histogram overlay plots showing F4/80 expression on the cell surface of CD11b− (left plots) and CD11b+ (right plots) Mϕ derived from uninfected GM+/+ (upper plots) and GM−/− (lower plots) mice (shaded histogram, isotype control; open histogram, specific Ab staining). (C) Total numbers of CD11b− and CD11b+ Mϕ in GM+/+ mice and GM−/− mice at the indicated time points postinfection. (D) Representative gating strategy used to further separate CD11b− and CD11b+ Mϕ into CD11chigh (gate R5a and R6a) and CD11clow (gate R5b and R6b) subsets in GM+/+ mice (upper panels) and GM−/− mice (lower panels) at D0 (left panels) and 14 dpi (right panels). (E) Total numbers of CD11chigh (upper graphs) and CD11clow (lower graphs) subsets of CD11b− Mϕ (left graphs) and CD11b+ Mϕ (right graphs) in the lungs of GM+/+ mice and GM−/− mice at the indicated time points postinfection. Data represent mean ± SEM of 6 mice per experimental cohort assayed individually in two separate experiments (GM+/+ mice, solid black lines; GM−/− mice, dashed gray lines). * p < 0.05 by unpaired Student t test for comparisons between GM+/+ and GM−/− mice at the same time point.
In response to infection in GM+/+ mice, the numbers of CD11b− Mϕ increased by a total of 5 fold and peaked at 14 dpi (Figure 5C, left panel), whereas the numbers of CD11b+ Mϕ increased by a total of 37 fold and peaked at 21 dpi (Figure 5C, right panel). In contrast, the numbers of CD11b− Mϕ in infected GM−/− mice were significantly reduced at 7, 14, and 21 dpi. The numbers of CD11b+ Mϕ in infected GM−/− mice were also reduced at 7, 14, and 21 dpi; however this reduction reached statistical significance only at 7 dpi.
Prior studies identified decreased CD11c cell surface expression by lung macrophages in uninfected GM-CSF deficient mice (35, 36). Yet, whether both CD11b− and CD11b+ Mϕ are similarly affected, and whether decreased CD11c expression is observed on macrophages in the lungs of GM−/− mice during cryptococcal lung infection is unclear. Using a modified gating scheme in which lung macrophages were subdivided on the basis of high versus low CD11c expression (Figure 5D), we observed prominent reductions in the CD11chigh subsets of both CD11b− and CD11b+ Mϕ (Figure 5E).
The immunophenotype of CD11b− and CD11b+ Mϕ in the lungs of infected GM+/+ and GM−/− mice at 14 dpi was further assessed using an antibody panel that targeted I-Ab (MHC Class II), cell surface pathogen recognition receptors, and costimulatory molecules. Expression of I-Ab, CD204 (scavenger receptor A), CD205 (DEC-205), and CD206 (mannose receptor) was decreased in CD11b− Mϕ in the lungs of infected GM−/− mice relative to GM+/+ mice (Figure 6A). No difference in CD11b− Mϕ expression of CD40, CD80, or CD86 was observed, whereas PD-L2 expression was reduced relative to GM+/+ mice (data not shown). CD11b+ Mϕ expression of I-Ab and CD206 were diminished in GM−/− mice, whereas CD204 and CD205 expression did not differ (relative to GM+/+ mice; Figure 6B). No difference in CD11b+ Mϕ expression of CD40 and CD80 was identified, whereas CD86 was increased and PD-L2 decreased in GM−/− mice (relative to GM+/+ mice; data not shown).
Figure 6.

