

Abstract
Cytosolic lipid droplets (LDs) are present in most cell types, and consist of a core comprising neutral lipids, mainly triglycerides and sterol esters, surrounded by a monolayer of phospholipids. LDs are heterogeneous in their structure, chemical composition, and tissue distribution. LDs are coated by several proteins, including perilipins and other structural proteins, lipogenic enzymes, lipases and membrane-trafficking proteins. Five proteins of the perilipin (PLIN) family (PLIN1 (perilipin), PLIN2 (adipose differentiation-related protein), PLIN3 (tail-interacting protein of 47 kDa), PLIN4 (S3-12), and PLIN5 (myocardial lipid droplet protein)), are associated with LD formation. More recently, the CIDE family of proteins, hypoxia-inducible protein 2 (HIG2), and patanin-like phospholipase domain-containing 3 (PNPLA3) have also gained attention in hepatic LD biology. Evidence suggests that LD proteins are involved in the pathophysiology of fatty liver diseases characterized by excessive lipid accumulation in hepatocytes. This review article will focus on how hepatic LDs and their associated proteins are involved in the pathogenesis of three chronic liver conditions: hepatitis C virus infection, non-alcoholic fatty liver disease, and alcoholic liver disease.
Keywords: liver, lipids, lipid droplet, perilipin, steatosis, hepatitis, obesity, diabetes, alcohol, NAFLD, HCV
Introduction
Hepatocytes are parenchymal cells of the liver responsible for mobilizing lipids for energy and storing excess lipids in the form of lipid droplets (LDs), thus making the liver the primary organ responsible for lipid homeostasis. Hepatocellular accumulation of excess LDs is called hepatic steatosis. LDs are thought to develop as either a bud, bicelle or vesicle from the lipid bilayer of the endoplasmic reticulum1-3, an organelle with which LDs share key enzymes involved in lipid synthesis and lipolysis. After a putative maturation process, the final LD structure is comprised of a core of neutral lipids (primarily triacylglycerols (TGs) and cholesterol esters) and a membrane monolayer of phospholipids and sphingomyelin. LDs ultimately distinguish themselves as bioactive organelles in their own right by associating with LD-specific proteins whose primary roles are in maintaining LD homeostasis1 (Figure 1).
Figure 1. Biogenesis of lipid droplets (LDs).
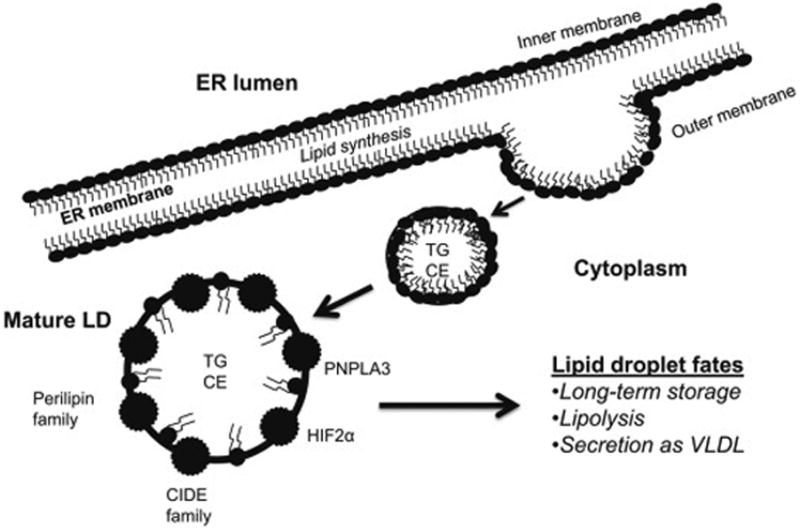
Neutral lipids are synthesized in the ER and stored in the LDs. The canonical model of LD formation shown in the figure posits that neutral lipids form a lens within the ER bilayer which then buds from the membrane and incorporates phospholipids from the cytosolic leaflet. Alternatively, the “bicelle model” of LD formation proposes that neutral lipids accumulate between the ER membrane leaflets instead of budding. The nascent LDs are excised from the ER membrane, and incorporate phospholipids from both cytosolic and luminal leaflets of the ER2. The latter model may explain how large unfolded proteins and viruses escape from the ER lumen into the cytoplasm. A “vesicular-budding model” proposes that small bilayer vesicles are tethered to the ER membrane and provide a platform for LD formation. Neutral lipids are synthesized and pumped from the ER into the vesicle bilayer, fill the intermembrane space, and the vesicular lumen eventually becomes a small inclusion inside the LDs3.
The predominant hepatocellular LD proteins are members of the perilipin family of proteins. There are five known perilipins (Perilipins 1-5 (PLIN1-5)) expressed in hepatocytes. Each perilipin is thought to have a distinct role, but few have been studied specifically in hepatocytes. More recently, the non-perilipin LD associated proteins hypoxia-inducible protein 2 (HIG2), the cell death-inducing DFFA-like effector (CIDE) family of proteins, and patanin-like phospholipase domain-containing 3 (PNPLA3/adiponutrin) have been discovered to have roles in hepatocellular lipid biology as well. HIG2 is a target of hypoxia-inducible factor 1 (HIF1), co-localizes with PLIN2 and PLIN3, and may be a marker of hepatic hypoxia4. The CIDE family of proteins includes CIDEA, CIDEB, and CIDEC/fat-specific protein 27 (Fsp 27)5. As their name implies, CIDE proteins promote cell death when upregulated, however, they associate with the LD membrane and, as a result, have roles in cellular lipid homeostasis. Finally, PNPLA3 is a protein with LD, ER and cytoplasmic distribution6. PNPLA3 has TG hydrolase and transacylase activity7 but its requirement for LD homeostasis remains controversial. A sequence variant of the PNPLA3 gene (rs738409 C>G; “G allele”) encodes an isoleucine-to-methionine substitution at position 148 and this particular single-nucleotide polymorphism (SNP) has gained significant attention since initially being associated with risk of hepatic steatosis in patients with non-alcoholic fatty liver disease (NAFLD)8.
