

Abstract
The multicomponent analysis of human breast milk (BM) by metabolic profiling is a new area of study applied to determining milk composition, and is capable of associating BM composition with maternal characteristics, and subsequent infant health outcomes. A multiplatform approach combining HPLC‐MS and ultra‐performance LC‐MS, GC‐MS, CE‐MS, and 1H NMR spectroscopy was used to comprehensively characterize metabolic profiles from seventy BM samples. A total of 710 metabolites spanning multiple molecular classes were defined. The utility of the individual and combined analytical platforms was explored in relation to numbers of metabolites identified, as well as the reproducibility of the methods. The greatest number of metabolites was identified by the single phase HPLC‐MS method, while CE‐MS uniquely profiled amino acids in detail and NMR was the most reproducible, whereas GC‐MS targeted volatile compounds and short chain fatty acids. Dynamic changes in BM composition were characterized over the first 3 months of lactation. Metabolites identified as altering in abundance over lactation included fucose, di‐ and triacylglycerols, and short chain fatty acids, known to be important for infant immunological, neurological, and gastrointestinal development, as well as being an important source of energy. This extensive metabolic coverage of the dynamic BM metabolome provides a baseline for investigating the impact of maternal characteristics, as well as establishing the impact of environmental and dietary factors on the composition of BM, with a focus on the downstream health consequences this may have for infants.
Keywords: Breast milk, Metabolomics, Metabonomics, MS, NMR
Abbreviations
- AMDIS
automated mass spectrometry deconvolution and identification system
- BM
breast milk
- FID
free induction decay
- HMO
human milk oligosaccharides
- IS
internal standard
- MFE
molecular feature extraction
- MTBE
methyl tert‐butyl ether
- OPLS‐DA
orthogonal partial least square discriminant analysis
- PCA
principal component analysis
- PLS‐DA
partial least square discriminant analysis
- QC
quality control
- SCFA
short chain fatty acids
- TSP
3‐trimethylsilyl propionic acid
- TMS
tetramethylsilane
- UPLC‐MS
ultra‐performance LC‐MS
1. Introduction
Understanding breast milk (BM) composition and how it changes over lactation is of importance in our continuing efforts to find the best way to feed babies when maternal milk is unavailable, in terms of understanding how to advise mothers, and in designing human milk substitutes 1. Breast milk is a complex biofluid; in addition to containing nutrients for infant growth and development, many chemical and biologically active factors are present whose purpose is not to provide nutrition, but to promote healthy infant development. For example, choline 2 and taurine 3 are essential factors needed for neurodevelopment, contributing toward membrane biosynthesis and neurotransmission.
Analysis of BM composition is complicated by many factors, including the changing nutritional composition of BM over the course of a feed, throughout the day, in response to maternal diet, as well as over the course of lactation 4. Additionally, the presence of large macromolecules and the ultrastructure of BM, which is micellar, mean analytical techniques must be preceded by sample preparation methods to reduce the complexity of BM prior to analysis. To date, studies on milk composition have primarily focused on macronutrient content, investigating total fat, protein, and lactose, or on analysis of a single component such as human milk oligosaccharides (HMO) 5.
Here, we set out to apply metabonomic technologies, including 1H NMR spectroscopy and MS coupled with GC, HPLC, ultra‐performance LC (UPLC), and CE, to characterize the micronutrient composition of BM using a sample volume smaller than traditional analytical techniques allow 6.
Very little previous research using metabonomics to analyze BM has been carried out. Only four studies using 1H NMR spectroscopy to profile global BM composition were identified using a search in PubMed 7, 8, 9, 10. Previous studies on BM composition using MS have predominantly investigated the lipid content 11 or structures of HMO 5. Recently, one study has combined some of these high throughput techniques, HPLC‐MS and GCMS, using a single phase extraction to widen the global metabolite profile obtainable from BM 12. The reluctance to implement a multiplatform approach in BM analysis may arise from the fact that each analytical technique normally requires different preprocessing steps, thus increasing the amount of sample volume and preparation time required. We therefore aimed to establish simple sample preparation protocols for the preanalytical preparation of BM samples for multiplatform metabonomic techniques, thus reducing the total volume of milk and preparation time required for achieving a comprehensive coverage of the milk metabolome.
Once the analytical protocol was established, we used a set of 70 BM samples, collected from 57 mothers, to (i) compare the relative sensitivity, and complementarity of the various analytical platforms, (ii) describe the global metabolic profile of BM and, (iii) ascertain the capability of these methods to explore the temporal change that occurs over the first three months of lactation. LC‐MS, GC‐MS, and 1H NMR spectroscopy were used to characterize hydrophobic metabolites while polar metabolites were analyzed using 1H NMR spectroscopy and CE‐MS. To ensure reproducibility of the extraction protocols and analytical procedures, evaluation of analytical platforms was undertaken using metabolites chosen to cover the full range of the spectral profiles for each technique, taking into consideration the RSD.
2. Materials and methods
2.1. Samples
The studies which these samples were collected for were approved by the UK National Research Ethics Service (ref 08/H07114/3 and 10/H0713/5). The mother's written, informed, consent was obtained. Seventy milk samples were obtained from 57 mothers, who delivered healthy term‐born infants. Predominantly hindmilk (collected postfeed) samples were obtained between 2 and 80 days postbirth, by manual expression. Samples were collected into collection bottles and aliquoted into 1.5 mL microcentrifuge tubes. Samples were frozen as soon as possible after collection and stored at –80°C until analysis. Before analysis, samples were defrosted at room temperature and vortexed vigorously to resuspend the milk fat globules.
2.2. Sample pretreatment
2.2.1. Dual phase extraction
A modified Folch extraction procedure was employed in order to prepare the samples for analysis 13. Two milliliters of chloroform/methanol in a 2:1 ratio was added to 400 μL of whole milk. This mixture was vortexed and 600 μL of purified water was added. Samples were then centrifuged for 10 min at 16 000 × g.
This method produces two phases and metabolites split into the different phases according to their polarity. Both phases were separated and evaporated to dryness using a speed vacuum concentrator (Eppendorf, Hamburg, Germany). Once dry, the phases were frozen at –80°C until analysis.
2.2.2. Single phase extraction
A single phase extraction method previously published 12 was also used to prepare BM samples for analysis using HPLC‐MS and GC‐MS. This involved mixing 50 μL of BM with 350 μL of methanol: methyl tert‐butyl ether (MTBE) in a 1:1 ratio. This mixture was then vortexed for 1 min to precipitate proteins. Vitamin E acetate was used as internal standard (IS), at a concentration of 25 mg/L.
A flowchart describing the analysis of the different fractions of BM using the different analytical platforms in order to characterize as many BM metabolites as possible is presented in Fig. 1. Sample preparation methods were selected according to the idiosyncrasies of each of the analytical techniques used, as well as instrumental availability. To optimize the HPLC‐MS method, particularly with respect to metabolite recovery, as we observed recovery of certain metabolites to be split across the aqueous and organic phase when using the dual phase extraction, we subsequently evaluated the single phase extraction, which is capable of extracting both moderately polar and apolar compounds simultaneously. Samples were analyzed in a single randomized run to minimize batch effects for all analytical techniques.
Figure 1.
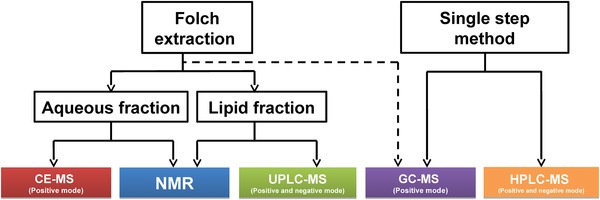
Workflow displaying the different metabonomic techniques selected to analyze the aqueous and lipid fraction of BM extractions.
2.3. 1H NMR sample preparation
The aqueous and lipid extracts were prepared using the modified Folch technique described above from 63 BM samples. The aqueous phase dried extract was reconstituted in 600 μL of D2O phosphate buffer, pH 7.2, with 3‐trimethylsilyl propionic acid (TSP) as an IS. The organic phase dried extract was reconstituted in 600 μL of deuterated chloroform with tetramethylsilane (TMS) as an IS. The reconstituted phases were transferred to 5 mm capillary tubes for 1H NMR spectroscopic analysis.
A quality control (QC) sample was prepared by pooling 50 μL of each of the samples; this QC sample was aliquoted and analyzed every ten samples.
2.4. CE‐MS sample preparation
Twenty‐one individual BM samples were prepared using the modified Folch method outlined above. The dried aqueous extract corresponding to 200 μL of BM was resuspended in 200 μL of 20 mM of formic acid solution and centrifuged to remove any particulate matter. The supernatant was transferred to a vial ready for injection into the CE‐MS instrument.
A QC sample was prepared by pooling 20 μL of each of the samples and an aliquot was analyzed every six samples.
2.5. UPLC‐MS lipid fraction sample preparation
Twenty one individual BM samples were prepared using the modified Folch method outlined above. The dried lipid extract corresponding to 200 μL of BM was resuspended in 200 μL of 2:1:1 isopropanol/water/acetonitrile (IPA:H2O:ACN), and centrifuged to remove any particulate matter. The supernatant was transferred to a vial, ready for injection into the ultra‐performance LC‐MS (UPLC‐MS) instrument.
Fifty microliters of each sample was also pooled and analyzed as a QC sample every six samples.
2.6. HPLC‐MS sample preparation
Thirty‐six BM samples were prepared using the methanol‐MTBE single phase outlined above.
QC samples were obtained after pooling 50 μL of each sample that was analyzed at the beginning and at the end of the sequence, and after every five samples to check system stability and performance of the analysis.
2.7. GC‐MS sample preparation
A single phase extraction was carried out on 200 μL of 18 BM samples, as outlined above. As previously described an aliquot of 150 μL, taken from the supernatant of the extraction, was transferred to a GC vial and evaporated to dryness in a speed vacuum concentrator 14. Methoxymation was carried out in the dark, at room temperature for 16 h after adding 10 μL of O‐methoxyamine hydrochloride in pyridine (15 mg/mL) to each GC vial, the resultant mixture was vortexed for 5 min. For derivatization, 20 μL of N,O‐Bis(trimethylsilyl)trifluoroacetamide with 1% trimethylchlorosilane was added to each sample and the solution was vortexed again for 5 min. Silylation was achieved after samples were heated in an oven for 1 h at 70°C. Finally, 100 μL of IS, heptane containing 10 ppm of C18:0 methyl ester, was added to each GC vial and vortexed for 2 min before GC analysis. Fifty microliters of each sample was also pooled and analyzed as a QC sample at the beginning and at the end of the sequence, and after every five samples to check system stability and performance of the analysis.