Immunophenotype and activation profile of CD11b− and CD11b+ Mϕ in GM+/+ and GM−/− mice with cryptococcal lung infection. (A–B) CD11b− (A) and CD11b+ Mϕ (B) were identified by FCA within lung leukocytes derived from GM+/+ and GM−/− mice at 14 dpi and evaluated for expression of I-Ab (MHC Class II), CD204 (scavenger receptor A), CD205 (Dec-205), and CD206 (mannose receptor). Upper panels display representative histogram overlays (shaded histogram, isotype control; open histogram, specific Ab staining) and lower panels show bar graphs of delta mean fluorescent intensity ± SEM of 5–6 mice per experimental cohort assayed individually in two separate experiments (black bars; GM+/+ mice; gray bars, GM−/− mice). (C) RNA was isolated from adherence-enriched total lung macrophages obtained from GM+/+ and GM−/− mice at 14 and 28 dpi. The level of mRNA expression of genes encoding for Arginase 1 (Arg1), Resistin like molecule alpha (Relmα, also termed Fizz; Retnla), CD206 (mannose receptor; Mrc1), and inducible nitric oxide synthase (iNOS; iNOS) were assessed by qRT-PCR. Expression levels were normalized to GAPDH and expressed relative to the mean expression of Mϕ obtained from GM+/+ mice at 14 dpi; data represent the mean ± SEM of 5 mice per experimental cohort. * p < 0.05 by unpaired Student t test for comparisons between GM+/+ and GM−/− mice. (D) CD11b− and CD11b+ Mϕ were isolated by fluorescent cell sorting from pooled lung leukocytes derived from cohorts of GM+/+ and GM−/− mice (n=3) at 14 dpi. Transcript levels of genes encoding for Arginase 1, Relmα, and iNOS were assessed by qRT-PCR. Results were normalized to GAPDH and displayed as mRNA expression relative to CD11b− Mϕ obtained from GM+/+ mice.
Macrophage activation was further assessed by gene expression profiling performed on adherence-enriched total lung macrophage preparations obtained from individual GM+/+ and GM−/− mice at 14 and 28 dpi. The reduced expression of Th2 cytokines in GM−/− mice (Figure 1B) led us to first evaluate gene expression of three proteins, Arginase 1, resistin-like-molecule-alpha (Relmα, also referred to as Fizz1), and CD206, associated with alternative macrophage activation (Figure 6C). Consistent with this hypothesis, qRT-PCR analysis showed that transcript levels of all three genes were decreased in macrophages obtained from GM−/− versus GM+/+ mice (Figure 6C). Expression of iNOS, which is associated with classical macrophage activation, was unchanged at 14 dpi, but significantly increased at 28 dpi (relative to GM+/+ mice; Figure 6C). Lastly, we determined whether these patterns of gene expression differed between CD11b− and CD11b+ Mϕ by performing qRT-PCR analysis on each individual sub-population after their isolation by fluorescence-activated cell sorting (using gating strategies described in Figure 5A) from pooled samples of lung leukocytes obtained from GM+/+ and GM−/− mice at 14 dpi. Expression of genes encoding for arginase and Relmα was decreased in both CD11b− and CD11b+ Mϕ isolated from GM−\− mice, whereas iNOS was increased (relative to GM+\+ mice; Figure 5D). Taken together, our studies of macrophage numbers, immunophenotype, and gene expression demonstrate that GM-CSF promotes substantial accumulation and alternative activation of CD11b− and CD11b+ macrophages in the lungs of mice with cryptococcal lung infection.
GM-CSF promotes localization of CD11c-expressing cells into the alveolar regions of lung inflammation in mice with cryptococcal lung infection
Our findings identified that GM-CSF mediated some immune effects commonly associated with cryptococcal clearance (enhanced Th17 responses and the accumulation of activated CD11b+ DC and Mϕ) as well as effects often associated with persistence (enhanced Th2 responses, decreased Th1 responses, and alternative macrophage activation) (26, 27, 30, 37). To further elucidate the effects of GM-CSF on control of cryptococcal lung infection, we assessed microanatomic features of the infection using lung sections obtained from infected GM+/+ and GM−/− mice at 14 and 28 dpi by light microscopy (Figure 7). In the lungs of GM+/+ mice obtained at 14 dpi, we observed cryptococci located in alveolar regions encompassed by patchy infiltrates comprised of numerous mononuclear cells, eosinophils, and larger cells with abundant cytoplasm (Figure 7A, B). At 28 dpi, these infiltrates had coalesced into loosely-formed foci of alveolar granulomas in which most fungal organisms were contained intracellularly within large foamy macrophages (Figure 7E, F). In contrast, examination of lung sections obtained from infected GM−/− mice at 14 dpi revealed alveolar spaces containing numerous Cryptococci, but relatively few leukocytes (Figure 7C, D). At 28 dpi, dense bronchovascular infiltrates containing small mononuclear cells were identified adjacent to airways, whereas scarce inflammatory cells and numerous “uncontained” cryptoccoci were observed in the alveolar spaces (Figure 7G, H).
Figure 7.

