

Abstract
The pulmonary epithelium serves as a barrier to prevent access of the inspired luminal contents to the subepithelium. In addition, the epithelium dictates the initial responses of the lung to both infectious and noninfectious stimuli. One mechanism by which the epithelium does this is by coordinating transport of diffusible molecules across the epithelial barrier, both through the cell and between cells. In this review, we will discuss a few emerging paradigms of permeability changes through altered ion transport and paracellular regulation by which the epithelium gates its response to potentially detrimental luminal stimuli. This review is a summary of talks presented during a symposium in Experimental Biology geared toward novel and less recognized methods of epithelial barrier regulation. First, we will discuss mechanisms of dynamic regulation of cell-cell contacts in the context of repetitive exposure to inhaled infectious and noninfectious insults. In the second section, we will briefly discuss mechanisms of transcellular ion homeostasis specifically focused on the role of claudins and paracellular ion-channel regulation in chronic barrier dysfunction. In the next section, we will address transcellular ion transport and highlight the role of Trek-1 in epithelial responses to lung injury. In the final section, we will outline the role of epithelial growth receptor in barrier regulation in baseline, acute lung injury, and airway disease. We will then end with a summary of mechanisms of epithelial control as well as discuss emerging paradigms of the epithelium role in shifting between a structural element that maintains tight cell-cell adhesion to a cell that initiates and participates in immune responses.
Keywords: lung, epithelial barrier, permeability, transport, ions
the lung epithelium is a mucosal surface composed of ciliated cells, mucous-producing cells, and undifferentiated basal cells that forms the interface between the lumen and the parenchyma from the nasal passage to the alveoli. It is the initial site of contact for all inspired substances and therefore acts as both a physical and an immunological barrier that protects the subepithelial tissue.
The physical barrier, comprised of the mucociliary apparatus, secreted antimicrobial substances, and the intercellular junctions, prevents inhaled toxins and pathogens from contacting the subepithelial tissue. The mucociliary apparatus traps foreign particles and removes them from the respiratory tract. Defects in this apparatus can lead to recurrent infections, as seen in patients with cystic fibrosis (104), chronic obstructive pulmonary disease (COPD) (14, 79), and ciliary dyskinesis (18, 19). Secreted antimicrobial substances including enzymes, protease inhibitors, and antimicrobial peptides further contribute to the immune response of the epithelial cell (134). Epithelial secretions contain enzymes such as lysozyme that have antimicrobial effects against both Gram-positive and Gram-negative bacteria (40, 76) as well as protease inhibitors, including serine protease inhibitor, secretory leukocyte protease inhibitor, and elafin, which reduce the effects of proteases that are produced by pathogens. In addition, surface antimicrobial peptides like human β-defensins further protect against numerous pathogens, including bacteria and viruses (116, 176).
The intercellular junctions form adhesive forces that connect neighboring cells and separate the external environment from the subepithelial tissue. There are three main types of intercellular junctions: the tight junctions, the adherens junctions, and the desmosomes (137). The tight junctions are located more apically compared with the other junctions and are multiprotein complexes that consist of transmembrane proteins, including claudins, occludins, and junctional adhesion molecules, as well as scaffolding proteins like the zonula occludens (ZO). These junctions form a continuous circumferential ring around the epithelial cells, which regulates the paracellular transport of ions and certain molecules (63, 157). The adherens junctions, composed of E-cadherin and several proteins from the catenin family, including α-catenin, β-catenin, and p120, anchor the actin cytoskeleton and mediate cell-to-cell adhesion (41, 63, 78). Together, the tight and adherens junctions form the apicoadherens junctions, which separate the apical and basolateral membranes and block the movement of integral membrane proteins between surfaces to maintain epithelial cell polarity (169). The desmosomes, adhesive junctions located on the lateral cell membrane, link to the intermediate filaments and help protect against mechanical stress. Inhaled toxins such as cigarette smoke damage these intercellular junctions, resulting in a leaky, hyperproliferative, and abnormally differentiated epithelium (22, 68, 73, 130, 150, 191).
It is increasingly recognized that the epithelium helps to maintain homeostasis in the lung, and disruption of the epithelial barrier can lead to the development of inflammation and diseases such as COPD and acute lung injury (ALI). Moreover, novel studies demonstrating that restoration of the epithelial barrier through transfer of mitochondria from bone marrow-derived stromal cells to epithelial cells can protect against ALI further establish the importance of the epithelial barrier (77). In this review, we will further discuss how the airway epithelium dynamically regulates its response to luminal stimuli by altering ion transport and paracellular permeability.
The Lung Epithelium: Insights into the Role of Regulation of Paracellular Permeability in Epithelial Signaling
The role of cell-cell adhesion in modulating epithelial signaling pathways.
The integrity of the physical barrier and epithelial cell polarity affect the interaction of cytokines and growth factors with their receptors, thereby modulating the immunological response of the epithelium environmental stimuli. In this respect, the epithelium also acts as an immunological barrier. Subtle changes in paracellular permeability can impact ligand interaction with its receptor. Moreover, certain cytokines are prevented from interacting with their cytokine receptors because of segregation to different sides of the apicoadherens junctional complex. Damage to the epithelium can affect the polarized distribution of cell surface receptors, allowing for increased receptor-ligand interaction and the activation of signaling transduction cascades and secretory activity (73, 171, 200). One specific example of such regulation is the epidermal growth factor (EGF), which regulates migration, proliferation, and differentiation following injury and enhances epithelial repair (186). In normal human airways, EGF receptor (EGFR) expression is limited and isolated to the basolateral membrane (187); however, expression is increased in response to cigarette smoke (165, 188), mechanical stress (139), and inhaled noxious substances like bleomycin (110). In a homeostatic state, the EGF ligand is located on the apical surface and therefore separated from its receptor on the basolateral surface. Moreover, the apically located growth factor heregulin is separated from its receptor erbB2/3 (also known as HER2/3), which is located on the basolateral membrane. These growth factors, therefore, only have access to their receptors when the epithelium is damaged, allowing for rapid targeted repair of the epithelial injury (200). The binding of heregulin to the erbB2/3 receptor leads to cell proliferation, differentiation, and mucous secretion (193, 201, 209), which allows the damaged epithelium to protect and repair itself. Therefore, although the epithelium expresses both the growth factors and their receptors, it can regulate the interaction such that the effects occur only at times of injury, and one mechanism by which the epithelium does this is by regulating paracellular permeability.
Dynamic regulation of paracellular permeability.
The intercellular junctions do not statically resist entry to all substances but act as dynamic structures that can open or close in response to both physiological and pathological stimuli (157, 167). By tightening cell-cell adhesion in response to mechanical stimuli such as shear stress or chemical environmental exposures such as inhaled particulate matter, the epithelia dynamically regulates receptor-ligand interactions (Fig. 1) (171). For example, airflow over the epithelium during breathing creates shear stress, which can lead to changes in barrier properties. Low, physiological levels of shear stress are generated during normal tidal volume breathing but can increase by several times during exercise, coughing, and bronchospasm. Exposure to shear stress leads to actin rearrangement and barrier enhancement (173). In addition, in response to both physiological levels of shear stress as well as pathological stimuli like particulate matter, pollution formed from the mixture of solid particles and liquid droplets found in air, there is increased membrane localization of septin 2 that decreases paracellular permeability by altering cortical actin rearrangement (171). Septin 2 is a member of the family of GTP-binding proteins that function in cytoskeletal arrangement and cell polarization. Membrane localization of septin 2 allows for increased interaction with actin, which regulates the rearrangement of cortical actin and enhances barrier function. Physiological levels of shear stress also lead to a selective decrease in the abundance of the apical water channel aquaporin 5 (172, 173), which helps to modulate the paracellular permeability in response to the stress. Aquaporin 5 stabilizes the microtubules (172) and antagonizes cell-to-cell adhesion (173), thereby affecting paracellular permeability. Thus the epithelium is able to dynamically regulate its response to experienced luminal stimuli.
Fig. 1.
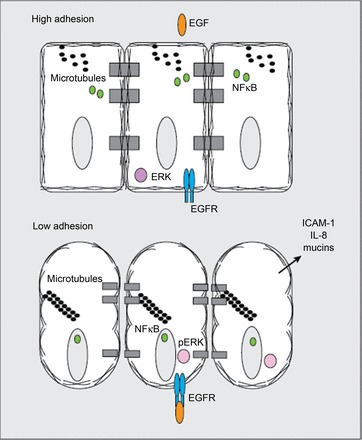
The epidermal growth factor receptor (EGFR) is restricted to the basal surface of the airway epithelial cells. This figure serves to highlight how the integrity of the apicoadherens junction can regulate epithelial cell signaling. Under conditions of low paracellular permeability created by the cytoskeleton and cell-cell contacts, there is segregation of EGF ligand and receptor, thereby inhibiting constant cellular signaling. In conditions of increased paracellular permeability, EGF can bind its receptor and activate its pathway, where phosphorylated ERK serves as readout of EGFR activation. Thus the epithelial barrier can modulate epithelial signaling pathways.
In summary, the airway epithelium is the first line of defense and prevents inspired substances from contacting the subepithelial tissue. The epithelium acts as both a physical and immunological barrier that continuously responds to both physiological and pathological stimuli in the environment and dynamically regulates its barrier in response to these stimuli. The apicoadherens junctions separate the apical and basolateral membranes, and therefore membrane integrity can affect cell signaling and the defenses mounted in response to environmental exposures. By tightly coordinating epithelial responses to environmental exposures and altering epithelial signaling, secreted products, and ion and water flux in response to stimuli, the epithelium is able to provide a dynamic barrier at the interface between the lung parenchyma and the luminal environment. Noxious stimuli such as chronic tobacco exposure, infections, particulate matter, and air pollution can lead to a breakdown in this regulatory response, leading to chronic epithelial activation, which can further propagate the damage, resulting in a leaky barrier. In the following sections, we will discuss in more detail how the tight junction proteins, ion transport, and EGFR contribute to the dynamic epithelial barrier.
The Lung Epithelium: Insights in Ion Transport Across the Barrier
Interactions between tight junctions and ion transporters to maintain lung fluid balance.
Control of airspace fluid volume and composition in the alveolus is primarily determined by transcellular ion transport, which requires a polarized epithelium with intact tight junctions. However, paracellular ion, macromolecule, and water transport through intact tight junctions also makes a significant contribution to lung fluid balance (115, 199). Furthermore, the effects of both bacterial and viral infections are quite complex, with data suggesting that a multitude of ion channels are altered in response to infection, and there is potential for creation of new channels by a host of viruses, termed viroporins. The detailed discussions of these effects are beyond the scope of this review. During the resolution of pulmonary edema, clearance of fluid from the airspaces is largely dependent on a transepithelial sodium concentration gradient driven by Na+/K+-ATPase. Apical sodium conductance through epithelial sodium channel (ENaC) is central to fluid clearance, but chloride and potassium channels also make important contributions (13, 37, 98). Apical-to-basolateral chloride transport may be particularly important because the maximal rate of sodium and water transport from the airspaces appears to be limited by the concomitant transport of chloride in many studies (9, 43, 122). Because experimental data suggest that a sizable fraction of transepithelial chloride transport occurs through the paracellular route in the alveolar epithelium (87), regulated paracellular ion transport could influence alveolar fluid clearance rates by this mechanism. In other words, changes in the selectivity and magnitude of paracellular ion conductance may influence rates of alveolar fluid clearance, either by limiting sodium leak or increasing chloride currents. The mechanisms for the regulation of paracellular transport in the lung and the potential interrelationship with transcellular transport are not fully understood, but recent work has begun to describe some of the major determinants of paracellular permselectivity in the alveolar epithelium. These studies have also provided some insight into deeper associations between the paracellular and transcellular routes of transport that is the regulation of transcellular transporters by cell junction proteins. One area of interest is the potential association between Na+/K+-ATPase and tight junction claudins.
Sodium/potassium ATPase as a regulator of cell junctions.
Sodium/potassium ATPase is made up of catalytic (α) and regulatory (β) subunits. Additional regulatory subunits of the FXYD family, such as the γ-subunit of Na+/K+-ATPase, may also associate with α/β-heterodimers and affect function. Pump activity in polarized epithelial cells is essential for lung fluid balance, and increased pump activity is associated with increased rates of vectorial fluid clearance across the alveolar epithelium (4, 107). Recent work has uncovered an additional role for Na+/K+-ATPase in tight junction formation and maintenance in epithelia (194). For example, the β-subunit of Na+/K+-ATPase associates with PDZ domain-containing proteins and localizes to tight junctions (109, 141). Although the β-subunit has been reported to function as a cell adhesion molecule, enzymatic pump activity of Na+/K+-ATPase is required for tight junction formation in Madin-Darby canine kidney (MDCK) cells (144). The mechanism for this function appears to be through Na+/K+-ATPase-mediated activation of RhoA signaling and cytoskeletal remodeling at the periphery of epithelial cells. Na+/K+-ATPase may also be required for the maintenance of tight junctions. In monolayers of retinal pigment epithelial cells and in pancreatic ductal cell (HPAF-II) monolayers with established tight junctions, inhibition of Na+/K+-ATPase with ouabain increased paracellular permeability to ions and macromolecules (142, 143). However, the junction-promoting effects of Na+/K+-ATPase may be context or cell-type specific because ouabain has also been reported to increase expression of claudins-4 and -1 in MDCK cells in association with decreased paracellular macromolecule permeability (97). Although the mechanisms for the regulation of tight junction formation and function by Na+/K+-ATPase are not fully understood, it appears that Na+/K+-ATPase can modulate tight junction assembly and function.