2.8. 1H NMR spectroscopic analysis
1H NMR spectra were acquired using a Bruker DRX 600 MHz spectrometer (Bruker Biospin, Karlsruhe, Germany), operating at 300 K, using the standard 1‐dimensional NMR pulse sequence [relaxation delay‐90°‐t1‐90°‐tm‐90°‐acquire‐FID]. The water peak was suppressed by selective irradiation during the relaxation delay of 2 s and mixing time (tm) of 100 ms; t1 was fixed to 2 μs. The 90° pulse length was adjusted to approximately 10 μs. A total of eight dummy scans and 64 scans were recorded into 32 k data points with a spectral width of 20 ppm. An exponential function corresponding to a line broadening of 0.3 Hz was applied to each free induction decay (FID) prior to Fourier transformation 15.
2.9. 1H NMR spectroscopy data treatment
The resulting 1H NMR spectra were digitalized and imported into Matlab 8.3 (MathWorks Inc. USA). The spectra were automatically corrected for phase and baseline distortions. Peaks were referenced to the TSP signal at δ 0.0 for aqueous samples, whereas the lipid extracts were referenced to the TMS signal at δ 0.0. The region containing the water resonance was removed in the aqueous fraction. Spectra were normalized to the total area and modeled using a Matlab script developed in‐house 16. Significant metabolites were obtained after investigating correlations over correlation coefficients values higher than 0.7.
2.10. CE‐MS analysis
CE‐MS TOF experiments were performed using an Agilent 7100 CE system coupled to an Agilent 6224 Accurate‐Mass TOF spectrometer system (Agilent Technologies, Wilmington, USA). The coupling was equipped with an electrospray source. The CE unit was operated by 3D‐CE ChemStation Rev B.04.03 (Agilent Technologies, Wilmington, USA), and MS data were acquired using a MassHunter WorkStation Acquisition B.02.01 (Agilent Technologies, Wilmington, USA) in centroid mode with a data acquisition rate of five spectra per second. Separations were carried out in a fused‐silica capillary (Agilent) with an internal diameter of 50 μm and a total length of 60 cm in normal polarity with a background electrolyte containing 0.1 mol/L formic acid solution. Before each analysis the capillary was conditioned with a first flush of 300 s of 2 M ammonium hydroxide, a second flush of 240 s of deionized water and a third flush of 300 s of run buffer. The capillary temperature was maintained at 25˚C. Samples were injected hydrodynamically at 50 mBar for 3 s and were carried out in a stacking manner. A voltage of 25 kV was applied for 20 min. The sheath liquid was delivered at a flow rate of 0.6 mL/min with split 1:100, comprising of methanol/water (70% v/v) and contained 1.0 mmol/l formic acid. The source that delivers auxiliary liquid was set at 6 μL/min.
The capillary voltage was set at 4 kV and the nitrogen gas flow rate, which was 200°C, was 10 L/min. The nebulizer was set to 10 psi 30 s after injection. The fragmentor and octupole voltage were set to 150 and 750 V, respectively. A solution containing 10 mL water, 90 mL methanol, 100 μL formic acid, with two reference masses; 1, 600 μL of 5 mmol/L purine, and 600 μL of 2.5 mmol/L HP 0921 that was infused directly into the ion source. The reference mass spectra were collected simultaneously with the analytical data and used for accurate mass correction and highly accurate mass measurement in the MS system. A mass acquisition range of 50–1700 m/z was used.
The samples were analyzed in a randomized run with a QC sample analyzed at the beginning of the run, every six samples, and at the end of the sequence.
2.11. CE‐MS data treatment
Data obtained by CE‐MS were cleaned of background noise and unrelated ions by the molecular feature extraction (MFE) tool in MassHunter (B.04.00, Agilent). The molecular feature extraction is capable of creating a list of all possible components represented by the full TOF mass spectral data and migration times. This was obtained using the following parameters: target data type of small molecules (chromatographic); peaks with height ≥ 200 counts; Peak spacing tolerance = 0.0025; m/z, ± 7.0 ppm; isotope model = common organic molecular; limited assigned change = 2. Alignment was performed first on all samples with QC samples, and later only for the samples under investigation. Corrections applied for the alignment were 2% migration time correction, and ±10 ppm mass correction. Data were normalized using total area normalization. The filtering and alignment of the data were performed with GeneSpring version 12.6 (Agilent) selecting features in the range of 0.5–20 min. Filtering was performed by eliminating features present in the QC samples with a coefficient of variation above 30%. SIMCA P+ 13.0 software (Umetrics, Umeå, Sweden) was then used for multivariate analysis. Variables with a |t‐statistic| ≥ 1.96 (z‐score, corresponding to the 97.5 percentile) 17 were considered significant.
2.12. UPLC‐MS lipid profiling
BM samples were run on Acquity UPLC system (Waters Ltd., Elstree, UK) coupled to a Xevo‐G2 quantitative QTOF mass spectrometer (Waters MS Technologies, Ltd., Manchester, UK). Chromatographic conditions have been previously established 18. Separation was performed using a CSH C18 (1.7 μm, 2.1 × 100 mm) column (Waters Corporation, Milford, USA) at 55ºC. The mobile phase consisted of 0.1% formic acid v/v and 10 mmol/L ammonium formate in 60:40 v/v ACN/H2O (A) and 0.1% formic acid v/v and 10 mmol/L ammonium formate in 90:10 v/v IPA/ACN (B). Flow rate was 0.4 mL/min. The injection volume was 5 μL.
MS was performed on a Xevo‐G2 QTOF mass spectrometer (Waters Ltd., Manchester, UK) in both positive and negative ion electrospray (ESI+ and ESI–) mode. The MS parameters were: capillary voltage, 1.5 kV; sample cone voltage, 30 V; source temperature 120ºC; desolvation temperature and gas flow, 600ºC and 1000 L/h, respectively; cone gas flow, 50 L/h and scan range 50–2000 m/z. For mass accuracy, a LockSpray interface was used with a 2 ng/μL leucine enkephalin (555.2645 amu) solution at 10 μL/min as the lock mass. Data were collected in centroid mode with lock mass scans collected every 30 s and averaged over three scans to perform mass correction.
A QC sample, prepared by combining equal aliquots of all the samples, was used to ensure system suitability and stability. The QC sample was injected at regular intervals throughout the analytical run. This QC samples was also used to condition the column (ten injections) and to obtain structural information, using an automated MS/MS method (data‐dependent acquisition).
2.13. UPLC‐MS data treatment
Data acquisition and analysis was performed using MassLynx software version 4.1. Raw data files were converted to NetCDF format. Data were then processed (noise filtered, aligned, and normalized) using the freely available software package XCMS in R. Features detected in individual samples were grouped across the sample set and the minimum fraction filter applied to exclude groups that contained features not found in > 50% of the samples, in each of the sample groups (QC sample and study samples). Further filtering was performed by eliminating features present in the QC sample with a coefficient of variation above 30%. Data were normalized using the median fold change 19. SIMCA P+ 13.0 (Umetrics, Umeå, Sweden) was then used for multivariate analysis. Variables with a |t‐statistic| ≥ 1.96 (z‐score, corresponding to the 97.5 percentile) 17 were considered significant.
2.14. HPLC‐MS single phase analysis
A HPLC system (1200 series, Agilent Technologies, Waldbronn, Germany) coupled to an Agilent QTOF (6520) with EI source was used to perform the global profiling analyses on a single phase, as previously published 20, further information on the analysis acquisition parameters for HPLC‐MS can be found in the SI.
The total run time was 60 min. QC samples were injected at the beginning and at the end of the analysis and after every five study samples.
2.15. HPLC‐MS single phase data treatment
MassHunter Qualitative Analysis Software (Agilent®, version B.05.00) was used to clean background noise from the spectral data and the MFE algorithm was used to export molecular entities. The output generated an MFE file containing a list of all molecular entities in the full TOF mass spectral data for each sample. Primary data treatment (alignment and filtering) was performed in mass profiler professional B.12.1 Agilent® software. Signals in the HPLC chromatogram corresponding to masses with a RSD less than 20% based on 10 independent replicates of a BM pool were retained for metabolic characterization. In addition, features with abundance less than 105 (positive mode) and 104 (negative mode), were discarded. SIMCA P+ 13.0 (Umetrics, Umeå, Sweden) was then used for multivariate analysis. Variables with a |t‐statistic| ≥ 1.96 (z‐score, corresponding to the 97.5 percentile) 17 were considered significant.
2.16. GC‐MS analysis
A GC instrument (Agilent 7890A) coupled to mass spectrometer (Agilent 5975C) was used to perform the global profiling analyses on a single phase 20, further information on the analysis acquisition parameters for GC‐MS can be found in the SI.
QC samples were injected at the beginning and at the end of the analysis and after every five study samples.
2.17. GC‐MS data treatment
The Agilent MSD ChemStation Software was used to acquire data. In order to perform retention index (RI) correction a match score was assigned between the experimental FAME mixture® analyzed and theoretical RI values based on RI values contained in the Fiehn RTL library 21. Retention index comparison of spectra with the Fiehn RTL library was performed in automated mass spectrometry deconvolution and identification system (AMDIS) software v.2.69 for peak detection and deconvolution. The targets obtained and confirmed from retention index search were used to build a bespoke library after correcting retention times. NIST library was also used for complementary identification.
An IS peak was used to examine the quality of the chromatograms acquired by total ion chromatogram. Metabolites present in the GC‐MS profiles were identified before multivariate analysis. Only compounds present in > 70% of all samples (QC samples and study samples) were maintained for further statistical analysis. SIMCA P+ 13.0 software (Umetrics, Umeå, Sweden) was then used for multivariate analysis. Variables with a |t‐statistic| ≥ 1.96 (z‐score, corresponding to the 97.5 percentile) 17 were considered significant.
2.18. Metabolite identification
Metabolites were identified from 1H NMR spectra by employing the online database HMDB, using chemical shift information from the 1 and 2 dimensional experiments collected. Peaks that proved difficult to assign were assigned by statistical total correlation spectroscopy, using an in‐house Matlab script 16. Confirmation of metabolite identity was obtained using spiked in standards, where necessary.
Metabolites detected using CE‐MS were tentatively identified by comparing high mass accuracy data (with less than 5 ppm tolerance) against standards analyzed for comparison, as well as against information contained in HMDB. Isotopic distribution was also used for particular chemical formula confirmation, and standards were injected, when available, in case different formulas corresponded to the same mass.
