

Abstract
Background
The epithelial-mesenchymal transition (EMT) has been shown to be involved in the process of invasion and metastasis of prostate cancer. SIRT1 is the mammalian homologue of the silent information regulator 2 (Sir2) gene, and is abnormally expressed in prostate cancer cells. Therefore, it is hypothesized that SIRT1 mediates the invasion/metastatic ability of prostate cancer via EMT regulation. This study thus investigated the effect of SIRT1 gene on the invasion and migration of prostate cancer cell line PC-3 via the small interference RNA (siRNA) against SIRT1.
Material/Methods
SiRNA construct was transfected into PC-3 cells, which were tested for the cell migration and invasion ability by scratch assay and Transwell migration assay, respectively. Expression levels of vimentin, E-cadherin, and N-cadherin were further quantified by Western blotting and RT-PCR.
Results
Both mRNA and protein levels of SIRT1 were depressed after siRNA transfection, along with weakened migration and invasion ability of PC-3 cells. Elevated E-cadherin and suppressed N-cadherin and vimentin were observed in those transfected cells.
Conclusions
The silencing of SIRT1 gene in PC-3 cells can suppress the movement, migration, and invasion functions of prostate cancer cells, possibly via the down-regulation of mesenchymal markers vimentin and N-cadherin accompanied with up-regulation of epithelial marker N-cadherin, thus reversing the EMT process.
MeSH Keywords: Cellulose 1,4-beta-Cellobiosidase; Precursor Cells, B-Lymphoid; Sirtuin 1
Background
Prostate cancer is a common malignant tumor in men; its occurrence and mortality have been increasing in recent years, largely due to the distal metastasis of tumors [1,2]. The mechanism underlying the distal metastasis of prostate cancer has not yet been fully defined [3,4]. The study of the molecular mechanism of distal metastasis of prostate cancer, therefore, can benefit clinical interventions for slowing or even inhibiting tumor invasion [5,6]. SIRT1, which is the mammalian homologue of silent information regulator 2 (Sir2) gene, participates in a wide array of cellular processes, including proliferation, apoptosis, and invasion, via non-histone deacetylation such as DNA repair protein P300, P73 NF-κB, tumor suppressor protein FOXO transcriptional factor, and P53 [7,8]. It can also maintain the silencing status of chromatin and stability of chromosome via histone modification, such as the de-acetylation at H3K9, H1K26, and H4K16 loci [9,10]. A dual-side role of SIRT1 gene in tumor has been suggested because it can either facilitate tumor growth via up-regulating oncogenes and epithelial-mesenchymal transition (EMT), or inhibit the tumor progression via suppressing angiogenesis and inflammation. In colorectal and prostate cancer, the expression profile of SIRT1 gene has an elevated expression level, whereas in other tumors, such as bladder cancer, malignant glioma, or ovary cancer tissues, it has shown a lower level than normal tissues.
EMT is known to play an important role in tumor development and invasion. It has been reported that SIRT1 plays an important role in EMT of prostate cancer cells, but its precise mechanism remains unclear. Some studies have reported elevated expression of E-cadherin after SIRT1 gene silencing, thus increasing the cell adhesion and suppressing tumor invasion, possibly via inhibiting the Wnt-signaling pathway [11,12]. This study constructed an SIRT1-interference vector, which was transfected into prostate cancer cell line PC-3, to observe the effect of SIRT1 on the invasion and migration of prostate cancer cells, along with the role of SIRT1 gene in EMT.
