

Abstract
Objective
To determine whether neurophysiological mechanisms indicating cortical excitability, long‐term potentiation (LTP)‐like plasticity, GABAergic and glutamatergic function are altered in patients with anti‐N‐methyl‐d‐aspartate receptor (NMDAR) encephalitis and whether they can be helpful as markers of diagnostic assessment, disease progression, and potentially therapy response.
Methods
Neurophysiological characterizations of patients with NMDAR encephalitis (n = 34, mean age: 28 ± 11 years; 30 females) and age/gender‐matched healthy controls (n = 27, 28.5 ± 10 years; 25 females) were performed using transcranial magnetic stimulation‐derived protocols including resting motor threshold, recruitment curve, intracortical facilitation, short intracortical inhibition, and cortical silent period. Paired associative stimulation (PAS) was applied to assess LTP‐like mechanisms which are mediated through NMDAR. Moreover, resting state functional connectivity was determined using functional magnetic resonance imaging.
Results
PAS‐induced plasticity differed significantly between groups (P = 0.0056). Cortical excitability, as assessed via motor‐evoked potentials after PAS, decreased in patients, whereas it increased in controls indicating malfunctioning of NMDAR in encephalitis patients. Lower PAS‐induced plasticity significantly correlated with the modified Rankin Scale (mRS) (r = −0.41; P = 0.0031) and was correlated with lower functional connectivity within the motor network in NMDAR encephalitis patients (P < 0.001, uncorrected). Other neurophysiological parameters were not significantly different between groups. Follow‐up assessments were available in six patients and demonstrated parallel improvement of PAS‐induced plasticity and mRS.
Interpretation
Assessment of PAS‐induced plasticity may help to determine NMDAR dysfunction and disease severity in NMDAR encephalitis, and might even aid as a sensitive, noninvasive, and well‐tolerated “electrophysiological biomarker” to monitor therapy response in the future.
Clinical Trial Registration
ClinicalTrials.gov: Identifier: NCT01865578
Introduction
Anti‐N‐methyl‐d‐aspartate receptor (NMDAR) encephalitis is the most common antibody‐mediated autoimmune encephalitis following a characteristic clinical course including psychiatric features, amnesia, epileptic seizures, and abnormal movements.1 Autoantibodies against the NR1 subunit of the NMDAR are found in serum and cerebrospinal fluid (CSF).2 They interfere with synaptic function by specific downregulation of neuronal membrane NMDAR.3 Antibody detection guides early diagnostics, and most patients benefit profoundly from rapid initiation of immunotherapy. However, understanding of the neurophysiological changes underlying altered clinical and cognitive function in the human brain remains incomplete, and lack of disease markers for prognosis and treatment monitoring often delays appropriate therapy. Both better understanding of neurophysiological underpinnings and noninvasive, objective, and widely available methods for determination of clinical severity are urgently needed.
Transcranial magnetic stimulation (TMS) is a noninvasive technique to assess human cortical neurophysiology in health and disease. Most commonly used TMS parameters are resting motor threshold (RMT), recruitment curve (RC), intracortical facilitation (ICF), short intracortical inhibition (SICI), and cortical silent period (CSP). They allow assessment of different neurotransmitter systems including predominant glutamatergic (ICF and paired associative stimulation [PAS]), GABA(A)ergic (SICI, motor‐evoked potentials [MEP], and CSP with low intensity), and GABA(B)ergic (SICI and CSP with high intensity) functions, confirmed in pharmacological studies.4 For example, cortical excitability and the paradigms of SICI and ICF were conversely changed by the NMDAR antagonist memantine.4, 5 However, no TMS parameter is solely mediated by a single neurotransmitter but rather by a combination of several neurotransmitters to a different degree.
One of the main factors underlying learning and memory formation at the cellular level is long‐term potentiation (LTP).6, 7 In humans, LTP‐like cortical plasticity can be assessed using peripheral electric stimulation and subsequent TMS. This so‐called PAS8 is now a widely used protocol to noninvasively investigate rapid‐onset cortical plasticity in healthy subjects and neurological patients.9, 10, 11, 12 Importantly, it has been shown to predominantly reflect NMDAR function,8, 13, 14 whereas MEP and motor threshold (MT) are relatively independent from NMDAR function.4, 8, 13, 15, 16, 17
Thus, PAS is an exciting candidate for assessing the underlying neurophysiological dysfunction and related clinical disability in NMDAR encephalitis. In the present study, we therefore aimed to determine (1) whether there are differences in PAS between NMDAR encephalitis patients and healthy controls, (2) whether other TMS‐derived neurophysiological parameters which are not mediated through NMDAR are altered, (3) whether neurophysiological parameters change throughout the course of disease, and (4) whether neurophysiological parameters correlate with other markers of disease activity such as a disease severity questionnaire (modified Rankin Scale, mRS) and resting state functional connectivity.
Material and Methods
Experimental design and subjects
In this parallel design trial, 34 patients (mean age 28.03 ± 10.85 years; 30 females, range: 18–68 years) with NMDAR encephalitis and 27 age‐ and sex‐matched healthy controls (mean age 28.48 ± 9.76 years; 25 females, range: 21–60 years) were enrolled. Six encephalitis patients received repeated measurement for follow‐up analyses. Participants underwent TMS assessments reported in accordance to the checklist for assessing methodological quality for TMS studies18 (Table S1). PAS protocol was defined as primary outcome. Secondary outcomes were other TMS measurements such as MEP, CSP, ICF, and SICI as well as exploratory correlation analyses of TMS‐derived neurophysiological parameters with clinician‐based rating of disease activity (mRS) and functional magnetic resonance imaging–based resting state functional connectivity.
Both groups were screened for contraindication for TMS assessments (see below), handedness indexed by Edinburgh Handedness Inventory,19 and the mRS, which runs from 0 to 6 scoring from complete health to severe symptoms to death.20 Despite limited capability to detect subtle clinical changes, the mRS was used in this study to allow comparability with previous studies in NMDAR encephalitis.1
Eligibility was defined as follows: patients and controls – (1) age above 18 years; (2) no contraindications for TMS such as a head injury or surgery, implanted medical devices, and any metal in the head; (3) no seizure in the last 6 months. Only controls – (4) no centrally active medication and (5) no medical, psychiatric, or neurological disorders. Only patients – (6) diagnosis of NMDAR encephalitis based on the characteristic clinical picture with signs of encephalitis (epileptic seizures, reduced levels of consciousness, and cognitive or mood changes), exclusion of alternative disorders (in particular viral causes), evidence of brain inflammation (MRI abnormalities, CSF inflammation, or positive biopsy/autopsy), and CSF IgG antibodies against the NMDAR at the time of diagnosis.21, 22
The study was approved by the Charité University Hospital Institutional Review Board. All participants gave their written informed consent. Patients were recruited from the Department of Neurology of Charité University Hospital and the Charité outpatient Center for Autoimmune Encephalitis and Paraneoplastic Neurological Syndromes. The trial was registered at ClinicalTrials.gov (identifier: NCT01865578).