Alveolar inflammation is diminished in GM−/− mice with cryptococcal lung infection. (A–H) Representative lungs sections obtained from GM+/+ mice (A,B,E,F) and GM−/− mice (C,D,G,H) at 14 (A–D) and 28 days (E–F) after intratracheal injection with C. neoformans strain 52D. Sections were H&E stained and examined by light microscopy at 200X magnification (A,C,E,G) or 400X magnification (B,D,F,H). Sections obtained from GM+/+ mice at 14 days post infection (dpi) viewed under low magnification (A) demonstrated early formation of alveolar infiltrates; higher magnification images (B) showed that most cryptococci (block arrowheads) are surrounded by numerous immune cells of varying size and morphology. In contrast, lung sections obtained from GM−/− mice at 14 dpi (C,D) revealed a paucity of immune cells and numerous “uncontained” cryptococci (open arrowheads) within alveolar regions. At 28 dpi, lung sections from GM+/+ mice (E, F) revealed extensive loose granulomatous alveolar infiltrates comprised of numerous large cells with macrophage morphology; many containing intracellular cryptococci (block arrowheads). Lung sections obtained from GM−/− mice at 28 dpi viewed under low magnification (G) demonstrated the presence of numerous small mononuclear cells within dense bronchovascular infiltrates (BVI) adjacent to airways (AW), whereas high magnification images (H) continued to show lack of immune cells within alveolar regions harboring abundant “uncontained” cryptococci (open arrowheads).
Our assessment by light microscopy of the microanatomic characteristics of cryptococcal lung infection showed a reduction in immune infiltrates at the site of infection in alveolar spaces in GM−/− mice (relative to GM+/+ mice; Figure 7). Total reductions in lung DC and Mϕ numbers were also identified (by FCA, Figures 3 and 5). We next compared the anatomic distribution and relative abundance of lung DC and Mϕ in GM+/+ versus GM−/− mice using CD11c antibody staining and fluorescent microscopy (Figure 8). Numerous CD11c+ cells were identified within early alveolar infiltrates in the lungs of GM+/+ mice at 14 dpi (Figure 8A and inset). In contrast, few CD11c+ cells were identified in the lungs of infected GM−/− mice at this time point (Figure 8B). At 28 dpi, numerous large CD11c+ cells displaying Mϕ phenotype were identified within established alveolar infiltrates in the lungs of GM+/+ mice (Figure 8C and inset), whereas CD11c+ cells were still absent in the alveolar regions of GM−/− mice (Figure 8D). Rather, lungs from infected GM−/− mice displayed dense accumulations of small CD11c-negative cells within bronchovascular infiltrates. These data, in conjunction with those obtained by light microscopy (Figure 7), show that GM-CSF promotes the localization of lung DC and Mϕ to sites of alveolar infection.
Figure 8.
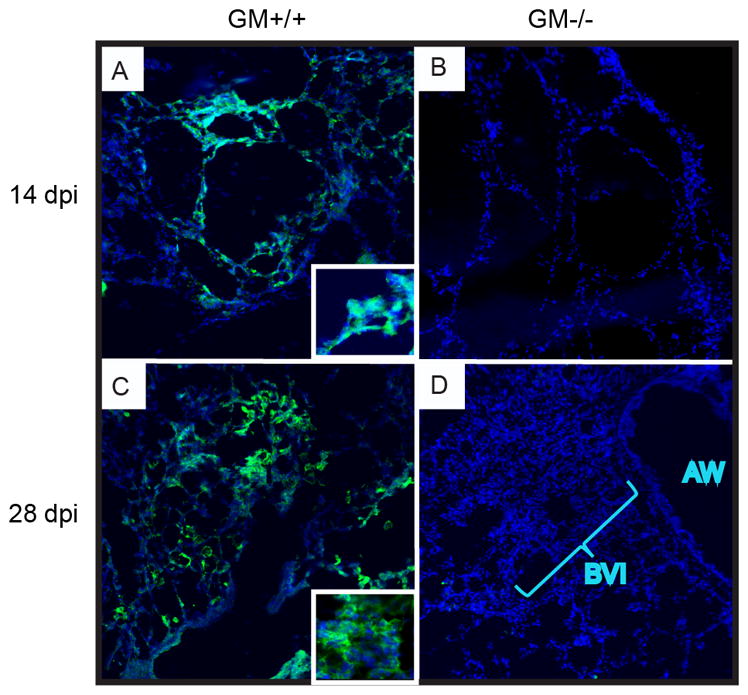
CD11c-expressing cells are diminished within alveolar regions of GM−/− mice with cryptococcal lung infection. (A–D) Representative lungs sections obtained from GM+/+ (A,C) and GM−/− (B,D) mice at 14 (A,B) and 28 (C,D) dpi. Sections were stained using anti-CD11c antibody (green) to identify DC and macrophages, whereas nuclei were stained with DAPI (blue). Sections were examined by fluorescent microscopy at 200X magnification. Numerous large CD11c+ cells were identified in the alveolar regions of GM+/+ mice at 14 (A) and 28 (C) dpi. At higher magnification (insets), these cells displayed abundant interdigitating and irregular cytoplasm suggestive of DC and macrophage morphology. In contrast, few CD11c+ cells were observed in GM−/− mice (B and D); rather dense bronchovascular infiltrates (BVI) of small CD11c− cells were seen adjacent to airways (AW) (D).
