

Abstract
Spinal cord injury (SCI) results in loss of movement, sensibility, and autonomic control at the level of the lesion and at lower parts of the body. Several experimental strategies have been used in attempts to increase endogenous mechanisms of neuroprotection, neuroplasticity, and repair, but with limited success. It is known that glucose-dependent insulinotropic peptide (GIP) and its receptor (GIPR) can enhance synaptic plasticity, neurogenesis, and axonal outgrowth. However, their role in the injury has never been studied. The aim of this study was to evaluate the changes in expression levels of both GIP and GIPR in acute and chronic phases of SCI in rats. Following SCI (2 to 24 h after damage), the rat spinal cord showed a lesion in which the epicenter had a cavity with hemorrhage and necrosis. Furthermore, the lesion cavity also showed ballooned cells 14 and 28 days after injury. We found that SCI induced increases in GIPR expression in areas neighboring the site of injury at 6 h and 28 days after the injury. Moreover, higher GIP expression was observed in these regions on day 28. Neuronal projections from the injury epicenter showed an increase in GIP immunoreactivity 24 h and 14 and 28 days after SCI. Interestingly, GIP was also found in progenitor cells at the spinal cord canal 24 h after injury, whereas both GIP and GIPR were present in progenitor cells at the injury epicenter 14 days after in SCI animals. These results suggest that GIP and its receptor might be implicated with neurogenesis and the repair process after SCI.
Key words: : glucose-dependent insulinotropic peptide (GIP), glucose-dependent insulinotropic peptide receptor (GIPR), nestin-positive cells, spinal cord injury (SCI)
Introduction
Spinal cord injury (SCI) disrupts ascending and descending axonal pathways and causes cellular destruction, inflammation, and demyelination, leading to loss of movement, sensibility, and autonomic control at the level of the lesion and below.1,2 Experimental models of contusion-, compression-, and crush-induced SCI in rats show good correlation with the pathophysiology of SCI in humans.3 Substantial tissue loss occurs after SCI originating a fluid-filled cavity in the center of the spinal cord at the site of injury that may even get larger over time, which can lead to further tissue damage.4,5
Despite the extremely limited regeneration of neurons in the adult central nervous system (CNS), there is some level of spontaneous recovery of sensorimotor function observed in spinal cord injured patients. This recovery must result from structural remodeling of spared axons and/or dendrites within the damaged systems, which can be regulated by endogenous mechanisms.6-8
Glucose-dependent insulinotropic peptide (GIP) is a member of the vasoactive intestinal peptide (VIP)/secretin/glucagon family of gastrointestinal regulatory polypeptides, and, like other members of this family, it has been associated with increased neuroplasticity.9,10 The GIP peptide and its receptor (GIPR) are ubiquitously distributed in the mammalian CNS,9,11-14 and homozygous Gipr-/-mice show impaired axonal regeneration after nerve crush13 and a reduced amount of proliferating cells in the adult hippocampus.9 We hypothesized that GIP and GIPR could be of particular importance in the context of SCI, due to their putative functions on synaptic plasticity,10,15 neurogenesis,9,10 and axonal outgrowth.13,16 Here, we describe dynamic changes in GIP and GIPR expression in the spinal cord of rats following injury by compression and describe in detail the pattern of expression of GIP and its receptor in the neural stem/progenitor cells in this injured tissue.
Methods
Subjects
Adult male Wistar rats (10 weeks old, 270–300 g body weight), obtained from the animal house of the Department of Pharmacology, Federal University of Santa Catarina (UFSC, Florianópolis, Brazil) were used throughout the experiments. Animals were housed in a room maintained at a constant temperature (22 ± 2°C) and humidity (60–80%), under a 12/12 h light/dark cycle and with food and water available ad libitum. All procedures were approved by the UFSC Ethics Committee (approval no. 23080.008708/2010-41) and were in agreement with the National Institutes of Health Animal Care Guidelines (NIH Publications no. 80-23).
Surgical procedure
SCI was induced as previously described17 with the following modifications: animals were previously treated with a single dose of wide spectrum antibiotic (oxytetracyclinchloridrate, 300 mg/kg, intramuscular). For surgery, rats were anesthetized with a 1:1 mixture of xylazine (10 mg/kg) and ketamine (70 mg/kg) given i.p. A 2-cm incision was made in the dorsal midline and the vertebral body was exposed. The paraspinal muscles were dissected from the T9 to T12 spinous processes and moved laterally with the aid of a forceps. After removal of the spinous process at the T10–T11 vertebral level, a small hole (1.5 mm diameter) at the T11 level was made with the help of a dental drill. This hole allowed the visualization of the spinal cord with the intact dura and the insertion of an embolectomy catheter (2-French Fogarty; Lemaitre Catheters, Burlington, VT).
Before each surgical procedure, the catheter diameter was calibrated to 3.0 mm with the aid of a caliper and a syringe pre-filled with distilled water. Following calibration, the catheter was inserted and advanced cranially for 1 cm, so that the center of the balloon rested at the level of the T10 vertebra. Then, the balloon was gently inflated up to a diameter of 3 mm, maintained during 1 min, deflated, and slowly removed. Muscles and skin were sutured in anatomical layers. Sham-operated animals were submitted to the same protocol; however, the catheter was not inserted into the epidural space. During the first 15 to 20 days post-SCI, urinary bladders were manually emptied twice daily, until micturition was restored.
Animal care procedures before and after surgery followed protocols previously decribed18 as well as the MASCIS (Multicenter Animal Spinal Cord Injury Study; PMID 8863191). Rats were divided into three experimental groups: naïve (no surgical intervention), sham-operated (surgical intervention, without catheter insertion), and SCI (surgical intervention with catheter insertion and inflation). SCI and sham-operated groups were euthanized at different times following surgery, and accounted for different phases of post-surgery recovery: 1) early phase of recovery (2, 6, and 24 h); 2) intermediate phase of recovery (14 days); and 3) late phase of recovery (28 days).