Material and Methods
Material
Prostate cancer cell line PC-3 cells were purchased from the Chinese Academy of Sciences Cell Bank, and were cultured in medium containing 10% fetal calf serum RPMI1640/F12 (purchased from Gibco, USA). Human prostate fibroblasts PrEC (purchased from Clonetics Corporation, USA) were cultured in prostatic epithelial growth medium PrECGM (purchased from Beyotime, China). RIRA cell lysate was purchased from Beyotime (China). Cells were cultured at 37°C, 5% CO2 incubator, using 0.25% trypsin for digestion and passage (purchased from Beyotime, China). During cell logarithmic growth phase, we took the appropriate concentration to seed in a 96-well culture plate or 25 cm2 flasks. Empty vector, Scrambled siRNA, and SIRT1 siRNA were all purchased from Jima Biotechnology Company (Shanghai); rabbit anti-human SIRT1 monoclonal antibody (dilution 1:2000) was purchased from Abcam Company (USA); rabbit anti-human E-cadherin, fluctuations in protein (Vimentin) and N-cadherin monoclonal antibodies were all purchased from Abgent (USA); fluorescence quantitative polymerase chain reaction kit was purchased from Qiagen (Germany); and the reverse transcription kit was purchased from Takara (Japan).
Small interference RNA (siRNA) transfection
SiRNA was synthesized based on human SIRT1 (Gen Bank No. NM_012238) RNAi target sequence (Forward: 5′-GAAGU UGACC UCCUCA UUGUdT dT-3′; Reverse: 5′-ACAAU GAGGA GGUCA ACUUC dTdT-3′). PC-3 cells (Cell bank of Chinese Academy of Science, China) were seeded into a 6-well plate and transfected by 20 μM siRNA with the help of Lipofectamine 2000 reagents (Invitrogen, USA) following manual instructions.
Groups
Both blank control (blank vector transfection) and negative control (20 μM scramble siRNA) groups were processed in parallel; 72 hours after transfection, cells were collected for further experiments.
Scratch assay
Cells (5×105 per well) were seeded into a 6-well plate until reaching 90% confluence. A scratch was made in each well using 10-μL pipette tips. After PBS washing (3 times), the width of the scratch was measured under an inverted microscope at different time points after transfection. Measurement was repeated 3 times.
Transwell assay
Cells at log-phase were fasted for 12 hours in serum-free medium. After rinsing, cells were re-suspended into 6×105 per mL. We added 0.1 mL cell suspensions into 24-well Transwell chambers. The lower chamber was filled with 0.5 mL DMEM medium containing fetal bovine serum (FBS, Gibco, USA). After 24-hour incubation, cells were stained by 0.1% crystal violet and rinsed in PBS, followed by 33% acetic acid and then inverted under a microscope (×200) microporous membrane lower cell, and 10 randomly selected fields were used to calculate the average.
Clonal formation assay
Cells at log-phase were digested by 0.25% pepsin and serially diluted into cell suspensions, which were inoculated into culture dishes for 10-day incubation. After the formation of visible colonies, cells were rinsed in PBS and fixed in methanol for 15 min. Giemsa buffer was used to stain the cells for 10 min, followed by air drying. The number of colonies containing more than 10 individual cells was determined under low magnification for calculating colony formation rate. Measurement was repeated 3 times.
Western blotting
PCR products were analyzed by agarose gel electrophoresis. Measurement was repeated 3 times and represented by relative expression: GAPDH gray value gene and the gray value ratio. We took the logarithmic growth phase cells and washed them in PBS. Using 1% NP-40 lysis buffer to process lysis, cells were extracted for total protein, and were separated in SDS-PAGE and transferred onto a PVDF membrane. After blocking by 5% defatted milk powder, monoclonal antibodies against SIRT1, vimentin, E-cadherin, and N-cadherin (1:1000, Abcam, USA) were applied for overnight incubation. After rinsing in TBST, secondary antibody (1:2000, Abcam, USA) was added for further 1-hour incubation. An ECL chromogenic kit was used to develop the membrane, which was exposed in a dark room. The image was captured and analyzed by Quantity One software.
RT-PCR
Total RNA was extracted by Trizol kit and was used as the template for synthesizing cDNA in a reverse transcription kit (Takara, Japan). RT-PCR was performed in a fluorescent quantitative PCR kit (QIAGEN, Germany) using specific primers for vimentin, E-cadherin, and N-cadherin (Table 1). Amplification products were separated by agarose gel electrophoresis and quantified by ultraviolet spectrometer. All experiments were performed in triplicates against a GAPDH housekeeping gene.
Table 1.