Measurement of cortical excitability via TMS
Measurements were performed using a Bistim2 stimulator and a figure‐of‐eight coil with 140 mm diameter (Magstim Company LTDA, Whitland, Dyfed, UK). Participants were seated in a comfortable chair. To record MEP, Ag/AgCl electrodes (ADinstruments, Colorado Springs, CO, USA) were placed over the first dorsal interosseous muscle (FDI), and a ground electrode was placed over the subjects’ forearm. Recordings were processed through Signal 4.05 software (Cambridge Electronic Design, Cambridge, UK) with a bandpass filter of 100 Hz to 10 kHz. Offline analyses were made with the Signal 4.05 software. TMS was applied over the hemisphere contralateral to the dominant hand. Initially, head measurements were taken to identify the location of the motor cortex (using the nasion, inion, and vertex as reference, then calculating individual 20% of the distance between both tragus spots to identify M1). Then, the TMS coil was held tangentially over M1 with an angle of 45° with respect to the sagittal line of the head. The exact hotspot was determined by eliciting the most stable and highest MEP amplitudes over the FDI. The hotspot was marked with a pen on each of the subjects’ heads to secure coil location and stability. Between each TMS pulse we set an interval of approximately 10 sec to avoid habituation.
First, RMT was assessed by using the lowest stimulator output at which three of the five trails had minimum amplitude of 50 μV.23, 24 This methodology results (together with the relatively young age of our cohort) in lower RMT than in other published studies where the stimulator output is set to result in 100 μV amplitudes. For eliciting MEP, the intensity of 130% of the individual RMT was chosen. This normalization helps to consider the well‐known interindividual variances in RMT. RC was assessed using the intensities of 130%, 140%, and 150% of the individual RMT.25 For the statistical analyses, we calculated the mean of 10 MEP per intensity. With these MEPs of different intensities, we calculated the ratios (130/150, and 130/140 and 140/150 for all subjects) and determined the RC.
CSP was assessed using stimulator output intensity at 110% and 140% of the individual RMT.26 During CSP measurement, patients had to perform isometric voluntary muscle contraction with approximately 10–20% of maximal force controlled visually via EMG. For data analyses, relative duration (duration of the beginning of the last MEP until the beginning of the next MEP) of the silent period was collected of 10 CSP. The ratio of 110/140 was calculated as it reflects the individual change best. Therefore, comparing results of patients and controls with this ratio, the pure differences between those groups were most robust. These single‐pulse measurements took approximately 10–15 min.
Moreover, paired‐pulse stimulation was applied using SICI and ICF.27, 28 SICI was performed using an interstimulus interval (ISI) of 3 msec and ICF with an ISI of 10 msec. For both paired‐pulse measures, the first (conditioning) stimulus was set to 70% of the individual MT, and the second (test) stimulus was set to the individual MEP intensity (130% of MT). Ten recordings of each TMS assessment protocol were randomly elicited (mixed with single‐pulse test MEP). Offline analyses included measures of peak‐to‐peak amplitude and the integral of all MEPs. Paired‐pulse measurements took approximately 7 min.
Paired associative stimulation
For evaluation of NMDAR‐specific function, the PAS protocol was applied, as none of the single‐pulse TMS parameter has a similarly strong linkage to NMDAR function. Before and immediately after the PAS protocol, 10 MEP at individual intensity (130% of RMT) were applied. Furthermore, 15 min after the protocol, 10 MEP were performed for follow‐up analyses. As described before,8, 10 we used paired stimulation of the ulnar nerve (electrical stimulation with 300% of individual sensory threshold) and the hotspot of the ipsilateral FDI over the motor cortex. The interstimulus interval was set at 25 msec, which has been proven to be robust for inducing increased cortical excitability.8 A total of 131 paired pulses were administered. In order to secure an equal state of attention during PAS protocol, participants were instructed to stay awake, voluntarily relax the tested hand, and to count the number of electrical ulnar nerve stimulations.
Resting state functional connectivity
Resting state functional connectivity data acquisition and analysis were performed as described previously in NMDAR encephalitis patients.29 Briefly, an echo‐planar imaging sequence (voxel size 3.4 mm isotropic, TR = 2250 msec, TE = 30 msec, 260 volumes) and a three‐dimensional 1 mm isotropic magnetization prepared rapid acquisition gradient echo sequences were acquired on a Siemens (Erlangen, Germany) Tim Trio 3T scanner. Analysis was performed with independent component analysis (ICA) and dual regression using FSL (www.fmrib.ox.ac.uk/fsl/). After preprocessing, the sensorimotor network was identified using temporal concatenation ICA as implemented in Multivariate Exploratory Linear Optimized Decomposition into Independent Component (FSL MELODIC).
Statistical analyses
Statistical analyses were performed using STATA (v11.0, College Station, TX, USA). The dependent variables were TMS measurements. First, data were tested for normal distribution using Skewness/Kurtosis test. If data were not normally distributed, log transformation was performed. Comparisons between groups (patients vs. healthy controls) used the individual increase or decrease in a TMS parameter compared to baseline MEPs (SICI/MEP; ICF/MEP) or between MEP with different intensities (CSP: CSP110%/CSP140%; RC: 150%/130% of MEP). With those calculations the comparison is most precise since it abolishes effects due to different RMT intensity and also small shifts of the TMS coil during the course of assessment.
Analysis of variance (ANOVA) was performed for TMS analyses with the following factors: TMS measurements (dependent variable, i.e., PAS, ICF, etc.), independent variable of group (NMDAR encephalitis vs. controls), time (if applicable) (i.e., MEP before and after PAS protocol), and interaction analysis of group and time. Afterward, analyses were repeated with subgroups including only patients without centrally active medication and mRS > 0. Patients with mRS = 0 were excluded to distinguish whether differences in RMT related to functional impairment from NMDAR encephalitis (as per definition: 0 = not functionally impaired). Furthermore, post hoc t‐tests were used to compare group differences. To correlate score on mRS and TMS measures, Pearson's correlation was used. All data are reported as mean ± standard error of the mean, and P‐values for statistical significance were set to P < 0.05, unless stated otherwise.
Exploratory group analysis of resting state functional connectivity data was carried out using dual regression and nonparametric permutation testing (FSL randomise; 5000 permutations) with an exploratory threshold (threshold‐free cluster enhancement: P < 0.001, uncorrected). Individual MEP changes (before and after PAS) were included as covariate of interest to investigate the correlation between PAS and motor network functional connectivity.29, 30, 31, 32
Results
In total, 34 patients with NMDAR encephalitis and 27 healthy matched controls were recruited (Table 1). Baseline comparisons did not reveal significant differences between groups regarding age (t‐test: P = 0.85), gender (NMDAR encephalitis: 30 female/4 male vs. controls: 25 female/2 male), and handedness (NMDAR encephalitis: 30 right/4 left handed vs. controls: 27 right/0 left handed). No subject reported any side effects previously considered in TMS applications such as seizures, headache, neck pain, or reduced concentration.
Table 1.