UPLC‐MS metabolite annotation was achieved by searching m/z values against online databases including METLIN, Lipidmaps, and HMDB with the mass error set to 10 ppm. In order to identify the lipid species, MS/MS data were inspected to obtain fragmentation patterns and the presence of head groups, increasing confidence in the identity of the metabolite assigned.
Tentative identification of metabolites detected by HPLC‐MS was undertaken by comparing accurate masses against the online university database, CEU‐mass mediator (http://ceumass.eps.uspceu.es/mediator/), which uses KEGG, METLIN, and Lipidmaps databases, with the mass error set to 10 ppm. Results were curated using MassHunter to compare potential hits against the experimental isotopic pattern distribution and retention time prediction.
For GC‐MS data, compounds were identified by comparing the experimental mass fragmentation patterns with those available in the NIST 08 and Fiehn RTL mass spectral libraries. Also the AMDIS software v.2.69 was used for automatic peak identification and deconvolution 22.
2.19. Analytical performance
In order to assess the reliability of the methods for comparing differences among metabolites the RSD was calculated for each of the analytical techniques using a set of signals from between 5 and 10 metabolites, selected so as to representatively and evenly cover the full metabolite profile as far as possible. The metabolites chosen, together with their % RSD values are provided in the Supporting Information Table 1.
2.20. Multivariate analysis
Differences among samples were evaluated using SIMCA P+ 13.0 multivariate modelling software (Umetrics, Sweden), which was used to plot data and perform multivariate analysis using pattern recognition techniques including principal components analysis (PCA), partial least squares projections to latent structures‐discriminant analysis (PLS‐DA), and orthogonal partial least squares projections to latent structures‐discriminant analysis (OPLS‐DA). These modeling techniques were carried out on the data obtained from the different analytical platforms to establish systematic differences in BM composition over the first three months of lactation.
For the purpose of analysis, samples were split into three time groups; samples collected on days 1–5 postbirth, days 6–10 postbirth, and more than 10 days postbirth, until day 80.
2.21. Reagents
All standards, chemicals, reagents, and solvents used are listed in the Supporting Information.
3. Results
3.1. Profile characterization
Each analytical platform generated a set of metabolites present in human breast milk, some of which were unique to that platform and some of which were detected by two or more platforms. The total coverage of the BM metabolome from the combined techniques consisted of 710 identified metabolites using a pooled BM sample. The structural identities of some metabolites were validated experimentally with the remainder identified based on matching of retention times and m/z ratios from databases, 15% of the identifications were validated experimentally. Seventy BM samples, collected between birth and three months postnatal age, were analyzed using multiplatform analytical techniques; HPLC‐MS, UPLC‐MS, GC‐MS, CE‐MS, and1H NMR. Metabolites present in the profiles of each of these techniques were, as far as possible, assigned (Supporting Information Fig. 1, and Supporting Information Tables 2–4).
3.2. 1H NMR spectroscopy profile characterization
Analyzing the aqueous fraction of BM using 1H NMR spectroscopy allowed the identification of 52 metabolites. In the spectra of the organic extracts 14 peaks were identified, however, these peaks do not always correspond to specific molecules, but represent lipid classes or distinct proton environs within the lipid. Similarly, in the aqueous phase there is much overlap between peaks, especially from sugar groups, meaning identification of the HMO are not definitive but relate to the proton environment. The aqueous and lipid fractions are displayed in Fig. 2A and B, respectively with metabolites identified outlined in Tables 1 and 2. The main metabolite classes identified from the aqueous fraction included; monosaccharides, disaccharides, HMO, amino acids, and their derivatives, choline containing molecules, intermediates of the tricarboxylic acid cycle, ketones, and short chain fatty acids.
Figure 2.
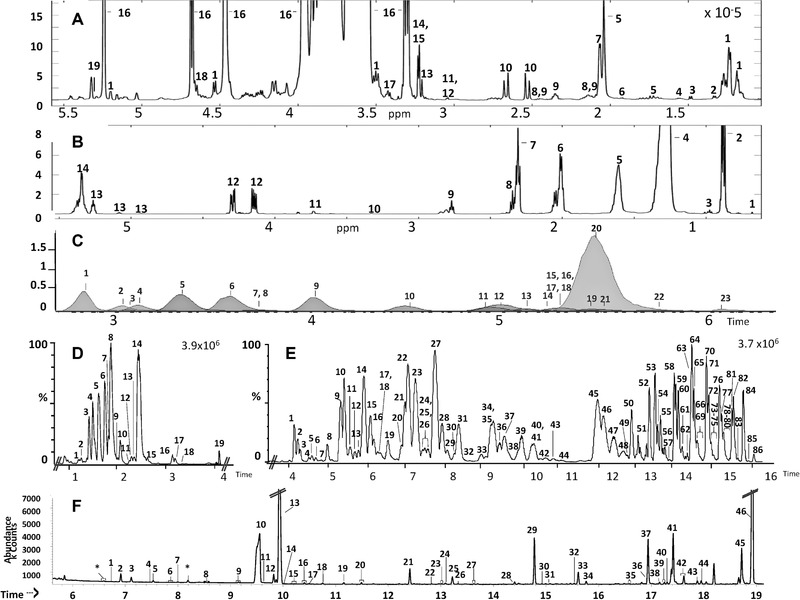
Profiles of BM analyzed via: (A) Median 1H NMR spectrum of aqueous fraction of BM, 1 = Fucose, 2 = Lactate, 3 = Alanine, 4 = Butyrate, 5 = N‐acetyl glutamine, 6 = Acetate, 7 = N‐acetyl neuraminic acid, 8 = Glutamine, 9 = Glutamate, 10 = Citrate, 11 = Creatine, 12 = Creatinine, 13 = Choline, 14 = Phosphocholine, 15 = Glycerophosphocholine, 16 = Lactose, 17 = Taurine, 18 = Glucose, 19 = Galactose. (B) Median 1H NMR spectrum of lipid fraction of BM 1 = Cholesterol, 2 = Terminal CH3, 3 = Omega 3 terminal CH3, 4 = Saturated CH2, 5 = CH 2‐CH2‐COOH, 6 = Unsaturated fatty acids CH 2‐CH = CH, 7 = CH 2‐COOH, 8 = Docosahexaenoic acid, = CH‐CH 2‐CH 2‐COOH, 9 = Polyunsaturated fatty acid, = CH‐CH2‐CH = , 10 = Phosphatidylcholine, ‐N(CH 3)3, 11 = Phospholipid, 12 = Glyceryl C1,3, 13 = Glyceryl C2, 14 = Unsaturated fatty acids, ‐CH = CH‐. (C) Aqueous fraction of BM analysed using CE‐MS in positive ionization mode; 1 = Creatinine, 2 = Lysine, 3 = Nicotinamide, 4 = Arginine, 5 = Carnitine, 6 = Acetyl‐L Carnitine, 7 = Cytosine, 8 = Cytidine, 9 = Creatine, 10 = Alanine, 11 = 5‐amino Valeric acid/valine, 12 = N‐methyl‐L‐Valine/leucine/isoleucine, 13 = Serine, 14 = Tryptophan, 15 = Threonine, 16 = Glycyl‐glycine, 17 = Methionine, 18 = Citrulline, 19 = Phenylalanine, 20 = Glutamic acid, 21 = Tyrosine, 22 = Cystine, 23 = Aspartic acid. (D) UPLC‐MS; lipid fraction of BM analysed in negative ionization mode, tentative identifications are shown in Supporting Information Table 4. (E) UPLC‐MS; lipid fraction analyzed using positive mode, tentative identifications are shown in Supporting Information Table 4. (F) GC‐MS single phase assignments from 6–19 min 1 = Pyruvic acid, 2 = Lactic acid (Standard confirmed), 3 = Glycolic acid, 4 = Valine 1, 5 = Alanine 1, 6 = 2‐Hydroxybutyric acid, 7 = 2‐Furoic acid, 8 = Isoleucine 1, 9 = Valine 2, 10 = Urea (Standard confirmed), 11 = Benzoic acid, 12 = Caprylic acid, 13 = Glycerol (Standard confirmed), 14 = Phosphoric acid, 15 = Proline 2 (Standard confirmed), 16 = Glycine, 17 = Succinic acid, 18 = Glyceric acid, 19 = Serine 2 (Standard confirmed), 20 = Threonine 2, 21 = Capric acid, 22 = Malic acid, 23 = Adipic acid, 24 = Threitol, 25 = Pyroglutamic acid, 26 = Glutamic acid 1, 27 = Creatinine, 28 = Glutamic acid 2 (Standard confirmed), 29 = Lauric acid, 30 = Lyxose 1/Lyxosylamine 1, 31 = Lyxose 2/Lyxosylamine 2/Ribose, 32 = Xylitol, 33 = Fucose 1, 34 = Fucose 2, 35 = Citric acid (Standard confirmed), 36 = Hippuric acid 2, 37 = Myristic acid, 38 = Tagatose 1/Sorbose 2/Sorbose 1/Fructose 1, 39 = Tagatose 2/Fructose 2/Fructose 1, 40 = Galactose 1/ Mannose 1/Allose 1/Gluconic acid lactone 1, 41 = Glucose 1/Talose 1 (Standard confirmed), 42 = Altrose 2/Mannose 2/Glucose 2/Allose 2/Talose 2, 43 = Mannitol/Sorbitol, 44 = 1‐Hexadecanol, 45 = Palmitoleic acid, 46 = Palmitic acid.
Table 1.