The specific mechanisms that determine the fate of an individual LD remain elusive. We and others9 speculate that there are pools of LDs with distinct lipid and protein composition that are either targeted for lipolysis, secretion in the form of very low density lipoprotein (VLDL), or long-term lipid storage making these organelles highly regulated and integrated within the hepatocellular machinery. Hepatic steatosis, therefore, ultimately results from dysregulation of this homeostatic process. This review will summarize the evidence to date of the mechanisms of LD accumulation with specific attention to the roles of LD proteins in hepatocellular biology in three common conditions: hepatitis C, NAFLD, and alcoholic liver disease (ALD).
Hepatitis C
Chronic liver disease due to hepatitis C virus (HCV) infection affects approximately 185 million people worldwide. Infection with HCV causes chronic hepatic inflammation, fibrosis, cirrhosis, and in some cases hepatocellular carcinoma10. HCV is a single-stranded positive-sense RNA virus whose structure and life cycle have largely been unraveled. The polyprotein product of the RNA virus is comprised of structural and non-structural components. The structural components include the HCV core protein and two envelope glycoproteins (E1 and E2). The non-structural (NS) components are NS2, NS3, NS4A, NS4B, NS5A and NS5B, most of which are involved in RNA replication through intrinsic protease or polymerase activity. Both the life cycle and infectivity of HCV are dependent on host lipid metabolism. In fact, HCV circulates as a component of host VLDL, engages the host's low density lipoprotein (LDL) receptor for hepatocellular uptake, and alters cellular lipid homeostasis to promote its own replication and release from cells11. Insodoing, HCV has direct effects on the host's serum lipid profile and hepatocellular lipid content. The following sections summarize current understanding of the mechanisms by which HCV promotes hepatocellular LD accumulation.
Intracellular viral assembly and LD association
HCV enters hepatocytes primarily via the LDL receptor after which the virus hones to the endoplasmic reticulum (ER) and forms an ER-derived complex adjacent to LDs. The complex is comprised of HCV viral proteins enveloped by a membranous lipid bilayer, referred to as the membranous web. The HCV polyprotein is anchored to the ER by core protein and from there its components gain access to LDs11 (Figure 2). These interactions with cellular LDs serve to regulate LD pool size through effects on lipid storage, oxidation, and secretion.
Figure 2. Interaction of HCV and LDs.
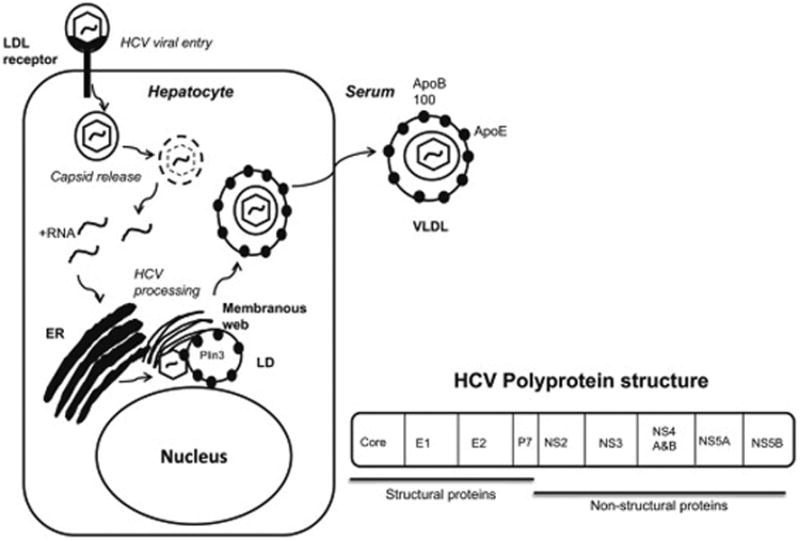
The replication of HCV RNA and viral particle production requires lipid droplets (LDs). Newly synthesized or uncoated viral genomic RNA is translated to a viral polyprotein followed by a cleaving polyprotein. Mature non-structural proteins form a complex with host factors. The positive strand of genomic RNA is enclosed with the core proteins, associated with non-structural proteins. The viral core protein is translocated to the LDs. It has been proposed that envelope proteins, e.g. NS2 and p7, determine the recruitment of viral protein from the LDs to the ER membrane. VLDL binds to viral particles before or after budding. The viral core protein increases the synthesis and storage of triglycerides and cholesterol esters in the LDs through interactions with host factors.