Evaluation of motor behavior
Basso, Beattie, and Bresnahan (BBB) open-field locomotion scale scoring was performed to evaluate the motor behavior of the SCI and sham-operated groups.19 Rats were placed individually in an open field. The 22-point (0–21) BBB scale was used to assess hindlimb locomotor recovery including joint movements, stepping ability, coordination, and trunk stability. A score of 21 indicates that an animal has consistent coordination and stability. The early stage of recovery shows paralysis and absence of limb movements and is given a score of 0. The animals were evaluated by two blinded researchers every 2 days from day 0 to day 28 following surgery in 5-min-long sessions.
Histopathology and immunohistochemistry
Sections 4-mm thick were obtained from paraffin-embedded rat spinal cords and immunohistochemistry detection was performed using polyclonal rabbit anti-GIPR antibody (1:300, kind gift of T.J. Kieffer, University of British Columbia, Canada), monoclonal rabbit anti-GIP antibody (1:250, LifeSpan BioSciences, Seattle, WA), or monoclonal mouse anti-nestin antibody (1:100, Millipore/Chemicon, Billerica, MA). Following quenching of endogenous peroxidase with 1.5% hydrogen peroxide in methanol (v/v) for 20 min, high-temperature antigen retrieval was performed by immersing the slides in a holder containing 10 mM trisodium citrate buffer pH 6.0, which in turn was immersed in a water bath at 95–98°C for 45 min, as previously described.14
The sections were incubated overnight in a humid chamber at room temperature with the primary antibodies described above. Then, slides were processed using the appropriate biotinylated secondary antibody and the streptavidin-horseradish peroxidase (HRP) complex (Vector Laboratories, Burlingame, CA) following the manufacturer's instructions. Subsequently, sections were developed with DAB (3,3'-diaminobenzidine) (DakoCytomation, Carpinteria, CA) in chromogen solution for an identical period and were further counterstained with Harris's hematoxylin. For double staining immunofluorescence reaction, sections were processed as described above, but the primary antibodies (anti-GIP/anti-nestin or anti-GIPR/anti-nestin) were simultaneously incubated overnight at 2–8°C. Next, slides were processed using chicken anti-rabbit Alexa Fluor 568 and goat anti-mouse Alexa Fluor 488 (1:300, Invitrogen, Carlsbad, CA), during 2 h at room temperature. After incubation, sections were rinsed with phosphate buffered saline (PBS) and coversliped with aqueous mounting media (Vectashield; Vector Laboratories, Burlingame, CA). Tissues from rats in control and SCI groups were placed on the same slide and processed under the exact same conditions. Negative control experiments included omission of primary antibody as well as substitution of the primary antibody by equivalent dilutions of non-immune rabbit or mouse immunoglobulin (IgG) serum, using the same staining protocol.
Imaging and image analysis
Relative quantification of GIP and GIPR immunostaining was performed as previously described.20 Two to three images from each section were captured using a Sight DS-5M-L1 digital camera (Nikon, Melville, NY) connected to an optical microscope (Eclipse 50i; Nikon) at 400x magnification. A threshold optical density (OD) that best discriminated staining from the background was obtained using the NIH ImageJ 1.36b imaging software (NIH, Bethesda, MD). Thresholds were determined using the Threshold Colour Plugins tool in NIH Image where images were tested at different thresholds to select the optimal contrast between positive staining and background staining. This threshold was record using the Macro Plugins tool in NIH Image and applied for all images. Total pixel intensity was determined and data were expressed as OD. The immunofluorescence samples were examined in a confocal microscope (DMI6000 B Microscope; Leica Microsystems, Wetzlar, Lahn-Dill, Germany). Data are shown as the average value from images of areas neighboring the site of injury.
RNA extraction and quantitative real-time PCR analysis
Spinal cord tissue from sham-operated or injured-animals were homogenized in 0.1 mL TRIzol© (Invitrogen), and ribonucleic acid (RNA) extraction was performed according to manufacturer's instructions. Purity and integrity of RNA were determined by the 260/280 nm absorbance ratio. Only preparations with ratios >1.8 were used in real-time polymerase chain reaction (RT-PCR) experiments. One μg RNA was used for complementary deoxyribonucleic acid (cDNA) synthesis using the Super-Strand III Reverse Transcriptase kit (Invitrogen). Expression of genes of interest was analyzed by quantitative PCR (qPCR) on an Applied Biosystems 7500 RT–PCR system using the Power SYBR kit (Applied Biosystems). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as endogenous control. The following primers pairs were used: GIP forw: 5′-CTCTTTGCCCAAGAGCCTCA- 3′, GIP rev: 5′-ATCAGAAGGTCCCTCAGCACA-3′, GIPR forw: 5′-TTCCCAGAAAGATGTGTCTCC-3′, and GIPR rev: 50 -CCCACCCCATAAAGAAGCT-3′. Cycle threshold (Ct) values were used to calculate fold changes in gene expression using the 2-ΔCt method. In all cases, reactions were performed in 15 μL reaction volumes.
Statistical analysis
Data are presented as mean ± standard error of mean (SEM). The statistical significance between groups was assessed by unpaired Student's t test or Mann-Whitney U test. P values <0.05 (p < 0.05) were considered to be indicative of significance. Data were analyzed using GraphPad Prism 4 software (San Diego, CA).
Results
Impaired locomotor activity and damage after SCI
Evaluation of motor behavior showed a BBB score of 0 for both hindlimbs of animals submitted to SCI on day 2 after surgery. This score increased gradually over time, reaching a score of 8 at 28 days post-SCI, characterizing a moderate paraplegia (Fig. 1). Spinal cords of injured rats analyzed at early stages post-trauma (2, 6, or 24 h) showed large cavity areas, marked necrosis, and hemorrhagic processes (Supplementary Fig. 1; see online supplementary material at http//:www.liebertpub.com). These histopathological alterations decreased over time. At 14 and 28 days after SCI, the gray matter of spinal cords was replaced by cavity areas and surrounded by ballooned cells, morphological findings that are consistent with neurodegeneration (Supplementary Fig. 2; see online supplementary material at http//:www.liebertpub.com). Sham-operated animals did not show changes in spinal cord cytoarchitecture or gray matter morphology (Supplementary Fig. 1 and 2).
FIG. 1.