Peak Assignments for 1H NMR Spectrum from the aqueous and lipid fraction of human BM
| # | Compound name | 1D /2D | Group | Assignment | 1H (δ) | 13C (δ) | Multiplicity | Metabolite class |
|---|---|---|---|---|---|---|---|---|
| 1 | 2‐Oxoglutarate | 2D | 2.42, 2.99 | 33.4, 38.6 | (t) | Citrate cycle | ||
| 2 | 2'‐Fucosyllactose | 2D |
|
|
|
|
HMO | |
| 3 | 3'‐Fucosyllactose | 2D |
|
|
|
|
HMO | |
| 4 | 3’‐Sialyllactose | 2D | Neu5Ac(α2‐3) |
|
|
|
HMO | |
| 5 | 6’‐Sialyllactose | 2D | Neu5Ac(α2‐6) |
|
|
|
HMO | |
| 6 | Lacto‐N‐neoDiFucohexaose | 2D | Fuc(α1‐3)GlcNAc |
|
|
|
HMO | |
| 7 |
|
2D | Gal(β1‐4)GlcNAc |
|
|
|
HMO | |
| 8 | Lactodifucotetraose | 2D |
|
|
|
|
HMO | |
| 9 | Lacto‐N‐difucohesaose I |
|
|
|
|
HMO | ||
| 10 | Lacto‐N‐difucohesaose II |
|
|
|
|
HMO | ||
| 11 | Lacto‐N‐fucopentaose II | Fuc(α1‐4) |
|
|
|
HMO | ||
| 12 | Lacto‐N‐fucopentaose III |
|
|
|
|
HMO | ||
| 13 | LNFP V |
|
|
|
|
HMO | ||
|
|
|
|
|||||
| 14 | Lacto‐N‐neotetraose | CH‐1 | 4.35 | 104.6 | HMO | |||
| 15 | Lacto‐N‐tetraose |
|
|
|
HMO | |||
| 16 | Acetate | 1D | 1.92 | (s) | Organic acid | |||
| 17 | Acetone | 1D | 2.22 | (s) | Ketone | |||
| 18 | Alanine | 1D, 2D | 1.48 | 19.0 | (d) | Amino Acid | ||
| 19 | Aspartate | 1D, 2D | 2.71, 2.8 | 39.3, 39.4 | (dd), (dd) | Amino Acid | ||
| 20 | Butyrate | 1D | 0.89, 1.55 | (t), (m) | Short chain fatty acids | |||
| 21 | Caprate | 1D, 2D | 1.27 | (m) | Short chain fatty acids | |||
| 22 | Caprylate | 1D | 1.27 | (m) | Short chain fatty acids | |||
| 23 | Carnitine | 1D, no 2D | 3.25 | (s) | Amino acid derivative | |||
| 24 | Choline | 1D, 2D | 3.18 | 58.4 | (s) | Cholines | ||
| 25 | Citrate | 1D, 2D | 2.53, 2.66 | 48.7, 48.7 | (dd) | Citrate cycle | ||
| 26 | Creatine | 1D | 3.04 | (s) | Organic Acid | |||
| 27 | Creatine phosphate | 1D | (s) | Organic Acid | ||||
| 28 | Creatinine | 1D | 3.048 | (s) | Organic Acid | |||
| 29 | Ethanolamine | 1D, 2D | 3.14 | 44.18 | (t) | Other | ||
| 30 | Formate | 1D | 8.46 | (s) | Organic acid | |||
| 31 | Fucose | 1D, 2D | 1.18, 1.23, 3.42, 4.15, 4.53, 5.2 | (d), (d), (m), (m), (d), (d) | Monosaccharide | |||
| 32 | Galactose | 2D | 5.28 | 95 | (d) | Monosaccharide | ||
| 33 | Glucose | 1D, 2D | CH‐1 | 4.65 | 98.82 | (d) | Monosaccharide | |
| 34 | Glutamate | 1D | 2.13, 2.35 | (m), (m) | Amino Acid | |||
| 35 | Glutamine | 1D | 2.13, 2.44 | (m), (m) | Amino Acid | |||
| 36 | Lactate | 1D, 2D | 1.33 | 22.9 | (d) | Organic acid | ||
| 37 | Lactose | 1D, 2D | 3.3, 3.54‐3.97, 4.46, 4.68, 5.24 | (t), (multiple peaks), (d), (d), (d) | Disaccharide | |||
| 38 | Isoleucine | 1D | 0.92 | (t) | Amino Acid | |||
| 39 | Leucine | 1D | 0.96, 1.7 | (t), (m) | Amino Acid | |||
| 40 | Lysine | 2D | 3.01 | 42.1 | (t) | Amino Acid | ||
| 41 | Methanol | 1D, 2D | 3.37 | 51.4 | (s) | Other | ||
| 42 | Methionine | 2D | 2.1 | 17.0 | (m) | Amino Acid | ||
| 43 | N‐Acetylneuraminic acid | 1D | 2.06 | (s) | Monosaccharide | |||
| 44 | N‐Acetylglucosamine | 1D | 2.04, 5.2 | (s), (d) | Monosaccharide | |||
| 45 | O‐Acetylcarnitine | 1D | (s) | Amino acid derivative | ||||
| 46 | O‐Phosphocholine | 1D | 3.2 | (s) | Cholines | |||
| 47 | sn‐Glycero‐3‐phosphocholine | 1D, 2D | 3.22 | 56.7 | (s) | Cholines | ||
| 48 | Succinate | 1D, 2D | 2.4 | 3.68 | (s) | Citrate cycle | ||
| 49 | Taurine | 2D | 3.41 | 38.3 | (t) | Amino acid derivative | ||
| 50 | Tyrosine | 2D | 6.83, 7.13 | 118.3, 133.2 | (m), (m) | Amino Acid | ||
| 51 | Urea | 1D | 5.78 | Other | ||||
| 52 | Valine | 1D | 0.99, 1.02 | (dd) | Amino Acid |
Table 2.
Peak assignments for 1H NMR spectrum from the lipid fraction of BM
| Peak | Proton | Chemical | Multiplicity |
|---|---|---|---|
| number | shift(s) (δ) | ||
| 1 | Terminal ‐CH3 | 0.88 | (t) |
| 2 | Terminal ‐CH3 in ω3 fatty acids | 0.97 | (t) |
| 3 | ‐CH2‐ | 1.25 | (s) |
| 4 | ‐CH2‐CH2‐COOH | 1.61 | (s) |
| 5 | ‐CH2‐CH=CH‐ | 2.01 | (q) |
| 6 | ‐CH2‐COOH | 2.30 | (t) |
| 7 | =CH‐CH2‐CH2‐COOH | 2.34 | (t) |
| 8 | =CH‐CH2‐CH= | 2.77 | (t) |
| 9 | ‐N(CH3)3 Phosphatydylcholine | 3.33 | (s) |
| 10 | Glyceryl, C1, 3, 2 | 4.14, 4.29, 5.26 | (m), (m), (m) |
| 11 | ‐CH=CH‐ | 5.34 | (m) |
3.3. CE‐MS profile characterization
Analyzing the aqueous fraction of BM using CE‐MS yielded 23 identified metabolites from the electropherogram; Fig. 2C displays an extracted ion chromatogram of a representative sample. Metabolites identified were mainly amino acids and their derivatives, organic acids and nicotinamide, glycylglycine and cytosine/dine, Table 3.
Table 3.
Identification of metabolites from the aqueous fraction of BM using CE‐MS
| Metabolite | Metabolite class | Formula | [M+H]+ (Da) | Experimental mass (Da) | Mass error (ppm) | Migration time (min) | |
|---|---|---|---|---|---|---|---|
| 1 | Creatinine | Organic Acid | C4H7N3O | 114.0662 | 114.0663 | 0.9 | 2.8 |
| 2 | Lysine | Amino acid | C6H14N2O2 | 147.1128 | 147.1127 | –0.7 | 3 |
| 3 | Nicotinamide | Vitamin | C6H6N2O | 123.0553 | 123.0552 | –0.8 | 3.1 |
| 4 | Arginine | Amino acid | C6H14N4O2 | 175.119 | 175.119 | 0.0 | 3.1 |
| 5 | Carnitine | Amino acid derivative | C7H15NO3 | 162.1125 | 162.1125 | 0.0 | 3.4 |
| 6 | Acetyl‐l‐carnitine | Amino acid derivative | C9H17NO4 | 204.123 | 204.1232 | 1.0 | 3.7 |
| 7 | Cytosine | Nucleotide | C4H5N3O | 112.0505 | 112.0504 | –0.9 | 3.7 |
| 8 | Cytidine | Nucleoside | C9H13N3O5 | 244.0928 | 244.0929 | 0.4 | 3.7 |
| 9 | Creatine | Organic Acid | C4H9N3O2 | 132.0768 | 132.0769 | 0.8 | 4.2 |
| 10 | Alanine | Amino acid | C3H7NO2 | 90.055 | 90.0551 | 8.7 | 4.5 |
| 11 | 5‐Aminovaleric acid/Valine | Amino acid | C5H11NO2 | 118.0863 | 118.0864 | 0.8 | 4.9 |
| 12 | n‐Methyl‐l‐Valine/Leucine/Isoleucine | Amino acid | C6H13NO2 | 132.1019 | 132.102 | 0.8 | 5 |
| 13 | Serine | Amino acid | C3H7NO3 | 106.0499 | 106.0499 | 0.0 | 5.2 |
| 14 | Tryptophan | Amino acid | C11H12N2O2 | 205.0972 | 205.0971 | –0.5 | 5.2 |
| 15 | Threonine | Amino acid | C4H9NO3 | 120.0655 | 120.0654 | –0.8 | 5.3 |
| 16 | Glycylglycine | Dipeptide | C4H8N2O3 | 133.0608 | 133.0609 | 0.8 | 5.2 |
| 17 | Methionine | Amino acid | C5H11NO2S | 150.0583 | 150.0582 | –0.7 | 5.3 |
| 18 | Citrulline | Amino acid | C6H13N3O3 | 176.103 | 176.1027 | –1.7 | 5.4 |
| 19 | Phenylalanine | Amino acid | C9H11NO2 | 166.0863 | 166.0861 | –1.2 | 5.4 |
| 20 | Glutamic acid | Amino acid | C5H9NO4 | 148.0604 | 148.0603 | –0.7 | 5.5 |
| 21 | Tyrosine | Amino acid | C9H11NO3 | 182.0812 | 182.0811 | –0.5 | 5.6 |
| 22 | Cystine | Amino acid | C6H12N2O4S2 | 241.0312 | 241.0311 | –0.4 | 5.8 |
| 23 | Aspartic acid | Amino acid | C4H7NO4 | 134.0448 | 134.0445 | –2.2 | 6.1 |
3.4. UPLC‐MS lipid fraction profile characterization
The lipid fraction of BM, obtained from the dual phase extraction, analyzed using UPLC‐MS in both negative and positive ionization mode was analyzed. The resulting chromatograms were characterized, allowing the tentative identification of 105 metabolites (Supporting Information Table 4). The base peak chromatogram for a representative sample in negative and positive ionization mode is presented in Fig. 2D and E, respectively, showing regions of a representative chromatogram with metabolites with the highest intensity identified. In negative ionization mode, the main metabolites identified were saturated fatty acids, monounsaturated fatty acids and polyunsaturated fatty acids while in positive ionization mode the main classes of lipid identified were triacylglycerols and phospholipids.