Discussion
Accumulating evidence identifies GM-CSF deficiency as a risk factor for the development of cryptococcal infections in humans (6, 7). Our prior study (17) and current findings show that control of cryptococcal lung infection is impaired in GM-CSF deficient mice. Our thorough temporal and spatial immunophenotyping of this response yielded the following observations in infected GM−/− mice (relative to GM+/+ mice): 1) Th2 and Th17 responses are decreased whereas Th1 responses are increased; 2) accumulation of CD11b+ DC and Mϕ are decreased whereas the numbers of Ly-6Chigh monocytes are unchanged or increased; 3) CD11b+ DC activation and alternative macrophage activation are decreased; and 4) localization of DC and Mϕ at sites of alveolar infection are decreased. Thus, the current study provides important evidence that the net positive effect of GM-CSF on control of cryptococcal lung infection results from a complex interplay between local immune processes affecting T cell polarization and the in situ differentiation, accumulation, activation, and alveolar localization of lung DC and Mϕ.
The most striking finding in the lungs of GM−/− mice with cryptococcal infection was the pronounced reduction of CD11b+ DC and macrophages, especially the subset of cells expressing the highest amounts of CD11c. This observation supports the previous findings of Paine et al who reported decreased CD11c expression on lung Mϕ obtained from GM−/− mice (36) and that of Guth et al who showed that a subset of fluorescently-labeled cells obtained from the bone marrow of GM−/− mice gained expression of CD11c following their intratracheal injection and subsequent recovery from the lungs of GM+/+ mice (35). In a related study, Guilliams et al showed that perinatal intrapulmonary GM-CSF administration to uninfected GM−/− mice restored high CD11c expression and alveolar Mϕ development from fetal monocytes (34). This group suggested that Mϕ present in the lungs of uninfected GM−/− mice are not “bona fide” Mϕ but rather a monocyte-Mϕ intermediate; our data supports this conclusion. Furthermore, whereas their studies were performed in naïve mice, our studies performed in infected mice suggest that the maturation defect observed in the absence of GM-CSF persists over time and despite a considerable microbial burden.
The consequence of this reduction in fully differentiated CD11c+ DC or Mϕ was most notable at the site of infection within the alveolar spaces of GM-CSF deficient mice. Loose granulomatous inflammation was prevalent in GM+/+ mice and we observed numerous mature appearing DC and Mϕ surrounding and containing individual Cryptococci. In contrast, alveolar inflammation was markedly reduced in infected GM-CSF deficient mice and numerous “free” Cryptococci were identified. Collectively, these findings demonstrate that GM-CSF promotes the accumulation of lung DC and Mϕ to the site of infection within the alveolar compartment. These reductions in alveolar infiltrates identified in mice with cryptococcal lung infection resemble those observed in GM-CSF deficient mice infected with Mycobacterium tuberculosis (40) suggesting a common mechanism leading to the increased susceptibility to pulmonary pathogens observed in GM−/− mice and patients with acquired GM-CSF deficiency.
Whereas alveolar inflammation was reduced in infected GM-CSF deficient mice, we identified dense mononuclear infiltrates surrounding bronchovascular structures that appeared anatomically distinct from the infected alveolar compartment. We believe it likely that these bronchovascular infiltrates contain an abundance of recruited monocytes that have failed to fully differentiate into CD11b+ DC or Mϕ in the absence of GM-CSF. In support of this hypothesis, data obtained by FCA revealed that the percentage and number of their Ly-6Chigh monocyte precursors were either equivalent or increased within the lungs and peripheral blood (percentage data only) of GM−/− mice (relative to infected GM+/+ mice) showing that the accumulation of these cells in response to infection is GM-CSF independent. These findings, as well as our prior studies (23, 24) and those of Guth et al (discussed above; (35)), suggest that the impaired accumulation of CD11b+ DC and Mϕ in GM−/− mice is attributable to their failure to differentiate from Ly-6Chigh monocytes within the infected pulmonary microenvironment.