Claudins as regulators of paracellular permeability.
Differential expression of the claudin family of tight junction proteins is a fundamental regulatory mechanism of paracellular permselectivity. Claudins are transmembrane proteins that are necessary and sufficient for tight junction strand formation (51). The spatially restricted expression of specific claudins along the course of the nephron exemplifies their permselectivity function (7). Moreover, point mutations in certain claudins result in clinically important changes in ion transport, and substantial experimental evidence supports a direct role for claudins in charge-selective paracellular ion conductance (6, 102, 195). Claudins on adjacent cells form paracellular pores with an ∼3-Angstrom diameter that function as relatively high-conductance, ungated ion channels (101, 145, 197). Claudins also influence paracellular macromolecule transport, and individual claudins appear to convey different properties to this charge-independent, lower-conductance pathway (167, 168). A leading hypothesis is that individual claudin strands within a tight junction establish the ion pore pathway, whereas the persistence or continuity of claudin strands within the membrane influences the macromolecule leak pathway (167). Because tight junctions consist of multiple layers of claudin-based strands that undergo continuous turnover, both conductance pathways can exist simultaneously. Three claudins, claudin-3, claudin-4, and claudin-18.1, are abundantly expressed in the alveolar epithelium although other claudins are expressed at low levels (95).
Lung-specific claudin-18.1 is a requirement for the alveolar epithelial permeability barrier.
Claudin-18.1 is of particular interest because it is the only known lung-specific tight junction protein, and it is the most abundantly expressed claudin in type 1 alveolar epithelial cells (AECs) (95). The role of claudin-18 in alveolar epithelial barrier function has recently been examined in knockout (KO) mice. Claudin-18 does not appear to be required for prenatal lung development; however, upon conversion to air breathing, alveolarization is abnormal in claudin-18 KO mice (96). Claudin-18-null mice have a threefold increase in alveolar epithelial permeability to macromolecules. There is a similar increase in macromolecule permeability in cultured primary AECs from KO mice and with siRNA knockdown of claudin-18 in wild-type cells (96). These data indicate that claudin-18 provides a macromolecule permeability barrier in the alveolar epithelium and suggest that the loss of this function results in abnormal alveolar homeostasis and impaired alveolarization. Interestingly, although macromolecule permeability is increased, claudin-18-null mice do not develop pulmonary edema (100). The reason for this may be that alveolar epithelial sodium and chloride transport is increased. The basal alveolar fluid clearance rate in claudin-18-null mice is approximately twice that of wild-type littermates. Examination of the expression levels of ion transporters in whole lung revealed a twofold increase in the β1-subunit of Na+/K+-ATPase and increased pump activity but no change or a slight decrease in the α-, β-, and γ-subunits of ENaC at the mRNA level. Because overexpression of Na+/K+-ATPase results in higher rates of alveolar fluid transport in animal models, the increase in β1-subunit expression may account for most of the increase in alveolar fluid clearance in claudin-18-null mice.
Chloride transport and alveolar fluid clearance in claudin-18-null mice.
As mentioned above, sodium transport in the lung can be limited by the cotransport of chloride, so increases in the levels of chloride transporters might be expected to facilitate higher rates of sodium and fluid transport. It is notable that cystic fibrosis transmembrane conductance regulator (CFTR) mRNA expression is increased by approximately fourfold in claudin-18 KO mice, but whether CFTR inhibition decreases alveolar fluid clearance in this context has not been reported. In addition to CFTR, several other regulators of chloride transport show increased expression in the lungs of claudin-18-null mice, including Na-K-2Cl cotransporter (NKCC1), CLC2, and SLC26A9. It is possible that these data point to a larger role for chloride transport in alveolar fluid clearance than has been previously appreciated. For example, chloride-dependent sodium transport into the airspaces appears to be an important mechanism for edema formation during hydrostatic stress (183). In the nephron, chloride-dependent sodium absorption is a mechanism for increased fluid retention and hypertension. This mechanism depends on the coupling of sodium transport to chloride transport via NKCC1. Although NKCC1 transport in a basolateral-to-apical direction is well established, transport in an apical-to-basolateral direction (or bidirectional transport) is possible (183). Therefore, the observed increase in chloride transporters in claudin-18-null mice is consistent with compensation of fluid clearance facilitated by maximizing chloride-limited sodium transport. Further investigation is required, but increased NKCC1 expression in claudin-18-null mice might also support the hypothesis that apical-to-basolateral chloride-dependent sodium transport is a mechanism for alveolar fluid clearance. In summary, the loss of claudin-18 results in increased paracellular permeability in the absence of increased lung water. Higher rates of fluid transport in these mice may be due to increased expression of the β-subunit of Na+/K+-ATPase along with chloride transporters. The mechanisms for the upregulation of ion transporters in the context of claudin-18 deficiency are not known, but one possibility is coregulation of cell junction constituents and ion transporters.
Functional implications of increased claudin-4 expression in claudin-18-null mice.
Claudin-4 mRNA and protein levels are increased fourfold in claudin-18 KO mice. Claudin-4 plays a distinct role in transepithelial ion transport and junction formation and may partly compensate for the loss of claudin-18. Claudin-4 acts as a relative sodium barrier and is also specifically upregulated during epithelial repair (72, 196, 207). In human and murine lungs, higher claudin-4 expression is associated with higher rates of alveolar fluid clearance (82, 152, 207), which is suggestive of a protective or reparative role for this claudin. Claudin-4 may also play a unique role in transepithelial ion transport. For example, in zebra fish, the claudin-4 ortholog, claudin-b, associates with Na+/K+-ATPase to affect decreased paracellular sodium transport in low-ion-concentration conditions (92, 93). In mouse studies, the loss of claudin-4 results in decreased alveolar fluid clearance and more severe pulmonary edema with injury (82, 207). These data are consistent with the hypothesis that lung epithelial tight junctions containing high levels of claudin-4 support higher rates of alveolar fluid clearance. This may be because claudin-4 forms high-resistance junctions that limit sodium conductance, resulting in a larger transepithelial sodium gradient. Alternatively, claudin-4 may play a role in the regulation of Na+/K+-ATPase, but further studies are needed. In claudin-18-null mice, higher levels of claudin-4 may partly explain the observed increase in alveolar fluid clearance.
Functional association between claudins and transcellular ion transport.
Taken together, available data suggest a functional association between tight junction claudins and transcellular ion transport. The hypothesis that claudin-4 and Na+/K+-ATPase have a cooperative role in transepithelial sodium transport in the lung requires further study, but it is clear that claudin-4 is necessary for normal barrier function and maximal rates of fluid clearance. It is also clear that claudin-18 forms a unique permeability barrier in the alveolar epithelium and is a requirement for alveolar homeostasis. The loss of claudin-18 results in a compensatory increase in claudin-4 along with numerous changes in transcellular ion transporters in association with increased rates of alveolar fluid clearance. The adaptation to chronic barrier leak in claudin-18-null mice may provide novel insights into the mechanisms of alveolar ion and water transport regulation in diseases characterized by epithelial barrier dysfunction.
The Lung Epithelium: The Role of Two-Pore-Domain Potassium Channels in Lung Epithelium
Transcellular ion homeostasis in the lung epithelium.
Maintenance of the integrity and the homeostatic properties of the alveolar capillary barrier plays a vital role in pulmonary health and disease (111). Through separation of the air- and blood-filled spaces in the lung, the alveolar epithelium is strategically positioned to interact closely with both environments and to regulate the electrolyte and water content of the alveolar spaces. In this section, we will briefly review 1) the major ion-transport mechanisms regulating the composition of the alveolar lining fluid (ALF) under physiological conditions, and 2) the role of stretch-activated ion channels (SACs) in the development of hyperoxia (HO), mechanical stretch-induced ALI, and acute respiratory distress syndrome (ARDS), with particular emphasis on epithelial barrier function and inflammatory cytokine secretion.
Maintaining tight control over the intra-alveolar environment requires complex synchronization of multiple epithelial ion-transport processes and a dynamic balance between continuous secretion, and reabsorption of Na+ and Cl− determines the composition of the ALF (131). When an increase in alveolar fluid absorption is required, as in the immediate postnatal period or in conditions associated with pulmonary edema like pneumonia and congestive heart failure (136, 205), Na+ is internalized via apical amiloride-sensitive ENaC channels and cyclic nucleotide-gated cation channels in both alveolar type I (AT I) and type II (AT II) cells (33, 36, 37, 45, 74). Internalized Na+ is then secreted into the interstitial space via the basolateral Na/K ATPase (4, 31). To maintain intracellular electroneutrality, apical Cl− absorption occurs mainly via CFTR channels or paracellular pathways (36). In combination, these electrolyte shifts create an osmotic gradient for water molecules to move from the alveolar into the interstitial space either via aquaporin channels or by diffusion (1, 88, 177, 198).
In contrast, when increased production of ALF is required, as part of physiological ALF turnover and aging, to maintain surfactant homeostasis, or in response to infectious or environmental pollution stimuli (67, 81, 106, 120), Cl− anions are actively secreted into the alveolar space mainly via apical CFTR channels, ClC channels, and Ca-dependent Cl− channels (35, 50, 69, 108). An active driving force for Cl− secretion is maintained by basolateral Na/K/2Cl cotransporters, K-Cl cotransporters, HCO3/Cl exchangers (5, 91, 151, 175, 190), as well as basolateral inwardly and outwardly rectifying Cl− channels (10, 69). Na+ ions are thought to follow Cl− ions from the interstitial into the alveolar space, but the molecular mechanisms underlying Na+ secretion are still not fully understood.
Similarly, basolateral K+ channels play an important role in maintaining ALF by creating a favorable electrochemical gradient for Na+ absorption and Cl− secretion, by restoring intracellular electroneutrality, and by stabilizing the resting membrane potential (112). A variety of K+ channels have been described in both AT I and AT II cells, including voltage-gated (Kv), ATP-dependent (KATP), inwardly rectifying (Kir), Ca2+-activated (KCa) K+ channels (8, 57), and small- and large-conductance (BK) K+ channels (11, 113). In addition to homeostatic processes, K+ channels also appear to be involved in epithelial wound healing, cell migration, cell proliferation, and cytokine secretion in in vitro and in vivo models of ALI/ARDS (8, 149, 161, 162, 192), and large-conductance BKCa channels may act as oxygen sensors in the lung epithelium (80).
Further complexity is added to this system when accounting for the intricate interactions between these ion channels and transporter proteins. For example, CFTR can downregulate ENaC expression (158); inhibition of KATP channels can inhibit ENaC currents (90); activation of KATP channels can increase ENaC and CFTR currents (98); and inhibition of KCa and Kv channels can inhibit ENaC and CFTR currents (17, 62). There are numerous limitations in the study of epithelial ion-transport processes; however, more recent approaches employing siRNA and shRNA knockdown of specific ion channels (163), and the broader availability of KO mice deficient in specific ion channels (32, 124) have significantly facilitated research in this field. Nevertheless, conditional KO models for specific epithelial ion channels, where a particular ion channel is absent from AT I or AT II cells only, are not always readily available.
The role of ENaC channels in lung fluid balance.
The ENaC channels are composed of four subunits (α,β, γ, and δ) coded for by the SCNN1A, SCNN1B, SCNN1G, and the SCNN1D genes, respectively. The α-subunit is required to form a functional ENaC channel, and the β- and γ-subunits amplify the activity of the channel. In mice, homozygous KO of the α-ENaC subunit leads to retention of fetal lung liquid, respiratory distress, and ultimately death (74). In humans, mutations in the ENaC subunit genes SCNN1A, SCNN1B, and SCNN1G can lead to autosomal recessive pseudohypoaldosteronism (PHA) characterized by life-long salt wasting, hyperkalemia, and dehydration (25). Children who have PHA type 1 with mutations in the genes encoding the α- and β-subunit have an increased volume of fluid in the lungs because of problems with fluid absorption (84), leading to recurrent respiratory problems. There are, however, case reports of preterm infants with a single homozygous mutation in the α-ENaC subunit who did not have immediate postnatal difficulty with lung fluid clearance, indicating that the α-ENaC may not be pivotal in the clearance of lung fluid at early stages of lung maturation (75).
Transforming growth factor (TGF)-β has been shown to be important in the regulation of ENaC activity, which has important implications for lung fluid balance. Peters et al. (135) demonstrated that TGF-β can drive the abnormal internalization of the ENaC complex, leading to a reduction in this complex at the lung epithelial cell surface, resulting in reduced sodium and fluid absorption (135). These data suggest a unique TGF-β-dependent mechanism to regulate ion and fluid transport in the lung and potentially a new pathological mechanism of ALI and ARDS.