Locomotor activity of rats following SCI. The degree of motor incapability was assessed every 2 days during 28 days after SCI, following criteria established by Basso, Beattie, and Bresnahan.19 *P < 0.05 in Mann-Whitney U test, comparing SCI and sham-operated groups. Data are represented as mean ± SEM; n = 8–10/group. SCI, spinal cord injury; SEM, standard error of mean.
Glucose-dependent insulinotropic peptide (GIP) and its receptor (GIPR) are up-regulated after SCI
Spinal cord gray matter neurons of naïve (Fig. 2A, C, and E) or sham-operated (Fig. 2B, D, and F) rats exhibited cytoplasmic GIPR-positive staining, mainly in motor neurons (Fig. 2). Surprisingly, areas neighboring the site of lesion in SCI animals showed a higher density of GIPR-immunoreactivity at 6 h (p = 0.0220) and 28 days (p = 0.0416) after injury when compared with sham-operated rats (Fig. 3A, C, F, and G). Although no significant changes in immunohistochemistry relative quantification were seen at other time-points, statistical analyses showed a trend to increase GIPR expression at 24 h after injury (p = 0.0574; Fig. 3G).
FIG. 2.
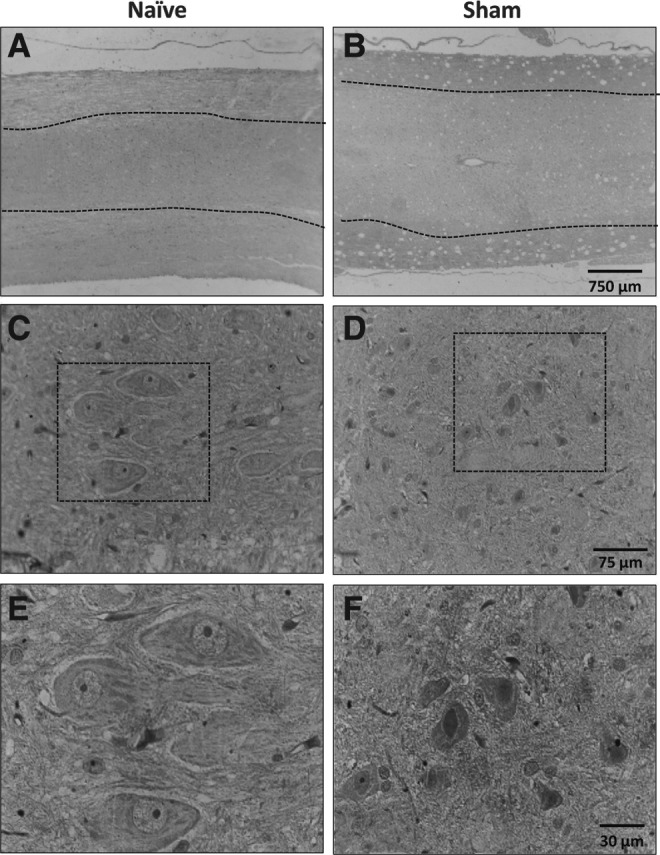
Analysis of GIPR expression in spinal cords of control groups. Representative images of GIPR immunostaining in spinal cord of naïve (A) or sham-operated (B) animals; gray matter is delimitated by dashed lines, with white matter on the surroundings. (C and D) Magnified images of the gray matter show moderate staining in cells morphologically compatible with neurons. (E and F) Magnified images of areas highlighted in C and D, where it is possible to observe the cytoplasmic staining for GIPR. GIPR, glucose-dependent insulinotropic peptide receptor. (A, B original magnification 20×; C, D original magnification 100×; E, F original magnification 400×.)
FIG. 3.

Analysis of GIPR expression in areas neighboring the site of injury. Representative image of GIPR immunostaining in spinal cord of sham-operated (A) or SCI animals 2 h (B), 6 h (C), 24 h (D), 14 days (E), and 28 days (F) after surgery. (G) Semi-quantitative analysis of GIPR levels in areas neighboring the site of injury. Data are shown as mean ± SEM, n = 5 animals/group. Results are represented as arbitrary units of optical density. *P ≤ 0.05 in Student's t test comparing SCI with the corresponding sham-operated groups. GIPR, glucose-dependent insulinotropic peptide receptor; SCI, spinal cord injury; SEM, standard error of mean. (Original magnification 400×.)
In naïve and control groups, GIPR immunostaining (Fig. 4) was found predominately in neuronal cell bodies, which were completely destroyed in the lesion epicenter (Supplementary Figs. 1 and 2). Therefore, digital computer-assisted semi-quantitative analysis of GIPR expression in the injury epicenter was impaired by a wide range of heterogeneity in histological features including hemorrhage and liquefactive necrosis among groups at different time-points following injury (Supplementary Figs. 1 and 2). Regardless, qualitative evaluation of GIPR immunostaining showed that lesion epicenter presented a large number of GIPR-positive balloon cells in chronic stages of SCI (14 and 28 days) (Fig. 4). To confirm immunohistochemistry relative quantification, we performed RT-PCR analysis in two experimental points (6 h and 28 days after SCI). Supplementary Figure 3A (see online supplementary material at http//:www.liebertpub.com) shows that GIPR RNA levels were increased in spinal cord tissue of injured-animals both 6 h and 28 days after injury, when compared with sham-operated rats.
FIG. 4.
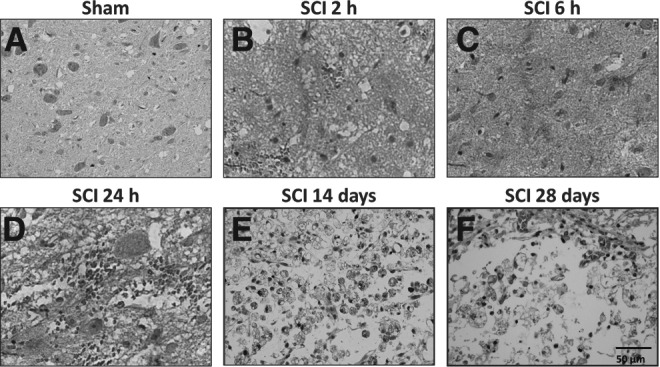
Analysis of GIPR expression in the injury epicenter. Representative images of GIPR immunostaining in the spinal cord of sham-operated (A) or SCI animals assessed 2 h (B), 6 h (C), 24 h (D), 14 days (E), or 28 days (F) after surgery. The injury epicenter of SCI animals assessed 14 days (E) or 28 days (F) (chronic phase) showed cytoplasmic granular staining in ballooned cells, which are morphologically compatible with degenerated neurons. GIPR, glucose-dependent insulinotropic peptide receptor; SCI, spinal cord injury. (Original magnification 400×.)