Primer sequence.
| Target gene | Sequence (5′-3′) | Fragment length (bp) |
|---|---|---|
| SIRT1 | Forward: CTCCT CTCTT GACTC GCCAT | 134 |
| Reverse: CGGTG TAACC CCTCC AAGTC | ||
| GAPDH | Forward: GGGAA ACTGT GGCG TGAT | 165 |
| Reverse: AAAGG TGGAG GAGTG GGT | ||
| E-cadherin | Forward: CTTCA ATCCC ACCAC G | 184 |
| Reverse: AAATG CCATC GTTGT TC | ||
| Vimentin | Forward: TCGCC AACTA CATCG ACAAG | 339 |
| Reverse: AGGGT GTTTT CGGCT TC | ||
| N-cadherin | Forward: CCACG CCGAG CCCCA GTATC | 232 |
| Reverse: CCCCC AGTCG TTCAG GTAA TCA |
Statistical analysis
SPSS 19.0 software was used to process all collected data, and those that fit normal distribution are presented as mean ± standard deviation (SD). Multiple-group comparison was performed by analysis of variance (ANOVA), followed by LSD test for between-group comparison. Statistical significance was defined when p<0.05.
Results
Transfection efficiency
Using RT-PCR and Western blotting, we found significant suppression of SIRT1 gene expression in transfected PC-3 cells (Figure 1), suggesting the silencing of endogenous gene expression by siRNA.
Figure 1.
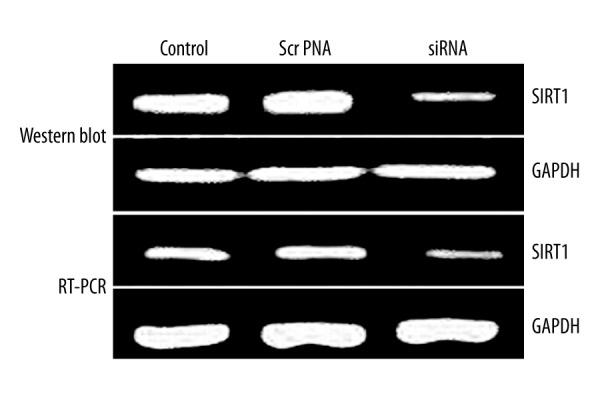
SIRT1 gene expression in PC-3 cells.
PC-3 cells mobility and migration
By the scratch assay, we found depressed mobility of siRNA-transfected PC-3 cells, compared to those in blank control and scrambled RNA-transfected cells (Figure 2), suggesting the inhibition of migration ability after silencing SIRT1 gene.
Figure 2.
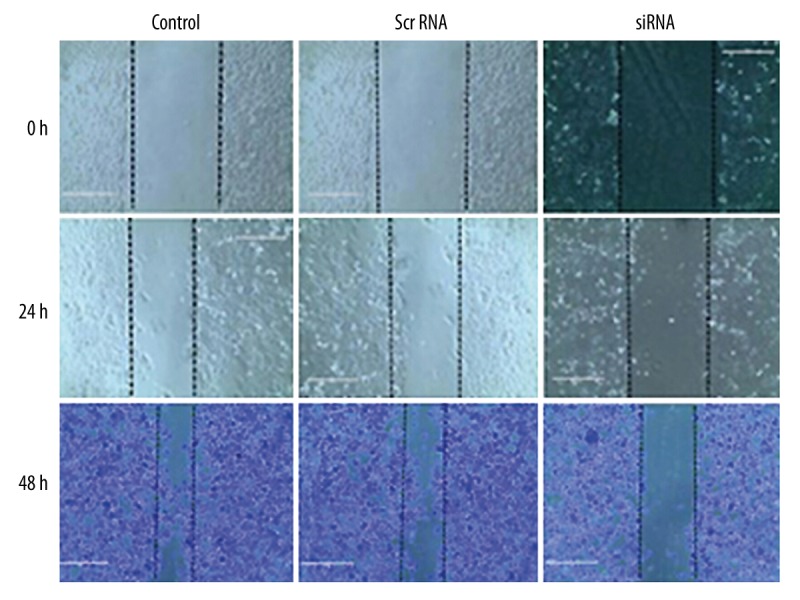
Transfected PC cells scratch test results.