Baseline characteristics of all patients and healthy controls
| Patients | Gender | Age (years) | Disease duration (months) | Handedness | Stimulated hemisphere | mRS | ICU treatment | Past immunosuppression | Medication at stimulation | NMDAR ab titer at TMS measurement | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Centrally active | Noncentrally active | CSF | Serum | |||||||||
| 1 | Female | 18 | 33 | Right | Left | 1 | Yes | MP, IVIG, PE, Ritux, Aza | – | – | n.d. | 1000 |
| 2 | Female | 22 | 11 | Right | Left | 1 | Yes | MP, PE, IVIG | Levetiracetam | – | Neg. | Neg. |
| 3 | Female | 46 | 26 | Left | Right | 1 | No | MP, Ritux, PE | Gabapentin | – | Neg. | 100 |
| 4 | Female | 21 | 14 | Right | Left | 3 | Yes | Pred, MMF | – | MMF; MP | 1 | 32 |
| 5 | Male | 33 | 19 | Right | Left | 3 | Yes | PE, Pred | Amantadin; Escitalopram | – | 10 | 100 |
| 6 | Female | 40 | 9 | Right | Left | 2 | Yes | Ritux, Pred, Cyclo | Levetiracetam | Enoxaparin sodium | Neg. | Neg. |
| 7 | Female | 32 | 43 | Right | Left | 2 | Yes | Pred, PE, Aza | Levetiracetam | MMF; Ranitidine; l‐Thyroxine | Neg. | Neg. |
| 8 | Female | 18 | 10 | Right | Left | 2 | Yes | MP, IVIG, PE | Levetiracetam; Valproic acid; Quetiapine | Esomeprazole; Pred | 3 | 10 |
| 9 | Female | 31 | 72 | Right | Left | 2 | Yes | MP, IA, Cyclo | Clonazepam; Phenobarbital; Levetiracetam | – | Neg. | Neg. |
| 10 | Female | 26 | 46 | Right | Left | 1 | No | Pred | – | – | Neg. | Neg. |
| 11 | Female | 23 | 44 | Left | Right | 1 | Yes | Pred, MMF | – | – | 3 | 100 |
| 12 | Female | 25 | 18 | Right | Left | 1 | Yes | Ritux, Pred, IVIG, PE | Pregabalin | Pantoprazole, Metoprolol | 3 | Neg. |
| 13 | Female | 37 | 9 | Right | Left | 2 | No | Pred | – | Pred | Neg. | Neg. |
| 14 | Female | 25 | 33 | Right | Left | 2 | No | Pred, PE | – | – | 3 | Neg. |
| 15 | Female | 46 | 57 | Right | Left | 1 | No | MP | – | – | 32 | Neg. |
| 16 | Female | 25 | 15 | Right | Left | 3 | Yes | MP, IVIG, PE, IA, Ritux | Levetiracetam; Lacosamid | – | 10 | 32 |
| 17 | Female | 21 | 4 | Right | Left | 2 | Yes | PE, MP, Ritux | Venlafaxine; Valproic acid | – | 10 | 100 |
| 18 | Male | 68 | 3 | Right | Left | 3 | No | Pred, IVIG, Ritux | Valproic acid | Ramipril; Amlodipine; Hydrochlorothiazide; Pantoprazole; Pred; Sitagliptin | 1 | 100 |
| 19 | Female | 18 | 4 | Right | Left | 2 | No | Pred, PE, Ritux | Lacosamid | Pred | 100 | 10 |
| 20 | Female | 22 | 35 | Left | Right | 1 | Yes | Pred, IVIG, PE, IA, Ritux | – | – | 10 | 100 |
| 21 | Female | 22 | 8 | Right | Left | 3 | Yes | PE, IVIG, Ritux, Cyclo, Pred | Quetiapine | – | 32 | 320 |
| 22 | Male | 31 | 80 | Right | Left | 3 | No | – | Valproic acid | Marihuana | 3 | 1 |
| 23 | Female | 18 | 5 | Right | Left | 2 | Yes | Pred, IVIG, PE, Ritux | Valproic acid; Levetiracetam | – | 32 | 10 |
| 24 | Female | 27 | 68 | Right | Left | 1 | Yes | IVIG, MP | Levetiracetam | – | 1 | Neg. |
| 25 | Female | 25 | 35 | Right | Left | 3 | Yes | MP, PE, IVIG, Cyclo, Ritux, MTX | Valproic acid | Aspirin; Folinic acid; MTX | n.d. | 10 |
| 26 | Female | 37 | 4 | Right | Left | 3 | No | MP, IA | – | l‐Thyroxine, Cetirizine | 3 | 100 |
| 27 | Female | 18 | 46 | Right | Left | 0 | Yes | MP, IVIG | – | – | 3 | 10 |
| 28 | Female | 37 | 7 | Right | Left | 1 | No | Pred, Ritux, PE | – | – | Neg. | 100 |
| 29 | Female | 18 | 11 | Right | Left | 2 | Yes | MP, Ritux, PE | Levetiracetam | l‐Thyroxine | 32 | 1000 |
| 30 | Female | 26 | 50 | Left | Right | 1 | Yes | Pred, IVIG | – | – | n.d. | 10 |
| 31 | Female | 18 | 11 | Right | Left | 1 | Yes | Pred, IVIG, Ritux | Levetiracetam | l‐Thyroxine | 10 | 100 |
| 32 | Male | 38 | 5 | Right | Left | 1 | Yes | PE, Pred | Mirtazapine; Risperdal; Biperiden; Oxcarbazepine; Valproic acid | – | 3 | 32 |
| 33 | Female | 22 | 6 | Right | Left | 1 | Yes | PE, Pred, IVIG, Ritux | Quetiapine; Topiramate | Ritux; PE | 10 | 100 |
| 34 | Female | 19 | 23 | Right | Left | 1 | Yes | Pred, IVIG, PE | Sertraline | Folinic acid; MTX | n.d. | 100 |
| Healthy controls | ||||||||||||
| 1 | Female | 25 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 2 | Female | 26 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 3 | Female | 35 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 4 | Female | 25 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 5 | Female | 24 | N/A | Right | Left | 0 | N/A | N/A | – | l‐Thyroxine | N/A | N/A |
| 6 | Male | 35 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 7 | Female | 56 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 8 | Female | 23 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 9 | Female | 23 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 10 | Female | 23 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 11 | Female | 28 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 12 | Female | 22 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 13 | Male | 60 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 14 | Female | 21 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 15 | Female | 24 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 16 | Female | 24 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 17 | Female | 21 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 18 | Female | 26 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 19 | Female | 25 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 20 | Female | 21 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 21 | Female | 26 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 22 | Female | 30 | N/A | Right | Left | 0 | N/A | N/A | – | l‐Thyroxine | N/A | N/A |
| 23 | Female | 33 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 24 | Female | 22 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 25 | Female | 23 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 26 | Female | 28 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
| 27 | Female | 40 | N/A | Right | Left | 0 | N/A | N/A | – | – | N/A | N/A |
mRS, modified Rankin Scale; ICU, intensive care unit; NMDAR, N‐methyl‐d‐aspartate receptor; TMS, transcranial magnetic stimulation; CSF, cerebrospinal fluid; N/A, not applicable; n.d., not determined; Neg., negative; IA, immunoadsorption; PE, plasma exchange; Pred, prednisolone; Ritux, rituximab; MP, methylprednisolone; IVIG, i.v. immunoglobulins; Cyclo, cyclophosphamide; Aza, azathioprine; MMF, mycophenolate mofetil; MTX, methotrexate.