The role of two-pore-domain potassium channels in ALI and ARDS.
The role of SACs in the development of HO and mechanical stretch-induced ALI/ARDS and the consequences of SAC dysregulation on epithelial barrier function and inflammatory cytokine secretion is of particular interest (181, 182). Both in vitro and in vivo studies have demonstrated that mechanical forces can alter transepithelial ion transport, which results in a net decrease in alveolar fluid clearance (50, 146, 204) and is directly associated with an increased length of mechanical ventilation and hospital mortality in patients with ALI/ARDS (70, 181).
The term SACs refers to a variety of ion channels, including Na+ channels such as ENaC, Cl− channels such as the ClC family, Ca2+ channels such as the transient receptor potential vanilloid family, and numerous K+ channels including two-pore domain potassium (K2P) channels, KATP and Kir channels (23, 126). SAC channels are ubiquitously expressed in body tissues including the heart, kidneys, genitourinary tract, gastrointestinal tract, leukocytes, and great blood vessels (179, 202, 203) and regulate a variety of cellular functions including the resting membrane potential, cell volume, gene expression, cell differentiation, and cell-contraction processes (16, 39, 154).
The K2P channel family consists of 15 members and is divided into 6 subfamilies: TWIK, THIK, TREK, TASK, TALK, and TRESK (156). A unique property of K2P channels is that they can be constitutively open, creating so-called K+ leak currents that maintain the negative resting membrane potential of a cell (12, 127), and, in tissues other than the lung, K2P channels have been shown to act as mechanosensors and mechanotransducers (212). For this review, we will focus specifically on the role of TREK-1 in ALI/ARDS.
Regulation and function of TREK-1 channels in models of ALI/ARDS.
TREK-1 expression was first detected in mouse, rat, and human AECs (163) but also appears to be expressed in airway epithelial cells (213). Preliminary data show that TREK-1-deficient cultured mouse AECs and human bronchial epithelial cells appear protected from a HO- and TNF-α-induced loss of transepithelial electrical resistance, suggesting a role for TREK-1 in epithelial barrier (Schwingshackl A, unpublished observations). In addition, stimulation of TREK-1-deficient mouse and human epithelial cells with TNF-α decreased IL-6 and RANTES secretion and increased in monocyte chemotactic protein-1 (MCP-1) secretion (Fig. 2). In contrast, keratinocyte chemokine/IL-8 secretion remained unchanged between TREK-1-deficient and control cells. Whereas several TNF-α-activated signaling pathways, including p38 kinase, PKC, and c-JNK1/2/3 were altered in TREK-1-deficient AECs, decreased IL-6 secretion from TREK-1-deficient AECs was related to impaired phosphorylation of the PKC isoform-θ after TNF-α stimulation, and increased MCP-1 secretion appeared unrelated to the increased phosphorylation of JNK1/2/3 in TREK-1-deficient AECs. The specific molecular signaling mechanisms underlying these alterations in cytokine secretion is presently being investigated.
Fig. 2.
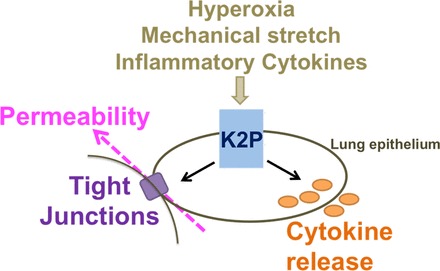
We propose that hyperoxia, mechanical stretch, and inflammatory cytokines alter the regulation of two-pore domain potassium (K2P) channel function. This can then impact on cytokine secretion and epithelial barrier function, and this dysregulation of these proteins can contribute to the pathogenesis of acute lung injury downstream.
On the basis of in vitro findings, it was anticipated that TREK-1 KO mice would be protected from HO- and mechanical stretch-induced lung injury. However, exposure of TREK-1 KO mice to 24 h of HO resulted in increased lung injury, as evidenced by gross lung anatomy, hematoxylin and eosin staining of lung sections (increased inflammatory cell infiltration and hemorrhage), increased lung injury scoring, decreased lung compliance, and increased macrophage and neutrophil accumulation in the bronchoalveolar lavage (BAL) fluid of TREK-1 KO mice (164). Interestingly, exposure of TREK-1 KO mice to HO plus mechanical ventilation resulted in no further injury compared with HO exposure alone (164).
Similar to in vitro studies (163), in vivo models show that HO exposure is a weak stimulus for cytokine secretion, and only HO plus mechanical ventilation increased IL-6, MCP-1, and TNF-α levels in the BAL fluid of both wild-type and TREK-1 KO mice. In accordance with in vitro findings, there were decreased amounts of IL-6 in HO plus mechanically ventilated TREK-1 KO mice (161). However, in contrast to in vitro findings (162), MCP-1 levels were decreased in HO plus mechanically ventilated TREK-1 KO mice, whereas TNF-α levels were unchanged between control and TREK-1 KO mice (164). Therefore, in this model, alterations in cytokine secretion between TREK-1 KO and wild-type mice were unlikely the cause for the observed increase in lung injury in the TREK-1 KO mice. Nevertheless, the contributions of cytokines such as IL-6, IL-1β, and TNF-α to the development of ALI/ARDS in other animal models and in human patients with ARDS are well documented (118, 119). The proinflammatory environment associated with the secretion of these mediators promotes neutrophil-, macrophage-, T cell-, and epithelium-mediated alveolar injury, causes loss of alveolar barrier function, and decreases alveolar surfactant levels (61, 94, 119, 185, 206). Similar to in vitro studies, the specific molecular mechanisms underlying these alterations in BAL cytokine levels in in vivo models remain to be determined. In addition, it will be important to investigate the cellular sources for these inflammatory mediators and quantify their relative contributions. Furthermore, it is of major importance to determine the time course of cytokine secretion in these ARDS models. It is quite possible that, although after 24 h of HO exposure and 4 h of mechanical ventilation BAL IL-6 and MCP-1 levels were decreased in TREK-1-deficient mice, these cytokines were increased at earlier time points and contributed to the observed lung injury. Of note, whereas TNF-α is a known proinflammatory cytokine in the early stages of ARDS, in chronic pulmonary inflammation, it may actually exert anti-inflammatory, beneficial effects and promote physiological wound healing (117).
Interestingly, alveolar barrier function appeared uncompromised in the mouse ARDS model because BAL protein levels were unchanged between control and TREK-1-deficient mice (164). HO alone caused only a minor disruption in alveolar barrier function, whereas the combination of HO and mechanical ventilation resulted in significant BAL protein accumulation in both mouse types. The protective effect of TREK-1 deficiency observed in vitro on TNF-α- and HO-induced loss of barrier function was not seen in the whole animal model. Whether a close interaction between TREK-1 and epithelial tight junction proteins, as seen in cultured AECs (unpublished observations), also exists in vivo remains to be confirmed.
One explanation for the increased lung damage observed in HO-exposed TREK-1 KO mice could be an increase in AT I and AT II cell apoptosis in these animals, as evidenced by TUNEL staining and corroborated by increased poly(ADP-ribose) polymerase-1 cleavage, one of the distal signaling molecules of the caspase cascade (164). Another possible explanation could be a decrease in surfactant protein C detected in TREK-1-deficient mice after prolonged HO exposure, potentially making lungs more vulnerable to HO- and mechanical stretch-induced injury (164). The molecular mechanisms linking TREK-1 deficiency to epithelial apoptosis and decreased surfactant production are presently unknown. As described above, we have observed substantial alterations in p38, PKC, and JNK phosphorylation in TREK-1-deficient epithelial cells after TNF-α stimulation (161, 162), but whether these kinases are involved in apoptosis or surfactant production needs to be determined. Another question that needs to be addressed in future studies is whether K+ flux through TREK-1 channels is required for the observed in vitro and in vivo effects or whether TREK-1 could exert K+-independent functions and act as a regulatory molecule rather than an ion channel, as reported, for example, in CFTR (159).
In conclusion, whereas in vitro epithelial TREK-1 deficiency appeared protective against the inflammatory stimuli responsible for the development of ALI/ARDS, in an in vivo model of ALI/ARDS, TREK-1 deficiency resulted in increased lung damage. The mechanisms accounting for these differences are presently unknown. Conditional TREK-1-deficient mice should give us insights into the molecular mechanisms responsible for the observed lung damage and help us resolve the discrepancies between in vitro and in vivo models. Once TREK-1 signaling pathways are delineated, the development of TREK-1-activating or -inhibiting agents may steer us toward new therapeutic approaches in ALI/ARDS. As with all known ion-channel modulators, drug-specificity issues will likely constitute future challenges, especially because K2P channels are quite ubiquitously expressed in the body tissues.
The Lung Epithelium: NRG-1-HER2/3 Signaling in Epithelial Barrier Regulation
HER family signaling: background.
Receptor tyrosine kinases, such as members of the human epidermal growth factor receptor (HER) family, and their ligands serve as important signaling molecules that regulate the epithelial response to extracellular signals and environmental stimuli. HER family signaling has been shown to regulate epithelial migration, growth, and differentiation as well as epithelial barrier function in numerous tissue beds including skin (184), intestine (38), cornea (123, 211, 215), and the lung (30). Identification of mutations of HER family genes as pathogenic in certain epithelial cancers (85, 180) has led to the successful deployment of targeted tyrosine kinase inhibitors (TKIs) aimed at inhibiting these receptors, particularly in breast and lung cancer (24, 105). However, recent data demonstrate that these receptors regulate pulmonary epithelial barrier function in nontransformed pulmonary epithelia, particularly in the context of inflammatory lung injury (46, 47).
The HER family is a complex signaling network consisting of four transmembrane receptors, HER1 or the EGFR, HER2, HER3, and HER4 and ten known ligands. The HER family is also referred to as the ErbB family after it was discovered that the avian erytheroblastosis virus gene encoded a mutant form of EGFR/HER1 (64, 65, 138). Members of the HER family are expressed on the basolateral surface of airway and AT I and II epithelial cells. The four HER receptors all share a similar structure including an extracellular ligand-binding domain, a single transmembrane domain, and a cytoplasmic tyrosine kinase domain (209). HER receptors exist in quiescent cells as single “closed” monomers. Ligand binding induces a conformational change to an “open” configuration, allowing for homo- or heterodimerization, leading to phosphorylation of intracellular tyrosine residues and activation of the kinase domain. This allows for docking of cytoplasmic proteins that facilitate initiation of downstream signaling. Certain structural distinctions influence how specific monomers interact to form signaling competent dimer pairs. HER2 has no ligand yet, under basal conditions, exists in a receptive open confirmation, making it the preferred binding partner of other HER receptors. In contrast, HER3 has at least four different ligands yet does not have a functioning kinase and partners with other receptors to influence signaling. Although it does not directly phosphorylate other proteins, HER3 interacts with other intracellular proteins and signaling pathways such as the phosphatidylinositol 3 kinase pathway (103). Additionally, in cancer cells, HER3-dependent signaling is a resistance mechanism against HER2-targeted TKI therapy (52). Broadly, ligands are organized depending on which receptors they bind and activate. EGF, TGF-α, and amphiregulin bind EGFR/HER1 exclusively, whereas β-cellulin, heparin-binding EGF (HB-EGF) and epiregulin can bind EGFR/HER1 or HER4. The final group of ligands includes the neuregulins (NRG-1–4). The four NRG proteins all share a similar EGF-like domain, which is sufficient for receptor activation yet differs in tissue distribution structurally, which can influence signaling, NRG-1 being the isoform most commonly studied.
HER family signaling is a complex, multilayered network influenced by factors such as dimerization partner, cell type (e.g., cancer vs. untransformed), and ligand. (148). For example, activated EGFR phosphorylates αCbl and Grb2 but only when activated in partnership with HER2, as opposed to other HER family members (58, 129). Additionally, whereas several ligands can bind and activate different HER receptors, each ligand elicits specific intracellular responses depending on cell type (e.g., transformed vs. nontransformed) and the availability of other HER receptors. In HER2-deficient cells, NRG-1 can induce EGFR/HER3 dimerization and activation, but, when HER2 is available, NRG-1 preferentially induces HER2/3 heterodimers, eliciting specific intracellular signaling patterns (46, 58).
Accumulating data suggest that the HER family is important in regulating epithelial barrier function in the lung. Activation of EGFR with TGF-α leads to epithelial proliferation, spreading, and motility (99, 153) (86) and enhances wound closure in cultured AECs (86). Additionally, epithelial injury leads to shedding of HER family ligands, and wound closure is delayed after exposure to an EGFR inhibitor. Similarly, NRG-1 shedding and HER2 activation are also observed in a scratch model of wound closure, and blockade of HER2 impedes healing following wounding (200). Notably, exogenous NRG-1 increases epithelial permeability similar to a monolayer of epithelial cells (46). These data suggest that HER activation can enhance, improve, or worsen epithelial barrier function, possibly depending on the continuity of the barrier at the time of receptor activation.
NRG-1-HER2/3: regulation of the pulmonary epithelial barrier.