Cell migration and invasion ability
In Transwell cell migration assay, siRNA-transfected cells had lower migrating and invasive cell numbers compared to controlled cells (Figure 3), 48-hour healing rates for cells in group C, D, and E were 14.83±1.81%, 15.01±1.92% and 89.36±9.17%, respectively, which suggests the inhibition of cell migration ability after SIRT1 gene silencing.
Figure 3.

PC-3 cell migration (A) and invasion (B) numbers. * p<0.05 compared to control group; # p<0.05 compared to scramble RNA group.
Clonal formation ability of PC-3 cells
Compared to the control and scramble RNA group, siRNA-transfected PC-3 cells had significantly lower number of colony formation. The number of colony-forming cells in group D and E were 231±4.6% and 86±2.7% (p<0.05, Figure 4).
Figure 4.

Clonal formation assay.
EMT marker protein expression
We further checked the expression level of EMT markers and found the elevation of epithelial marker. PT-PCR amplification products were analyzed by agarose gel electrophoresis, with repeated measurements 3 times to indicate the relative expression of GAPDH gray value gene and the gray value ratio, and then we compared them with the negative control group. The results show that relative expression of E-cadherin number in group E was 1.36, and Vimentin and N-cadherin relative expressions were 0.66 and 0.31, respectively. E-cadherin was expressed in siRNA-transfected PC-3 cells (p<0.05, Figure 5), which also had suppressed expression of mesenchymal markers, including vimentin and N-cadherin (p<0.05, Figure 5). Western blot results showed that after the inhibition of PC-3 cells SIRT1 gene, E-cadherin expression in group E was increased (P<0.05) to 1.73 times that of the negative control group. Vimentin and N-cadherin were decreased (P<0.05) to 0.72 and 0.51 times the negative control group, consistent with PT-PCR results (Figure 6).
Figure 5.
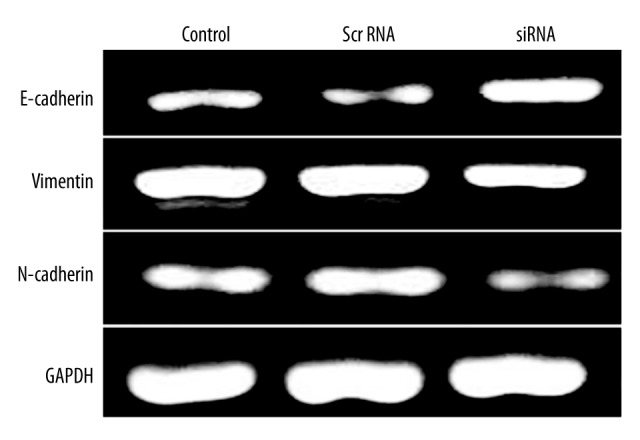
RT-PCR for EMT marker protein expression levels.
Figure 6.
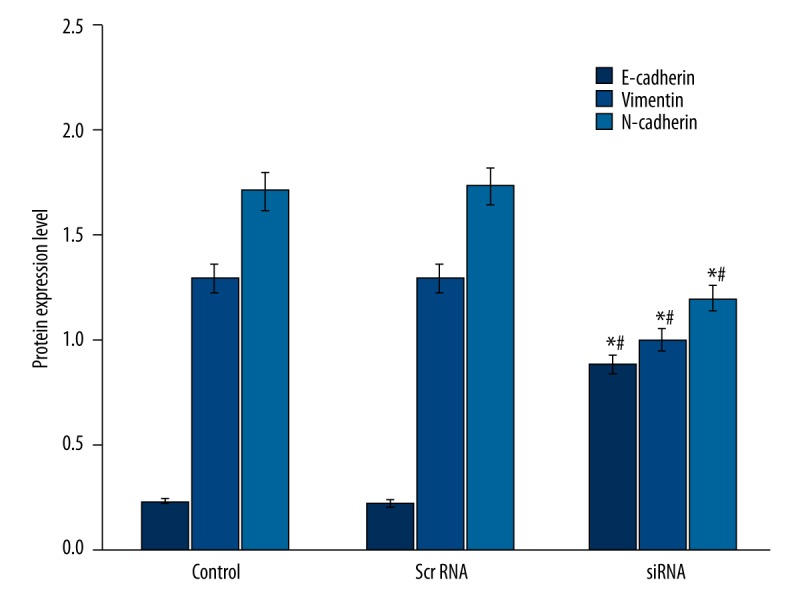
Western blotting for EMT markers in PC-3 cells. * p<0.05 compared to control group; # p<0.05 compared to scramble RNA group.