The effects of HCV on lipid storage are predominantly through the direct interactions of HCV proteins with the LD membrane. The interaction of core protein with the ER membrane-associated enzyme diacylglycerol acyltransferase 1 (DGAT 1) is required for the binding of HCV core protein to LDs. DGAT 1 is a rate-limiting enzyme in TG synthesis from diacylglycerol and promotes core LD-binding likely through its regulation of HCV assembly. Core protein remains associated with the ER membrane in DGAT1 inhibited cells but localizes to LD upon DGAT1 overexpression12. Non-structural HCV proteins also play a role in LD binding. NS3 is present in the LD fractions of control-treated cells but not in cells treated with a DGAT1 inhibitor12. The hydrophobic amino acid residues of NS4B's alpha-helices help HCV target LDs, and mutations of these residues prevent virus replication13. In cells with replicating virions, NS5A is found on LDs and in association with HCV replication complexes14. These data demonstrate that LDs are central to HCV's designs on the human host.
HCV and LD proteins
NS5A binds LDs through several protein-protein interactions. Namely, NS5A forms a complex with HCV core protein and DGAT-114; associates with the Rab GTPase, Rab18, which facilitates recruitment of NS5A to LDs15; and binds PLIN3 (or tail-interacting protein of 47kD (TIP47)) via its amphipathic helix on the surface of LDs16. PLIN3 is an LD protein whose association with LDs increases in HCV-replicon cells. The interaction between NS5A and PLIN3 is required for HCV replication, as there is reduced PLIN3 binding to NS5A and a near absence of colony formation in HCV replicon cells transfected with mutant NS5A16. Although other perilipin proteins are expressed in the livers of HCV patients17, the role of these proteins in promoting HCV infection is unknown.
The roles of CIDE proteins, HIG2, and PNPLA3 in HCV infection are also not well understood. CIDEB may be important for hepatocyte differentiation and is required for HCV infection of hepatocytes in vitro likely due to promotion of endosomal fusion18. HIG2 is upregulated in patients with HCV19, but the specific role of this LD protein in HCV has not been directly examined. The PNPLA3 “G allele” is associated with hepatic steatosis and risk of progression in HCV in some populations20. The specific interaction between this gene product and the HCV viral machinery, however, is unknown.
Clinical implications of HCV hepatic steatosis
About 70% of HCV patients develop hepatic steatosis; and the presence of steatosis correlates with both HCV genotype and disease progression21. Patients with HCV genotype 3 have the highest prevalence of hepatic steatosis21 likely due to the direct effects of the HCV virus on host cellular lipid machinery discussed above. Patients with genotype 1 also have a high prevalence of hepatic steatosis, but in these patients, the development of hepatic steatosis correlates with BMI and insulin resistance21, leading many to suggest that it is the development of the metabolic syndrome rather than the virus itself that promotes hepatocellular lipid accumulation. In fact, Adinolfi et al. demonstrated that steatosis is associated with visceral obesity in 76% of patients with genotype 1 or 2 infection (or mixed); while 70% of genotype 3a patients are normal weight21. These differences may simply reflect an association between host genetic background and geographic distribution of viral genotypes; or may result from yet undefined viral factors that impact lipid biology.
Non-alcoholic fatty liver disease (NAFLD)
NAFLD is the hepatic manifestation of the “metabolic syndrome”. NAFLD is diagnosed by the presence of excess hepatocellular LD accumulation in the absence of other causes of chronic liver disease. NAFLD affects up to one-third of the population22, making this a significant public health issue worldwide. Despite its prevalence, there is no consensus on the primary drivers of disease progression. NAFLD can progress from hepatic steatosis to steatohepatitis, cirrhosis and hepatocellular carcinoma. Hepatic steatosis in NAFLD is at least partially due to pertubations of lipid synthetic and oxidative pathways. More recently, the role of LDs and their associated proteins has become an active area of investigation in NAFLD.
NAFLD and mechanisms of LD accumulation
Upregulation of lipogenic pathways, impairment of mitochondrial fatty acid oxidation, and inhibition of TG secretion all contribute to the development of hepatic steatosis in NAFLD. A comprehensive discussion of these pathways is beyond the scope of the current review but several key factors are thought to play a role. Increased de novo lipogenesis significantly contributes to the development of hepatic steatosis in NAFLD patients. The transcription factors sterol regulatory binding protein 1 (SREBP1) and carbohydrate responsive element binding protein (ChREBP) are key regulators of lipogenic genes increased in both patients23 and rodent models of NAFLD; and deficiency of these transcription factors ameliorates hepatic steatosis in experimental models24, 25. Defects of fatty acid mitochondrial oxidation are also evident in NAFLD patients and likely result from both direct and indirect effects on mitochondrial structure and function26. Finally, the contribution of VLDL secretion to hepatic steatosis in NAFLD is unclear, as it has been shown to be both reduced27 and increased28 in NAFLD patients.
NAFLD and LD proteins
Impairment of the pathways listed above have a net effect of hepatocellular LD accumulation, and an associated increase of both perilipin and non-perilipin proteins. Of the perilipins, PLIN2 is most upregulated in the livers of rodents29 and humans with NAFLD17. PLIN2 promotes TG accumulation30, inhibits fatty acid oxidation31, and impairs glucose tolerance29, 32-34. PLIN2 knock-out mice are protected from hepatic steatosis when fed a high fat diet34, 35, a diet that induces NAFLD. When bred with obesity-prone leptin deficient mice, these mice have reduced steatosis despite some compensation of PLIN3 for PLIN232. Our group specifically examined the role of hepatic PLIN2 with anti-sense oligonucleotide (ASO) knockdown of hepatic PLIN2 in wildtype mice fed a high-fat diet33 and leptin-deficient mice30. PLIN2-ASO reduced hepatic TG and reduced genes involved in fatty acid metabolism30, 33. Notably, reduction of steatosis was also associated with an upregulation of fibrosis genes, suggesting that hepatic PLIN2 may protect the liver from lipotoxicity33 despite its impairment of glucose homeostasis30.