We identified complex effects of GM-CSF on DC and Mϕ activation that might be viewed as both favorable and unfavorable. In the absence of GM-CSF, CD11b+ DC expressed less CD80 and a trend towards less MHC Class II relative to GM+/+ mice. Similarly, expression of MHC Class II, CD204, and CD205 were decreased in CD11b− and CD11b+ Mϕ when GM-CSF was absent. These findings show that GM-CSF promotes the expression of numerous important markers associated with antigen presentation and phagocytosis that would be expected to improve fungal clearance. However, GM-CSF deficiency was also associated with reduced Mϕ expression of CD206, Arginase 1, and Relmα and increased expression of iNOS. Thus, GM-CSF expression was linked with alternative Mϕ activation, which might be expected to impair fungal clearance. Although the myeloid cells in GM−/− mice express iNOS (likely in response to the observed increase in IFNγ), their inability to fully differentiate and mobilize to infected alveolar spaces likely limits their fungicidal capabilities. The degree to which GM-CSF affects these activation profiles directly (by signaling through the GM-CSF receptor; reviewed in (9)) or indirectly (through changes in the cytokine or surfactant milieu) is uncertain and warrants additional investigation in future studies.
Our data also identifies similarly complex effects of GM-CSF on T cell polarization responses in this model. In infected GM−/− mice, production of IL-4, IL-5, IL-10, and IL-13 by lung leukocytes was reduced as were the percentage and number of CD4+ T cells staining for intracellular IL-5 and IL-13. These results confirm and extend findings from our initial study (17) and provide clear and convincing evidence that GM-CSF promotes Th2 responses in this model. In contrast, Th1 responses appeared increased in the absence of GM-CSF and we believe this may be attributable to reduced counter-regulation by IL-10, as we have recently shown that blockade of IL-10 signaling increases IFNγ production by CD4+ T cells in mice with cryptococcal lung infection (27). Note that based on results of prior studies (26, 38), we might have expected that the decreased Th2 response and increased Th1 responses observed in GM−/− mice (relative to GM+/+ mice) would have decreased the fungal burden in the lung, which was not the case. Yet, it remains possible that the increased Th1/Th2 balance observed in GM−/− mice had some favorable effect on fungal clearance which was nonetheless insufficient to overcome the more pronounced impairments observed in myeloid cells in the absence of GM-CSF. In addition, Th17 responses were not increased in parallel with Th1 responses, but rather were decreased in GM−/− mice, demonstrating that these responses can be differentially regulated and also offering another potential explanation for the beneficial effect of GM-CSF as we have shown that IL-17 provides modest protection against progressive cryptococcal infection in this model (39).
In summary, our findings reinforce a net protective role for GM-CSF in host defenses against cryptococcal lung infection. Despite promoting maladaptive Th2 responses, our results show that GM-CSF exerts a favorable local effect within the infected pulmonary microenvironment to promote the differentiation, accumulation, activation, and alveolar localization of lung DC and Mϕ. Our collective findings show that GM-CSF critically contributes to the formation of loose granulomatous inflammation that controls fungal burdens in this model of persistent cryptococcal lung infection. These findings advance our understanding of the role of GM-CSF in antifungal host defenses and help explain the susceptibility to cryptococcal infections in patients with acquired anti GM-CSF antibodies.
Supplementary Material
Acknowledgments
We would like to acknowledge Valerie R. Stolberg for her assistance with fluorescence microscopy.
The abbreviations used are
- AF
autofluorescent
- AM
alveolar macrophage
- C.
Cryptococcus
- dpi
days post infection
- DC
dendritic cells
- ExM
exudate macrophages
- FCA
flow cytometry analysis
- GM-CSF
granulocyte-monocyte colony stimulating factor
- GM+/+ mice
wild-type C57BL/6 mice capable of producing GM-CSF
- GM−/− mice
GM-CSF deficient mice
- i.t
intratracheal
- Mϕ
macrophages
- PAP
pulmonary alveolar proteinosis
Footnotes
Supported by a Career Development Award-2 (J.J.O), Merit Review Awards (J.J.O. and M.A.O.) from the Biomedical Laboratory Research & Development Service, Department of Veterans Affairs and an NIH T32 Training Grant (T32 HL007749; B.J.M., trainee; L.M.N., trainee).