Recent evidence has identified the NRG-1-HER2/3 system as activated in and a critical modulator of pulmonary epithelial barrier function in ALI and ARDS. ALI and ARDS are syndromes marked by a hyperinflammatory state with an increase in cytokines such as IL-6 and IL-8 that are felt to participate in disease pathogenesis. In particular, the cytokine IL-1β has been implicated as a central mediator of ALI, both in animal models and humans (53, 71, 114, 132, 133, 140, 174). IL-1β is increased in the lavage of patients with ARDS, and exogenous IL-1β leads to increased epithelial permeability in an epithelial monolayer as well as in animal models (89). Therapeutically, strategies to block IL-1β signaling are protective in bleomycin and ventilator-induced models of lung injury (53, 132) (49). Epithelial NRG-1-HER2 signaling has been recently identified as activated by IL-1β and central in the pathogenesis of epithelial injury and permeability induced by IL-1β. In cultured airway and alveolar cells, IL-1β-mediated increases in epithelial permeability are HER2 dependent (46). IL-1β induces shedding of NRG-1 and HER2 activation without evidence of EGFR activation. The protease ADAM17 has been identified as the sheddase responsible for NRG-1 shedding. Pharmacological (inhibitors) or genetic (siRNA) strategies to inhibit ADAM17, NRG-1, or HER2 significantly attenuate IL-1β-mediated changes in epithelial barrier function. Hence, an overall pathway of IL-1β-ADAM17-NRG-1-HER2/3 leading to increased epithelial permeability has emerged as a new important regulator of epithelial barrier function in ALI.
NRG-1-HER2/3 signaling also participates in animal models of lung injury. Bleomycin-induced ALI is associated with both increased BAL NRG-1, indicative of surface shedding as well as pulmonary HER2 activation. Pharmacological ADAM17 inhibition attenuates NRG-1 cleavage, pulmonary HER2 phosphorylation, and indices of injury in mice exposed to bleomycin. Bleomycin serves as a model of both acute and chronic fibrotic lung injury, and targeting HER2 is protective in the fibrotic phase as well as the acute phase of lung injury following bleomycin. Use of an inhibitor that prevents HER2 dimerization with HER3 prevented both HER2/3 activation as well as fibrosis at 21 days after bleomycin exposure (44). Similarly, transgenic mice lacking pulmonary epithelial HER3 are protected from bleomycin-induced fibrosis (125). Although in these studies indices of acute injury were not specifically studied, there was suggestion of protection in alveolar leak early following bleomycin. Mechanistically, alterations in alveolar epithelial permeability have been linked to a profibrotic response in the lung (2, 3, 15, 178), possibly providing a link between early acute injury marked by compromised alveolar epithelial barrier function and delayed fibrotic injury.
Proteases are critical participants in the HER family signaling, as they translate external signals into HER receptor activation through shedding of surface ligands. Numerous agonists that induce epithelia barrier dysfunction do so via protease-mediated HER ligand shedding, leading to receptor activation. Examples of this include IL-1β, mechanical stretch, and hypertonic stress. In particular, ADAM17 has been identified as the sheddase responsible for cleaving numerous HER family ligands and has been linked to disease marked by inflammation and increased permeability (21, 27, 29, 55, 211). In vitro, blocking ADAM17 reduces IL-1β-induced epithelial permeability through blocking NRG-1 shedding and subsequent HER2 activation. In vivo, intratracheal instillation of the ADAM17 inhibitor attenuated bleomycin-induced ALI, including protein leak. This was associated with decreased NRG-1 lavage levels and HER2 activation. Whereas ADAM17 KO mice display embryonic lethality, mice deficient in the endogenous inhibitor of ADAM17, tissue inhibitor of metalloprotease-3, have delayed resolution of lung injury following bleomycin administration, supporting the importance of this sheddase in ALI, although the role of HER ligands and receptors was not specifically investigated.
Perhaps the most compelling data indicating that the HER family participates in epithelial barrier disruption in the lung is that patients with ARDS have evidence of active ligand shedding in the lung. TGF-α is elevated in lavage from patients, suggesting pathway activation. Similarly, increased NRG-1 lavage levels have been detected from patients with ARDS. As a biomarker, NRG-1 levels were significantly correlated with inflammation as measured by inflammatory cytokines. Notably, NRG-1 elevations were associated with worse outcomes as measured by ventilator-free days. Plasma NRG-1 is also increased in patients with ARDS, suggesting that the source of NRG-1 leading to HER2 activation could be extrapulmonary.
NRG-1-HER2/3 signaling: mechanisms regulating epithelial cell-cell adhesion and permeability.
Knowledge regarding the mechanisms by which HER2 and the other HER receptors influence epithelial permeability is incomplete, but several pathways have been outlined. HER proteins modulate cell-cell adhesion proteins of the tight junction and the adherens junction. EGFR alters claudin expression and spatial rearrangement of claudins and ZO-1 (28, 189). HER2 has also been linked to the tight junction proteins ZO-1 and occludin (54, 56), as well as integrin-β4 in regulating cell-cell adhesion (66). Recent data suggest that the adherens junction protein β-catenin is a required intermediary influencing HER2-mediated barrier function in the lung. Phosphorylation at tyrosine Y-654 of β-catenin has been linked to altered barrier function. NRG-1-mediated HER2 activation leads to β-catenin Y-654 phosphorylation as well as disruption of β-catenin-E-cadherin interaction at the cell junction in pulmonary epithelial cells (48). This is associated with impaired E-cadherin-mediated cell-cell adhesion. Finally, loss of β-catenin via shRNA knockdown results in a significant attenuation in NRG-1- HER2/3-induced barrier dysfunction. Together, these data suggest that NRG-1-HER3-HER2 activation leads to β-catenin tyrosine phosphorylation, disruption of adherens junction, and alteration of the pulmonary epithelial barrier (Fig. 3).
Fig. 3.
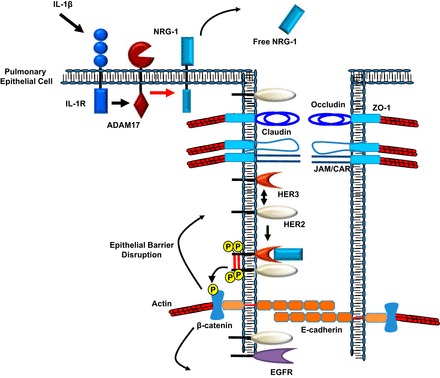
Cytokine activation can lead to increased free neuregulin 1 (NRG-1). Initial disruption of the epithelial barrier allows for NRG-1- human epidermal growth factor receptor 3 (HER3)-HER2 activation. The activation of this complex can lead to β-catenin tyrosine phosphorylation, which further propagates disruption of adherens junction, and alteration of the pulmonary epithelial barrier. ZO, zonula occludens; JAM, junctional adhesion molecules; CAR, coxsackie and adenovirus receptor.
Additionally, processes inherent in barrier functions such as proliferation and cell migration suggest other pathways by which HER proteins might influence the epithelial barrier. The GTPases Rho and Rac have both been linked to HER family signaling. In a breast cancer epithelial cell line, HER2-dependent Rac activation was required for NRG-1-mediated cell migration (208). Rac activation following NRG-1 is likely via HER2-dependent P-Rex1 activation, as downregulation of P-Rex1 inhibited Rac activation and cell migration following NRG-1 (121). Similarly, EGFR activation leads to Rac activation and Rac-dependent cell migration (34). During epithelial wound closure, Rho activation follows EGFR activation, and, in a corneal epithelial wound model, Rho inhibition decreased both cell migration and proliferation, resulting in delayed wound closure (210).
Proteins involved in formation of tight junctions and adherens junctions are also mobilized after HER family activation. EGFR activation alters expression levels of claudins and the cellular localization of claudins and ZO-1 (28, 189). NRG-1-HER2 mediated changes in epithelial permeability are also associated with alterations in cellular localization of the tight junction proteins ZO-1 and occludin (54, 56). Integrin-β4, an adhesion molecule that regulates cell-cell junctions (66) and epithelial proliferation (26), amplifies HER2 activation in epithelial cells to activate STAT and disrupt cell-cell-adhesion (60). Previous reports have suggested a structural and functional link between the adherens junction protein β-catenin and HER2. Structurally, HER2 colocalizes to the basolateral plasma membrane via binding to the PDZ domain of erbin (20). Erbin has been identified as a binding partner for the p120 catenin protein p0071 as well as direct binding partner for β-catenin (59, 147). In gastric and breast cancer epithelial cells, TGF-α induces EGFR-dependent HER2 activation, β-catenin phosphorylation, and β-catenin-HER2 association although the role of HER2 in β-catenin phosphorylation was not specifically studied (83, 128, 160, 170). Functionally, HER2 and β-catenin have both been linked to breast cancer pathogenesis. In murine breast cancer models utilizing a constitutively active HER2, β-catenin signaling was activated and required for tumor initiation and metastasis (155). Blockade of HER2 using trastuzumab blocked both HER2 activation and β-catenin phosphorylation, as well as β-catenin-dependent transcription of surviving breast cancer cells (214).
Conclusion
The airway epithelium is a physical and immunological barrier that sits at the interface between the lumen and the lung parenchyma and is the initial site of contact for all inspired substances. It is constantly exposed to both physiological and pathological stimuli and dynamically regulates its barrier. This dynamic regulation occurs via changes in cell-cell adhesion attributable to altered cytoskeletal and junctional protein arrangement and expression, as well as changes in transcellular and paracellular ion permeability in response to these stimuli to help maintain homeostasis. The ion channels on the apical and basolateral cell surfaces and the junctional complexes, which directly connect adjacent epithelial cells, play a key role in controlling the movement of substances from the lumen into the subepithelial space both via paracellular and transcellular transport. The junctions also separate the apical and basolateral cell surface, which maintains the specialized function of each cell surface, controls the interaction between various cytokines, growth factors, and their receptors, and modulates the extent and location of inflammatory response to stimuli. Exposure to noxious stimuli such as mechanical stretch/trauma, HO, infection, cigarette smoke, or particulate matter damages the epithelial barrier, resulting in a loss of compartmentalization and a hyperproliferative, inflammatory state that is seen in many pulmonary diseases.
The junction proteins and ion channels interact to maintain lung fluid balance. Initial studies indicate that changes in the levels of the tight junction claudins can alter fluid balance. However, further work needs to be done to characterize the compensatory shifts in junction proteins that occur in response to physiological and pathological stimuli to maintain tissue homeostasis.
As outlined earlier, stretch activated ion channels play a role in epithelial barrier function. TREK-1 has been linked to the pathogenesis of ALI/ARDS; however, there are conflicting results in in vitro and in vivo models of ALI/ARDS. Future studies delineating the role of TREK-1 in ALI/ARDS could potentially lead to the development of new targeted therapies for this disease. The ENaC complex is another ion channel shown to be important in lung fluid balance, but better defining the role of the individual subunits in fluid balance as well as identifying other pathways that contribute to the trafficking of this complex will be important in further understanding the pathogenesis of diseases such as ARDS.
Although it is hypothesized that changes in the physical barrier in response to chronic stimulation by both physiological and pathological stimuli precipitate the immunological changes that lead to diseases such as COPD, ALI, and ARDs, the sequence and time course of these changes are yet to be determined. As discussed in this review, the epithelium dynamically regulates its response to a variety of stimuli to promote repair and regeneration; however, we do not understand mechanisms of dysregulation of this dynamic repair process. Future studies should further explore the link between altered transcellular and paracellular transport and the activation of the inflammatory cascades that result in chronic epithelial barrier dysfunction and injury. In addition, much attention has been devoted to pathways that mediate lung inflammation and injury; however, pathways that protect against lung injury are not well studied, and therefore few have been detailed in the literature. It is critical to note that Islam et al. (77) demonstrated that both epithelial injury and the downstream consequences of disease can be reversed by targeting the epithelium, which was done using mitochondrial transfer (77). However, this raises a future directive to, not only identify pathways involved in damage, but also investigate protective mechanisms. Recently, the apelin-APJ pathway has been shown to be protective against ARDS/ALI, but more work needs to be done to identify other pathways that protect against lung inflammation and injury (42). Our future directive is not to simply define and dissect disrupted molecular pathways in the epithelium but target and manipulate protective mechanisms to alter the course of disease. In conditions such as COPD, where there is a long lead time before the onset of disease, there is ample opportunity to prevent the development by manipulating epithelial targets. In conditions such as ARDS, we need to fine-tune mechanisms to target the epithelium to ameliorate the severity of disease and amplify repair and resolution.
As discussed earlier in this review, the maintenance of ion and fluid homeostasis in the lung is complex and requires the dynamic interaction between numerous ion channels and transporter proteins. Studies have started to explore the connections between paracellular and transcellular transport in maintaining fluid balance and ion homeostasis. However, our knowledge regarding this subject is still in its infancy. We have already identified certain transporters, such as aquaporin 5, which have roles in dictating both transcellular water transport and paracellular permeability as outlined in The Lung Epithelium: Insights into the Role of Regulation of Paracellular Permeability in Epithelial Signaling. Further defining the contribution of these transporters to the pathogenesis of different pulmonary diseases could potentially lead to the development of novel therapeutics. Indeed, Ivacaftor, a drug approved to treat patients with cystic fibrosis who have specific mutations in the CFTR protein, is one such example, which could have reaches into noncystic fibrosis lung diseases as well.