GIP-immunoreactivity was also increased in areas neighboring the site of injury in SCI animals compared with sham-operated rats, only when assessed 28 days after surgery (Fig. 5). No significant increase in GIP expression was observed in spinal cords of sham-operated animals during the corresponding time-course of SCI (Fig. 5G). Relative quantification at the injury epicenter showed that GIP levels were substantially higher in the spinal cord of SCI animals 24 h and 14 and 28 days after surgery when compared with sham-operated groups (Fig. 6). RT-PCR analysis confirms that GIP RNA levels were significantly increased in spinal cord tissue of injured animals at 28 days (Supplementary Fig. 3B).
FIG. 5.

Analysis of GIP expression in areas neighboring the site of injury. Representative images of GIP immunoreactivity in the spinal cord of sham-operated (A) or SCI animals assessed 2 h (B), 6 h (C), 24 h (D), 14 days (E), or 28 days (F) after surgery. (G) Semi-quantitative analysis of GIP levels in areas neighboring the site of injury. Each column represents the group mean and vertical lines represent the SEM of data obtained from five animals. Results are represented as arbitrary units of optical density. *P ≤ 0.05 in Student's t test comparing SCI with the corresponding sham-operated groups. GIP, glucose-dependent insulinotropic peptide; SCI, spinal cord injury; SEM, standard error of mean. (Original magnification 400×.)
FIG. 6.

Analysis of GIP expression in the injury epicenter. Representative images of GIP immunostaining in the spinal cord of sham-operated (A) or SCI animals assessed 2 h (B), 6 h (C), 24 h (D), 14 days (E), or 28 days (F) after surgery. The injury epicenter of SCI animals show granular staining in the cytoplasm and in extensions of neuronal cells. (G) Semi-quantitative assessment of GIP in the neuronal extensions present in the injury epicenter or in the gray matter of sham-operated animals. Data are shown as mean ± SEM, n = 5 animals/group. Results are represented as arbitrary units of optical density. *P ≤ 0.05 in Student's t test comparing SCI with corresponding sham-operated groups. GIP, glucose-dependent insulinotropic peptide; SCI, spinal cord injury; SEM, standard error of mean. (Original magnification 400×.)
SCI induces neural stem/progenitor cell proliferation in the spinal cord canal and injury epicenter
Figure 7 shows the immunoreactivity for nestin (marker for neural stem/progenitor cells) in the spinal cord canal of SCI animals at different post-surgery stages. Relative quantification showed that nestin immunoreactivity was higher in areas adjacent to the spinal cord canal in SCI animals 24 h and 14 days after injury (Fig. 7A, D, E, and G), but not at 2 and 6 h or 28 days (Fig. 7A–C, and F–G). Interestingly, only at 14 days after surgery the spinal cord of SCI animals expressed nestin in the injury epicenter (Fig. 8E and G). On the other hand, SCI groups evaluated at 2 h, 6 h, 24 h, and 28 days after injury did not express substantially nestin immunoreactivity in the injury epicenter (Fig. 8A–D and F–G).
FIG. 7.

Detection of neural progenitors in the medullary canal of the injured spinal cord. Representative images of nestin immunostaining in ependymal cells of the spinal cord canal of sham-operated (A) or SCI animals assessed 2 h (B), 6 h (C), 24 h (D), 14 days (E), or 28 days (F) after surgery. (G) Semi-quantitative analysis of nestin immunostaining. Data are shown as mean ± SEM, n = 5 animals/group. Results are represented as arbitrary units of optical density. *P ≤ 0.05 in Student's t test comparing SCI with corresponding sham-operated groups. With arrows highlight positive-stained cells. SCI, spinal cord injury; SEM, standard error of mean. (Original magnification 400×.)
FIG. 8.

Detection of neural progenitors in the epicenter of injured spinal cord. Representative images of nestin immunostaining in ependymal cells of the medullary canal of sham-operated (A) or SCI animals assessed 2 h (B), 6 h (C), 24 h (D), 14 days (E), or 28 days (F) after surgery. (G) Semi-quantitative analysis of nestin immunostaining. Data are shown as mean ± SEM, n = 5 animals/group. Results are represented as arbitrary units of optical density. *P ≤ 0.05 in Student's t test comparing SCI with corresponding sham-operated groups. White arrows highlight positive-stained cells. SCI, spinal cord injury; SEM, standard error of mean. (Original magnification 400×.)
Nestin-positive cells co-localize with GIP and GIPR in the spinal cord canal and injury epicenter
Immunofluorescent double-staining using anti-nestin and anti-GIP or anti-GIPR antibodies revealed that ependymal cells of the spinal cord canal stained for nestin were also positive for GIP (Fig. 9 D–F), but not for GIPR (Fig. 9 A–C), 24 h after SCI. On the other hand, progenitor cells present at the injury epicenter showed double staining for nestin and both GIP and GIPR on day 14 after surgery (Fig. 9 G–L).
FIG. 9.