3.5. HPLC‐MS single phase profile characterization
Characterization of the chromatogram resulting from HPLC‐MS analysis of a pooled sample from the single phase extraction was done as thoroughly as possible (Supporting Information Fig. 1) (adapted from a previously published paper, copyright granted 20). This resulted in the tentative identification of 289 individual compounds in positive mode ionization, and 126 metabolites in negative ionization mode, of which 23 were in common, giving a total of 392 individual metabolites, Supporting Information Tables 2 and 3. As described in previous publications fatty acids, glycerophospholipids, sphingolipids, sterol lipids, monoacylglycerols (MG), diacylglycerols (DG), triacylglycerols (TG), and cholesteryl esters were observed 12, 23 [24–26].
3.6. GC‐MS single phase profile characterization
A pooled sample of BM was analyzed in a single phase by GC‐MS and metabolites identified. The total compound chromatogram of the extraction, displayed in Fig. 2F, highlights a range of molecules including; amino acids, fatty acids, organic acids, hexose, and pentose sugars, TCA intermediates, cholesterol, and disaccharides. A total of 56 metabolites were identified, displayed in Table 4 (reproduced with permission from Villaseñor et al.) 12.
Table 4.
Compounds identified in BM by GC‐MS, (data adapted from Villaseñor et al. 12)
| No. | Metabolite class | Metabolite | Target ion (Da) | Retention time |
|---|---|---|---|---|
| 1 | Organic acid | Pyruvic acid | 174 | 6.76 |
| 2 | Organic acid | Lactic acid (standard confirmed) | 147 | 6.92 |
| 3 | Organic acid | Glycolic acid | 147 | 7.13 |
| 4 | Amino acid | Valine 1 | 72 | 7.43 |
| 5 | Amino acid | Alanine 1 | 116 | 7.53 |
| 6 | Organic acid | 2‐Hydroxybutyric acid | 147 | 7.87 |
| 7 | Aromatic homomonocyclic compounds | 2‐Furoic acid | 125 | 7.99 |
| 8 | Amino acid | Isoleucine 1 | 86 | 8.57 |
| 9 | Amino acid | Valine 2 | 144 | 9.14 |
| 10 | Aliphatic acyclic compounds | Urea (standard confirmed) | 147 | 9.54 |
| 11 | Aromatic homomonocyclic compounds | Benzoic acid | 179 | 9.62 |
| 12 | Fatty acids | Caprylic acid | 201 | 9.81 |
| 13 | Sugar alcohols | Glycerol (standard confirmed) | 147 | 9.92 |
| 14 | Nonmetal oxoanionic Compounds | Phosphoric acid | 299 | 10 |
| 15 | Amino acid | Proline 2 (standard confirmed) | 142 | 10.28 |
| 16 | Amino acid | Glycine | 174 | 10.39 |
| 17 | Carboxylic acids | Succinic acid | 147 | 10.48 |
| 18 | Sugar acids | Glyceric acid | 189 | 10.74 |
| 19 | Amino acid | Serine 2 (standard confirmed) | 204 | 11.14 |
| 20 | Amino acid | Threonine 2 | 218 | 11.48 |
| 21 | Fatty acids | Capric acid | 229 | 12.39 |
| 22 | Organic acids | Malic acid | 233 | 12.8 |
| 23 | Organic acids | Adipic acid | 111 | 13 |
| 24 | Sugar alcohols | Threitol | 217 | 13.06 |
| 25 | Pyrrolidines | Pyroglutamic acid | 156 | 13.21 |
| 26 | Amino acid | Glutamic acid 1 | 174 | 13.33 |
| 27 | Lactams | Creatinine | 115 | 13.62 |
| 28 | Amino acid | Glutamic acid 2 (Standard confirmed) | 246 | 14.37 |
| 29 | Fatty acids | Lauric acid | 257 | 14.75 |
| 30 | Sugar acids | Lyxose 1/Lyxosylamine 1 | 103 | 14.89 |
| 31 | Sugar acids/monosaccharides | Lyxose 2/Lyxosylamine 2/Ribose | 103 | 15.06 |
| 32 | Sugar alcohols | Xylitol | 217 | 15.47 |
| 33 | Monosaccharides | Fucose 1 | 117 | 15.59 |
| 34 | Monosaccharides | Fucose 2 | 117 | 15.73 |
| 35 | Carboxylic acids | Citric acid (standard confirmed) | 273 | 16.57 |
| 36 | Amino acids and derivatives | Hippuric acid 2 | 105 | 16.88 |
| 37 | Fatty acids | Myristic acid | 117 | 16.91 |
| 38 | Monosaccharides | Tagatose 1/Sorbose 2/Sorbose 1/Fructose 1 | 103 | 17.12 |
| 39 | Monosaccharides | Tagatose 2/Fructose 2/Fructose 1 | 103 | 17.24 |
| 40 | Monosaccharides | Galactose 1/Mannose 1/Allose 1/Gluconic acid lactone 1 | 205 | 17.28 |
| 41 | Monosaccharides | Glucose 1/Talose 1 (standard confirmed) | 319 | 17.4 |
| 42 | Monosaccharides | Altrose 2/Mannose 2/Glucose 2/Allose 2/Talose 2 | 319 | 17.54 |
| 43 | Fatty alcohols | Mannitol/sorbitol | 319 | 17.87 |
| 44 | Fatty alcohols | 1‐Hexadecanol | 299 | 17.95 |
| 45 | Fatty acids | Palmitoleic acid | 311 | 18.68 |
| 46 | Fatty acids | Palmitic acid | 117 | 18.88 |
| 47 | Monosaccharides | N‐acetyl‐D‐mannosamine 1/N‐acetyl‐D‐mannosamine 2 | 319 | 19.19 |
| 48 | Cyclic alcohols | Myo‐inositol (standard confirmed) | 318 | 19.32 |
| 49 | Fatty acid esters | Methyl stearate (internal standard) | 74 | 19.66 |
| 50 | Fatty acids | Heptadecanoic acid | 327 | 19.81 |
| 51 | Fatty acids | Linoleic acid | 75 | 20.42 |
| 52 | Fatty acids | Oleic acid | 339 | 20.48 |
| 53 | Fatty acids | Stearic acid (standard confirmed) | 341 | 20.69 |
| 54 | Fatty acids | Arachidic acid | 369 | 22.37 |
| 55 | Disaccharides | Sucrose | 361 | 24.1 |
| 56 | Disaccharides | Lactose 1 | 361 | 24.46 |
| 57 | Disaccharides | Trehalose/maltose 1/maltose 2 | 361 | 24.91 |
| 58 | Disaccharides | Galactinol 1 | 204 | 26.3 |
| 59 | Steroids and steroid derivatives | Cholesterol | 129 | 27.64 |
Numbers after identification correspond to the number of trimethylsilyl groups found on the molecule after derivatization.
3.7. Core and unique in BM metabolome signatures: Comparison of platform capabilities in metabolite coverage
With regard to analytical platforms that identified aqueous metabolites, some overlap between platforms was noted, but each platform contributed an additional unique set of metabolites. For example, GC‐MS and 1H NMR generated signals for 21 common metabolites, including sugars, amino acids, fatty acids, and tricarboxylic acids, of which five were also in common with the CE‐MS generated profile (alanine, creatinine, glutamate, isoleucine, and valine). Both 1H NMR and GC‐MS characterized several sugars including galactose, glucose, and lactose, while GC‐MS additionally profiled several polyols (mannitol, sorbitol, myo‐inositol, threitol, xylitol). GC‐MS uniquely detected numerous medium to long chain fatty acids, whereas HMO and short chain fatty acids were profiled by 1H NMR spectroscopy. CE‐MS gave the most comprehensive amino acid profile, and both 1H NMR and CE‐MS characterized several carnitines. The UPLC‐MS profiles, in both positive and negative ionization mode of the lipid fraction yielded data from sphingomyelins, phosphocholines, phosphoethanolamines, and di‐ and triglycerides. It was observed that some high abundance metabolites from the Folch extraction were detected in both the polar and nonpolar fractions, indicating that this method would not be the ideal choice for quantitation since distribution of metabolites across two compartments makes calculation of total concentration less accurate.
3.8. Analytical performance; RSD
Prior to analysis, system stability and performance were assessed using QC samples. Additionally, QC samples were analyzed every few samples throughout the run to ensure good instrumental performance over the run. This allows for the repetition of sample analysis if necessary. Data quality was also evaluated prior to the multivariate analysis, by building a PCA model including samples and QC samples, which cluster into the center of the plot. Moreover, data arising from the QCs were used to filter instrumental noise from the MS data of the breast milk samples.
The RSD was calculated for each of the methods, and was used to establish the reproducibility of the methods used (Supporting Information Table 1). All of the methods were found to be robust and reproducible. Of all the methods, 1H NMR of the aqueous BM fraction was found to be the most reproducible, with mean RSD value of 6.4% and range of 0.1–24%, while GC‐MS gave a similar mean value of 7.65% (1.8–18.6%). All the LC‐MS methods also showed good reproducibility across the selected metabolites, with mean values of 8.15% (4.6–15%), for the single phase HPLC extraction method and mean RSDs of 7.95 and 13.56% for the positive and negative mode of the Folch extracted samples, respectively (ranges, 3.2–18.3% and 2.7–26.6%). The mean RSD for the CE‐MS metabolites was the weakest in terms of reproducibility, 24.18%, but was still within the accepted cut off value of 30% RSD (12.7–29.1%).
3.9. Multiplatform assessment of metabolite alterations in BM over time
The multiplatform approach was applied to a set of BM samples in order to identify which metabolites were altered in concentration over lactation between the three different groups; days 1–5 postbirth (group 1), 6–10 days postbirth (group 2) and >10 days postbirth (group 3). First, PCA models, provided in Supporting Information Fig. 2, model details shown in Table 5, show that differentiation of samples over time according to inherent similarity in chemical composition was apparent only for 1H NMR and CE‐MS analysis of the aqueous fraction, and for the models constructed from the single phase extraction method run using HPLC‐MS, PCA R 2X values; HPLC‐MS single phase extraction R 2X = 0.457, 1H NMR of the aqueous fraction R 2X = 0.406, and GC‐MS R 2X = 0.337 CE‐MS R 2X = 0.333, ranked in descending order of strength, gave relatively strong models defining temporal changes in BM composition. PLS‐DA models comparing the three time groups against one another were computed for each of the analytical methods used; these models displayed a gradual change in the composition of BM in relation to postnatal age (data not shown) affecting the quality of the models.
Table 5.