In summary, the airway epithelium is a dynamic barrier that constantly responds to luminal stimuli and coordinates its response to maintain homeostasis in the lung. Breakdown in this coordinated response can lead to a myriad of lung diseases.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
K.B., J.H.F., and V.K.S. prepared figures; K.B., J.A.F., A.S., J.H.F., and V.K.S. drafted manuscript; V.K.S. edited and revised manuscript; V.K.S. approved final version of manuscript.
REFERENCES
- 1.Ablimit A, Hasan B, Lu W, Qin W, Wushouer Q, Zhong N, Upur H. Changes in water channel aquaporin 1 and aquaporin 5 in the small airways and the alveoli in a rat asthma model. Micron 45: 68–73, 2013. [DOI] [PubMed] [Google Scholar]
- 2.Adamson IY, Hedgecock C, Bowden DH. Epithelial cell-fibroblast interactions in lung injury and repair. Am J Pathol 137: 385–392, 1990. [PMC free article] [PubMed] [Google Scholar]
- 3.Adamson IY, Young L, Bowden DH. Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol 130: 377–383, 1988. [PMC free article] [PubMed] [Google Scholar]
- 4.Adir Y, Welch LC, Dumasius V, Factor P, Sznajder JI, Ridge KM. Overexpression of the Na-K-ATPase α2-subunit improves lung liquid clearance during ventilation-induced lung injury. Am J Physiol Lung Cell Mol Physiol 294: L1233–L1237, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Al-Bazzaz FJ, Hafez N, Tyagi S, Gailey CA, Toofanfard M, Alrefai WA, Nazir TM, Ramaswamy K, Dudeja PK. Detection of Cl-HCO3- and Na+-H+ exchangers in human airways epithelium. JOP 2: 285–290, 2001. [PubMed] [Google Scholar]
- 6.Al-Haggar M, Bakr A, Tajima T, Fujieda K, Hammad A, Soliman O, Darwish A, Al-Said A, Yahia S, Abdel-Hady D. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: unusual clinical associations and novel claudin16 mutation in an Egyptian family. Clin Exp Nephrol 13: 288–294, 2009. [DOI] [PubMed] [Google Scholar]
- 7.Angelow S, Ahlstrom R, Yu AS. Biology of claudins. Am J Physiol Renal Physiol 295: F867–F876, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bardou O, Trinh NT, Brochiero E. Molecular diversity and function of K+ channels in airway and alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 296: L145–L155, 2009. [DOI] [PubMed] [Google Scholar]
- 9.Basset G, Crone C, Saumon G. Fluid absorption by rat lung in situ: pathways for sodium entry in the luminal membrane of alveolar epithelium. J Physiol 384: 325–345, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger J, Richter K, Clauss WG, Fronius M. Evidence for basolateral Cl- channels as modulators of apical Cl- secretion in pulmonary epithelia of Xenopus laevis. Am J Physiol Regul Integr Comp Physiol 300: R616–R623, 2011. [DOI] [PubMed] [Google Scholar]
- 11.Bernard K, Bogliolo S, Soriani O, Ehrenfeld J. Modulation of calcium-dependent chloride secretion by basolateral SK4-like channels in a human bronchial cell line. J Membr Biol 196: 15–31, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Berrier C, Pozza A, de Lacroix de Lavalette A, Chardonnet S, Mesneau A, Jaxel C, le Maire M, Ghazi A. The purified mechanosensitive channel TREK-1 is directly sensitive to membrane tension. J Biol Chem 288: 27307–27314, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berthiaume Y, Matthay MA. Alveolar edema fluid clearance and acute lung injury. Respir Physiol Neurobiol 159: 350–359, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhowmik A, Chahal K, Austin G, Chakravorty I. Improving mucociliary clearance in chronic obstructive pulmonary disease. Respir Med 103: 496–502, 2009. [DOI] [PubMed] [Google Scholar]
- 15.Bitterman PB. Pathogenesis of fibrosis in acute lung injury. Am J Med 92: 39S–43S, 1992. [DOI] [PubMed] [Google Scholar]
- 16.Bittner S, Ruck T, Schuhmann MK, Herrmann AM, Moha ou Maati H, Bobak N, Gobel K, Langhauser F, Stegner D, Ehling P, Borsotto M, Pape HC, Nieswandt B, Kleinschnitz C, Heurteaux C, Galla HJ, Budde T, Wiendl H, Meuth SG. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat Med 19: 1161–1165, 2013. [DOI] [PubMed] [Google Scholar]
- 17.Boiko N, Kucher V, Eaton BA, Stockand JD. Inhibition of neuronal degenerin/epithelial Na+ channels by the multiple sclerosis drug 4-aminopyridine. J Biol Chem 288: 9418–9427, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boon M, De Boeck K, Jorissen M, Meyts I. Primary ciliary dyskinesia and humoral immunodeficiency—is there a missing link? Respir Med 108: 931–934. [DOI] [PubMed] [Google Scholar]
- 19.Boon M, Jorissen M, Proesmans M, De Boeck K. Primary ciliary dyskinesia, an orphan disease. Eur J Pediatr 172: 151–162. [DOI] [PubMed] [Google Scholar]
- 20.Borg JP, Marchetto S, Le Bivic A, Ollendorff V, Jaulin-Bastard F, Saito H, Fournier E, Adélaïde J, Margolis B, Birnbaum D. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol 2: 407, 2000. [DOI] [PubMed] [Google Scholar]
- 21.Borrell-Pages M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J 22: 1114–1124, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burke WM, Roberts CM, Bryant DH, Cairns D, Yeates M, Morgan GW, Martin BJ, Blake H, Penny R, Zaunders JJ, Breit SN. Smoking-induced changes in epithelial lining fluid volume, cell density and protein. Eur Respir J 5: 780–784, 1992. [PubMed] [Google Scholar]
- 23.Buxton IL, Heyman N, Wu YY, Barnett S, Ulrich C. A role of stretch-activated potassium currents in the regulation of uterine smooth muscle contraction. Acta Pharmacol Sin 32: 758–764, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carr LL, Finigan JH, Kern JA. Evaluation and treatment of patients with non-small cell lung cancer. Med Clin North Am 95: 1041–1054, 2011. [DOI] [PubMed] [Google Scholar]
- 25.Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet 12: 248–253, 1996. [DOI] [PubMed] [Google Scholar]
- 26.Chapman HA, Li X, Alexander JP, Brumwell A, Lorizio W, Tan K, Sonnenberg A, Wei Y, Vu TH. Integrin alpha6beta4 identifies an adult distal lung epithelial population with regenerative potential in mice. J Clin Invest 121: 2855–2862, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J Biol Chem 282: 33714–33724, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Chen SP, Zhou B, Willis BC, Sandoval AJ, Liebler JM, Kim KJ, Ann DK, Crandall ED, Borok Z. Effects of transdifferentiation and EGF on claudin isoform expression in alveolar epithelial cells. J Appl Physiol 98: 322–328, 2005. [DOI] [PubMed] [Google Scholar]
- 29.Colon AL, Menchen LA, Hurtado O, De Cristobal J, Lizasoain I, Leza JC, Lorenzo P, Moro MA. Implication of TNF-alpha convertase (TACE/ADAM17) in inducible nitric oxide synthase expression and inflammation in an experimental model of colitis. Cytokine 16: 220–226, 2001. [DOI] [PubMed] [Google Scholar]
- 30.Crosby LM, Waters CM. Epithelial repair mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol 298: L715–L731, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crump RG, Askew GR, Wert SE, Lingrel JB, Joiner CH. In situ localization of sodium-potassium ATPase mRNA in developing mouse lung epithelium. Am J Physiol Lung Cell Mol Physiol 269: L299–L308, 1995. [DOI] [PubMed] [Google Scholar]
- 32.De Lisle RC. Disrupted tight junctions in the small intestine of cystic fibrosis mice. Cell Tissue Res 355: 131–142, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Demaio L, Tseng W, Balverde Z, Alvarez JR, Kim KJ, Kelley DG, Senior RM, Crandall ED, Borok Z. Characterization of mouse alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol 296: L1051–L1058, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dise RS, Frey MR, Whitehead RH, Polk DB. Epidermal growth factor stimulates Rac activation through Src and phosphatidylinositol 3-kinase to promote colonic epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 294: G276–G285, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Dobbs LG, Johnson MD. Alveolar epithelial transport in the adult lung. Respir Physiol Neurobiol 159: 283–300, 2007. [DOI] [PubMed] [Google Scholar]
- 36.Eaton DC, Chen J, Ramosevac S, Matalon S, Jain L. Regulation of Na+ channels in lung alveolar type II epithelial cells. Proc Am Thorac Soc 1: 10–16, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol 71: 403–423, 2009. [DOI] [PubMed] [Google Scholar]
- 38.Egan LJ, de Lecea A, Lehrman ED, Myhre GM, Eckmann L, Kagnoff MF. Nuclear factor-κB activation promotes restitution of wounded intestinal epithelial monolayers. Am J Physiol Cell Physiol 285: C1028–C1035, 2003. [DOI] [PubMed] [Google Scholar]
- 39.Eisenhut M, Wallace H. Ion channels in inflammation. Pflügers Arch 461: 401–421, 2011. [DOI] [PubMed] [Google Scholar]
- 40.Ellison RT 3rd, Giehl TJ. Killing of Gram-negative bacteria by lactoferrin and lysozyme. J Clin Invest 88: 1080–1091, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Falk MM. Adherens junctions remain dynamic. BMC Biol 8: 34, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan XF, Xue F, Zhang YQ, Xing XP, Liu H, Mao SZ, Kong XX, Gao YQ, Liu SF, Gong YS. The apelin-APJ axis is an endogenous counter-injury mechanism in experimental acute lung injury. Chest. In press. [DOI] [PubMed] [Google Scholar]
- 43.Fang X, Song Y, Hirsch J, Galietta LJ, Pedemonte N, Zemans RL, Dolganov G, Verkman AS, Matthay MA. Contribution of CFTR to apical-basolateral fluid transport in cultured human alveolar epithelial type II cells. Am J Physiol Lung Cell Mol Physiol 290: L242–L249, 2006. [DOI] [PubMed] [Google Scholar]
- 44.Faress JA, Nethery DE, Kern EF, Eisenberg R, Jacono FJ, Allen CL, Kern JA. Bleomycin-induced pulmonary fibrosis is attenuated by a monoclonal antibody targeting HER2. J Appl Physiol 103: 2077–2083, 2007. [DOI] [PubMed] [Google Scholar]
- 45.Fesenko EE, Kolesnikov SS, Lyubarsky AL. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature 313: 310–313, 1985. [DOI] [PubMed] [Google Scholar]
- 46.Finigan JH, Faress JA, Wilkinson E, Mishra RS, Nethery DE, Wyler D, Shatat M, Ware LB, Matthay MA, Mason R, Silver RF, Kern JA. Neuregulin-1-human epidermal receptor-2 signaling is a central regulator of pulmonary epithelial permeability and acute lung injury. J Biol Chem 286: 10660–10670, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finigan JH, Mishra R, Vasu VT, Silveira LJ, Nethery DE, Standiford TJ, Burnham EL, Moss M, Kern JA. Bal neuregulin-1 is elevated in acute lung injury and correlates with inflammation. Eur Respir J 41: 396–401, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finigan M, Warm J. Ventilatory considerations in cardiac patients. Nurs Clin North Am 7: 541–548, 1972. [PubMed] [Google Scholar]
- 49.Frank JA, Pittet JF, Wray C, Matthay MA. Protection from experimental ventilator-induced acute lung injury by IL-1 receptor blockade. Thorax 63: 147–153, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Fronius M, Clauss WG, Althaus M. Why do we have to move fluid to be able to breathe? Front Physiol 3: 146, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Furuse M, Sasaki H, Fujimoto K, Tsukita S. A single gene product, claudin-1 or -2, reconstitutes tight junction strands and recruits occludin in fibroblasts. J Cell Biol 143: 391–401, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sanchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, Manning HC, Chang J, Arteaga CL. Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc Natl Acad Sci USA 108: 5021–5026, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, Ryffel B, Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest 117: 3786–3799, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Giri S, Poindexter KM, Sundar SN, Firestone GL. Arecoline induced disruption of expression and localization of the tight junctional protein ZO-1 is dependent on the HER 2 expression in human endometrial Ishikawa cells. BMC Cell Biol 11: 53, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gloushankova NA. Changes in regulation of cell-cell adhesion during tumor transformation. Biochemistry 73: 742–750, 2008. [DOI] [PubMed] [Google Scholar]