Discussion
In tumor migration and invasion process, EMT is an effective way for epithelial derived tumor cells to obtain the migrating ability. EMT under physiological conditions was transferred to mesenchymal cells via epithelial cells intracellular signaling pathway, and biomolecules, such as Vimentin, N-cadherin, and E-cadherin, were all changed in its expression approach accordingly, as supported by both in vivo and in vitro studies showing the important role of EMT in primary invasion and secondary metastasis of prostate cancer [11,12]. The study of molecular mechanisms underlying EMT process, therefore, is of great importance in searching for markers of tumor metastasis. The molecular markers of EMT mainly include the elevated expression of mesenchymal markers vimentin and N-cadherin, along with suppressed expression of epithelial marker E-cadherin, accompanied by potentiated cell migration ability [13–15]. Belonging to the cadherin family, E-cadherin is mainly expressed in epithelial-derived cell surface and exerts its intracellular adhesion function via its specific binding on both cytoskeleton proteins and cytoplasmic concatemers. N-cadherin regulation of prostate cancer metastasis and activation were reported to be related to PI3K/Akt signaling pathway [16]. The down-regulation of E-cadherin compromised the adhesion between cells, making them easier to detach from the basal matrix. In prostate cancer tissues, E-cadherin is believed to be closely related with degree of tumor differentiation [16,17].
N-cadherin also belongs to the cadherin family but exerts an effect that is the opposite of that of E-cadherin. It is a surface marker for non-epithelial-derived cells and plays an important role in mediating cell adhesion between muscular tissues, fibroblasts, and neural tissues. Previous studies have shown the correlation between cell migration/invasion abilities and the over-expression of N-cadherin plus down-regulation of E-cadherin [15,16]. Mainly expressed in mesenchymal tissues, vimentin can affect the formation of cytoskeleton and thus cell shape, and has been indicated in the metastasis/invasion of prostate cancer [15,17]. SIRT1 participates in various cellular processes, including differentiation and apoptosis, via its de-acetylation modification of gene transcription. The elevated expression of SIRT1 occurs in prostate cancer cells, probably due to the potency of its inhibition on tumor-suppressor gene p53 via de-acetylation. An in vitro study revealed the cell cycle arrest and reversing of drug resistance of tumor cells by siRNA for silencing SIRT1. This study constructed an SIRT1 interfering vector, which was transfected into prostate cancer PC-3 cells for further observing the effect of siRNA-SIRT1 in EMT process and expression of EMT markers.
Our results showed depressed SIRT1 expression levels after transfecting SIRT1-siRNA. Both scratch and clonal formation assays showed suppressed cell migration ability of SIRT1-silencing PC-3 cells. Transwell migration assay indicated fewer migrated and invasive cells after SIRT1 gene silencing. In summary, this study demonstrated the inhibition of PC-3 cell migration and invasion ability by the specific silencing of SIRT1 gene. Western blotting and RT-PCR studies showed elevated E-cadherin along with suppressed N-cadherin and vimentin after SIRT1 gene silencing. In tumor cells, E-cadherin can prevent the metastasis by stabilization of cell-to-cell adhesion. For example, the infiltration of pancreatic cancer is dependent on the low expression of E-cadherin. The knockout of E-cadherin gene in PC-3 cells remarkably potentiates the invasiveness, with increased number of cells with features of hepatocytes.