We next examined the role of hepatic PLIN3 in a NAFLD mouse model. PLIN3 is upregulated in the livers of mice fed a high-fat diet. We demonstrated that PLIN3 ASO reduces hepatic TG and improves insulin sensitivity36 To our knowledge, this is the only study of PLIN3 hepatic reduction in experimental NAFLD.
PLIN5 is increased in the livers of fatty liver dystrophic (fld) mice37 In a study by Wang, et al, plin5 knock-out mice were protected from hepatic steatosis due to increased lipolysis and fat oxidation and like PLIN2, PLIN5 is suspected to play a protective role against lipotoxicity. To date, there have been no studies examining the liver-specific role of PLIN5 in vivo, but in hepatoma cells, PLIN5 binds adipose-triglyceride lipase (ATGL) and the ATGL activator, α-β-hydrolase domain-containing 5 (ABHD5)38 thus providing a mechanistic link between PLIN5 and lipolysis.
Among the non-perilipin proteins, CIDEA and CIDEC appear to have a role in fatty liver. CIDEA is increased in fld mice, and adenoviral overexpression of SREBP1 in wildtype hepatocytes results in increased CIDEA expression37. CIDEA is also upregulated in CIDEC-deficient mice and adenoviral delivery of short hairpin RNA CIDEA reduces hepatic steatosis in these CIDEC-deficient mice39. Like CIDEA, hepatic CIDEC is increased in fld mice37 and in leptin-deficient ob/ob mice40. Overexpression of CIDEC promotes steatosis while CIDEC knock-down reduces steatosis in ob/ob mice lacking hepatic PPARγ40. The contribution of CIDE proteins to the pathogenesis of NAFLD remains an active area of study.
The role of HIG2 in NAFLD is an emerging area of investigation. Inactivation of hepatic von Hippel-Lindau protein causes hepatic steatosis, inflammation and upregulation of Hif genes, a phenotype ameliorated by HIG2 disruption41, 42. HIG2 may also regulate hepatic PLIN2 expression42 and prevent LD lipolysis43, thus providing a link between cellular oxygenation status and LD formation.
The role of the Pnpla3 “G allele” has been investigated in several experimental models of NAFLD, but the in vivo function of its protein product remains elusive. Pnpla3 knock-out mouse models show no specific hepatic phenotype44, 45, while a recent Pnpla3 “G allele” knock-in demonstrates that this allele confers increased risk of hepatic steatosis and accumulation of larger hepatocellular LDs possibly through sequestration of comparative gene identification 58 (CGI), a co-factor required for activation of ATGL46. However, this potential mechanism has not been supported because CGI is not increased in the LDs of Pnpla3 transgenic mice.46
LD proteins in human NAFLD
In humans with NAFLD, several hepatic LD proteins are upregulated. PLIN1 is upregulated in steatohepatitis due to NAFLD (non-alcoholic steatohepatitis, NASH)17, 47; but PLIN1 protein is not typically expressed in normal hepatocytes17. Moreover, the distribution of hepatic PLIN1 in NAFLD patients differs from that of PLIN2 and PLIN3 in that it surround mostly large LDs17, 47. PLIN2 is upregulated in human NAFLD17, 47-49 and appears to be a more reliable marker of hepatic LDs. PLIN2 is present in both hepatocytes and hepatic stellate cells (a fibrogenic cell type) and surrounds LDs of varying sizes47, 49. PLIN2 LD protein expression also correlates with oxidative damage reflected in ballooned hepatocytes47. A polymorphism of the PLIN2 gene was recently reported50 providing further insight into the role of this protein. A minor allele of this missense polymorphism Ser251Pro disrupts a PLIN2 alpha-helix and causes reduced plasma TG and VLDL levels50, thus giving indirect evidence of the effects of this polymorphism on hepatic lipid homeostasis.
PLIN3 upregulation has been observed in human steatotic livers17, 48. To date, there have been no human studies examining hepatic PLIN4 or PLIN5; and the role of the non-perilipin LD proteins HIG2 and CIDE family remains unclear in human NAFLD. Genome wide association studies have associated the PNPLA3 “G allele” variant with both serum ALT and hepatic fat content8 in NAFLD patients. Although the biologic role of the PNPLA3 “G allele” is debated, the independent association of this variant with NAFLD risk and progression has been established in numerous studies51. To date, however, incorporation of PNPLA3 genetic testing is not a part of routine care of NAFLD patients.
Alcoholic Liver Disease (ALD)
ALD is the most common cause of liver failure worldwide and accounts for 44% of all liver related deaths in the United States. Similar to both NAFLD and HCV, hepatic steatosis is a key histologic feature and develops in over 90% of heavy drinkers. Only 15% of heavy drinkers with hepatic steatosis develop progressive liver disease52 suggesting that there are additional factors that mediate liver disease in heavy drinkers. Due to the limitations of existing ALD rodent and cellular models, the pleiotropic effects of alcohol on hepatocellular LD formation are incompletely understood. Nevertheless, steatogenic effects of chronic alcohol have been shown to result from upregulation of lipid synthetic and storage proteins, downregulation of fatty acid oxidation enzymes and impairment of LD secretory mechanisms.