- 56.Gon Y, Matsumoto K, Terakado M, Sekiyama A, Maruoka S, Takeshita I, Kozu Y, Okayama Y, Ra C, Hashimoto S. Heregulin activation of ErbB2/ErbB3 signaling potentiates the integrity of airway epithelial barrier. Exp Cell Res 317: 1947–1953, 2011. [DOI] [PubMed] [Google Scholar]
- 57.Gonzalez C, Baez-Nieto D, Valencia I, Oyarzun I, Rojas P, Naranjo D, Latorre R. K+ channels: function-structural overview. Compr Physiol 2: 2087–2149, 2012. [DOI] [PubMed] [Google Scholar]
- 58.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16: 1647–1655, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gujral Taranjit S, Karp Ethan S, Chan M, Chang Bryan H, MacBeath G. Family-wide investigation of PDZ domain-mediated protein-protein interactions implicates β-catenin in maintaining the integrity of tight junctions. Chem Biol 20: 816–827, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo W, Pylayeva Y, Pepe A, Yoshioka T, Muller WJ, Inghirami G, Giancotti FG. Beta 4 integrin amplifies ErbB2 signaling to promote mammary tumorigenesis. Cell 126: 489–502, 2006. [DOI] [PubMed] [Google Scholar]
- 61.Halbertsma FJ, Vaneker M, Scheffer GJ, van der Hoeven JG. Cytokines and biotrauma in ventilator-induced lung injury: a critical review of the literature. Neth J Med 63: 382–392, 2005. [PubMed] [Google Scholar]
- 62.Han DY, Nie HG, Gu X, Nayak RC, Su XF, Fu J, Chang Y, Rao V, Ji HL. K+ channel openers restore verapamil-inhibited lung fluid resolution and transepithelial ion transport. Respir Res 11: 65, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta 1778: 660–669, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayman MJ, Beug H. Identification of a form of the avian erythroblastosis virus erb-B gene product at the cell surface. Nature 309: 460–462, 1984. [DOI] [PubMed] [Google Scholar]
- 65.Hayman MJ, Ramsay GM, Savin K, Kitchener G, Graf T, Beug H. Identification and characterization of the avian erythroblastosis virus erbB gene product as a membrane glycoprotein. Cell 32: 579–588, 1983. [DOI] [PubMed] [Google Scholar]
- 66.Hintermann E, Yang N, O'Sullivan D, Higgins JM, Quaranta V. Integrin alpha6beta4-erbB2 complex inhibits haptotaxis by up-regulating E-cadherin cell-cell junctions in keratinocytes. J Biol Chem 280: 8004–8015, 2005. [DOI] [PubMed] [Google Scholar]
- 67.Hobi N, Siber G, Bouzas V, Ravasio A, Perez-Gil J, Haller T. Physiological variables affecting surface film formation by native lamellar body-like pulmonary surfactant particles. Biochim Biophys Acta 1838: 1842–1850, 2014. [DOI] [PubMed] [Google Scholar]
- 68.Hogg JC, Timens W. The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol 4: 435–459, 2009. [DOI] [PubMed] [Google Scholar]
- 69.Hollenhorst MI, Richter K, Fronius M. Ion transport by pulmonary epithelia. J Biomed Biotechnol 2011: 174306, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holter JF, Weiland JE, Pacht ER, Gadek JE, Davis WB. Protein permeability in the adult respiratory distress syndrome. Loss of size selectivity of the alveolar epithelium. J Clin Invest 78: 1513–1522, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoshino T, Okamoto M, Sakazaki Y, Kato S, Young HA, Aizawa H. Role of proinflammatory cytokines IL-18 and IL-1beta in bleomycin-induced lung injury in humans and mice. Am J Respir Cell Mol Biol 41: 661–670, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hou J, Gomes AS, Paul DL, Goodenough DA. Study of claudin function by RNA interference. J Biol Chem 281: 36117–36123, 2006. [DOI] [PubMed] [Google Scholar]
- 73.Humlicek AL, Manzel LJ, Chin CL, Shi L, Excoffon KJ, Winter MC, Shasby DM, Look DC. Paracellular permeability restricts airway epithelial responses to selectively allow activation by mediators at the basolateral surface. J Immunol 178: 6395–6403, 2007. [DOI] [PubMed] [Google Scholar]
- 74.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet 12: 325–328, 1996. [DOI] [PubMed] [Google Scholar]
- 75.Huppmann S, Lankes E, Schnabel D, Buhrer C. Unimpaired postnatal respiratory adaptation in a preterm human infant with a homozygous ENaC-alpha unit loss-of-function mutation. J Perinatol 31: 802–803. [DOI] [PubMed] [Google Scholar]
- 76.Ibrahim HR, Aoki T, Pellegrini A. Strategies for new antimicrobial proteins and peptides: lysozyme and aprotinin as model molecules. Curr Pharm Des 8: 671–693, 2002. [DOI] [PubMed] [Google Scholar]
- 77.Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18: 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jamora C, Fuchs E. Intercellular adhesion, signalling and the cytoskeleton. Nat Cell Biol 4: E101–E108, 2002. [DOI] [PubMed] [Google Scholar]
- 79.Jansen HM, Sachs AP, van Alphen L. Predisposing conditions to bacterial infections in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 151: 2073–2080, 1995. [DOI] [PubMed] [Google Scholar]
- 80.Jovanovic S, Crawford RM, Ranki HJ, Jovanovic A. Large conductance Ca2+-activated K+ channels sense acute changes in oxygen tension in alveolar epithelial cells. Am J Respir Cell Mol Biol 28: 363–372, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Joyce-Brady M, Hiratake J. Inhibiting glutathione metabolism in lung lining fluid as a strategy to augment antioxidant defense. Curr Enzym Inhib 7: 71–78, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kage H, Flodby P, Gao D, Kim YH, Marconett CN, DeMaio L, Kim KJ, Crandall ED, Borok Z. Claudin 4 knockout mice: normal physiological phenotype with increased susceptibility to lung injury. Am J Physiol Lung Cell Mol Physiol 307: L524–L536, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kanai Y, Ochiai A, Shibata T, Oyama T, Ushijima S, Akimoto S, Hirohashi S. c-erbB-2 gene product directly associates with beta-catenin and plakoglobin. Biochem Biophys Res Commun 208: 1067–1072, 1995. [DOI] [PubMed] [Google Scholar]
- 84.Kerem E, Bistritzer T, Hanukoglu A, Hofmann T, Zhou Z, Bennett W, MacLaughlin E, Barker P, Nash M, Quittell L, Boucher R, Knowles MR. Pulmonary epithelial sodium-channel dysfunction and excess airway liquid in pseudohypoaldosteronism. N Engl J Med 341: 156–162, 1999. [DOI] [PubMed] [Google Scholar]
- 85.Kern JA, Robinson RA, Gazdar A, Torney L, Weiner DB. Mechanisms of p185HER2 expression in human non-small cell lung cancer cell lines. Am J Respir Cell Mol Biol 6: 359–363, 1992. [DOI] [PubMed] [Google Scholar]
- 86.Kheradmand F, Folkesson HG, Shum L, Derynk R, Pytela R, Matthay MA. Transforming growth factor-α enhances alveolar epithelial cell repair in a new in vitro model. Am J Physiol 267: L728–L738, 1994. [DOI] [PubMed] [Google Scholar]
- 87.Kim KJ, Cheek JM, Crandall ED. Contribution of active Na+ and Cl- fluxes to net ion transport by alveolar epithelium. Respir Physiol 85: 245–256, 1991. [DOI] [PubMed] [Google Scholar]
- 88.King LS, Yasui M. Aquaporins and disease: lessons from mice to humans. Trends Endocrinol Metab 13: 355–360, 2002. [DOI] [PubMed] [Google Scholar]
- 89.Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest 107: 1529–1536, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Konstas AA, Bielfeld-Ackermann A, Korbmacher C. Sulfonylurea receptors inhibit the epithelial sodium channel (ENaC) by reducing surface expression. Pflügers Arch 442: 752–761, 2001. [DOI] [PubMed] [Google Scholar]
- 91.Kunzelmann K, Schreiber R, Kmit A, Jantarajit W, Martins JR, Faria D, Kongsuphol P, Ousingsawat J, Tian Y. Expression and function of epithelial anoctamins. Exp Physiol 97: 184–192, 2012. [DOI] [PubMed] [Google Scholar]
- 92.Kwong RW, Kumai Y, Perry SF. Evidence for a role of tight junctions in regulating sodium permeability in zebrafish (Danio rerio) acclimated to ion-poor water. J Comp Physiol B 183: 203–213, 2013. [DOI] [PubMed] [Google Scholar]
- 93.Kwong RW, Perry SF. The tight junction protein claudin-b regulates epithelial permeability and sodium handling in larval zebrafish, Danio rerio. Am J Physiol Regul Integr Comp Physiol 304: R504–R513, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lacherade JC, Van De Louw A, Planus E, Escudier E, D'Ortho MP, Lafuma C, Harf A, Delclaux C. Evaluation of basement membrane degradation during TNF-α-induced increase in epithelial permeability. Am J Physiol Lung Cell Mol Physiol 281: L134–L143, 2001. [DOI] [PubMed] [Google Scholar]
- 95.LaFemina MJ, Rokkam D, Chandrasena A, Pan J, Bajaj A, Johnson M, Frank JA. Keratinocyte growth factor enhances barrier function without altering claudin expression in primary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 299: L724–L734, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.LaFemina MJ, Sutherland KM, Bentley T, Gonzales LW, Allen L, Chapin CJ, Rokkam D, Sweerus KA, Dobbs LG, Ballard PL, Frank JA. Claudin-18 deficiency results in alveolar barrier dysfunction and impaired alveogenesis in mice. Am J Respir Cell Mol Biol 51: 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Larre I, Lazaro A, Contreras RG, Balda MS, Matter K, Flores-Maldonado C, Ponce A, Flores-Benitez D, Rincon-Heredia R, Padilla-Benavides T, Castillo A, Shoshani L, Cereijido M. Ouabain modulates epithelial cell tight junction. Proc Natl Acad Sci USA 107: 11387–11392, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leroy C, Prive A, Bourret JC, Berthiaume Y, Ferraro P, Brochiero E. Regulation of ENaC and CFTR expression with K+ channel modulators and effect on fluid absorption across alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 291: L1207–L1219, 2006. [DOI] [PubMed] [Google Scholar]
- 99.Leslie CC, McCormick-Shannon K, Shannon JM, Garrick B, Damm D, Abraham JA, Mason RJ. Heparin-binding EGF-like growth factor is a mitogen for rat alveolar type II cells. Am J Respir Cell Mol Biol 16: 379–387, 1997. [DOI] [PubMed] [Google Scholar]
- 100.Li G, Flodby P, Luo J, Kage H, Sipos A, Gao D, Ji Y, Beard LL, Marconett CN, DeMaio L, Kim YH, Kim KJ, Laird-Offringa IA, Minoo P, Liebler JM, Zhou B, Crandall ED, Borok Z. Knockout mice reveal key roles for claudin 18 in alveolar barrier properties and fluid homeostasis. Am J Respir Cell Mol Biol 51: 210–222, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li J, Zhuo M, Pei L, Rajagopal M, Yu AS. Comprehensive cysteine-scanning mutagenesis reveals Claudin-2 pore-lining residues with different intrapore locations. J Biol Chem 289: 6475–6484, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liang GH, Weber CR. Molecular aspects of tight junction barrier function. Curr Opin Pharmacol 19C: 84–89, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu J, Nethery D, Kern JA. Neuregulin-1 induces branching morphogenesis in the developing lung through a P13K signal pathway. Exp Lung Res 30: 465–478, 2004. [DOI] [PubMed] [Google Scholar]
- 104.Livraghi A, Randell SH. Cystic fibrosis and other respiratory diseases of impaired mucus clearance. Toxicol Pathol 35: 116–129, 2007. [DOI] [PubMed] [Google Scholar]
- 105.Loi S, de Azambuja E, Pugliano L, Sotiriou C, Piccart MJ. HER2-overexpressing breast cancer: time for the cure with less chemotherapy? Curr Opin Oncol 23: 547–558, 2011. [DOI] [PubMed] [Google Scholar]
- 106.Londino JD, Lazrak A, Jurkuvenaite A, Collawn JF, Noah JW, Matalon S. Influenza matrix protein 2 alters CFTR expression and function through its ion channel activity. Am J Physiol Lung Cell Mol Physiol 304: L582–L592, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Looney MR, Sartori C, Chakraborty S, James PF, Lingrel JB, Matthay MA. Decreased expression of both the α1- and α2-subunits of the Na-K-ATPase reduces maximal alveolar epithelial fluid clearance. Am J Physiol Lung Cell Mol Physiol 289: L104–L110, 2005. [DOI] [PubMed] [Google Scholar]
- 108.MacVinish LJ, Cope G, Ropenga A, Cuthbert AW. Chloride transporting capability of Calu-3 epithelia following persistent knockdown of the cystic fibrosis transmembrane conductance regulator, CFTR. Br J Pharmacol 150: 1055–1065, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Madan P, Rose K, Watson AJ. Na/K-ATPase beta1 subunit expression is required for blastocyst formation and normal assembly of trophectoderm tight junction-associated proteins. J Biol Chem 282: 12127–12134, 2007. [DOI] [PubMed] [Google Scholar]