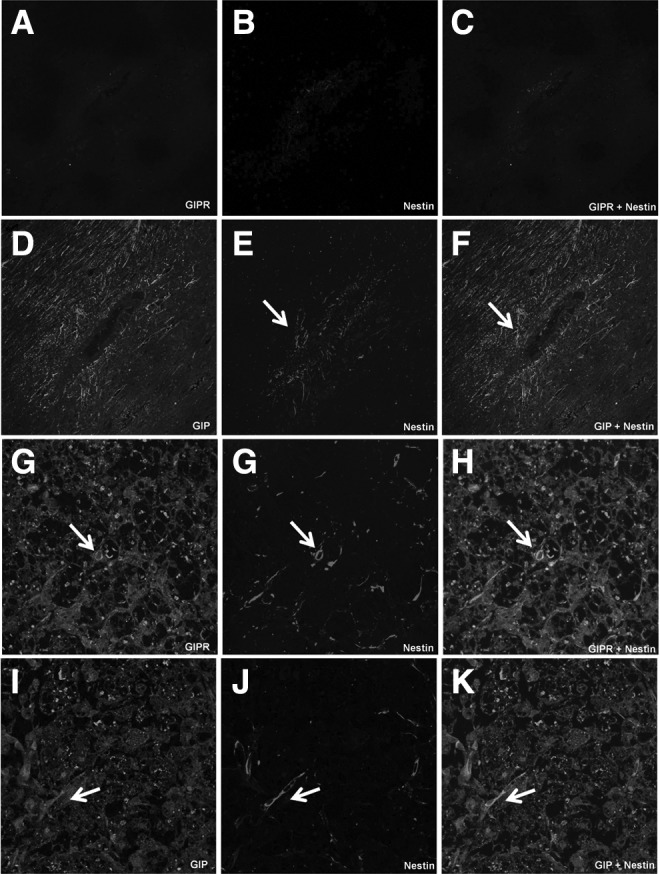
Detection of GIP and GIPR in nestin-positive neuronal progenitors of spinal cord injured-rats. Nestin-positive neuronal progenitors of SCI animals assessed 24 h after surgery co-express GIP (D–F) but not GIPR (A–C). Nestin-positive neuronal progenitors of SCI animals assessed 14 days after surgery co-express GIPR (G–I) and GIP (J, L, and M). White arrows highlight positive-stained cells. GIP, glucose-dependent insulinotropic peptide; GIPR, glucose-dependent insulinotropic peptide receptor; SCI, spinal cord injury. (Original magnification 400×.)
Discussion
Neurons of the adult CNS are incapable of recovering axonal and dendritic connections after injury. An interruption within the communication between healthy neurons and a cascade of events can lead to neuronal degeneration and cell death. In this context, CNS injuries result in immediate neuronal and glial cell death, followed by an insidious late pathological process. The challenge in treating spinal cord-injured patients is intimately associated with the long period during which secondary injuries can occur, including several vascular, cellular, and biochemical events that result in the progressive loss of neurons, axonal demyelination, and the establishment of an unfavorable environment.21,22 Therapeutic strategies to compensate for the loss of neurons and stimulate the reconstruction of disrupted neuronal circuits are used to stimulate the proliferation of endogenous stem/progenitor cells, promoting neuronal differentiation and maturation and facilitating neurite outgrowth and synaptic integration (neurogenesis).4,5,23-25 It is known that GIP and its receptor (GIPR) are widely expressed in nervous tissue, always co-localized with neuronal markers,12 and that GIP and its receptor are related to neurogenesis,9,10 axonal outgrowth,13,16 and other neuroprotective functions. However, the roles of GIP and GIPR in SCI recovery are still not completely understood. Here, we described temporal and regional expression of GIP and GIPR in injured spinal cord of rats submitted to compression-induced traumatic SCI. GIP and GIPR immunostaining in spinal cord were associated with morphological changes during acute (2, 6, and 24 h) and chronic (14 and 28 day) phases of this traumatic SCI model.
Several studies have demonstrated that insulin and incretins have other noble functions beyond the control of cellular energy metabolism.26-29 Insulin is an anabolic hormone essential for the maintenance of glucose homeostasis, and important for cellular growth and differentiation.30 Recent studies have revealed that activation of neuronal insulin receptors results in a decrease in amyloid beta-induced synapto-toxicity, suggesting that insulin may play a role in neuroprotection.27,31 Wu and colleagues investigated insulin's neuroprotective effects in the treatment of SCI, and found that systemic administration of insulin has anti-apoptotic and anti-inflammatory effects on the pathogenesis of SCI.32 Insulin acts on specific membrane receptors, which share great structural and functional similarities with insulin-like growth factor 1 (IGF-1) receptor.33 Several studies have reported that IGF-1 attenuates the damage associated with SCI, and reduces the stress-induced tissue swelling and electrophysiological imbalance of the spinal cord.34-36 An important effect of IGF-1 is its involvement with the control of axonal growth, because IGF-1 specifically enhances the extent and rate of murine corticospinal motor neuron axonal outgrowth in culture.37
Interestingly, the incretins GIP and GLP are positive modulators of the effects of insulin, expression of IGF-1 receptor, neuronal plasticity, neurogenesis, and neuroprotection.38-41 In the present study, we evaluated for the first time the pattern of GIP and GIPR expression in the spinal cord of rats submitted to SCI. Spinal cord samples collected 6 h (acute phase) or 28 days (chronic phase) after surgery showed increased GIPR levels in neurons neighboring the site of injury when compared with gray matter neurons of sham-operated animals. Moreover, prominent GIPR immunostaining was observed in balloon cells at the site of injury in the chronic phase, which is in agreement with the presence of degenerated neurons. However, the temporal pattern of GIPR expression at the site of injury was not quantified due to the large morphological differences observed in the injury epicenter when comparing the acute and chronic phases of SCI. On the other hand, we found changes in the GIP immunoreactivity in neurons adjacent to the site of injury only at day 28 after surgery, when compared with the gray matter of sham-operated animals. Semi-quantitative assessment of GIP immunostaining in the injury epicenter showed that GIP was up-regulated 24 h as well as 14 and 28 days after SCI compared with corresponding sham-operated groups. Therefore, up-regulation of GIP and its receptor after SCI could be involved in neuronal regeneration.
Experimental therapeutic interventions suggest that residual regeneration of CNS neurons can be stimulated if injured axons are exposed to growth-favorable environmental conditions.42-45 Several studies have shown that cell genesis and synaptic plasticity of stem cells can be influenced by stress, enriched environments, and physical exercise.46-50 In fact, it has been demonstrated that spinal cord progenitor cells are sensitive to tissue injury.51 The reaction of these cells following injury involves division, migration, and maturation of neural precursors.52 When CNS injury causes acute demyelination, progenitor cells can proliferate and differentiate into mature myelinating oligodendrocytes.53-55 Therefore, stem cells can contribute to cell replacement and regeneration following injury to the CNS.