The role of de novo lipogenesis in LD accumulation in ALD has been explored in several studies. Srebp1c (an isoform of Srebp) is activated in experimental alcohol steatosis; and its downstream fatty acid synthetic gene targets are upregulated in hepatoma cells and in mice chronically fed a 27.5% calorie content alcohol diet53. Although up-regulation of Srebp1c appears to be involved in ethanol-induced lipid accumulation, the physiologic effects of this up-regulation may be modest at best as the contribution of de novo lipogenesis to hepatic lipid accumulation in response to ethanol is estimated to be less than 5%54. These data suggest that changes in lipogenic gene and protein expression in response to ethanol may not necessarily translate into measureable physiologic endpoints and may be dependent on the duration and amount of alcohol consumption and the availability of other dietary macronutrients.
There is substantial experimental evidence of the adverse effects of alcohol on fatty acid oxidation and resultant LD accumulation, and there are likely several mechanisms involved. For example, alcohol has direct and indirect toxic effects on hepatic mitochondria and its components that ultimately impair the mitochondria's ability to oxidize fatty acids; and alcohol inhibits AMPK and PPAR α, key signaling pathways involved in fatty acid oxidation55.
Alcohol impairs VLDL secretion. VLDL levels may be reduced by host immune response to acetaldehyde VLDL adducts, as alcoholic patients have higher serum IgG titers than non-alcoholics against VLDL; and ApoB-containing lipoproteins from these alcoholic patients are more prone to acetaldehyde adduct formation than non-alcoholic patients56. To our knowledge, however, this study has not been replicated in patients with and without ALD.
ALD and LD proteins
PLIN2 is the most prominent LD protein seen in alcoholic fatty liver 57, 58, and it is overexpressed in humans with alcoholic steatohepatitis17. In experimental alcoholic steatosis, PLIN2 associated LDs temporally increase and have a zone two predominance59. We observed this temporal correlation between hepatic PLIN2 upregulation and the development of steatosis; and additionally observed a correlation between PLIN2 upregulation and the onset of glucose intolerance in alcohol-fed mice60. We were also the first to demonstrate that PLIN2 is required for the development of steatosis in mice chronically fed alcohol57. However, the exact mechanism by which alcohol regulates PLIN2 requires further investigation. To our knowledge, the role of other perilipin LD proteins has not been directly examined in either human or experimental ALD. Moreover, although the PNPLA3 I148M polymorphism is associated with ALD risk and progression61, 62; the cellular role of this protein in experimental ALD has not been established.
Conclusion
Hepatocellular LDs and their associated proteins are key mediators of cellular lipid homeostasis and their biology is an emerging area of investigation in chronic liver diseases. Both perilipin and non-perilipin LD proteins can be modulated to promote lipid storage and utilization. The remarkable ability of these proteins to be regulated by both host and foreign proteins suggests that these proteins can be targeted for therapeutic purposes. However, more research is needed to examine the optimal balance between hepatic steatosis prevention and permissive steatosis, as the latter may be required to prevent cellular exposure to toxic lipid metabolites. In addition, the mechanisms underlying differential regulation of perilipin LD proteins in the context of hepatic steatotic diseases is not well understood. Exploration of these and other areas will greatly advance our understanding of hepatic steatosis, the earliest pathologic stage of the most common liver diseases worldwide.
Highlights.
We review the major types of hepatic lipid droplet proteins, including perilipin and non-perilipin family members
We review theoretical models of hepatic lipid droplet formation
We discuss the evidence to date for a biologic role of lipid droplet proteins in three common hepatic steatotic diseases: Hepatitis C, non-alcoholic fatty liver disease, and alcoholic liver disease
Acknowledgments
R.M. Carr is supported by National Institutes of Health grant K08-AA021424, P30-DK-50306 pilot and feasibility grant from the Penn Diabetes Research Center, and Robert Wood Johnson Foundation Harold Amos Medical Faculty Development Award 71586. R.S. Ahima is supported by National Institutes of Health grant P01-DK049210.