- 110.Madtes DK, Busby HK, Strandjord TP, Clark JG. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am J Respir Cell Mol Biol 11: 540–551, 1994. [DOI] [PubMed] [Google Scholar]
- 111.Maina JN, West JB. Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol Rev 85: 811–844, 2005. [DOI] [PubMed] [Google Scholar]
- 112.Mall M, Wissner A, Schreiber R, Kuehr J, Seydewitz HH, Brandis M, Greger R, Kunzelmann K. Role of K (V)LQT1 in cyclic adenosine monophosphate-mediated Cl- secretion in human airway epithelia. Am J Respir Cell Mol Biol 23: 283–289, 2000. [DOI] [PubMed] [Google Scholar]
- 113.Manzanares D, Gonzalez C, Ivonnet P, Chen RS, Valencia-Gattas M, Conner GE, Larsson HP, Salathe M. Functional apical large conductance, Ca2+-activated, and voltage-dependent K+ channels are required for maintenance of airway surface liquid volume. J Biol Chem 286: 19830–19839, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mato N, Fujii M, Hakamata Y, Kobayashi E, Sato A, Hayakawa M, Ohto-Ozaki H, Bando M, Ohno S, Tominaga S, Sugiyama Y. Interleukin-1 receptor-related protein ST2 suppresses the initial stage of bleomycin-induced lung injury. Eur Respir J 33: 1415–1428, 2009. [DOI] [PubMed] [Google Scholar]
- 115.Matthay MA. Resolution of pulmonary edema. Thirty years of progress. Am J Respir Crit Care Med 189: 1301–1308, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McCray PB Jr, Bentley L. Human airway epithelia express a beta-defensin. Am J Respir Cell Mol Biol 16: 343–349, 1997. [DOI] [PubMed] [Google Scholar]
- 117.McGrath EE, Lawrie A, Marriott HM, Mercer P, Cross SS, Arnold N, Singleton V, Thompson AA, Walmsley SR, Renshaw SA, Sabroe I, Chambers RC, Dockrell DH, Whyte MK. Deficiency of tumour necrosis factor-related apoptosis-inducing ligand exacerbates lung injury and fibrosis. Thorax 67: 796–803, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Meduri GU, Annane D, Chrousos GP, Marik PE, Sinclair SE. Activation and regulation of systemic inflammation in ARDS: rationale for prolonged glucocorticoid therapy. Chest 136: 1631–1643, 2009. [DOI] [PubMed] [Google Scholar]
- 119.Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R, Leeper K. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 107: 1062–1073, 1995. [DOI] [PubMed] [Google Scholar]
- 120.Moliva JI, Rajaram MV, Sidiki S, Sasindran SJ, Guirado E, Pan XJ, Wang SH, Ross P Jr, Lafuse WP, Schlesinger LS, Turner J, Torrelles JB. Molecular composition of the alveolar lining fluid in the aging lung. Age (Dordr) 36: 9633, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Montero JC, Seoane S, Ocana A, Pandiella A. P-Rex1 participates in neuregulin-ErbB signal transduction and its expression correlates with patient outcome in breast cancer. Oncogene 30: 1059–1071, 2011. [DOI] [PubMed] [Google Scholar]
- 122.Mutlu GM, Adir Y, Jameel M, Akhmedov AT, Welch L, Dumasius V, Meng FJ, Zabner J, Koenig C, Lewis ER, Balagani R, Traver G, Sznajder JI, Factor P. Interdependency of beta-adrenergic receptors and CFTR in regulation of alveolar active Na+ transport. Circ Res 96: 999–1005, 2005. [DOI] [PubMed] [Google Scholar]
- 123.Nakamura Y, Sotozono C, Kinoshita S. The epidermal growth factor receptor (EGFR): role in corneal wound healing and homeostasis. Exp Eye Res 72: 511–517, 2001. [DOI] [PubMed] [Google Scholar]
- 124.Namiranian K, Brink CD, Goodman JC, Robertson CS, Bryan RM Jr. Traumatic brain injury in mice lacking the K channel, TREK-1. J Cereb Blood Flow Metab 31: e1–e6, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nethery DE, Moore BB, Minowada G, Carroll J, Faress JA, Kern JA. Expression of mutant human epidermal receptor 3 attenuates lung fibrosis and improves survival in mice. J Appl Physiol 99: 298–307, 2005. [DOI] [PubMed] [Google Scholar]
- 126.Nilius B, Honore E. Sensing pressure with ion channels. Trends Neurosci 35: 477–486, 2012. [DOI] [PubMed] [Google Scholar]
- 127.Noel J, Sandoz G, Lesage F. Molecular regulations governing TREK and TRAAK channel functions. Channels (Austin) 5: 402–409, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ochiai A, Akimoto S, Kanai Y, Shibata T, Oyama T, Hirohashi S. c-erbB-2 gene product associates with catenins in human cancer cells. Biochem Biophys Res Commun 205: 73–78, 1994. [DOI] [PubMed] [Google Scholar]
- 129.Olayioye MA, Graus-Porta D, Beerli RR, Rohrer J, Gay B, Hynes NE. ErbB-1 and ErbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol Cell Biol 18: 5042–5051, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Olivera DS, Boggs SE, Beenhouwer C, Aden J, Knall C. Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitro. Inhal Toxicol 19: 13–22, 2007. [DOI] [PubMed] [Google Scholar]
- 131.Olver RE, Strang LB. Ion fluxes across the pulmonary epithelium and the secretion of lung liquid in the foetal lamb. J Physiol 241: 327–357, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ortiz LA, Dutreil M, Fattman C, Pandey AC, Torres G, Go K, Phinney DG. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci USA 104: 11002–11007, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F 2nd, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 164: 1896–1903, 2001. [DOI] [PubMed] [Google Scholar]
- 134.Parker D, Prince A. Innate immunity in the respiratory epithelium. Am J Respir Cell Mol Biol 45: 189–201, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Peters DM, Vadasz I, Wujak L, Wygrecka M, Olschewski A, Becker C, Herold S, Papp R, Mayer K, Rummel S, Brandes RP, Gunther A, Waldegger S, Eickelberg O, Seeger W, Morty RE. TGF-beta directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci USA 111: E374–E383, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Planes C, Randrianarison NH, Charles RP, Frateschi S, Cluzeaud F, Vuagniaux G, Soler P, Clerici C, Rossier BC, Hummler E. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol Med 2: 26–37, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pohl C, Hermanns MI, Uboldi C, Bock M, Fuchs S, Dei-Anang J, Mayer E, Kehe K, Kummer W, Kirkpatrick CJ. Barrier functions and paracellular integrity in human cell culture models of the proximal respiratory unit. Eur J Pharm Biopharm 72: 339–349, 2009. [DOI] [PubMed] [Google Scholar]
- 138.Privalsky ML, Sealy L, Bishop JM, McGrath JP, Levinson AD. The product of the avian erythroblastosis virus erbB locus is a glycoprotein. Cell 32: 1257–1267, 1983. [DOI] [PubMed] [Google Scholar]
- 139.Puddicombe SM, Polosa R, Richter A, Krishna MT, Howarth PH, Holgate ST, Davies DE. Involvement of the epidermal growth factor receptor in epithelial repair in asthma. FASEB J 14: 1362–1374, 2000. [DOI] [PubMed] [Google Scholar]
- 140.Pugin J, Ricou B, Steinberg KP, Suter PM, Martin TR. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am J Respir Crit Care Med 153: 1850–1856, 1996. [DOI] [PubMed] [Google Scholar]
- 141.Rajasekaran AK, Rajasekaran SA. Role of Na-K-ATPase in the assembly of tight junctions. Am J Physiol Renal Physiol 285: F388–F396, 2003. [DOI] [PubMed] [Google Scholar]
- 142.Rajasekaran SA, Barwe SP, Gopal J, Ryazantsev S, Schneeberger EE, Rajasekaran AK. Na-K-ATPase regulates tight junction permeability through occludin phosphorylation in pancreatic epithelial cells. Am J Physiol Gastrointest Liver Physiol 292: G124–G133, 2007. [DOI] [PubMed] [Google Scholar]
- 143.Rajasekaran SA, Hu J, Gopal J, Gallemore R, Ryazantsev S, Bok D, Rajasekaran AK. Na,K-ATPase inhibition alters tight junction structure and permeability in human retinal pigment epithelial cells. Am J Physiol Cell Physiol 284: C1497–C1507, 2003. [DOI] [PubMed] [Google Scholar]
- 144.Rajasekaran SA, Palmer LG, Moon SY, Peralta Soler A, Apodaca GL, Harper JF, Zheng Y, Rajasekaran AK. Na,K-ATPase activity is required for formation of tight junctions, desmosomes, and induction of polarity in epithelial cells. Mol Biol Cell 12: 3717–3732, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Raleigh DR, Boe DM, Yu D, Weber CR, Marchiando AM, Bradford EM, Wang Y, Wu L, Schneeberger EE, Shen L, Turner JR. Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. J Cell Biol 193: 565–582, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ramsingh R, Grygorczyk A, Solecki A, Cherkaoui LS, Berthiaume Y, Grygorczyk R. Cell deformation at the air-liquid interface induces Ca2+-dependent ATP release from lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 300: L587–L595, 2011. [DOI] [PubMed] [Google Scholar]
- 147.Ress A, Moelling K. The PDZ protein erbin modulates beta-catenin-dependent transcription. Eur Surg Res 41: 284–289, 2008. [DOI] [PubMed] [Google Scholar]
- 148.Riese DJ 2nd, van Raaij TM, Plowman GD, Andrews GC, Stern DF. The cellular response to neuregulins is governed by complex interactions of the erbB receptor family. Mol Cell Biol 15: 5770–5776, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Roan E, Waters CM, Teng B, Ghosh M, Schwingshackl A. The 2-pore domain potassium channel TREK-1 regulates stretch-induced detachment of alveolar epithelial cells. PLoS One 9: e89429, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Roberts CM, Cairns D, Bryant DH, Burke WM, Yeates M, Blake H, Penny R, Shelley L, Zaunders JJ, Breit SN. Changes in epithelial lining fluid albumin associated with smoking and interstitial lung disease. Eur Respir J 6: 110–115, 1993. [PubMed] [Google Scholar]
- 151.Rock JR, O'Neal WK, Gabriel SE, Randell SH, Harfe BD, Boucher RC, Grubb BR. Transmembrane protein 16A (TMEM16A) is a Ca2+-regulated Cl- secretory channel in mouse airways. J Biol Chem 284: 14875–14880, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Rokkam D, Lafemina MJ, Lee JW, Matthay MA, Frank JA. Claudin-4 levels are associated with intact alveolar fluid clearance in human lungs. Am J Pathol 179: 1081–1087, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ryan RM, Mineo-Kuhn MM, Kramer CM, Finkelstein JN. Growth factors alter neonatal type II alveolar epithelial cell proliferation. Am J Physiol 266: L17–L22, 1994. [DOI] [PubMed] [Google Scholar]
- 154.Sasaki S, Yui N, Noda Y. Actin directly interacts with different membrane channel proteins and influences channel activities: AQP2 as a model. Biochim Biophys Acta 1838: 514–520, 2014. [DOI] [PubMed] [Google Scholar]
- 155.Schade B, Lesurf R, Sanguin-Gendreau V, Bui T, Deblois G, O'Toole SA, Millar EKA, Zardawi SJ, Lopez-Knowles E, Sutherland RL, Giguère V, Kahn M, Hallett M, Muller WJ. β-Catenin signaling is a critical event in erbb2-mediated mammary tumor progression. Cancer Res 73: 4474–4487, 2013. [DOI] [PubMed] [Google Scholar]
- 156.Schmidt C, Wiedmann F, Schweizer PA, Katus HA, Thomas D. [Cardiac two-pore-domain potassium channels (K2P): Physiology, pharmacology, and therapeutic potential]. Dtsch Med Wochenschr 137: 1654–1658, 2012. [DOI] [PubMed] [Google Scholar]
- 157.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 286: C1213–C1228, 2004. [DOI] [PubMed] [Google Scholar]
- 158.Schreiber R, Hopf A, Mall M, Greger R, Kunzelmann K. The first-nucleotide binding domain of the cystic-fibrosis transmembrane conductance regulator is important for inhibition of the epithelial Na+ channel. Proc Natl Acad Sci USA 96: 5310–5315, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Schreiber R, Nitschke R, Greger R, Kunzelmann K. The cystic fibrosis transmembrane conductance regulator activates aquaporin 3 in airway epithelial cells. J Biol Chem 274: 11811–11816, 1999. [DOI] [PubMed] [Google Scholar]
- 160.Schroeder JA, Adriance MC, McConnell EJ, Thompson MC, Pockaj B, Gendler SJ. ErbB-beta-catenin complexes are associated with human infiltrating ductal breast and murine mammary tumor virus (MMTV)-Wnt-1 and MMTV-c-Neu transgenic carcinomas. J Biol Chem 277: 22692–22698, 2002. [DOI] [PubMed] [Google Scholar]