PCA scores plots, white diamonds represent samples taken 1–5 days post‐birth, black squares 6–10 days, white stars >10 days, model characteristics are displayed below
| PCA models | R 2 | Components | Outliers | t[1] | t[2] | Scaling | |
|---|---|---|---|---|---|---|---|
| A | NMR aqueous | 0.406 | 2 | 3 | 0.27 | 0.128 | UV |
| B | CE‐MS; Positive mode | 0.333 | 1a | 0 | 0.23 | Par | |
| C | HPLC‐MS, | 0.457 | 2 | 2 | 0.37 | 0.08 | UV |
| D | NMR Lipid | 0.484 | 2 | 3 | 0.35 | 0.12 | UV |
| E | UPLC‐MS; Positive mode lipid | 0.567 | 2 | 0 | 0.39 | 0.17 | Par |
| F | UPLC‐MS; Negative mode lipid | 0.331 | 2 | 0 | 0.23 | 0.1 | UV |
| G | GCMS, Single phase | 0.337 | 2 | 1 | 0.19 | 0.14 | UV |
Second component was used for plotting purposes only.
For this reason, pairwise analysis using OPLS‐DA was selected as the most suitable method to establish which metabolites changed over time, comparing groups 1 versus 2, 1 versus 3 and 2 versus 3. As shown in Table 6, each technique yielded strong models indicating significant systematic differences in BM composition over time, with the exception of models corresponding to the NMR lipid phase and UPLC‐MS lipid phase in negative ionization mode, with these models showing a poor capability of prediction. Cross‐validated scores plots corresponding to the OPLS‐DA models can be found in the Supporting Information Figs. 3–5.
Table 6.
OPLS‐DA model values and descriptive information of models run to identify metabolites included in Table 7
| OPLS‐DA models | Pairwise model | R 2X | R 2Y | Q 2 | Components | Scaling | |
|---|---|---|---|---|---|---|---|
| A | NMR aqueous |
|
|
|
|
2 | UV |
| B | CE‐MS; Positive mode |
|
|
|
|
2 | Par |
| C | HPLC‐MS, single phase |
|
|
|
|
2 | UV |
| D | NMR Lipida |
|
|
|
|
2 | UV |
| E | Lipid UPLC‐MS; Positive mode |
|
|
|
|
2 | Par |
| F | Lipid UPLC‐MS; Negative modea |
|
|
|
|
2 | UV |
| G | GC‐MS, single phase |
|
|
|
|
2 | UV |
These models have a low predictive capability.
Metabolites identified from the OPLS‐DA models as altering between time groups are displayed in Table 7. Changes identified in BM composition over the first few days of lactation include an increase in the relative concentrations of di‐ and triacylglycerols along with a more sustained increase in lactose, several amino acids, and short and medium chain fatty acids. Conversely a time dependent decrease was observed in multiple HMO, several phosphocholines, and energy metabolites such as citrate and pyruvate.
Table 7.
Metabolites identified as changing in abundance in breast milk samples collected at different times postbirth
| Class | Metabolite | 1st 5 days versus day 6–10 | 1st 5 days versus day +10 | Day 6–10 versus day +10 |
|---|---|---|---|---|
| Amino acids | Alanine | A | ||
| Glutamine | A | A | ||
| Acids | 3,4 Hydroxymandelic acid | g | ||
| Butyrate | A | A | ||
| Citrate | a | a | a | |
| Taurine | fa | |||
| Pyruvic acid | g | |||
| Ceramides | Glucosyl Ceramide (36:2) | Fa | ||
| Ceramide (20:4) | c | |||
| PE‐Ceramide (38:2) | fa | |||
| PI‐Ceramide (40:0) | fa | |||
| PE‐Ceramide (38:2) | ||||
| Diacylglycerols | Diacylglycerol (24:0) | C | ||
| Diacylglycerol (26:0) | C | |||
| Diacylglycerol (28:2) | C | |||
| Diacylglycerol (30:2) | C | |||
| Diacylglycerol (34:0) | E | |||
| Diacylglycerol (34:1) | E | |||
| Diacylglycerol (34:3) | E | E | ||
| Diacylglycerol (40:7) | C | E | ||
| Diacylglycerol (44:10) | e | e | ||
| Prenol Lipids | Tocopherol | g | g | |
| Pyrimidine Nucleosides | N4‐Acetylcytidine | B | ||
| Amino Acids Derivatives | N‐Acetyl glutamine | a | a | a |
| Steroids and Steroid Derivatives | Cholesteryl stearate | fa | ||
| Cholesterol | da, g | |||
| Sugar alcohols | Glycerol | G | ||
| Phthalic Acid and Derivatives | Dioctyl phthalate | G | ||
| Fatty Acid Esters | Methyl Stearate | G | ||
| Fatty Acids | 11S‐hydroxy‐hexadecanoic acid | Fa | ||
| 9,12‐octadecadienoic acid | G | G | ||
| Arachidonic Acid | C | |||
| Caprylic acid | Fa | Fa | ||
| 3‐Hydroxycapric acid | Fa | |||
| Hexadecenoic acid | Fa | |||
| Myristic acid | g | |||
| Oleic acid | G | |||
| Palmitoleic acid | G | |||
| Lauric acid | G | |||
| Linoleic acid | G | G | ||
| Polyunsaturated FA | da | |||
| Terminal CH3 on omega 3 fatty acids | da | |||
| Saccharides | Fucose (from HMO) | a | a, g | a |
| d‐Glucosaminic acid (from HMO) | g | g | ||
| N‐Acetyl neuraminic acid (from HMO) | a | a | a | |
| Glucose | G | G | ||
| Lactose | A, G | A | A | |
| Gluconic acid lactone | G | |||
| Glycerophosphocholines | Glycerophosphocholine (18:0) | e | ||
| Glycerophosphocholine (34:0) | e | e | ||
| Glycerophosphocholine (32:0) | e | e | ||
| Glycerophosphocholine (32:1) | e | |||
| Glycerophosphocholine (30:0) | e | e | e | |
| Glycerophosphocholine (38:5) | fa | |||
| Glycerophosphocholine (34:2) | e | e | ||
| Glycerophosphocholine (36:1) | e | c | ||
| Glycerophosphocholine (36:2) | E | |||
| Glycerophosphocholine (36:3) | e | |||
| Glycerophosphocholine (36:4) | fa | |||
| Glycerophosphocholine (40:7) | fa | |||
| Glycerophosphocholine | A | A | ||
| Glycerophospho‐ethanolamines | Glycerophosphoethanolamine (40:6) | Fa | ||
| Glycerophosphoethanolamine (36:2) | E | |||
| Glycerophosphoethanolamine (42:3) | fa | |||
| Glycerophosphoethanolamine (44:2) | fa | |||
| Glycerophosphoserines | Glycerophosphoserine (30:3) | Fa | ||
| Glycerophosphoserine (18:1) | fa | |||
| Glycererophosphoglycerol | Glycerophosphoglycerols (44:7) | fa | ||
| Glycerphosphoinositols | Glycerophosphoinositol (35:2) | fa | ||
| Glycerophosphoinositol (32:0) | E | |||
| Glycerophosphoinositol (37:4) | fa | |||
| Monoacylglycerols | Monoacylglycerol (18:3) | b | b | |
| Sphingomyelins | Sphingomyelin (42:0) | Fa | ||
| Sphingomyelin (34:1) | e | e | ||
| Sphingomyelin (36:3) | c | |||
| Sphingomyelin (38:1) | e | |||
| Triacylglycerols | Triacylglycerol (34:0) | E | e | |
| Triacylglycerol (36:0) | E | e | ||
| Triacylglycerol (38:0) | E | e | ||
| Triacylglycerol (42:1) | E | e | ||
| Triacylglycerol (42:2) | E | e | ||
| Triacylglycerol (44:2) | E | e | ||
| Triacylglycerol (45:1) | Fa | |||
| Triacylglycerol (48:1) | e | fa | ||
| Triacylglycerol (48:4) | e | |||
| Triacylglycerol (48:5) | e | |||
| Triacylglycerol (50:2) | e | |||
| Triacylglycerol (51:2) | C | |||
| Triacylglycerol (51:7) | F | |||
| Triacylglycerol (52:2) | e | E | ||
| Triacylglycerol (55:9) | fa | |||
| Triacylglycerol (58:2) | c | |||
| Triacylglycerol (60:3) | c | c | c |
These alterations were identified in models possessing a low predictive capability.
Capitals relate to higher concentration in the later time group, while lower case letters refer to metabolites that were decreased in abundance in the later time point. NMR aqueous A, a; CE‐MS aqueous B, b; HPLC‐MS single phase C, c; NMR lipid D, d; UPLC‐MS positive E, e; UPLC‐MS negative F, f; GC‐MS single phase G, g.
4. Discussion
We have created the most comprehensive metabolic map of BM to date using four different analytical platforms and two sample preparation techniques, with absolute (15%) and tentative identification of 710 metabolites; previous studies have commonly identified between 50 and 100 metabolites. Additionally, we have demonstrated the utility of a multiplatform analytical technique to investigate questions of clinical and biological importance by reporting the changes in abundance of these metabolites over the first 3 months of lactation, comparing the utility of the different platforms.
In regard to previous studies employing metabonomic strategies to investigate breast milk composition, both Smilowitz et al. and Pratico et al. identified differences between samples based on the profiles of HMO secreted using 1H NMR spectroscopy 7, 9. Previous research has also employed MS to analyze human BM, these studies have primarily focused on either the lipid content 11, or diversity of HMO present in BM 5. In addition, a study optimizing sample treatment prior to analysis using MS focused on the first week of lactation to the third month of lactation 12.
The range of metabolites contained in BM means that no single analytic technique is capable of resolving the entire BM metabolome. We designed an analytical strategy to allow us to select the most appropriate analytical platforms to resolve as many BM metabolites as possible. Analysis of nonpolar metabolites in BM is essential. Lipids are one of the major energy sources in BM, contributing 40–55% of the total energy of BM 27, and are essential for neurodevelopment 28. Furthermore, short chain fatty acids (SCFA) are not only an important source of energy 29, but their presence in BM is essential for maturation of the gastrointestinal tract 30. Among the polar metabolites, many interesting and important classes of molecules are present such as digestible sugars, which represent a readily available energy source for the infant and are necessary for healthy growth; furthermore, HMO, notable for their absence in infant formula, contribute to establishing a healthy intestinal microbiome. In fact one of the most prominent changes observed in the time course analysis of BM were the decreasing concentrations of HMO over the study period.