Ependymal cells of the spinal canal are a source of adult spinal cord progenitor cells.56,57 Temporal and spatial pattern of proliferation, differentiation, and migration of neural progenitors as well as the potential function of these cells in spinal cord traumatic processes are the focus of much research. To identify the presence of progenitor cells in spinal cord of animals submitted to compression-induced traumatic SCI, histological sections from spinal cords of sham-operated and SCI animals were assessed at different experimental time-points to detect nestin, a neural stem/progenitor cell marker. We showed here that the number of nestin-positive cells was increased in the medullary canal and in the injury epicenter of SCI animals when assessed 24 h and 14 days after surgery, respectively. On the other hand, sham-operated animals evaluated at different moments after surgery showed no positive staining to nestin. These findings are in agreement with a previous study performed by Cizkova and colleagues, which showed that traumatic SCI results in a rapid induction of nestin expression in ependymal cells of the spinal cord canal.58 Our results also support an earlier study that described a significant increase in progenitor cells in the injury epicenter 7 days but not 28 days after surgery.59 The absence of progenitor cells in the injury epicenter of SCI animals 28 days after surgery may result from the process of cell differentiation, which leads to the loss of nestin expression. Two very interesting findings of our study are that: 1) GIP was detected in progenitor cells in the spinal cord canal during the acute phase of SCI; and 2) both GIP and GIPR are found in progenitor cells at the injury epicenter during the chronic phase in SCI animals. The presence of GIP and its receptor on progenitor cells suggests that the up-regulation of these molecules could be important for cell replacement after SCI.
Taken together, our results suggest that GIP and GIPR are associated with damage, proliferation (neurogenesis), and/or repair processes in the CNS (axonal growth and/or dendritic) through paracrine or autocrine mechanisms. Nevertheless, additional studies using different experimental models of axonal regeneration, dendritic growth, proliferation, and differentiation of neural progenitors are essential to improve our understanding of the neuroprotective role of GIP and its receptor, and of the possible mechanism involved in GIP-mediated CNS repair.
Supplementary Material
Acknowledgments
This work was supported by grants from the following Brazilian funding agencies: Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES). TLM and CPF received a research productivity grant from the Araucaria Research Foundation of Parana State, Brazil, and FAPERJ, respectively.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Lindvall O., and Kokaia Z. (2010). Stem cells in human neurodegenerative disorders—time for clinical translation? J. Clin. Invest. 120, 29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison D.J., and Ditor D.S. (2014). Immune dysfunction and chronic inflammation following spinal cord injury. Spinal Cord 53, 14–18 [DOI] [PubMed] [Google Scholar]
- 3.Kwon B.K., Oxland T.R., and Tetzlaff W. (2002). Animal models used in spinal cord regeneration research. Spine 27, 1504–1510 [DOI] [PubMed] [Google Scholar]
- 4.Vawda R., and Fehlings M.G. (2013). Mesenchymal cells in the treatment of spinal cord injury: current and future perspectives. Curr. Stem Cell Res. Ther. 8, 25–38 [DOI] [PubMed] [Google Scholar]
- 5.Wu Y., Satkunendrarajah K., and Fehlings M.G. (2014). Riluzole improves outcome following ischemia-reperfusion injury to the spinal cord by preventing delayed paraplegia. Neuroscience 265, 302–312 [DOI] [PubMed] [Google Scholar]
- 6.Franklin R.J., and Ffrench-Constant C. (2008). Remyelination in the CNS: from biology to therapy. Nat. Rev. Neurosci. 9, 839–855 [DOI] [PubMed] [Google Scholar]
- 7.Silver J., Schwab M.E., and Popovich P.G. (2014). Central nervous system regenerative failure: role of oligodendrocytes, astrocytes, and microglia. Cold Spring Harb. Perspect. Biol. 7, a020602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spejo A.B., and Oliveira A.L. (2014). Synaptic rearrangement following axonal injury: old and new players. Neuropharmacology 96(Pt A), 113-123 [DOI] [PubMed] [Google Scholar]
- 9.Nyberg J., Anderson M.F., Meister B., Alborn A.M., Strom A.K., Brederlau A., Illerskog A.C., Nilsson O., Kieffer T.J., Hietala M.A., Ricksten A., and Eriksson P.S. (2005). Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J. Neurosci. 25, 1816–1825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Figueiredo C.P., Pamplona F.A., Mazzuco T.L., Aguiar A.S., Jr., Walz R., and Prediger R.D. (2010). Role of the glucose-dependent insulinotropic polypeptide and its receptor in the central nervous system: therapeutic potential in neurological diseases. Behav. Pharmacol. 21, 394–408 [DOI] [PubMed] [Google Scholar]
- 11.Usdin T.B., Mezey E., Button D.C., Brownstein M.J., and Bonner T.I. (1993). Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology 133, 2861–2870 [DOI] [PubMed] [Google Scholar]
- 12.Nyberg J., Jacobsson C., Anderson M.F., and Eriksson P.S. (2007). Immunohistochemical distribution of glucose-dependent insulinotropic polypeptide in the adult rat brain. J. Neurosci. Res. 85, 2099–2119 [DOI] [PubMed] [Google Scholar]
- 13.Buhren B.A., Gasis M., Thorens B., Muller H.W., and Bosse F. (2009). Glucose-dependent insulinotropic polypeptide (GIP) and its receptor (GIPR): cellular localization, lesion-affected expression, and impaired regenerative axonal growth. J. Neurosci. Res. 87, 1858–1870 [DOI] [PubMed] [Google Scholar]