- 161.Schwingshackl A, Teng B, Ghosh M, Lim KG, Tigyi G, Narayanan D, Jaggar JH, Waters CM. Regulation of interleukin-6 secretion by the two-pore-domain potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 304: L276–L286, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Schwingshackl A, Teng B, Ghosh M, Waters CM. Regulation of monocyte chemotactic protein-1 secretion by the two-pore-domain potassium (K2P) channel TREK-1 in human alveolar epithelial cells. Am J Transl Res 5: 530–542, 2013. [PMC free article] [PubMed] [Google Scholar]
- 163.Schwingshackl A, Teng B, Ghosh M, West AN, Makena P, Gorantla V, Sinclair SE, Waters CM. Regulation and function of the two-pore-domain (K2P) potassium channel Trek-1 in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: L93–L102, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Schwingshackl A, Teng B, Makena P, Ghosh M, Sinclair SE, Luellen C, Balasz L, Rovnaghi C, Bryan RM, Lloyd EE, Fitzpatrick E, Saravia JS, Cormier SA, Waters CM. Deficiency of the two-pore-domain potassium channel TREK-1 promotes hyperoxia-induced lung injury. Crit Care Med 42: e692–e701, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shaykhiev R, Zuo WL, Chao I, Fukui T, Witover B, Brekman A, Crystal RG. EGF shifts human airway basal cell fate toward a smoking-associated airway epithelial phenotype. Proc Natl Acad Sci USA 110: 12102–12107, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Shen L, Weber CR, Raleigh DR, Yu D, Turner JR. Tight junction pore and leak pathways: a dynamic duo. Annu Rev Physiol 73: 283–309, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Shen L, Weber CR, Turner JR. The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. J Cell Biol 181: 683–695, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shin K, Fogg VC, Margolis B. Tight junctions and cell polarity. Annu Rev Cell Dev Biol 22: 207–235, 2006. [DOI] [PubMed] [Google Scholar]
- 170.Shomori K, Ochiai A, Akimoto S, Ino Y, Shudo K, Ito H, Hirohashi S. Tyrosine-phosphorylation of the 12th armadillo-repeat of beta-catenin is associated with cadherin dysfunction in human cancer. Int J Oncol 35: 517–524, 2009. [DOI] [PubMed] [Google Scholar]
- 171.Sidhaye VK, Chau E, Breysse PN, King LS. Septin-2 mediates airway epithelial barrier function in physiologic and pathologic conditions. Am J Respir Cell Mol Biol 45: 120–126, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sidhaye VK, Chau E, Srivastava V, Sirimalle S, Balabhadrapatruni C, Aggarwal NR, D'Alessio FR, Robinson DN, King LS. A novel role for aquaporin-5 in enhancing microtubule organization and stability. PLoS One 7: e38717, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Sidhaye VK, Schweitzer KS, Caterina MJ, Shimoda L, King LS. Shear stress regulates aquaporin-5 and airway epithelial barrier function. Proc Natl Acad Sci USA 105: 3345–3350, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Siler TM, Swierkosz JE, Hyers TM, Fowler AA, Webster RO. Immunoreactive interleukin-1 in bronchoalveolar lavage fluid of high-risk patients and patients with the adult respiratory distress syndrome. Exp Lung Res 15: 881–894, 1989. [DOI] [PubMed] [Google Scholar]
- 175.Simard CF, Bergeron MJ, Frenette-Cotton R, Carpentier GA, Pelchat ME, Caron L, Isenring P. Homooligomeric and heterooligomeric associations between K+-Cl- cotransporter isoforms and between K+-Cl- and Na+-K+-Cl- cotransporters. J Biol Chem 282: 18083–18093, 2007. [DOI] [PubMed] [Google Scholar]
- 176.Singh PK, Jia HP, Wiles K, Hesselberth J, Liu L, Conway BA, Greenberg EP, Valore EV, Welsh MJ, Ganz T, Tack BF, McCray PB Jr. Production of beta-defensins by human airway epithelia. Proc Natl Acad Sci USA 95: 14961–14966, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Singha O, Kengkoom K, Chaimongkolnukul K, Cherdyu S, Pongponratn E, Ketjareon T, Panavechkijkul Y, Ampawong S. Pulmonary edema due to oral gavage in a toxicological study related to aquaporin-1, -4 and -5 expression. J Toxicol Pathol 26: 283–291, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Sisson TH, Mendez M, Choi K, Subbotina N, Courey A, Cunningham A, Dave A, Engelhardt JF, Liu X, White ES, Thannickal VJ, Moore BB, Christensen PJ, Simon RH. Targeted injury of type II alveolar epithelial cells induces pulmonary fibrosis. Am J Respir Crit Care Med 181: 254–263, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Skryma R, Prevarskaya N, Gkika D, Shuba Y. From urgency to frequency: facts and controversies of TRPs in the lower urinary tract. Nat Rev Urol 8: 617–630, 2011. [DOI] [PubMed] [Google Scholar]
- 180.Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, Levin WJ, Stuart SG, Udove J, Ullrich A, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 244: 707–712, 1989. [DOI] [PubMed] [Google Scholar]
- 181.Slutsky AS, Ranieri VM. Ventilator-induced lung injury. N Engl J Med 370: 980, 2014. [DOI] [PubMed] [Google Scholar]
- 182.Smith LS, Zimmerman JJ, Martin TR. Mechanisms of acute respiratory distress syndrome in children and adults: a review and suggestions for future research. Pediatr Crit Care Med 14: 631–643, 2013. [DOI] [PubMed] [Google Scholar]
- 183.Solymosi EA, Kaestle-Gembardt SM, Vadasz I, Wang L, Neye N, Chupin CJ, Rozowsky S, Ruehl R, Tabuchi A, Schulz H, Kapus A, Morty RE, Kuebler WM. Chloride transport-driven alveolar fluid secretion is a major contributor to cardiogenic lung edema. Proc Natl Acad Sci USA 110: E2308–E2316, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Stoll S, Garner W, Elder J. Heparin-binding ligands mediate autocrine epidermal growth factor receptor activation in skin organ culture. J Clin Invest 100: 1271–1281, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Strieter RM, Kunkel SL. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J Investig Med 42: 640–651, 1994. [PubMed] [Google Scholar]
- 186.Takeyama K, Dabbagh K, Lee HM, Agusti C, Lausier JA, Ueki IF, Grattan KM, Nadel JA. Epidermal growth factor system regulates mucin production in airways. Proc Natl Acad Sci USA 96: 3081–3086, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Takeyama K, Fahy JV, Nadel JA. Relationship of epidermal growth factor receptors to goblet cell production in human bronchi. Am J Respir Crit Care Med 163: 511–516, 2001. [DOI] [PubMed] [Google Scholar]
- 188.Takeyama K, Jung B, Shim JJ, Burgel PR, Dao-Pick T, Ueki IF, Protin U, Kroschel P, Nadel JA. Activation of epidermal growth factor receptors is responsible for mucin synthesis induced by cigarette smoke. Am J Physiol Lung Cell Mol Physiol 280: L165–L172, 2001. [DOI] [PubMed] [Google Scholar]
- 189.Terakado M, Gon Y, Sekiyama A, Takeshita I, Kozu Y, Matsumoto K, Takahashi N, Hashimoto S. The Rac1/JNK pathway is critical for EGFR-dependent barrier formation in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 300: L56–L63, 2011. [DOI] [PubMed] [Google Scholar]
- 190.Tessier GJ, Traynor TR, Kannan MS, O'Grady SM. Mechanisms of sodium and chloride transport across equine tracheal epithelium. Am J Physiol 259: L459–L467, 1990. [DOI] [PubMed] [Google Scholar]
- 191.Thorley AJ, Tetley TD. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2: 409–428, 2007. [PMC free article] [PubMed] [Google Scholar]
- 192.Trinh NT, Prive A, Kheir L, Bourret JC, Hijazi T, Amraei MG, Noel J, Brochiero E. Involvement of KATP and KvLQT1 K+ channels in EGF-stimulated alveolar epithelial cell repair processes. Am J Physiol Lung Cell Mol Physiol 293: L870–L882, 2007. [DOI] [PubMed] [Google Scholar]
- 193.van der Geer P, Hunter T, Lindberg RA. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu Rev Cell Biol 10: 251–337, 1994. [DOI] [PubMed] [Google Scholar]
- 194.Van Driessche W, Kreindler JL, Malik AB, Margulies S, Lewis SA, Kim KJ. Interrelations/cross talk between transcellular transport function and paracellular tight junctional properties in lung epithelial and endothelial barriers. Am J Physiol Lung Cell Mol Physiol 293: L520–L524, 2007. [DOI] [PubMed] [Google Scholar]
- 195.Van Itallie CM, Anderson JM. The molecular physiology of tight junction pores. Physiology (Bethesda) 19: 331–338, 2004. [DOI] [PubMed] [Google Scholar]
- 196.Van Itallie CM, Fanning AS, Anderson JM. Reversal of charge selectivity in cation or anion-selective epithelial lines by expression of different claudins. Am J Physiol Renal Physiol 285: F1078–F1084, 2003. [DOI] [PubMed] [Google Scholar]
- 197.Van Itallie CM, Holmes J, Bridges A, Gookin JL, Coccaro MR, Proctor W, Colegio OR, Anderson JM. The density of small tight junction pores varies among cell types and is increased by expression of claudin-2. J Cell Sci 121: 298–305, 2008. [DOI] [PubMed] [Google Scholar]
- 198.Verkman AS. Role of aquaporins in lung liquid physiology. Respir Physiol Neurobiol 159: 324–330, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Verkman AS, Matthay MA, Song Y. Aquaporin water channels and lung physiology. Am J Physiol Lung Cell Mol Physiol 278: L867–L879, 2000. [DOI] [PubMed] [Google Scholar]
- 200.Vermeer PD, Einwalter LA, Moninger TO, Rokhlina T, Kern JA, Zabner J, Welsh MJ. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature 422: 322–326, 2003. [DOI] [PubMed] [Google Scholar]
- 201.Walker RA. The erbB/HER type 1 tyrosine kinase receptor family. J Pathol 185: 234–235, 1998. [DOI] [PubMed] [Google Scholar]
- 202.Wang JH, Thampatty BP. An introductory review of cell mechanobiology. Biomech Model Mechanobiol 5: 1–16, 2006. [DOI] [PubMed] [Google Scholar]
- 203.Watanabe H, Murakami M, Ohba T, Takahashi Y, Ito H. TRP channel and cardiovascular disease. Pharmacol Ther 118: 337–351, 2008. [DOI] [PubMed] [Google Scholar]
- 204.Waters CM, Ridge KM, Sunio G, Venetsanou K, Sznajder JI. Mechanical stretching of alveolar epithelial cells increases Na+-K+-ATPase activity. J Appl Physiol 87: 715–721, 1999. [DOI] [PubMed] [Google Scholar]
- 205.Wilson SM, Olver RE, Walters DV. Developmental regulation of lumenal lung fluid and electrolyte transport. Respir Physiol Neurobiol 159: 247–255, 2007. [DOI] [PubMed] [Google Scholar]
- 206.Wispe JR, Clark JC, Warner BB, Fajardo D, Hull WE, Holtzman RB, Whitsett JA. Tumor necrosis factor-alpha inhibits expression of pulmonary surfactant protein. J Clin Invest 86: 1954–1960, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wray C, Mao Y, Pan J, Chandrasena A, Piasta F, Frank JA. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am J Physiol Lung Cell Mol Physiol 297: L219–L227, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Yang C, Liu Y, Lemmon MA, Kazanietz MG. Essential role for Rac in heregulin beta1 mitogenic signaling: a mechanism that involves epidermal growth factor receptor and is independent of ErbB4. Mol Cell Biol 26: 831–842, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127–137, 2001. [DOI] [PubMed] [Google Scholar]
- 210.Yin J, Lu J, Yu FS. Role of small GTPase Rho in regulating corneal epithelial wound healing. Invest Ophthalmol Vis Sci 49: 900–909, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Yin J, Yu FS. ERK1/2 mediate wounding- and G-protein-coupled receptor ligands-induced EGFR activation via regulating ADAM17 and HB-EGF shedding. Invest Ophthalmol Vis Sci 50: 132–139, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhao F, Dong L, Cheng L, Zeng Q, Su F. Effects of acute mechanical stretch on the expression of mechanosensitive potassium channel TREK-1 in rat left ventricle. J Huazhong Univ Sci Technolog Med Sci 27: 385–387, 2007. [DOI] [PubMed] [Google Scholar]
- 213.Zhao KQ, Xiong G, Wilber M, Cohen NA, Kreindler JL. A role for two-pore K+ channels in modulating Na+ absorption and Cl- secretion in normal human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: L4–L12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Zhu H, Zhang G, Wang Y, Xu N, He S, Zhang W, Chen M, Liu M, Quan L, Bai J, Xu N. Inhibition of ErbB2 by Herceptin reduces survivin expression via the ErbB2-β-catenin/TCF4-survivin pathway in ErbB2-overexpressed breast cancer cells. Cancer Sci 101: 1156–1162, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Zieske JD, Takahashi H, Hutcheon AE, Dalbone AC. Activation of epidermal growth factor receptor during corneal epithelial migration. Invest Ophthalmol Vis Sci 41: 1346–1355, 2000. [PubMed] [Google Scholar]