In order to ensure our profiling techniques adequately covered these metabolite groups we chose UPLC‐MS for lipidomic analysis, GC‐MS for the analysis of volatile polar compounds, particularly SCFA. 1H NMR allowed broad coverage of compounds from aromatic to aliphatic compounds; it also gave some idea of the content of major lipid classes. We selected CE‐MS for the detection of ionizable polar compounds present in BM, particularly amino acids, for which it was the best method of detection. Amino acids have been demonstrated to be important cell signaling molecules, imbalances in particular amino acids, which are analytes of interest in infant nutrition studies, can lead to health problems, and are important in conditions such as phenylketonuria, and may exert other specific negative physiological effects 31, 32. 1H NMR, LC‐MS, and CE‐MS involve minimal sample preparation compared to GC‐MS and therefore are suitable for BM compositional screening of larger sample cohorts. The LOD for each of the different analytical platforms is well established. In regard to NMR spectroscopy (for a 600 MHz instrument), the LOD is in the nanomolecular range (10−9), while for MS, the LOD is in the range of 10−14 to 10−18 mol 33.
Each platform contributed a set of unique metabolites to the overall global profile. There was some overlap in metabolites identified by the different platforms, but insofar as this allows corroboration of the data, this may be regarded as a strength of the multiplatform analytical approach. Ultimately, given the complexity of BM composition, we have demonstrated that there is no single analytical platform capable of identifying all metabolites present in milk, and that each platform confers advantages for some chemical classes of molecule and deficiencies in others.
We conclude that a multiplatform approach maximizes the number of metabolites that can be identified in BM. Clearly, it is not often practical to use three or four separate analytical platforms to characterize every sample and thus careful consideration should be given to choice of platform. We found that the platforms used to assess the polar components of BM gave the most statistically robust results. CE‐MS was the least reproducible of all techniques in terms of RSD, but uniquely profiled amino acids in detail. For global profiling we would recommend at the very least that the platforms used should be capable of identifying both polar and nonpolar metabolites. If using a single platform for analysis, 1H NMR has the advantage of being highly reproducible and allowing coverage of a large range of metabolite classes including amino acids, sugars (simple through to complex HMO), lipid (SCFA), and metabolic intermediaries (e.g. TCA cycle metabolites). Both 1H NMR and MS can be employed in quantitative/semiquantitative analysis.
With regard to the two different sample treatment extraction methods, Folch extraction and single phase methanol/MTBE (50:50) extraction, the optimal method to use will depend on the analytical platform and analytes of interest. The Folch extraction is compatible with all the analytical platforms evaluated here, allowing a single extraction technique to be employed, and is therefore valuable when utilizing a multiplatform approach for screening BM. However, Folch extraction duplicates the number of analyses needed due to the splitting of polar and nonpolar phases, which must be analyzed independently. Another disadvantage of Folch extraction is that metabolites present in high concentrations do not necessarily partition exclusively into either one of the phases, therefore metabolite quantification based on this method can be unreliable for some compounds. This could partially account for the relatively poor performance of the Folch extracted UPLC‐MS models in distinguishing temporal changes in BM over the early lactation period. The single step method is unsuitable for use with 1H NMR as nonpolar metabolites are retained, which further obscure spectra due to overlapping signals.
In terms of numbers of metabolites, the single phase extraction followed by HPLC‐MS analysis gives the best profile but did not capture the HMO, amino acids, and sugars as well as some of the other techniques. Moreover, many of the metabolites identified via the LC‐MS methods need confirmation since the available databases are not adequate for absolute structural confirmation. 1H NMR analysis of the polar BM fraction gave the best insight into HMO and provided the most reproducible data in terms of stability of the analytical profile and capturing systematic compositional changes over time.
Beyond simply establishing a list of metabolites present in BM (although this is not unimportant), in order to answer questions of biological relevance it is necessary to show that the analytical platforms used can be employed to study relative changes in these metabolites between different milk samples. We used our analytical platforms to assess the temporal change in BM composition 34 over the first 3 months of lactation. This revealed temporal alterations in keeping with previous reports using established biochemical assays of BM composition.
Specifically, our findings confirm a number of previous reports in relation to the changes in BM composition over time. For example, Ilcol et al. demonstrated that glycerophosphocholine concentrations significantly increase in concentration from colostrum to mature milk 2, we find similarly. Tocopherol was decreased over the first week of lactation, but remained at constant concentrations after this; this is in agreement with reports showing a decrease in α‐tocopherol in early lactation 35. We also confirm previous reports of decreasing concentrations of citrate 36 and HMO 37, and increases in lactose 38, glutamine, and alanine 37 in mature milk. Carlson et al. previously described changes in amino acid concentrations during lactation 37, which also matched our observations. Our data demonstrate that the analytical techniques utilized have the power to elucidate temporal changes in breast milk in great detail, and to identify changes in hitherto unknown BM metabolites. For example, glycylglycine, a dipeptide, was detected in breast milk using CE‐MS, which has not to our knowledge been identified previously. This approach will provide a unique opportunity to understand how BM is biologically tailored to support human development.
Our work has identified many metabolites present in BM whose biological function has not been fully elucidated, an important avenue of future research. A limitation of the current study is that it focuses on the first three months of lactation and thus changes which occur later on in lactation need illumination. As well as this, this study was performed on a relatively small cohort size and a larger cohort would give greater confidence that results obtained are applicable to the population in general.
The composition of BM is complex and subject to the influence of many factors such as the time of day samples are collected, the time during the feed at which the sample is collected, time since the last feed and maternal diet. Ideally these factors require standardization to increase confidence in results obtained from future studies, however they are often difficult to control. Solely investigating BM composition reduces the complexity of the true relationship between the mother and the infant through BM composition. For example, factors present in BM are altered during digestion, and can take on new properties, including acting as antimicrobial agents 39.
BM contains a number of enzymes, which not only aid the digestion of milk, but also play important roles in gut development. The gut of the human infant has little lipase activity, relying on the presence of lipoprotein lipase and bile salt stimulated lipase for digestion of the tri‐ and diacylglycerols in BM 40. The relative changes in lipase concentrations in the BM over the course of lactation, is a possible cause of altered lipid profile. As well as this, factors such as HMO selectively encourage the growth of beneficial bacteria, orchestrating the development of the infant microbiome, important for the immune and metabolic development of the infant 41.
The change in BM composition over lactation is known to reflect the infant's needs, becoming more energy dense over lactation, with increasing carbohydrate and lipid concentrations to provide for the infants increased energy requirements. Total protein is seen to decrease, probably due to the decreasing concentration of factors such as immunoglobulins secreted into BM 42. This not only reflects the infants decreased requirement for them, as their own immune system becomes functional, but also reflects the increasing inability of the infant gut to absorb proteins, as gut closure occurs and the permeability of the gut to macromolecules decreases over the first 3 days of life 43.
In summary, we have shown the complementarity of various analytical technologies for characterizing BM composition and have identified their respective strengths and limitations. This comprehensive coverage of the metabolome should provide a baseline for further studies to promote understanding of the impact of maternal characteristics, environmental, and dietary factors on BM composition and inform the consequences this may have for infants. It will also assist in the fulfillment of the ethical imperative to design and produce the best nutrient support possible for babies without access to BM.
Supporting information
Table S1. The relative standard deviation (RSD) was calculated for each of the methods used to establish the reproducibility of the methods used.
Table S2. Compound characterization of breast milk profile by LC‐QTOF‐MS in positive mode. Tentative identification was performed on features with a RSD less than 20% and abundance above of 105 from 10 independent replicates of a pool of human milk. Compounds were grouped into a biochemical or lipid category; in this instance compounds were included within one main class: Fatty acyls (116), glycerolipids (131), glycerophospholipids (78), prenol lipids (6), sphingolipids (27), sterol lipids (32) and aminoacid (1) and then into two sub‐groups.
Table S3. Compound characterization of breast milk profile by LC‐QTOF‐MS in negative mode. Tentative identification was performed on features with a RSD less than 20% and abundance above of 105 from 10 independent replicates of a pool of human milk. Compounds were grouped into a biochemical or lipid category; in this instance compounds were included within one main class: Fatty acyls (116), glycerolipids (131), glycerophospholipids (78), prenol lipids (6), sphingolipids (27), sterol lipids (32) and aminoacid (1) and then into two sub‐groups.
Table S4. Metabolites tentatively identified in positive and negative ionization mode using UPLC‐MS.
Figure S1. (A) Total Compound Chromatogram in positive and (B) negative mode, acquired by LC‐QTOF‐MS. Metabolite identification in BM profile by classes of lipid. Extraction system [methanol: MTBE (50:50)].
Figure S2. OPLS‐DA cross validation scores plots, separated by time groups. White diamonds represent samples taken during days 1‐5 post‐birth; black squares show samples taken days 6‐10 post‐birth, model characteristics are displayed in Table 4.
Figure S3. OPLS‐DA cross validated scores plots, separated by time groups. White diamonds represent samples taken during days 1‐5 post‐birth, white stars show samples taken >10 days post‐birth.
Figure S4. OPLS‐DA cross validated scores plots separated by time groups. Black squares represent samples taken during days 6‐10 post‐birth, white stars show samples taken >10 days post‐birth.elps5505‐sup‐0003‐Figure2.tiff
Acknowledgments
The authors would like to acknowledge Dr Chris Gale for the provision of samples and Ms Suzan Jeffries for the collection of samples. N.J.A. is supported by a doctoral research fellowship from the Westminster Medical School Research Trust. M.A.L. and A.V. received a mobility grant from Santander bank. IGP is funded by a NIHR postgraduate research fellowship.
The authors have declared the following potential conflicts of interest: N.J.A. has received funding from Danone and Medela to attend an educational conference. M.J.H. has received funding from Danone to attend an educational conference. N.M. has received consultancy fees from Ferring Pharmaceuticals, and a speaker honorarium for an educational meeting funded by Nestle International in which they had no organizational involvement. The other authors have no relevant conflicts of interest to declare.
Colour Online: See the article online to view Fig. 1 in colour.