Footnotes
Conflict of interest: The authors have no conflicts to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brasaemle DL, Wolins NE. Packaging of fat: An evolving model of lipid droplet assembly and expansion. J Biol Chem. 2012;287:2273–2279. doi: 10.1074/jbc.R111.309088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ploegh HL. A lipid-based model for the creation of an escape hatch from the endoplasmic reticulum. Nature. 2007;448:435–438. doi: 10.1038/nature06004. [DOI] [PubMed] [Google Scholar]
- 3.Walther TC, Farese RV., Jr The life of lipid droplets. Biochim Biophys Acta. 2009;1791:459–466. doi: 10.1016/j.bbalip.2008.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gimm T, Wiese M, Teschemacher B, Deggerich A, Schodel J, Knaup KX, Hackenbeck T, Hellerbrand C, Amann K, Wiesener MS, Honing S, Eckardt KU, Warnecke C. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. Faseb J. 2010;24:4443–4458. doi: 10.1096/fj.10-159806. [DOI] [PubMed] [Google Scholar]
- 5.Inohara N, Koseki T, Chen S, Wu X, Nunez G. Cide, a novel family of cell death activators with homology to the 45 kda subunit of the DNA fragmentation factor. Embo J. 1998;17:2526–2533. doi: 10.1093/emboj/17.9.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruhanen H, Perttila J, Holtta-Vuori M, Zhou Y, Yki-Jarvinen H, Ikonen E, Kakela R, Olkkonen VM. Pnpla3 mediates hepatocyte triacylglycerol remodeling. J Lipid Res. 2014;55:739–746. doi: 10.1194/jlr.M046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase a2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 8.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Genetic variation in pnpla3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mashek DG, Khan SA, Sathyanarayan A, Ploeger JM, Franklin MP. Hepatic lipid droplet biology: Getting to the root of fatty liver. Hepatology. 2015 doi: 10.1002/hep.27839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, Barnes E. Global distribution and prevalence of hepatitis c virus genotypes. Hepatology. 2015;61:77–87. doi: 10.1002/hep.27259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheel TK, Rice CM. Understanding the hepatitis c virus life cycle paves the way for highly effective therapies. Nat Med. 2013;19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Herker E, Harris C, Hernandez C, Carpentier A, Kaehlcke K, Rosenberg AR, Farese RV, Jr, Ott M. Efficient hepatitis c virus particle formation requires diacylglycerol acyltransferase-1. Nat Med. 2010;16:1295–1298. doi: 10.1038/nm.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka T, Kuroda K, Ikeda M, Wakita T, Kato N, Makishima M. Hepatitis c virus ns4b targets lipid droplets through hydrophobic residues in the amphipathic helices. J Lipid Res. 2013;54:881–892. doi: 10.1194/jlr.M026443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camus G, Herker E, Modi AA, Haas JT, Ramage HR, Farese RV, Jr, Ott M. Diacylglycerol acyltransferase-1 localizes hepatitis c virus ns5a protein to lipid droplets and enhances ns5a interaction with the viral capsid core. J Biol Chem. 2013;288:9915–9923. doi: 10.1074/jbc.M112.434910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salloum S, Wang H, Ferguson C, Parton RG, Tai AW. Rab18 binds to hepatitis c virus ns5a and promotes interaction between sites of viral replication and lipid droplets. PLoS Pathog. 2013;9:e1003513. doi: 10.1371/journal.ppat.1003513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogt DA, Camus G, Herker E, Webster BR, Tsou CL, Greene WC, Yen TS, Ott M. Lipid droplet-binding protein tip47 regulates hepatitis c virus rna replication through interaction with the viral ns5a protein. PLoS Pathog. 2013;9:e1003302. doi: 10.1371/journal.ppat.1003302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Straub BK, Stoeffel P, Heid H, Zimbelmann R, Schirmacher P. Differential pattern of lipid droplet-associated proteins and de novo perilipin expression in hepatocyte steatogenesis. Hepatology. 2008;47:1936–1946. doi: 10.1002/hep.22268. [DOI] [PubMed] [Google Scholar]
- 18.Wu X, Lee EM, Hammack C, Robotham JM, Basu M, Lang J, Brinton MA, Tang H. Cell death-inducing dffa-like effector b is required for hepatitis c virus entry into hepatocytes. J Virol. 2014;88:8433–8444. doi: 10.1128/JVI.00081-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hagist S, Sultmann H, Millonig G, Hebling U, Kieslich D, Kuner R, Balaguer S, Seitz HK, Poustka A, Mueller S. In vitro-targeted gene identification in patients with hepatitis c using a genome-wide microarray technology. Hepatology. 2009;49:378–386. doi: 10.1002/hep.22677. [DOI] [PubMed] [Google Scholar]
- 20.Trepo E, Pradat P, Potthoff A, Momozawa Y, Quertinmont E, Gustot T, Lemmers A, Berthillon P, Amininejad L, Chevallier M, Schlue J, Kreipe H, Deviere J, Manns M, Trepo C, Sninsky J, Wedemeyer H, Franchimont D, Moreno C. Impact of patatin-like phospholipase-3 (rs738409 c>g) polymorphism on fibrosis progression and steatosis in chronic hepatitis c. Hepatology. 2011;54:60–69. doi: 10.1002/hep.24350. [DOI] [PubMed] [Google Scholar]
- 21.Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G. Steatosis accelerates the progression of liver damage of chronic hepatitis c patients and correlates with specific hcv genotype and visceral obesity. Hepatology. 2001;33:1358–1364. doi: 10.1053/jhep.2001.24432. [DOI] [PubMed] [Google Scholar]