Portions of this work have been presented previously at the International Conference of the American Thoracic Society, in Denver, CO, May 2011 and San Francisco, CA, May 2012.
Disclosures
None
References
- 1.Li SS, Mody CH. Cryptococcus. Proc Am Thorac Soc. 2010;7:186–196. doi: 10.1513/pats.200907-063AL. [DOI] [PubMed] [Google Scholar]
- 2.Park BJ, Wannemuehler KA, Marston BJ, Govender N, Pappas PG, Chiller TM. Estimation of the current global burden of cryptococcal meningitis among persons living with HIV/AIDS. AIDS. 2009;23:525–530. doi: 10.1097/QAD.0b013e328322ffac. [DOI] [PubMed] [Google Scholar]
- 3.Chen S, Sorrell T, Nimmo G, Speed B, Currie B, Ellis D, Marriott D, Pfeiffer T, Parr D, Byth K. Epidemiology and host- and variety-dependent characteristics of infection due to Cryptococcus neoformans in Australia and New Zealand. Australasian Cryptococcal Study Group. Clin Infect Dis. 2000;31:499–508. doi: 10.1086/313992. [DOI] [PubMed] [Google Scholar]
- 4.Choi YH, Ngamskulrungroj P, Varma A, Sionov E, Hwang SM, Carriconde F, Meyer W, Litvintseva AP, Lee WG, Shin JH, Kim EC, Lee KW, Choi TY, Lee YS, Kwon-Chung KJ. Prevalence of the VNIc genotype of Cryptococcus neoformans in non-HIV-associated cryptococcosis in the Republic of Korea. FEMS yeast research. 2010;10:769–778. doi: 10.1111/j.1567-1364.2010.00648.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitchell DH, Sorrell TC, Allworth AM, Heath CH, McGregor AR, Papanaoum K, Richards MJ, Gottlieb T. Cryptococcal disease of the CNS in immunocompetent hosts: influence of cryptococcal variety on clinical manifestations and outcome. Clin Infect Dis. 1995;20:611–616. doi: 10.1093/clinids/20.3.611. [DOI] [PubMed] [Google Scholar]
- 6.Rosen LB, Freeman AF, Yang LM, Jutivorakool K, Olivier KN, Angkasekwinai N, Suputtamongkol Y, Bennett JE, Pyrgos V, Williamson PR, Ding L, Holland SM, Browne SK. Anti-GM-CSF autoantibodies in patients with cryptococcal meningitis. J Immunol. 2013;190:3959–3966. doi: 10.4049/jimmunol.1202526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saijo T, Chen J, Chen SC, Rosen LB, Yi J, Sorrell TC, Bennett JE, Holland SM, Browne SK, Kwon-Chung KJ. Anti-granulocyte-macrophage colony-stimulating factor autoantibodies are a risk factor for central nervous system infection by Cryptococcus gattii in otherwise immunocompetent patients. MBio. 2014;5:e00912–00914. doi: 10.1128/mBio.00912-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor. Critical reviews in immunology. 2005;25:405–428. doi: 10.1615/critrevimmunol.v25.i5.50. [DOI] [PubMed] [Google Scholar]
- 9.Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, Lopez AF. The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood. 2009;114:1289–1298. doi: 10.1182/blood-2008-12-164004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitamura T, Tanaka N, Watanabe J, Uchida, Kanegasaki S, Yamada Y, Nakata K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med. 1999;190:875–880. doi: 10.1084/jem.190.6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanaka N, Watanabe J, Kitamura T, Yamada Y, Kanegasaki S, Nakata K. Lungs of patients with idiopathic pulmonary alveolar proteinosis express a factor which neutralizes granulocyte-macrophage colony stimulating factor. FEBS letters. 1999;442:246–250. doi: 10.1016/s0014-5793(98)01668-8. [DOI] [PubMed] [Google Scholar]
- 12.Trapnell BC, Carey BC, Uchida K, Suzuki T. Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Current opinion in immunology. 2009;21:514–521. doi: 10.1016/j.coi.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee YC, Chew GT, Robinson BW. Pulmonary and meningeal cryptococcosis in pulmonary alveolar proteinosis. Australian and New Zealand journal of medicine. 1999;29:843–844. doi: 10.1111/j.1445-5994.1999.tb00803.x. [DOI] [PubMed] [Google Scholar]