- 14.Figueiredo C.P., Bicca M.A., Latini A., Prediger R.D., Medeiros R., and Calixto J.B. (2011). Folic acid plus alpha-tocopherol mitigates amyloid-beta-induced neurotoxicity through modulation of mitochondrial complexes activity. J. Alzheimers Dis. 24, 61–75 [DOI] [PubMed] [Google Scholar]
- 15.Gault V.A., and Holscher C. (2008). Protease-resistant glucose-dependent insulinotropic polypeptide agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. J. Neurophysiol. 99, 1590–1595 [DOI] [PubMed] [Google Scholar]
- 16.Abankwa D., Kury P., and Muller H.W. (2002). Dynamic changes in gene expression profiles following axotomy of projection fibres in the Mammalian CNS. Mol. Cell. Neurosci. 21, 421–435 [DOI] [PubMed] [Google Scholar]
- 17.Vanicky I., Urdzikova L., Saganova K., Cizkova D., and Galik J. (2001). A simple and reproducible model of spinal cord injury induced by epidural balloon inflation in the rat. J. Neurotrauma 18, 1399–1407 [DOI] [PubMed] [Google Scholar]
- 18.Santos-Benito F.F., Munoz-Quiles C., and Ramon-Cueto A. (2006). Long-term care of paraplegic laboratory mammals. J. Neurotrauma 23, 521–536 [DOI] [PubMed] [Google Scholar]
- 19.Basso D.M., Beattie M.S., and Bresnahan J.C. (1995). A sensitive and reliable locomotor rating scale for open field testing in rats. J. Neurotrauma 12, 1–21 [DOI] [PubMed] [Google Scholar]
- 20.Figueiredo C.P., Antunes V.L., Moreira E.L., de Mello N., Medeiros R., Di Giunta G., Lobao-Soares B., Linhares M., Lin K., Mazzuco T.L., Prediger R.D., and Walz R. (2011). Glucose-dependent insulinotropic peptide receptor expression in the hippocampus and neocortex of mesial temporal lobe epilepsy patients and rats undergoing pilocarpine induced status epilepticus. Peptides 32, 781–789 [DOI] [PubMed] [Google Scholar]
- 21.Oyinbo C.A. (2011). Secondary injury mechanisms in traumatic spinal cord injury: a nugget of this multiply cascade. Acta neurobiol. Exper. (Wars) 71, 281–299 [DOI] [PubMed] [Google Scholar]
- 22.Kwon B.K., Tetzlaff W., Grauer J.N., Beiner J., and Vaccaro A.R. (2004). Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 4, 451–464 [DOI] [PubMed] [Google Scholar]
- 23.Ming G.L., and Song H. (2005). Adult neurogenesis in the mammalian central nervous system. Ann. Rev. Neurosci. 28, 223–250 [DOI] [PubMed] [Google Scholar]
- 24.Thuret S., Moon L.D., and Gage F.H. (2006). Therapeutic interventions after spinal cord injury. Nat. Rev. Neurosci. 7, 628–643 [DOI] [PubMed] [Google Scholar]
- 25.Fehlings M.G., Wilson J.R., and Cho N. (2014). Methylprednisolone for the treatment of acute spinal cord injury: counterpoint. Neurosurgery 61 (Suppl 1), 36–42 [DOI] [PubMed] [Google Scholar]
- 26.Unger J.W., Livingston J.N., and Moss A.M. (1991). Insulin receptors in the central nervous system: localization, signalling mechanisms and functional aspects. Prog. Neurobiol. 36, 343–362 [DOI] [PubMed] [Google Scholar]
- 27.Zhao W.Q., De Felice F.G., Fernandez S., Chen H., Lambert M.P., Quon M.J., Krafft G.A., and Klein W.L. (2008). Amyloid beta oligomers induce impairment of neuronal insulin receptors. F.A.S.E.B. 22, 246–260 [DOI] [PubMed] [Google Scholar]
- 28.Rotz M.E., Ganetsky V.S., Sen S., and Thomas T.F. (2014). Implications of incretin-based therapies on cardiovascular disease. Int. J. Clin. Pract. 69, 531–549 [DOI] [PubMed] [Google Scholar]
- 29.Kealy J., Bennett R., and Lowry J.P. (2014). Real-time effects of insulin-induced hypoglycaemia on hippocampal glucose and oxygen. Brain Res. 1598, 76–87 [DOI] [PubMed] [Google Scholar]
- 30.Saltiel A.R., and Kahn C.R. (2001). Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414, 799–806 [DOI] [PubMed] [Google Scholar]
- 31.De Felice F.G., Vieira M.N., Bomfim T.R., Decker H., Velasco P.T., Lambert M.P., Viola K.L., Zhao W.Q., Ferreira S.T., and Klein W.L. (2009). Protection of synapses against Alzheimer's-linked toxins: insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. U. S. A. 106, 1971–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X.H., Yang S.H., Duan D.Y., Cheng H.H., Bao Y.T., and Zhang Y. (2007). Anti-apoptotic effect of insulin in the control of cell death and neurologic deficit after acute spinal cord injury in rats. J. Neurotrauma 24, 1502–1512 [DOI] [PubMed] [Google Scholar]
- 33.Siddle K., Urso B., Niesler C.A., Cope D.L., Molina L., Surinya K.H., and Soos M.A. (2001). Specificity in ligand binding and intracellular signalling by insulin and insulin-like growth factor receptors. Biochem. Soc. Trans. 29, 513–525 [DOI] [PubMed] [Google Scholar]
- 34.Sharma H.S., Nyberg F., Gordh T., Alm P., and Westman J. (2000). Neurotrophic factors influence upregulation of constitutive isoform of heme oxygenase and cellular stress response in the spinal cord following trauma. An experimental study using immunohistochemistry in the rat. Amino Acids 19, 351–361 [PubMed] [Google Scholar]
- 35.Winkler T., Sharma H.S., Stalberg E., and Badgaiyan R.D. (2000). Neurotrophic factors attenuate alterations in spinal cord evoked potentials and edema formation following trauma to the rat spinal cord. Acta Neurochir. Suppl. 76, 291–296 [DOI] [PubMed] [Google Scholar]
- 36.Hwang D.H., Shin H.Y., Kwon M.J., Choi J.Y., Ryu B.Y., and Kim B.G. (2014). Survival of neural stem cell grafts in the lesioned spinal cord is enhanced by a combination of treadmill locomotor training via insulin-like growth factor-1 signaling. J. Neurosci. 34, 12788–12800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ozdinler P.H., and Macklis J.D. (2006). IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nature Neurosci. 9, 1371–1381 [DOI] [PubMed] [Google Scholar]