5 References
- 1. Menjo, A. , Mizuno, K. , Murase, M. , Nishida, Y. , Taki, M. , Itabashi, K. , Shimono, T. , Namba, K. , Acta Paediatr. 2009, 98, 380–384. [DOI] [PubMed] [Google Scholar]
- 2. Ilcol, Y. O. , Ozbek, R. , Hamurtekin, E. , Ulus, I. H. , J. Nutr. Biochem. 2005, 16, 489–499. [DOI] [PubMed] [Google Scholar]
- 3. Wharton, B. A. , Morley, R. , Isaacs, E. B. , Cole, T. J. , Lucas, A. , Arch. Dis. Childhood 2004, 89, F497–F498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Michaelsen, K. F. , Skafte, L. , Badsberg, J. H. , Jorgensen, M. , J. Pediatric Gastroenterol. Nutr. 1990, 11, 229–239. [DOI] [PubMed] [Google Scholar]
- 5. German, J. B. , Freeman, S. L. , Lebrilla, C. B. , Mills, D. A. , Nestle Nutr. Workshop Series Paediatr. Prog. 2008, 62, 205–218; discussion 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nicholson, J. K. , Lindon, J. C. , Holmes, E. , Xenobiotica 1999, 29, 1181–1189. [DOI] [PubMed] [Google Scholar]
- 7. Smilowitz, J. T. , O'Sullivan, A. , Barile, D. , German, J. B. , Lonnerdal, B. , Slupsky, C. M. , J. Nutr. 2013, 143, 1709–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marincola, F. C. , Noto, A. , Caboni, P. , Reali, A. , Barberini, L. , Lussu, M. , Murgia, F. , Santoru, M. L. , Atzori, L. , Fanos, V. , J. Mat. Fetal Neonatal Med. 2012, 25, 62–67. [DOI] [PubMed] [Google Scholar]
- 9. Pratico, G. , Capuani, G. , Tomassini, A. , Baldassarre, M. E. , Delfini, M. , Miccheli, A. , Nat. Product Res. 2014, 28, 95–101. [DOI] [PubMed] [Google Scholar]
- 10. O'Sullivan, A. , He, X. , McNiven, E. M. , Hinde, K. , Haggarty, N. W. , Lonnerdal, B. , Slupsky, C. M. , J. Pediatr. Gastroenterol. Nutr. 2013, 56, 355–363. [DOI] [PubMed] [Google Scholar]
- 11. Lindholm, E. S. , Strandvik, B. , Altman, D. , Moller, A. , Kilander, C. P. , Prostag. Leukotr. Ess. 2013, 88, 211–217. [DOI] [PubMed] [Google Scholar]
- 12. Villasenor, A. , Garcia‐Perez, I. , Garcia, A. , Posma, J. M. , Fernandez‐Lopez, M. , Andreas, N. J. , Modi, N. , Holmes, E. , Barbas, C. , Anal. Chem. 2014, 86, 8245–8252. [DOI] [PubMed] [Google Scholar]
- 13. Folch, J. , Lees, M. , Sloane Stanley, G. H. , J. Biol. Chem. 1957, 226, 497–509. [PubMed] [Google Scholar]
- 14. Garcia, A. , Barbas, C. , Methods Mol. Biol. 2011, 708, 191–204. [DOI] [PubMed] [Google Scholar]
- 15. Beckonert, O. , Keun, H. C. , Ebbels, T. M. , Bundy, J. , Holmes, E. , Lindon, J. C. , Nicholson, J. K. , Nat. Protocols 2007, 2, 2692–2703. [DOI] [PubMed] [Google Scholar]
- 16. Cloarec, O. , Dumas, M. E. , Craig, A. , Barton, R. H. , Trygg, J. , Hudson, J. , Blancher, C. , Gauguier, D. , Lindon, J. C. , Holmes, E. , Nicholson, J. , Anal. Chem. 2005, 77, 1282–1289. [DOI] [PubMed] [Google Scholar]
- 17. Fisher, R. A. , Statistical Methods for Research Workers, Oliver and Boyd, Edinburg h, UK: 1938. [Google Scholar]
- 18. G. Isaac, S. M. , Astarita G., Waters Appl. Note 2011. [Google Scholar]
- 19. Veselkov, K. A. , Vingara, L. K. , Masson, P. , Robinette, S. L. , Want, E. , Li, J. V. , Barton, R. H. , Boursier‐Neyret, C. , Walther, B. , Ebbels, T. M. , Pelczer, I. , Holmes, E. , Lindon, J. C. , Nicholson, J. K. , Anal. Chem. 2011, 83, 5864–5872. [DOI] [PubMed] [Google Scholar]
- 20. Villasenor, A. , Garcia‐Perez, I. , Garcia, A. , Posma, J. M. , Fernandez‐Lopez, M. , Nicholas, A. J. , Modi, N. , Holmes, E. , Barbas, C. , Anal. Chem. 2014, 86, 8245–8252. [DOI] [PubMed] [Google Scholar]
- 21. Halket, J. M. , Waterman, D. , Przyborowska, A. M. , Patel, R. K. , Fraser, P. D. , Bramley, P. M. , J. Exp. Bot. 2005, 56, 219–243. [DOI] [PubMed] [Google Scholar]
- 22. Zhang, J. , Koo, I. , Wang, B. , Gao, Q. W. , Zheng, C. H. , Zhang, X. , J. Chromatogr. A 2012, 1251, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jensen, R. G. , Lipids 1999, 34, 1243–1271. [DOI] [PubMed] [Google Scholar]
- 24. Blaas, N. , Schüürmann, C. , Bartke, N. , Stahl, B. , Humpf, H. U. , J. Agric. Food Chem. 2011, 59, 6018–6024. [DOI] [PubMed] [Google Scholar]
- 25. Tijerina‐Sáenz, A. , Innis, S. M. , Kitts, D. D. , Acta Paediatr. 2009, 98, 1793–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Crill, C. M. , Helms, R. A. , Nutr. Clin. Pract. 2007, 22, 204–213. [DOI] [PubMed] [Google Scholar]
- 27. Koletzko, B. , Rodriguez‐Palmero, M. , Demmelmair, H. , Fidler, N. , Jensen, R. , Sauerwald, T. , Early Hum. Dev. 2001, 65 (Suppl), S3–S18. [DOI] [PubMed] [Google Scholar]
- 28. Roncada, P. , Stipetic, L. H. , Bonizzi, L. , Burchmore, R. J. , Kennedy, M. W. , J. Proteomics 2013, 88, 47–57. [DOI] [PubMed] [Google Scholar]
- 29. Donohoe, D. R. , Garge, N. , Zhang, X. , Sun, W. , O'Connell, T. M. , Bunger, M. K. , Bultman, S. J. , Cell Metab. 2011, 13, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peng, L. , Li, Z. R. , Green, R. S. , Holzman, I. R. , Lin, J. , J. Nutr. 2009, 139, 1619–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Filiano, J. J. , Clin. Perinatol. 2006, 33, 411–479. [DOI] [PubMed] [Google Scholar]
- 32. Roth, E. , Druml, W. , Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 67–74. [DOI] [PubMed] [Google Scholar]
- 33. Holzgrabe, U. , Diehl, B , in: Holzgrabe U. (Ed.), NMR Spectroscopy in Pharmaceutical Analysis, Elsevier, Oxford: 2011. [Google Scholar]
- 34. Prosser, C. G. , Saint, L. , Hartmann, P. E. , Aust. J. Exp. Biol. Med. Sci. 1984, 62(Pt 2), 215–228. [DOI] [PubMed] [Google Scholar]
- 35. Chappell, J. E. , Francis, T. , Clandinin, M. T. , Early Hum. Dev. 1985, 11, 157–167. [DOI] [PubMed] [Google Scholar]
- 36. Morriss, F. H., Jr. , Brewer, E. D. , Spedale, S. B. , Riddle, L. , Temple, D. M. , Caprioli, R. M. , West, M. S. , Pediatrics 1986, 78, 458–464. [PubMed] [Google Scholar]
- 37. Carlson, S. E. , Adv. Pediatr. 1985, 32, 43–70. [PubMed] [Google Scholar]
- 38. Coppa, G. V. , Gabrielli, O. , Pierani, P. , Catassi, C. , Carlucci, A. , Giorgi, P. L. , Pediatrics 1993, 91, 637–641. [PubMed] [Google Scholar]
- 39. Liepke, C. , Zucht, H. D. , Forssmann, W. G. , Standker, L. , J. Chromatogr. B Biomed. Sci. Appl. 2001, 752, 369–377. [DOI] [PubMed] [Google Scholar]
- 40. Blackberg, L. , Hernell, O. , Eur. J. Biochem. 1981, 116, 221–225. [DOI] [PubMed] [Google Scholar]
- 41. German, J. B. , Dillard, C. J. , Ward, R. E. , Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 653–658. [DOI] [PubMed] [Google Scholar]
- 42. Mickleson, K. N. , Moriarty, K. M. , J. Pediatr. Gastroenterol. Nutr. 1982, 1, 381–384. [DOI] [PubMed] [Google Scholar]
- 43. Vukavic, T. , J. Pediatr. Gastroenterol. Nutr. 1984, 3, 700–703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The relative standard deviation (RSD) was calculated for each of the methods used to establish the reproducibility of the methods used.
Table S2. Compound characterization of breast milk profile by LC‐QTOF‐MS in positive mode. Tentative identification was performed on features with a RSD less than 20% and abundance above of 105 from 10 independent replicates of a pool of human milk. Compounds were grouped into a biochemical or lipid category; in this instance compounds were included within one main class: Fatty acyls (116), glycerolipids (131), glycerophospholipids (78), prenol lipids (6), sphingolipids (27), sterol lipids (32) and aminoacid (1) and then into two sub‐groups.
Table S3. Compound characterization of breast milk profile by LC‐QTOF‐MS in negative mode. Tentative identification was performed on features with a RSD less than 20% and abundance above of 105 from 10 independent replicates of a pool of human milk. Compounds were grouped into a biochemical or lipid category; in this instance compounds were included within one main class: Fatty acyls (116), glycerolipids (131), glycerophospholipids (78), prenol lipids (6), sphingolipids (27), sterol lipids (32) and aminoacid (1) and then into two sub‐groups.
Table S4. Metabolites tentatively identified in positive and negative ionization mode using UPLC‐MS.
Figure S1. (A) Total Compound Chromatogram in positive and (B) negative mode, acquired by LC‐QTOF‐MS. Metabolite identification in BM profile by classes of lipid. Extraction system [methanol: MTBE (50:50)].
Figure S2. OPLS‐DA cross validation scores plots, separated by time groups. White diamonds represent samples taken during days 1‐5 post‐birth; black squares show samples taken days 6‐10 post‐birth, model characteristics are displayed in Table 4.
Figure S3. OPLS‐DA cross validated scores plots, separated by time groups. White diamonds represent samples taken during days 1‐5 post‐birth, white stars show samples taken >10 days post‐birth.
Figure S4. OPLS‐DA cross validated scores plots separated by time groups. Black squares represent samples taken during days 6‐10 post‐birth, white stars show samples taken >10 days post‐birth.elps5505‐sup‐0003‐Figure2.tiff