With the suppression of E-cadherin expression, various metastatic and tumor stem cell markers, including urokinase plasminogen activator (uPA), CX-CR5, Notch1, and CD44. uPA can facilitate the tumor invasion/metastasis via the reformation of extracellular matrix (ECM) and basal membrane. The PI3K signaling pathway is known to play an important role in uPA-induced tumor invasion, and the inhibition of PI3K signaling can decrease uPA secretion, which is dependent on the PI3K/Akt signaling pathway [17–19]. N-cadherin monoclonal antibody can retard the transition of prostate cancer into hormone-independent type, significantly decreasing tumor invasiveness. N-cadherin can activate the PI3K/Akt signaling pathway way to increase the expression of mononuclear cell chemotactic factor 1 (MCP-1) to induce tumor angiogenesis for cancer metastasis. A complex can be formed consisting of PI3K, VEGFR-2, β-catenin, and E-cadherin to initiate downstream PI3K/Akt signaling pathway to participate in VEGF-induced pathway for endothelial cells adhesion and migration [16,19–22]. Therefore, the inhibition of SIRT1 expression in PC-3 cells may decrease the invasion and migration ability, and reverse the EMT process by down-regulating vimentin and N-cadherin, and up-regulating E-cadherin, possibly via the PI3K/Akt signaling pathway.
Conclusions
The inhibition of SIRT1 gene expression in prostate cancer cell line PC-3 can decrease the motility, invasiveness, and migration ability, leading to prostate cancer metastasis, and its mechanism may be associated with de-regulation of the expression levels of mesenchymal markers such as Vimentin and N-cadherin, and up-regulation of the expression epithelial markers E-cadherin.
Footnotes
Source of support: Supported by the Natural Scientific Research Funds of China (1, A possible mechanism of stathmin as a potential target for treatment of Phobia, No. 31300830; 2, Deep brain stimulation in the nucleus accumbens has a more important role in the treatment of heroin addiction, No. 81101022) and the Shanxi National Science Foundation Project (Effects of Stathmin on the survival of axotomized retinal ganglion cells in adult rats, No. 2014JQ4120)
References
- 1.Zhou ZW, Li XX, He ZX, et al. Induction of apoptosis and autophagy via sirtuin1 and PI3K/Akt/mTOR-mediated pathways by plumbagin in human prostate cancer cells. Drug Des Devel Ther. 2015;9:1511–54. doi: 10.2147/DDDT.S75976. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Wu D, Yu S, Jia L, et al. Orphan nuclear receptor TLX functions as a potent suppressor of oncogene-induced senescence in prostate cancer via its transcriptional co-regulation of the CDKN1A (p21(WAF1) (/) (CIP1)) and SIRT1 genes. J Pathol. 2015;236(1):103–15. doi: 10.1002/path.4505. [DOI] [PubMed] [Google Scholar]
- 3.Di Sante G, Pestell TG, Casimiro MC, et al. Loss of Sirt1 promotes prostatic intraepithelial neoplasia, reduces mitophagy, and delays PARK2 translocation to mitochondria. Am J Pathol. 2015;185(1):266–79. doi: 10.1016/j.ajpath.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilking MJ, Ahmad N. The role of SIRT1 in cancer: the saga continues. Am J Pathol. 2015;185(1):26–28. doi: 10.1016/j.ajpath.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Long Q, Xu J, Osunkoya AO, et al. Global transcriptome analysis of formalin-fixed prostate cancer specimens identifies biomarkers of disease recurrence. Cancer Res. 2014;74(12):3228–37. doi: 10.1158/0008-5472.CAN-13-2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baptista T, Graça I, Sousa EJ, et al. Regulation of histone H2A.Z expression is mediated by sirtuin 1 in prostate cancer. Oncotarget. 2013;4(10):1673–85. doi: 10.18632/oncotarget.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vanella L, Di Giacomo C, Acquaviva R, et al. Apoptotic markers in a prostate cancer cell line: effect of ellagic acid. Oncol Rep. 2013;30(6):2804–10. doi: 10.3892/or.2013.2757. [DOI] [PubMed] [Google Scholar]