Paired associative stimulation
All data were tested for normal distribution and – if not normally distributed – log transformed. MEP baseline values before PAS protocol did not differ significantly comparing both groups (unpaired, two‐sided t‐test: P = 0.46; Fig. 1A and B). MEP after PAS protocol was consistently decreased in the NMDAR encephalitis group by −10.2% (MEP pre: 0.53 ± 0.54 mV; MEP post: 0.48 ± 0.89 mV), whereas it was increased in the healthy control group by 26.1% (MEP pre: 0.63 ± 0.42 mV; MEP post: 0.76 ± 0.63 mV) (Fig. 1C). Testing all PAS results together, ANOVA with independent variables for time (MEP pre and post PAS protocol) and group (NMDAR encephalitis vs. healthy controls) showed a significant interaction analysis of time and group (F 1,55 = 8.35, P = 0.0056) indicating differential PAS effects on both groups (Fig. 1C). When expressing data as percentage of change relative to baseline MEP, post hoc t‐test confirmed a significant difference among both groups (unpaired, two‐sided t‐test: P = 0.043) (expressed as [(t 2 − t 1)/t 1] × 100).
Figure 1.
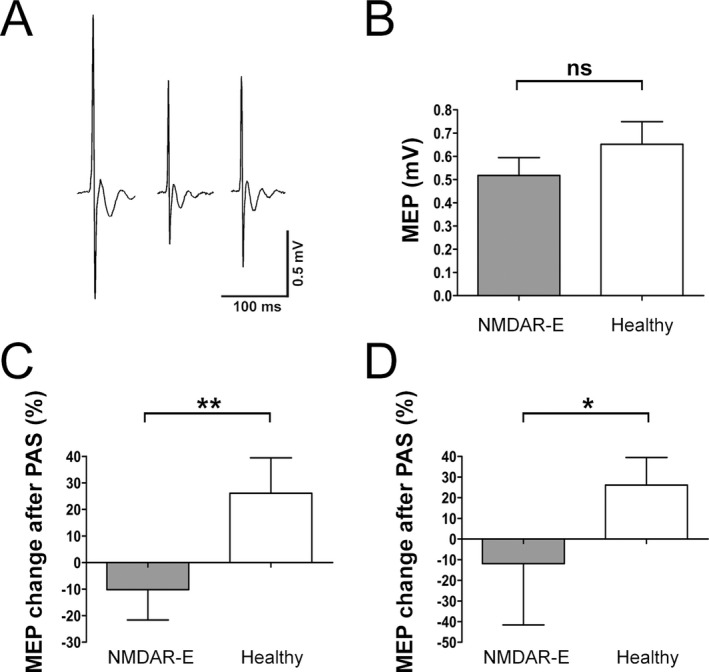
Paired associative stimulation (PAS) defines NMDAR (N‐methyl‐d‐aspartate receptor) dysfunction in encephalitis patients and correlates with disease severity. (A) Representative motor‐evoked potential (MEP) traces of PAS assessment at baseline (left), immediately after PAS (middle) and after 15 min (right). (B) Baseline MEP values before PAS were not different between patients with NMDAR encephalitis and healthy controls. (C) In contrast, MEP changes before and after PAS protocol decreased in the encephalitis group, whereas they increased in the control group. (D) Similar changes were observed in the subgroup of patients without centrally active medication.
The same analyses were repeated in the subgroup including only patients without centrally active medication and with mRS > 0 (n = 11). MEP after PAS protocol showed a decrease in the NMDAR encephalitis group of −11.93% (MEP pre: 0.76 ± 0.62 mV; MEP post: 0.45 ± 0.27 mV), whereas it increased in the healthy group by 26.1% (MEP pre: 0.63 ± 0.42 mV; MEP post: 0.76 ± 0.63 mV). ANOVA still showed a significant interaction of time and group (F 1,36 = 6.24, P = 0.0178) (Fig. 1D) indicating that the PAS protocol had differential effects on both groups. However, post hoc t‐test revealed a nonsignificant effect between both groups (unpaired, two‐sided t‐test: P = 0.199) (expressed as [(t 2 − t 1)/t 1] × 100).
Given that mainly levetiracetam is thought to have an effect on PAS measures (see Discussion), analyses were repeated in a subgroup of patients without levetiracetam medication and with mRS > 0 (n = 19). MEP after PAS protocol showed a decrease in the NMDAR encephalitis group of −16.80% (MEP pre: 0.52 ± 0.51 mV; MEP post: 0.32 ± 0.25 mV), whereas it increased in the healthy group by 26.1% (MEP pre: 0.63 ± 0.42 mV; MEP post: 0.76 ±0.63 mV). Similar to the whole group analysis, ANOVA showed a significant interaction of time and group (F 1,47 = 8.90, P = 0.0047), and post hoc t‐test revealed a significant change between both groups (unpaired, two‐sided t‐test: P = 0.040) (expressed as percentage of change relative to baseline MEP (%): [(t 2 − t 1)/t 1] × 100).
Other TMS‐derived neurophysiological assessments
Comparing patients with healthy controls (Fig. 2A), there was a significant difference in RMT (NMDAR encephalitis: 39.65 ± 7.95% stimulator output; controls: 34.81 ± 6.42%; unpaired, two‐sided t‐test: P = 0.015). Data for RMT were normally distributed. Subgroup analysis of 11 patients without centrally active medication and mRS > 0 compared to controls (Fig. 2B) showed no significant differences (NMDAR encephalitis: 36.55 ± 6.98%; controls: 34.81 ± 6.42%; unpaired, two‐sided t‐test: P = 0.45), suggesting that the higher RMT in patients may be related to functional impairment in NMDAR encephalitis or the effect of medication.
Figure 2.