- 22.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the american association for the study of liver diseases, american college of gastroenterology, and the american gastroenterological association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 23.Benhamed F, Denechaud PD, Lemoine M, Robichon C, Moldes M, Bertrand-Michel J, Ratziu V, Serfaty L, Housset C, Capeau J, Girard J, Guillou H, Postic C. The lipogenic transcription factor chrebp dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122:2176–2194. doi: 10.1172/JCI41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, Girard J, Postic C. Liver-specific inhibition of chrebp improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–2170. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 25.Yahagi N, Shimano H, Hasty AH, Matsuzaka T, Ide T, Yoshikawa T, Amemiya-Kudo M, Tomita S, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Harada K, Gotoda T, Nagai R, Ishibashi S, Yamada N. Absence of sterol regulatory element-binding protein-1 (srebp-1) ameliorates fatty livers but not obesity or insulin resistance in lep(ob)/lep(ob) mice. J Biol Chem. 2002;277:19353–19357. doi: 10.1074/jbc.M201584200. [DOI] [PubMed] [Google Scholar]
- 26.Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, Parker WD., Jr Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol. 1999;31:430–434. doi: 10.1016/s0168-8278(99)80033-6. [DOI] [PubMed] [Google Scholar]
- 27.Vedala A, Wang W, Neese RA, Christiansen MP, Hellerstein MK. Delayed secretory pathway contributions to vldl-triglycerides from plasma nefa, diet, and de novo lipogenesis in humans. J Lipid Res. 2006;47:2562–2574. doi: 10.1194/jlr.M600200-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology. 2014;146:726–735. doi: 10.1053/j.gastro.2013.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varela GM, Antwi DA, Dhir R, Yin X, Singhal NS, Graham MJ, Crooke RM, Ahima RS. Inhibition of adrp prevents diet-induced insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2008;295:G621–628. doi: 10.1152/ajpgi.90204.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai Y, Varela GM, Jackson MB, Graham MJ, Crooke RM, Ahima RS. Reduction of hepatosteatosis and lipid levels by an adipose differentiation-related protein antisense oligonucleotide. Gastroenterology. 2007;132:1947–1954. doi: 10.1053/j.gastro.2007.02.046. [DOI] [PubMed] [Google Scholar]
- 31.Magnusson B, Asp L, Bostrom P, Ruiz M, Stillemark-Billton P, Linden D, Boren J, Olofsson SO. Adipocyte differentiation-related protein promotes fatty acid storage in cytosolic triglycerides and inhibits secretion of very low-density lipoproteins. Arterioscler Thromb Vasc Biol. 2006;26:1566–1571. doi: 10.1161/01.ATV.0000223345.11820.da. [DOI] [PubMed] [Google Scholar]
- 32.Chang BH, Li L, Saha P, Chan L. Absence of adipose differentiation related protein upregulates hepatic vldl secretion, relieves hepatosteatosis, and improves whole body insulin resistance in leptin-deficient mice. J Lipid Res. 2010;51:2132–2142. doi: 10.1194/jlr.M004515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imai Y, Boyle S, Varela GM, Caron E, Yin X, Dhir R, Graham MJ, Ahima RS. Effects of perilipin 2 antisense oligonucleotide treatment on hepatic lipid metabolism and gene expression. Physiol Genomics. 2012;44:1125–1131. doi: 10.1152/physiolgenomics.00045.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McManaman JL, Bales ES, Orlicky DJ, Jackman M, MacLean PS, Cain S, Crunk AE, Mansur A, Graham CE, Bowman TA, Greenberg AS. Perilipin-2-null mice are protected against diet-induced obesity, adipose inflammation, and fatty liver disease. J Lipid Res. 2013;54:1346–1359. doi: 10.1194/jlr.M035063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang BH, Li L, Paul A, Taniguchi S, Nannegari V, Heird WC, Chan L. Protection against fatty liver but normal adipogenesis in mice lacking adipose differentiation-related protein. Mol Cell Biol. 2006;26:1063–1076. doi: 10.1128/MCB.26.3.1063-1076.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carr RM, Patel RT, Rao V, Dhir R, Graham MJ, Crooke RM, Ahima RS. Reduction of tip47 improves hepatic steatosis and glucose homeostasis in mice. Am J Physiol Regul Integr Comp Physiol. 2012;302:R996–1003. doi: 10.1152/ajpregu.00177.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall AM, Brunt EM, Chen Z, Viswakarma N, Reddy JK, Wolins NE, Finck BN. Dynamic and differential regulation of proteins that coat lipid droplets in fatty liver dystrophic mice. J Lipid Res. 2010;51:554–563. doi: 10.1194/jlr.M000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Granneman JG, Moore HP, Mottillo EP, Zhu Z, Zhou L. Interactions of perilipin-5 (PLIN5) with adipose triglyceride lipase. J Biol Chem. 2011;286:5126–5135. doi: 10.1074/jbc.M110.180711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou L, Park SY, Xu L, Xia X, Ye J, Su L, Jeong KH, Hur JH, Oh H, Tamori Y, Zingaretti CM, Cinti S, Argente J, Yu M, Wu L, Ju S, Guan F, Yang H, Choi CS, Savage DB, Li P. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged fsp27-deficient mice. Nat Commun. 2015;6:5949. doi: 10.1038/ncomms6949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, Gonzalez FJ. Hepatic steatosis in leptin-deficient mice is promoted by the ppargamma target gene fsp27. Cell Metab. 2008;7:302–311. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qu A, Taylor M, Xue X, Matsubara T, Metzger D, Chambon P, Gonzalez FJ, Shah YM. Hypoxia-inducible transcription factor 2alpha promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology. 2011;54:472–483. doi: 10.1002/hep.24400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rankin EB, Rha J, Selak MA, Unger TL, Keith B, Liu Q, Haase VH. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol. 2009;29:4527–4538. doi: 10.1128/MCB.00200-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DiStefano MT, Danai LV, Roth Flach RJ, Chawla A, Pedersen DJ, Guilherme A, Czech MP. The lipid droplet protein hypoxia-inducible gene 2 promotes hepatic triglyceride deposition by inhibiting lipolysis. J Biol Chem. 2015;290:15175–15184. doi: 10.1074/jbc.M115.650184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basantani MK, Sitnick MT, Cai L, Brenner DS, Gardner NP, Li JZ, Schoiswohl G, Yang K, Kumari M, Gross RW, Zechner R, Kershaw EE. Pnpla3/adiponutrin deficiency in mice does not contribute to fatty liver disease or metabolic syndrome. J Lipid Res. 2011;52:318–329. doi: 10.1194/jlr.M011205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen W, Chang B, Li L, Chan L. Patatin-like phospholipase domain-containing 3/adiponutrin deficiency in mice is not associated with fatty liver disease. Hepatology. 2010;52:1134–1142. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, Cohen JC, Hobbs HH. Pnpla3i148m knockin mice accumulate pnpla3 on lipid droplets and develop hepatic steatosis. Hepatology. 2015;61:108–118. doi: 10.1002/hep.27242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fujii H, Ikura Y, Arimoto J, Sugioka K, Iezzoni JC, Park SH, Naruko T, Itabe H, Kawada N, Caldwell SH, Ueda M. Expression of perilipin and adipophilin in nonalcoholic fatty liver disease; relevance to oxidative injury and hepatocyte ballooning. J Atheroscler Thromb. 2009;16:893–901. doi: 10.5551/jat.2055. [DOI] [PubMed] [Google Scholar]
- 48.Pawella LM, Hashani M, Eiteneuer E, Renner M, Bartenschlager R, Schirmacher P, Straub BK. Perilipin discerns chronic from acute hepatocellular steatosis. J Hepatol. 2014;60:633–642. doi: 10.1016/j.jhep.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 49.Straub BK, Gyoengyoesi B, Koenig M, Hashani M, Pawella LM, Herpel E, Mueller W, Macher-Goeppinger S, Heid H, Schirmacher P. Adipophilin/perilipin-2 as a lipid droplet-specific marker for metabolically active cells and diseases associated with metabolic dysregulation. Histopathology. 2013;62:617–631. doi: 10.1111/his.12038. [DOI] [PubMed] [Google Scholar]
- 50.Magne J, Aminoff A, Perman Sundelin J, Mannila MN, Gustafsson P, Hultenby K, Wernerson A, Bauer G, Listenberger L, Neville MJ, Karpe F, Boren J, Ehrenborg E. The minor allele of the missense polymorphism ser251pro in perilipin 2 (PLIN2) disrupts an alpha-helix, affects lipolysis, and is associated with reduced plasma triglyceride concentration in humans. Faseb J. 2013;27:3090–3099. doi: 10.1096/fj.13-228759. [DOI] [PubMed] [Google Scholar]
- 51.Dongiovanni P, Donati B, Fares R, Lombardi R, Mancina RM, Romeo S, Valenti L. Pnpla3 i148m polymorphism and progressive liver disease. World J Gastroenterol. 2013;19:6969–6978. doi: 10.3748/wjg.v19.i41.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O'Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- 53.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (srebp) J Biol Chem. 2002;277:29342–29347. doi: 10.1074/jbc.M202411200. [DOI] [PubMed] [Google Scholar]
- 54.Siler SQ, Neese RA, Hellerstein MK. De novo lipogenesis, lipid kinetics, and whole-body lipid balances in humans after acute alcohol consumption. Am J Clin Nutr. 1999;70:928–936. doi: 10.1093/ajcn/70.5.928. [DOI] [PubMed] [Google Scholar]
- 55.Hoek JB, Cahill A, Pastorino JG. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology. 2002;122:2049–2063. doi: 10.1053/gast.2002.33613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wehr H, Rodo M, Lieber CS, Baraona E. Acetaldehyde adducts and autoantibodies against vldl and ldl in alcoholics. J Lipid Res. 1993;34:1237–1244. [PubMed] [Google Scholar]
- 57.Carr RM, Peralta G, Yin X, Ahima RS. Absence of perilipin 2 prevents hepatic steatosis, glucose intolerance and ceramide accumulation in alcohol-fed mice. PLoS One. 2014;9:e97118. doi: 10.1371/journal.pone.0097118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mak KM, Ren C, Ponomarenko A, Cao Q, Lieber CS. Adipose differentiation-related protein is a reliable lipid droplet marker in alcoholic fatty liver of rats. Alcohol Clin Exp Res. 2008;32:683–689. doi: 10.1111/j.1530-0277.2008.00624.x. [DOI] [PubMed] [Google Scholar]
- 59.Orlicky DJ, Roede JR, Bales E, Greenwood C, Greenberg A, Petersen D, McManaman JL. Chronic ethanol consumption in mice alters hepatocyte lipid droplet properties. Alcohol Clin Exp Res. 2011;35:1020–1033. doi: 10.1111/j.1530-0277.2011.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carr RM, Dhir R, Yin X, Agarwal B, Ahima RS. Temporal effects of ethanol consumption on energy homeostasis, hepatic steatosis, and insulin sensitivity in mice. Alcohol Clin Exp Res. 2013;37:1091–1099. doi: 10.1111/acer.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, Rietschel M, Schafmayer C, Braun F, Hinrichsen H, Gunther R, Arlt A, Seeger M, Muller S, Seitz HK, Soyka M, Lerch M, Lammert F, Sarrazin C, Kubitz R, Haussinger D, Hellerbrand C, Broring D, Schreiber S, Kiefer F, Spanagel R, Mann K, Datz C, Krawczak M, Wodarz N, Volzke H, Hampe J. Genetic variation in the pnpla3 gene is associated with alcoholic liver injury in caucasians. Hepatology. 2011;53:86–95. doi: 10.1002/hep.24017. [DOI] [PubMed] [Google Scholar]
- 62.Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in pnpla3 is associated with alcoholic liver disease. Nat Genet. 2010;42:21–23. doi: 10.1038/ng.488. [DOI] [PubMed] [Google Scholar]