- 38.Salehi M., Aulinger B.A., and D'Alessio D.A. (2008). Targeting beta-cell mass in type 2 diabetes: promise and limitations of new drugs based on incretins. Endocrine Rev. 29, 367–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leen J.L., Izzo A., Upadhyay C., Rowland K.J., Dube P.E., Gu S., Heximer S.P., Rhodes C.J., Storm D.R., Lund P.K., and Brubaker P.L. (2011). Mechanism of action of glucagon-like peptide-2 to increase IGF-I mRNA in intestinal subepithelial fibroblasts. Endocrinology 152, 436–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Faivre E., Hamilton A., and Holscher C. (2012). Effects of acute and chronic administration of GIP analogues on cognition, synaptic plasticity and neurogenesis in mice. Eur. J. Pharmacol. 674, 294–306 [DOI] [PubMed] [Google Scholar]
- 41.Faivre E., Gault V.A., Thorens B., and Holscher C. (2011). Glucose-dependent insulinotropic polypeptide receptor knockout mice are impaired in learning, synaptic plasticity, and neurogenesis. J. Neurophysiol. 105, 1574–1580 [DOI] [PubMed] [Google Scholar]
- 42.Filous A.R., Tran A., Howell C.J., Busch S.A., Evans T.A., Stallcup W.B., Kang S.H., Bergles D.E., Lee S.I., Levine J.M., and Silver J. (2014). Entrapment via synaptic-like connections between NG2 Proteoglycan+ cells and dystrophic axons in the lesion plays a role in regeneration failure after spinal cord injury. J. Neurosci. 34, 16369–16384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee D.H., Luo X., Yungher B.J., Bray E., Lee J.K., and Park K.K. (2014). Mammalian target of rapamycin's distinct roles and effectiveness in promoting compensatory axonal sprouting in the injured CNS. J. Neurosci. 34, 15347–15355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu B., Park D., Ohtake Y., Li H., Hayat U., Liu J., Selzer M.E., Longo F.M., and Li S. (2015). Role of CSPG receptor LAR phosphatase in restricting axon regeneration after CNS injury. Neurobiol. Dis. 73, 36–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silver J., and Miller J.H. (2004). Regeneration beyond the glial scar. Nat. Rev. Neurosci. 5, 146–156 [DOI] [PubMed] [Google Scholar]
- 46.Nunez-Espinosa C., Ferreira I., Rios-Kristjansson J.G., Rizo-Roca D., Godoy M.D., Rico L.G., Rubi-Sans G., Torrella J.R., Pages T., Petriz J., and Viscor G. (2014). Effects of Intermittent Hypoxia and Light Aerobic Exercise on Circulating Stem Cells and Side Population, After Strenuous Eccentric Exercise in Trained Rats. Curr. Stem Cell Res. Ther. 10, 132–139 [DOI] [PubMed] [Google Scholar]
- 47.Monteiro B.M., Moreira F.A., Massensini A.R., Moraes M.F., and Pereira G.S. (2014). Enriched environment increases neurogenesis and improves social memory persistence in socially isolated adult mice. Hippocampus 24, 239–248 [DOI] [PubMed] [Google Scholar]
- 48.Amiri F., Jahanian-Najafabadi A., and Roudkenar M.H. (2014). In vitro augmentation of mesenchymal stem cells viability in stressful microenvironments: in vitro augmentation of mesenchymal stem cells viability. Cell Stress Chaperones 2, 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bechara R.G., and Kelly A.M. (2013). Exercise improves object recognition memory and induces BDNF expression and cell proliferation in cognitively enriched rats. Behav. Brain Res. 245, 96–100 [DOI] [PubMed] [Google Scholar]
- 50.Rizzi S., Bianchi P., Guidi S., Ciani E., and Bartesaghi R. (2011). Impact of environmental enrichment on neurogenesis in the dentate gyrus during the early postnatal period. Brain Res. 1415, 23–33 [DOI] [PubMed] [Google Scholar]
- 51.Obermair F.J., Schroter A., and Thallmair M. (2008). Endogenous neural progenitor cells as therapeutic target after spinal cord injury. Physiology 23, 296–304 [DOI] [PubMed] [Google Scholar]
- 52.Okano H., Sakaguchi M., Ohki K., Suzuki N., and Sawamoto K. (2007). Regeneration of the central nervous system using endogenous repair mechanisms. J. Neurochem. 102, 1459–1465 [DOI] [PubMed] [Google Scholar]
- 53.McTigue D.M., Horner P.J., Stokes B.T., and Gage F.H. (1998). Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J. Neurosci. 18, 5354–5365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Franklin R.J., Gilson J.M., and Blakemore W.F. (1997). Local recruitment of remyelinating cells in the repair of demyelination in the central nervous system. J. Neurosci. Res. 50, 337–344 [DOI] [PubMed] [Google Scholar]
- 55.Gensert J.M., and Goldman J.E. (1997). Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron 19, 197–203 [DOI] [PubMed] [Google Scholar]
- 56.Meletis K., Barnabe-Heider F., Carlen M., Evergren E., Tomilin N., Shupliakov O., and Frisen J. (2008). Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 6, e182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foret A., Quertainmont R., Botman O., Bouhy D., Amabili P., Brook G., Schoenen J., and Franzen R. (2010). Stem cells in the adult rat spinal cord: plasticity after injury and treadmill training exercise. J. Neurochem. 112, 762–772 [DOI] [PubMed] [Google Scholar]
- 58.Cizkova D., Nagyova M., Slovinska L., Novotna I., Radonak J., Cizek M., Mechirova E., Tomori Z., Hlucilova J., Motlik J., Sulla I., Jr., and Vanicky I. (2009). Response of ependymal progenitors to spinal cord injury or enhanced physical activity in adult rat. Cell. Mol. Neurobiol. 29, 999–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McTigue D.M., Wei P., and Stokes B.T. (2001). Proliferation of NG2-positive cells and altered oligodendrocyte numbers in the contused rat spinal cord. J. Neurosci. 21, 3392–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.