- 14.Sunderland WA, Campbell RA, Edwards MJ. Pulmonary alveolar proteinosis and pulmonary cryptococcosis in an adolescent boy. The Journal of pediatrics. 1972;80:450–456. doi: 10.1016/s0022-3476(72)80503-1. [DOI] [PubMed] [Google Scholar]
- 15.Punatar AD, Kusne S, Blair JE, Seville MT, Vikram HR. Opportunistic infections in patients with pulmonary alveolar proteinosis. J Infect. 2012;65:173–179. doi: 10.1016/j.jinf.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 16.Chen GH, Curtis JL, Mody CH, Christensen PJ, Armstrong LR, Toews GB. Effect of granulocyte-macrophage colony-stimulating factor on rat alveolar macrophage anticryptococcal activity in vitro. J Immunol. 1994;152:724–734. [PubMed] [Google Scholar]
- 17.Chen GH, Olszewski MA, McDonald RA, Wells JC, Paine R, 3rd, Huffnagle GB, Toews GB. Role of granulocyte macrophage colony-stimulating factor in host defense against pulmonary Cryptococcus neoformans infection during murine allergic bronchopulmonary mycosis. Am J Pathol. 2007;170:1028–1040. doi: 10.2353/ajpath.2007.060595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brummer E, Kamberi M, Stevens DA. Regulation by granulocyte-macrophage colony-stimulating factor and/or steroids given in vivo of proinflammatory cytokine and chemokine production by bronchoalveolar macrophages in response to Aspergillus conidia. J Infect Dis. 2003;187:705–709. doi: 10.1086/368383. [DOI] [PubMed] [Google Scholar]
- 19.Deepe GS, Jr, Gibbons R, Woodward E. Neutralization of endogenous granulocyte-macrophage colony-stimulating factor subverts the protective immune response to Histoplasma capsulatum. J Immunol. 1999;163:4985–4993. [PubMed] [Google Scholar]
- 20.Mandujano JF, D’Souza NB, Nelson S, Summer WR, Beckerman RC, Shellito JE. Granulocyte-macrophage colony stimulating factor and Pneumocystis carinii pneumonia in mice. Am J Respir Crit Care Med. 1995;151:1233–1238. doi: 10.1164/ajrccm/151.4.1233. [DOI] [PubMed] [Google Scholar]
- 21.Paine R, 3rd, Preston AM, Wilcoxen S, Jin H, Siu BB, Morris SB, Reed JA, Ross G, Whitsett JA, Beck JM. Granulocyte-macrophage colony-stimulating factor in the innate immune response to Pneumocystis carinii pneumonia in mice. J Immunol. 2000;164:2602–2609. doi: 10.4049/jimmunol.164.5.2602. [DOI] [PubMed] [Google Scholar]
- 22.Schelenz S, Smith DA, Bancroft GJ. Cytokine and chemokine responses following pulmonary challenge with Aspergillus fumigatus: obligatory role of TNF-alpha and GM-CSF in neutrophil recruitment. Med Mycol. 1999;37:183–194. doi: 10.1046/j.1365-280x.1999.00219.x. [DOI] [PubMed] [Google Scholar]
- 23.Osterholzer JJ, Chen GH, Olszewski MA, Curtis JL, Huffnagle GB, Toews GB. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6Chigh monocytes. J Immunol. 2009;183:8044–8053. doi: 10.4049/jimmunol.0902823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osterholzer JJ, Chen GH, Olszewski MA, Zhang YM, Curtis JL, Huffnagle GB, Toews GB. Chemokine receptor 2-mediated accumulation of fungicidal exudate macrophages in mice that clear cryptococcal lung infection. Am J Pathol. 2011;178:198–211. doi: 10.1016/j.ajpath.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osterholzer JJ, Curtis JL, Polak T, Ames T, Chen GH, McDonald R, Huffnagle GB, Toews GB. CCR2 mediates conventional dendritic cell recruitment and the formation of bronchovascular mononuclear cell infiltrates in the lungs of mice infected with Cryptococcus neoformans. J Immunol. 2008;181:610–620. doi: 10.4049/jimmunol.181.1.610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen GH, McNamara DA, Hernandez Y, Huffnagle GB, Toews GB, Olszewski MA. Inheritance of immune polarization patterns is linked to resistance versus susceptibility to Cryptococcus neoformans in a mouse model. Infect Immun. 2008;76:2379–2391. doi: 10.1128/IAI.01143-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murdock BJ, Teitz-Tennenbaum S, Chen GH, Dils AJ, Malachowski AN, Curtis JL, Olszewski MA, Osterholzer JJ. Early or late IL-10 blockade enhances Th1 and Th17 effector responses and promotes fungal clearance in mice with cryptococcal lung infection. J Immunol. 2014;193:4107–4116. doi: 10.4049/jimmunol.1400650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, et al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science (New York, NY. 1994;264:713–716. doi: 10.1126/science.8171324. [DOI] [PubMed] [Google Scholar]