- 8.Moore RL, Faller DV. SIRT1 represses estrogen-signaling, ligand-independent ERα-mediated transcription, and cell proliferation in estrogen-responsive breast cells. J Endocrinol. 2013;216(3):273–85. doi: 10.1530/JOE-12-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lovaas JD, Zhu L, Chiao CY, et al. SIRT1 enhances matrix metalloproteinase-2 expression and tumor cell invasion in prostate cancer cells. Prostate. 2013;73(5):522–30. doi: 10.1002/pros.22592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Byles V, Zhu L, Lovaas JD, et al. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene. 2012;31(43):4619–29. doi: 10.1038/onc.2011.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huffman DM. Editorial comment to inhibition of cortactin and SIRT1 expression attenuates migration and invasion of prostate cancer DU145 cells. Int J Urol. 2012;19(1):79–80. doi: 10.1111/j.1442-2042.2011.02915.x. [DOI] [PubMed] [Google Scholar]
- 12.Nakane K, Fujita Y, Terazawa R, et al. Inhibition of cortactin and SIRT1 expression attenuates migration and invasion of prostate cancer DU145 cells. Int J Urol. 2012;19(1):71–79. doi: 10.1111/j.1442-2042.2011.02888.x. [DOI] [PubMed] [Google Scholar]
- 13.Powell MJ, Casimiro MC, Cordon-Cardo C, et al. Disruption of a Sirt1-dependent autophagy checkpoint in the prostate results in prostatic intraepithelial neoplasia lesion formation. Cancer Res. 2011;71(3):964–75. doi: 10.1158/0008-5472.CAN-10-3172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung-Hynes B, Schmit TL, Reagan-Shaw SR, et al. Melatonin, a novel Sirt1 inhibitor, imparts antiproliferative effects against prostate cancer in vitro in culture and in vivo in TRAMP model. J Pineal Res. 2011;50(2):140–49. doi: 10.1111/j.1600-079X.2010.00823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drivalos A, Papatsoris AG, Chrisofos M, et al. The role of the cell adhesion molecules (integrins/cadherins) in prostate cancer. Int Braz J Urol. 2011;37(3):302–6. doi: 10.1590/s1677-55382011000300002. [DOI] [PubMed] [Google Scholar]
- 16.Nalla AK, Estes N, Patel J, et al. N-cadherin mediates angiogenesis by regulating monocyte chemoattractant protein-1 expression via PI3K/Akt signaling in prostate cancer cells. Exp Cell Res. 2011;317(17):2512–21. doi: 10.1016/j.yexcr.2011.07.024. [DOI] [PubMed] [Google Scholar]
- 17.Wang B, Hasan MK, Alvarado E, et al. NAMPT overexpression in prostate cancer and its contribution to tumor cell survival and stress response. Oncogene. 2011;30(8):907–21. doi: 10.1038/onc.2010.468. [DOI] [PubMed] [Google Scholar]
- 18.Kojima K, Fujita Y, Nozawa Y, et al. MiR-34a attenuates paclitaxel-resistance of hormone-refractory prostate cancer PC-3 cells through direct and indirect mechanisms. Prostate. 2010;70(14):1501–12. doi: 10.1002/pros.21185. [DOI] [PubMed] [Google Scholar]
- 19.Jung-Hynes B, Ahmad N. Role of p53 in the anti-proliferative effects of Sirt1 inhibition in prostate cancer cells. Cell Cycle. 2009;8(10):1478–83. doi: 10.4161/cc.8.10.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu K, Katiyar S, Witkiewicz A, et al. The cell fate determination factor dachshund inhibits androgen receptor signaling and prostate cancer cellular growth. Cancer Res. 2009;69(8):3347–55. doi: 10.1158/0008-5472.CAN-08-3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goyal J, Antonarakis ES. Clinical evaluation of abiraterone in the treatment of metastatic prostate cancer. Clin Med Insights Urol. 2013;(07):1–14. doi: 10.4137/CMU.S8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung-Hynes B, Nihal M, Zhong W, et al. Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition. J Biol Chem. 2009;284(6):3823–32. doi: 10.1074/jbc.M807869200. [DOI] [PMC free article] [PubMed] [Google Scholar]