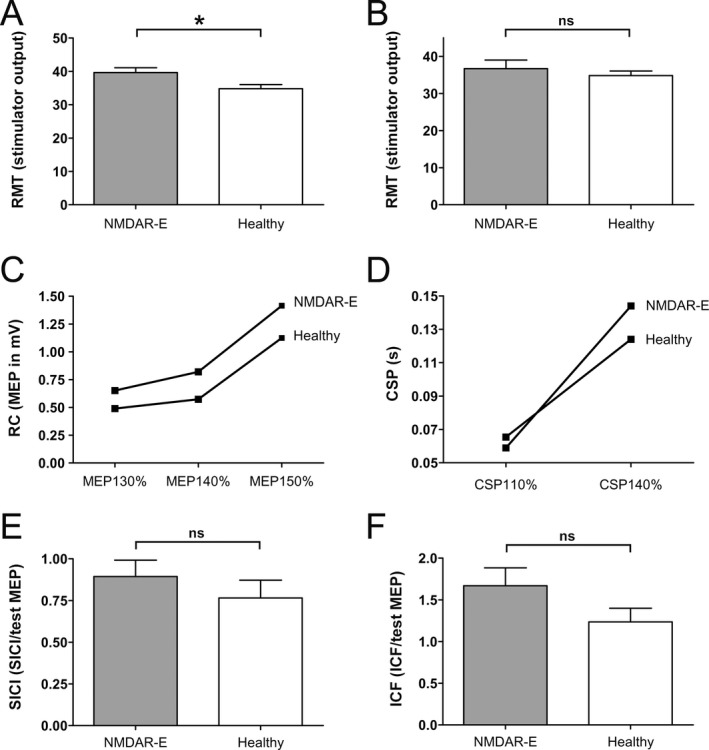
Characterization of transcranial magnetic stimulation (TMS) parameters in patients with NMDAR (N‐methyl‐d‐aspartate receptor) encephalitis. (A) Slightly increased resting motor threshold (RMT) in patients with NMDAR encephalitis versus healthy controls. (B) No difference in RMT in patients without centrally active medication. (C) Recruitment curves of NMDAR encephalitis patients and controls. Motor‐evoked potential (MEP) values with intensity of 130%, 140%, and 150% of individual RMT. (D) Cortical silent period (CSP) with 110% and 140% in patients and controls. No differences for short intracortical inhibition (SICI) (E) and intracortical facilitation (ICF) (F) between patients with NMDAR encephalitis and controls.
To exclude effects of interindividual RMT (which is highly variable also in healthy subjects), MEP measurements were routinely standardized at the intensity of 130% of individual RMT which is the well‐accepted gold standard. MEP at intensities of 130% and 150% of RMT were compared between groups revealing no significant differences (unpaired, two‐sided t‐test: 130%: P = 0.146; 150%: P = 0.1941). Analyzing the nonmedication subgroup, t‐tests were also not significantly different between groups (130%: P = 0.776; 150%: P = 0.068). In contrast, MEP with 140% (which is part of the RC analysis) showed a significant difference between NMDAR encephalitis patients and healthy controls (unpaired, two‐sided t‐test: P = 0.0073), whereas subgroup analyses revealed no significant differences (P = 0.1226). Similarly, the RC (t‐test: 150%/130%: P = 0.974; 140%/130%: P = 0.6432; 150%/140%: P = 0.591; Fig. 2C), the CSP with 110% and 140% intensity (110% t‐test: P = 0.351; 140% t‐test: P = 0.2336; Fig. 2D), and the paired‐pulse parameters of SICI and ICF (expressed as: ICF/MEP; SICI/MEP; t‐test: SICI: P = 0.192; ICF: P = 0.394; Fig. 2E and F) were not different between patients and controls.
Correlation analyses
PAS and mRS
Correlation analysis was performed in order to detect an association of mRS and changes of MEP after PAS in patients with NMDAR encephalitis and controls. Data showed a clear association of better functional scales with PAS changes (P = 0.0031, Pearson's correlation, r = −0.41; Fig. 3A). In contrast, there was no correlation between PAS changes and disease duration, arguing against improvement of PAS measurement simply due to longer duration from the acute phase of the encephalitis (P = 0.9464, Pearson's correlation, r = 0.01; Fig. 3B).
Figure 3.
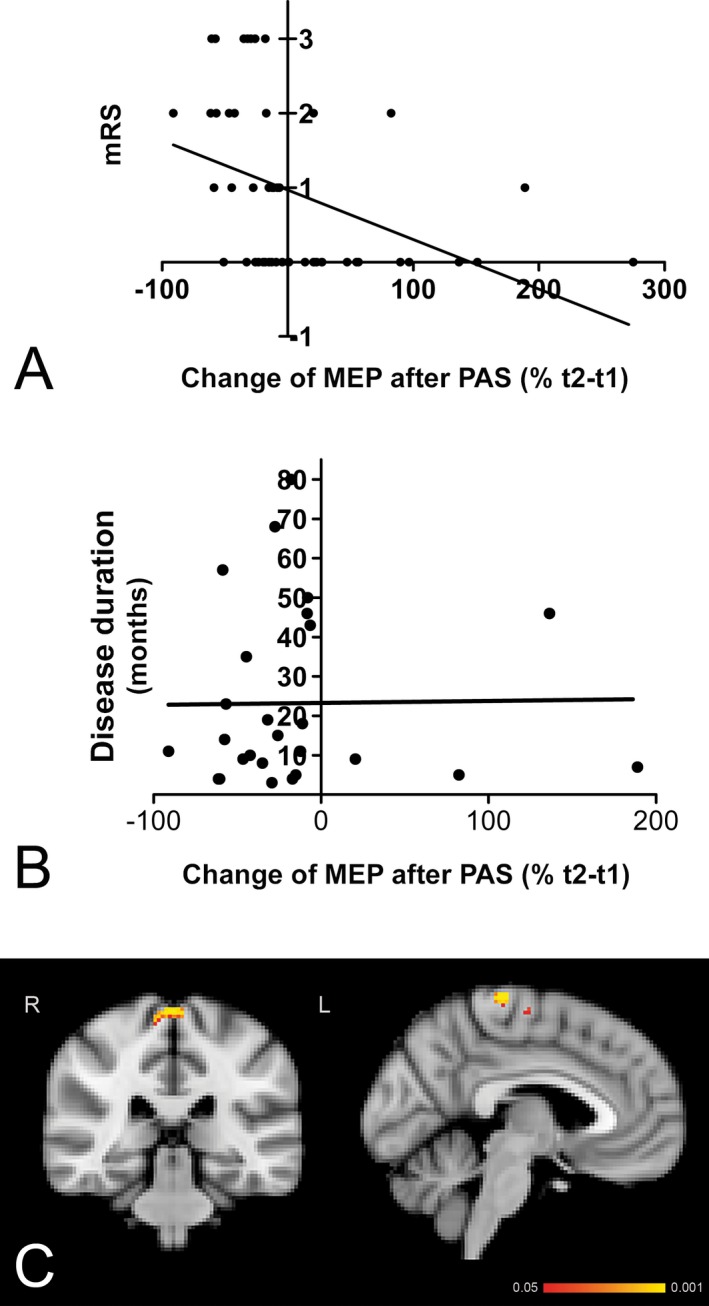
(A) Paired associative stimulation (PAS) changes correlate with disease severity as measured with the modified Rankin Scale (mRS). (B) In contrast, PAS changes did not correlate with the duration of disease. (C) Correlation of PAS‐induced plasticity with motor network functional connectivity. Resting state functional connectivity of the motor network with bilateral precentral gyrus correlated positively with PAS‐induced plasticity in NMDAR (N‐methyl‐d‐aspartate receptor) encephalitis patients.
PAS and resting state functional connectivity
Higher functional connectivity within the motor network was correlated with higher PAS‐induced plasticity in NMDAR encephalitis patients (P < 0.001, uncorrected), that is, higher functional connectivity between the motor network as a whole and bilateral precentral gyrus regions was associated with higher PAS‐induced plasticity (Fig. 3C).