- 29.Guerrero A, Jain N, Wang X, Fries BC. Cryptococcus neoformans variants generated by phenotypic switching differ in virulence through effects on macrophage activation. Infect Immun. 2010;78:1049–1057. doi: 10.1128/IAI.01049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murdock BJ, Huffnagle GB, Olszewski MA, Osterholzer JJ. Interleukin-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and gamma interferon production. Infect Immun. 2014;82:937–948. doi: 10.1128/IAI.01477-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Osterholzer JJ, Milam JE, Chen GH, Toews GB, Huffnagle GB, Olszewski MA. Role of dendritic cells and alveolar macrophages in regulating early host defense against pulmonary infection with Cryptococcus neoformans. Infect Immun. 2009;77:3749–3758. doi: 10.1128/IAI.00454-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiu Y, Zeltzer S, Zhang Y, Wang F, Chen GH, Dayrit J, Murdock BJ, Bhan U, Toews GB, Osterholzer JJ, Standiford TJ, Olszewski MA. Early induction of CCL7 downstream of TLR9 signaling promotes the development of robust immunity to cryptococcal infection. J Immunol. 2012;188:3940–3948. doi: 10.4049/jimmunol.1103053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osterholzer JJ, Olszewski MA, Murdock BJ, Chen GH, Erb-Downward JR, Subbotina N, Browning K, Lin Y, Morey RE, Dayrit JK, Horowitz JC, Simon RH, Sisson TH. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J Immunol. 2013;190:3447–3457. doi: 10.4049/jimmunol.1200604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, Deswarte K, Malissen B, Hammad H, Lambrecht BN. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guth AM, Janssen WJ, Bosio CM, Crouch EC, Henson PM, Dow SW. Lung environment determines unique phenotype of alveolar macrophages. American journal of physiology Lung cellular and molecular physiology. 2009;296:L936–946. doi: 10.1152/ajplung.90625.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paine R, 3rd, Morris SB, Jin H, Wilcoxen SE, Phare SM, Moore BB, Coffey MJ, Toews GB. Impaired functional activity of alveolar macrophages from GM-CSF-deficient mice. American journal of physiology Lung cellular and molecular physiology. 2001;281:L1210–1218. doi: 10.1152/ajplung.2001.281.5.L1210. [DOI] [PubMed] [Google Scholar]
- 37.Arora S, Olszewski MA, Tsang TM, McDonald RA, Toews GB, Huffnagle GB. Effect of cytokine interplay on macrophage polarization during chronic pulmonary infection with Cryptococcus neoformans. Infect Immun. 2011;79:1915–1926. doi: 10.1128/IAI.01270-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hernandez Y, Arora S, Erb-Downward JR, McDonald RA, Toews GB, Huffnagle GB. Distinct roles for IL-4 and IL-10 in regulating T2 immunity during allergic bronchopulmonary mycosis. J Immunol. 2005;174:1027–1036. doi: 10.4049/jimmunol.174.2.1027. [DOI] [PubMed] [Google Scholar]
- 39.Murdock BJ, Huffnagle GB, Olszewski MA, Osterholzer JJ. IL-17A enhances host defense against cryptococcal lung infection through effects mediated by leukocyte recruitment, activation, and IFN-gamma production. Infect Immun. 2013 doi: 10.1128/IAI.01477-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gonzalez-Juarrero M, Hattle JM, Izzo A, Junqueira-Kipnis AP, Shim TS, Trapnell BC, Cooper AM, Orme IM. Disruption of granulocyte macrophage-colony stimulating factor production in the lungs severely affects the ability of mice to control Mycobacterium tuberculosis infection. J Leukoc Biol. 2005;77:914–922. doi: 10.1189/jlb.1204723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.