Follow‐up analyses
Of the 34 enrolled patients with NMDAR encephalitis, six were available for follow‐up analysis (Table 2). Five patients showed clinical improvement measured with the mRS, all of these also showed improvement in the PAS protocol suggesting that TMS could potentially be helpful in the monitoring of clinical improvement. One patient was clinically unchanged at follow‐up after a prolonged clinical relapse; here no PAS improvement was detectable.
Table 2.
Follow‐up data
| Patient ID | Disease months Visit 1/2 | mRS Visit 1/2 | Antibody titer | PAS (% change) Visit 1/2 | Change of medication Visit 1 → 2 | |
|---|---|---|---|---|---|---|
| Serum Visit 1/2 | CSF Visit 1/2 | |||||
| 4 | 14/38 | 3/2 | 32/100 | 1/n.d. | −57.63/−13.95 (↑) | No |
| 7 | 42/66 | 2/1 | 0/0 | 0/0 | −6.47/+1.89 (↑) | LEV → Escitalopram |
| 10 | 46/61 | 1/1 | 32/0 | 1/0 | −8.25/−1.56 (↑) | No |
| 15 | 57/74 | 1/0 | 10/32 | 32/n.d. | −58.71/+101.28 (↑) | No |
| 17 | 5/13 | 2/1 | 100/100 | 10/n.d. | −39.61/−18.49 (↑) | VAL → LTG |
| 21 | 8/12 | 3/3 | 320/100 | 32/32 | −35.01/−43.88 (↓) | Quetiapin → No |
mRS, modified Rankin Scale; CSF, cerebrospinal fluid; PAS, paired associative stimulation; n.d., not determined; LTG, lamotrigin; VPA, valproate; LEV, levetiracetam.
Discussion
The present study demonstrated significant differences in PAS‐induced plasticity in patients with NMDAR encephalitis compared to healthy controls, namely decreases in patients and increases in controls. Given that PAS‐induced plasticity is dependent on NMDAR function, these findings for the first time provide in vivo neurophysiological evidence of malfunctioning of NMDAR capacity in a large sample of patients harboring pathogenic antibodies against the NMDAR. Moreover, brain dysfunction as indexed by PAS results were significantly correlated with disease severity and motor network functional connectivity. Follow‐up assessments demonstrated parallel improvement of PAS‐induced plasticity and mRS. No differences emerged between groups for markers of GABA(A/B)ergic function indicating specificity for dysfunction in glutamatergic‐based LTP‐like plasticity.
To the best of our knowledge, only one case report has documented TMS‐derived neurophysiological measurements in the acute phase of a woman with NMDAR encephalitis,33 reporting normal MEP amplitudes and latencies, as well as intact corticospinal tracts, consistent with our findings. Initially, the protocol of sensory afferent facilitation (protocol of a conditioning electrical stimulus applied to a peripheral nerve followed by a TMS pulse to the contralateral M1 which typically enhances MEP) showed an exaggerated response, but improved and normalized after patient's health condition increased and was stabilized while sensory afferent inhibition (protocol of an electrical stimulus applied to a peripheral nerve followed by a TMS pulse which typically suppresses MEP34) remained absent both times. This is consistent with our follow‐up data showing parallel improvement of PAS and mRS.
Resting state functional MRI has been shown to reveal characteristic alterations of functional connectivity in NMDAR encephalitis patients despite normal routine MRI.29 Specifically, it was shown that patients have reduced functional connectivity of the hippocampus with the default mode network that correlated with individual memory performance. Here, we found that motor network connectivity predicted individual PAS response in patients. This observation indicates that functional connectivity alterations might be even more widespread and include the motor network. Moreover, these findings may provide a link between electrophysiological and imaging markers of NMDAR dysfunction in encephalitis patients given that combination of these two complementary tools will advance studies of brain connectivity, although the exact interaction needs to be addressed in further studies.35
We further provide new safety data on single‐ and paired‐pulse TMS. TMS application included patients suffering from encephalitis‐related seizures in the past with the last seizure >6 months before. Antiepileptic drugs were not changed for the study. No patient experienced a seizure. Based on these data it seems ethically justified in prospective studies to also perform TMS in patients with acute NMDAR encephalitis as the probability of triggering seizures with single‐ and paired‐pulse TMS seems to be extremely low. Literature reviews support the notion that single‐ and paired‐pulse TMS are safe, most likely also in patients with epilepsy.36 Future studies should therefore include patients in the acute phase of NMDAR encephalitis as this will likely result in further clues on the predictability of clinical progression/remission, relapses, therapy responses, and prognosis.33
A potential confounding factor for TMS is the use of central nervous system (CNS)‐active medication. Previous studies showed an effect mainly for levetiracetam on the PAS protocol, whereas most drugs did not affect this measurement, such as tiagabine, diazepam, lamotrigine, piracetam, gabapentin, and topiramate.17 Reanalysis of our data using both a subgroup of patients lacking levetiracetam and patients lacking all CNS‐active medications lead to equal results confirming the significant PAS differences between NMDAR encephalitis patients and controls. When comparing all encephalitis patients with controls, we found significant differences in RMT which was not detectable in the subgroup without centrally active medication (including all drugs such as tiagabine, diazepam, lamotrigine, piracetam, gabapentin, topiramate, and levetiracetam). Even though there is somewhat conflicting data whether levetiracetam has large or subtle effects on single‐pulse TMS parameters,37, 38, 39 we conclude that the observed differences in RMT might partially depend on medication, while the observed PAS effect is most likely related to receptor changes attributable to NMDAR encephalitis. However, it is also possible that the subgroup without medication was too small to detect differences (n = 11), and larger patient sizes are needed to confirm the influence of the medication. Finally, it is possible that the subgroup required less medication because of more advanced clinical remission, even though mRS was not better in the subgroup. Another limitation is the fact that only six patients were available for follow‐up analyses. Since the prevalence of NMDAR encephalitis is relatively low, follow‐up studies with a bigger sample size would need several more years. However, the data of six patients suggest that PAS and mRS changed in the same way regarding disease improvement or deterioration. ICF did not show a significant difference among groups. One might have expected impaired ICF in NMDAR encephalitis patients since ICF is known to be (at least partially) mediated through NMDAR. However, ICF is not solely mediated by NMDAR, but also by GABAA receptors as indicated in studies with benzodiazepines4, 40, 41 as well as by non‐NMDA glutamate receptors.4, 42 In addition, ketamine, an NMDAR antagonist, showed no effects on ICF.16 PAS is known to be predominantly mediated via NMDAR, however, also other neurotransmitters with different dominance are likely involved. Despite contribution of NMDAR currents in both ICF and PAS, they are independent TMS parameters with different underlying mechanisms, which explain why only one of these two neurophysiological measurements can be altered.
It is an intriguing question as to whether TMS might not only be useful for early determination of disease severity and follow‐up monitoring in NMDAR encephalitis, but also whether therapeutic application can improve hippocampal function and synaptic plasticity. There is evidence from animal models that rTMS may increase the mRNA and protein expression of NMDAR.43 A human study confirmed that after‐effects of rTMS (theta burst stimulation) are linked to the function of the NMDAR.44 Moreover, animal and behavioral studies45, 46, 47, 48, 49 provide compelling evidence that rTMS alters synaptic plasticity of hippocampal areas. Thus, rTMS may be used in order to improve hippocampal function and increase neuronal NMDAR expression. Along these lines, prospective studies should evaluate its potential for therapeutic application in patients with NMDAR encephalitis.
Taken together, we show that (1) there was a significant difference in PAS between NMDAR encephalitis patients and healthy controls, (2) other TMS‐derived neurophysiological parameters which are not mediated through NMDAR were not altered, (3) neurophysiological parameters depend on the disease state, and (4) were significantly correlated with already established markers of disease activity such as mRS and resting state functional connectivity. Thus, measurement of PAS‐induced plasticity may help to detect subtle NMDAR dysfunction and disease severity in NMDAR encephalitis. It might even serve as a sensitive, widely available and noninvasive “neurophysiological biomarker” to support diagnostic and therapeutic decisions and to monitor therapy response in the future. In six available patients, follow‐up analyses demonstrated parallel improvement of PAS changes and mRS. If the findings are confirmed in larger follow‐up cohorts, TMS will be an objective way to monitor the disease course, to identify relapses, and to control therapy responses.
Author Contributions
M. S. V., A. F., and H. P. contributed to the study concept and design. M. S. V., C. F., L. H., B. J., F. P., A. F., and H. P. performed the data acquisition and analysis. M. S. V., C. F., A. F., and H. P. drafted the manuscript and figures.
Conflict of Interest
Dr. Flöel reports personal fees and received consulting fees from Schwabe, personal fees and received honoraria for oral presentations from Novartis, Böhringer‐Ingelheim, Souvenaid, grants from Deutsche Forschungsgemeinschaft and Bundesministerium für Bildung und Forschung, outside the submitted work. Dr. Harms received personal compensation from Biogen, Merck‐Serono, Roche, Genzyme, Diamed and Bayer for speaking and compensation from Novartis, Biogen, Roche and Genzyme as member of advisory board. He received travel support from TEVA, Novartis, Grifols and Biogen.
Supporting information
Table S1. Checklist for assessing the methodological quality of TMS reporting.
Acknowledgments
This study has been supported by grants from the German Academic Exchange Service (to H. P.; DAAD, D/10/43923), the German Research Foundation (DFG, to H. P.: PR 1274/2‐1; to A. F.: Fl‐379‐8/1, 379‐10/1, 379‐11/1, DFG‐Exc‐257; to F. P.: DFG‐Exc‐257), and the German Ministry for Education and Research (BMBF, to A. F.: FKZ 01EO0801, 01GY1144, 01GQ1424A, 01GQ1420B).
References
- 1. Titulaer MJ, McCracken L, Gabilondo I, et al. Treatment and prognostic factors for long‐term outcome in patients with anti‐NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 2013;12:157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gresa‐Arribas N, Titulaer MJ, Torrents A, et al. Antibody titres at diagnosis and during follow‐up of anti‐NMDA receptor encephalitis: a retrospective study. Lancet Neurol 2013;13:167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hughes EG, Peng X, Gleichman AJ, et al. Cellular and synaptic mechanisms of anti‐NMDA receptor encephalitis. J Neurosci 2010;30:5866–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paulus W, Classen J, Cohen LG, et al. State of the art: pharmacologic effects on cortical excitability measures tested by transcranial magnetic stimulation. Brain Stimul 2008;1:151–163. [DOI] [PubMed] [Google Scholar]
- 5. Schwenkreis P, Maier C, Pleger B, et al. NMDA‐mediated mechanisms in cortical excitability changes after limb amputation. Acta Neurol Scand 2003;108:179–184. [DOI] [PubMed] [Google Scholar]
- 6. Frantseva MV, Fitzgerald PB, Chen R, et al. Evidence for impaired long‐term potentiation in schizophrenia and its relationship to motor skill learning. Cereb Cortex 2008;18:990–996. [DOI] [PubMed] [Google Scholar]
- 7. Rioult‐Pedotti MS, Friedman D, Donoghue JP. Learning‐induced LTP in neocortex. Science 2000;290:533–536. [DOI] [PubMed] [Google Scholar]
- 8. Stefan K, Kunesch E, Cohen LG, et al. Induction of plasticity in the human motor cortex by paired associative stimulation. Brain 2000;123(Pt 3):572–584. [DOI] [PubMed] [Google Scholar]
- 9. List J, Duning T, Meinzer M, et al. Enhanced rapid‐onset cortical plasticity in CADASIL as a possible mechanism of preserved cognition. Cereb Cortex 2011;21:2774–2787. [DOI] [PubMed] [Google Scholar]
- 10. List J, Duning T, Kurten J, et al. Cortical plasticity is preserved in nondemented older individuals with severe ischemic small vessel disease. Hum Brain Mapp 2012;34:1464–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weise D, Schramm A, Stefan K, et al. The two sides of associative plasticity in writer's cramp. Brain 2006;129:2709–2721. [DOI] [PubMed] [Google Scholar]
- 12. Zeller D, aufm Kampe K, Biller A, et al. Rapid‐onset central motor plasticity in multiple sclerosis. Neurology 2010;74:728–735. [DOI] [PubMed] [Google Scholar]
- 13. Stefan K, Kunesch E, Benecke R, et al. Mechanisms of enhancement of human motor cortex excitability induced by interventional paired associative stimulation. J Physiol 2002;543:699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lucke C, Heidegger T, Rohner M, et al. Deleterious effects of a low amount of ethanol on LTP‐like plasticity in human cortex. Neuropsychopharmacology 2014;39:1508–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwenkreis P, Witscher K, Pleger B, et al. The NMDA antagonist memantine affects training induced motor cortex plasticity – a study using transcranial magnetic stimulation. BMC Neurosci 2005;6:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Di Lazzaro V, Oliviero A, Profice P, et al. Ketamine increases human motor cortex excitability to transcranial magnetic stimulation. J Physiol 2003;547:485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heidegger T, Krakow K, Ziemann U. Effects of antiepileptic drugs on associative LTP‐like plasticity in human motor cortex. Eur J Neurosci 2010;32:1215–1222. [DOI] [PubMed] [Google Scholar]
- 18. Chipchase L, Schabrun S, Cohen L, et al. A checklist for assessing the methodological quality of studies using transcranial magnetic stimulation to study the motor system: an international consensus study. Clin Neurophysiol 2012;123:1698–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Oldfield RC. The assessment and analysis of handedness: the Edinburgh inventory. Neuropsychologia 1971;9:97–113. [DOI] [PubMed] [Google Scholar]
- 20. Rankin J. Cerebral vascular accidents in patients over the age of 60. II. Prognosis. Scott Med J 1957;2:200–215. [DOI] [PubMed] [Google Scholar]
- 21. Dalmau J, Lancaster E, Martinez‐Hernandez E, et al. Clinical experience and laboratory investigations in patients with anti‐NMDAR encephalitis. Lancet Neurol 2010;10:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Prüss H, Dalmau J, Harms L, et al. Retrospective analysis of NMDA receptor antibodies in encephalitis of unknown origin. Neurology 2010;75:1735–1739. [DOI] [PubMed] [Google Scholar]
- 23. Matsunaga K, Uozumi T, Tsuji S, Murai Y. Age‐dependent changes in physiological threshold asymmetries for the motor evoked potential and silent period following transcranial magnetic stimulation. Electroencephalogr Clin Neurophysiol 1998;109:502–507. [DOI] [PubMed] [Google Scholar]
- 24. Zarkowski P, Navarro R, Pavlicova M, et al. The effect of daily prefrontal repetitive transcranial magnetic stimulation over several weeks on resting motor threshold. Brain Stimul 2009;2:163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kukke SN, Paine RW, Chao CC, et al. Efficient and reliable characterization of the corticospinal system using transcranial magnetic stimulation. J Clin Neurophysiol 2014;31:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Braune HJ, Fritz C. Transcranial magnetic stimulation‐evoked inhibition of voluntary muscle activity (silent period) is impaired in patients with ischemic hemispheric lesion. Stroke 1995;26:550–553. [DOI] [PubMed] [Google Scholar]
- 27. Salih F, Khatami R, Steinheimer S, et al. Inhibitory and excitatory intracortical circuits across the human sleep‐wake cycle using paired‐pulse transcranial magnetic stimulation. J Physiol 2005;565:695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schwenkreis P, Liepert J, Witscher K, et al. Riluzole suppresses motor cortex facilitation in correlation to its plasma level. A study using transcranial magnetic stimulation. Exp Brain Res 2000;135:293–299. [DOI] [PubMed] [Google Scholar]
- 29. Finke C, Kopp UA, Scheel M, et al. Functional and structural brain changes in anti‐N‐methyl‐d‐aspartate receptor encephalitis. Ann Neurol 2013;74:284–296. [DOI] [PubMed] [Google Scholar]
- 30. Paolini M, Keeser D, Ingrisch M, et al. Resting‐state networks in healthy adult subjects: a comparison between a 32‐element and an 8‐element phased array head coil at 3.0 Tesla. Acta Radiol 2015;56:605–613. [DOI] [PubMed] [Google Scholar]
- 31. Filippini N, MacIntosh BJ, Hough MG, et al. Distinct patterns of brain activity in young carriers of the APOE‐epsilon4 allele. Proc Natl Acad Sci USA 2009;106:7209–7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Beckmann CF, DeLuca M, Devlin JT, Smith SM. Investigations into resting‐state connectivity using independent component analysis. Philos Trans R Soc Lond B Biol Sci 2005;360:1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dou YH, Lai KL, Liao KK, Chen SP. Abnormal sensory‐motor integration in a patient with anti‐NMDA‐receptor encephalitis. J Neurol 2012;259:1490–1493. [DOI] [PubMed] [Google Scholar]
- 34. Bikmullina R, Baumer T, Zittel S, Munchau A. Sensory afferent inhibition within and between limbs in humans. Clin Neurophysiol 2009;120:610–618. [DOI] [PubMed] [Google Scholar]
- 35. Fox MD, Halko MA, Eldaief MC, Pascual‐Leone A. Measuring and manipulating brain connectivity with resting state functional connectivity magnetic resonance imaging (fcMRI) and transcranial magnetic stimulation (TMS). NeuroImage 2012;62:2232–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rossi S, Hallett M, Rossini PM, Pascual‐Leone A. Safety, ethical considerations, and application guidelines for the use of transcranial magnetic stimulation in clinical practice and research. Clin Neurophysiol 2009;120:2008–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sohn YH, Kaelin‐Lang A, Jung HY, Hallett M. Effect of levetiracetam on human corticospinal excitability. Neurology 2001;57:858–863. [DOI] [PubMed] [Google Scholar]
- 38. Reis J, Wentrup A, Hamer HM, et al. Levetiracetam influences human motor cortex excitability mainly by modulation of ion channel function – a TMS study. Epilepsy Res 2004;62:41–51. [DOI] [PubMed] [Google Scholar]
- 39. Solinas C, Lee YC, Reutens DC. Effect of levetiracetam on cortical excitability: a transcranial magnetic stimulation study. Eur J Neurol 2008;15:501–505. [DOI] [PubMed] [Google Scholar]
- 40. Mohammadi B, Krampfl K, Petri S, et al. Selective and nonselective benzodiazepine agonists have different effects on motor cortex excitability. Muscle Nerve 2006;33:778–784. [DOI] [PubMed] [Google Scholar]
- 41. Inghilleri M, Berardelli A, Marchetti P, Manfredi M. Effects of diazepam, baclofen and thiopental on the silent period evoked by transcranial magnetic stimulation in humans. Exp Brain Res 1996;109:467–472. [DOI] [PubMed] [Google Scholar]
- 42. Hwa GG, Avoli M. Excitatory postsynaptic potentials recorded from regular‐spiking cells in layers II/III of rat sensorimotor cortex. J Neurophysiol 1992;67:728–737. [DOI] [PubMed] [Google Scholar]
- 43. Wang F, Geng X, Tao HY, Cheng Y. The restoration after repetitive transcranial magnetic stimulation treatment on cognitive ability of vascular dementia rats and its impacts on synaptic plasticity in hippocampal CA1 area. J Mol Neurosci 2010;41:145–155. [DOI] [PubMed] [Google Scholar]
- 44. Huang YZ, Chen RS, Rothwell JC, Wen HY. The after‐effect of human theta burst stimulation is NMDA receptor dependent. Clin Neurophysiol 2007;118:1028–1032. [DOI] [PubMed] [Google Scholar]
- 45. Vlachos A, Muller‐Dahlhaus F, Rosskopp J, et al. Repetitive magnetic stimulation induces functional and structural plasticity of excitatory postsynapses in mouse organotypic hippocampal slice cultures. J Neurosci 2012;32:17514–17523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ma J, Zhang Z, Kang L, et al. Repetitive transcranial magnetic stimulation (rTMS) influences spatial cognition and modulates hippocampal structural synaptic plasticity in aging mice. Exp Gerontol 2014;58:256–268. [DOI] [PubMed] [Google Scholar]
- 47. Zhang Y, Mao RR, Chen ZF, et al. Deep‐brain magnetic stimulation promotes adult hippocampal neurogenesis and alleviates stress‐related behaviors in mouse models for neuropsychiatric disorders. Mol Brain 2014;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma J, Zhang Z, Su Y, et al. Magnetic stimulation modulates structural synaptic plasticity and regulates BDNF‐TrkB signal pathway in cultured hippocampal neurons. Neurochem Int 2012;62:84–91. [DOI] [PubMed] [Google Scholar]
- 49. Wang F, Geng X, Tao HY, Cheng Y. The restoration after repetitive transcranial magnetic stimulation treatment on cognitive ability of vascular dementia rats and its impacts on synaptic plasticity in hippocampal CA1 area. J Mol Neurosci 2009;41:145–155. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Checklist for assessing the methodological quality of TMS reporting.