

Abstract
Mammalian global genomic nucleotide excision repair requires lesion recognition by XPC, whose detailed binding mechanism remains to be elucidated. Here we have delineated the dynamic molecular pathway and energetics of lesion-specific and productive binding by the Rad4/yeast XPC lesion recognition factor, as it forms the open complex [Min, J. H., and Pavletich, N. P. (2007) Nature 449, 570–575; Chen, X., et al. (2015) Nat. Commun. 6, 5849] that is required for excision. We investigated extensively a cis-syn cyclobutane pyrimidine dimer in mismatched duplex DNA, using high-level computational approaches. Our results delineate a preferred correlated motion mechanism, which provides for the first time an atomistic description of the sequence of events as Rad4 productively binds to the damaged DNA.
Global genomic nucleotide excision repair (NER) is a key cellular defense mechanism against a broad range of DNA lesions, such as certain ones derived from polycyclic aromatic chemical pollutants, reactive oxygen species,3 and the ultraviolet (UV) components of sunlight.4 The latter are among the most prevalent of DNA lesions and are responsible for the majority of human skin cancers, including melanoma.5 The vital importance of NER is demonstrated in the human xeroderma pigmentosum (XP) diseases, caused by mutations in NER genes. The XP-C variant stems from mutations in the NER lesion recognition protein, XPC; these mutations cause extraordinary sensitivity to sunlight with a great proclivity for skin cancers at an early age. The failure to repair UV-derived lesions, predominantly cis-syn cyclobutane pyrimidine dimers (CPD) (Figure 1A), is responsible for this cancer-prone syndrome.5,6
Figure 1.
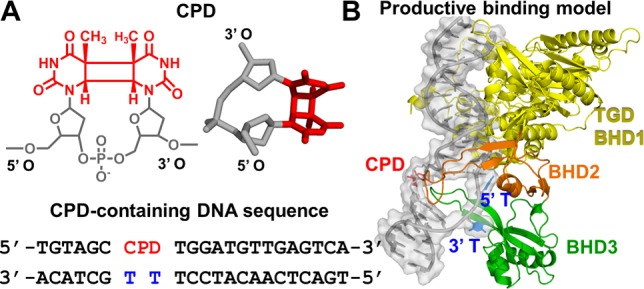
(A) CPD lesion (chemical and three-dimensional structures, the latter taken from the crystal structure of PDB entry 1T4I(34)) and DNA sequence context taken from the crystal structure of PDB entry 2QSG1. (B) Productive binding model of Rad4 and CPD-containing DNA, with the sequence shown in panel A, based on crystal structures of PDB entries 2QSG1 and 1T4I.34 The protein is shown as a cartoon, the CPD lesion as sticks, and the DNA as a cartoon with a surface overlay. Hydrogen atoms are not shown for the sake of clarity.
The XPC-RAD23B protein complex has been identified as the critical factor that recognizes NER-susceptible lesions7−9 in mammals: it subsequently recruits TFIIH, whose XPD helicase verifies the lesion.4,10−12 Other NER factors are then recruited to ultimately excise a 24–32-mer oligonucleotide containing the damage.9,13−15 When the CPD lesion contains normal partner bases adenine, the auxiliary proteins UV-DDB, composed of the heterodimer of DDB1 and DDB2, first recognize the damage site and then hand it off to XPC.4,16 However, when faced with mismatched partners, XPC can recognize the lesion site directly.15,17
How the broad range of DNA lesions differing greatly in chemical structure are recognized by XPC has been elucidated by an X-ray crystallographic structure of Saccharomyces cerevisiae Rad4-Rad23, the yeast ortholog of human XPC-RAD23B, in complex with a CPD-containing DNA duplex with mismatched thymine partners [Protein Data Bank (PDB18) entry 2QSG].1 The β-hairpin of the Rad4 BHD3 domain is inserted into the DNA helix from the major groove side while BHD2 contacts the minor groove, the CPD lesion (unresolved) is extruded and not contacted by the protein, and the two mismatched thymine partners are flipped out into Rad4 pockets. This structure, the productive open complex that correctly recruits TFIIH, strongly supports the proposal that the NER lesion recognition factor does not recognize the lesion itself, but the local distortions, dynamics, and destabilizations in the DNA associated with them.15,19−33 We have used molecular modeling and molecular dynamics (MD) simulations (detailed in the Supporting Information) to model unresolved residues in BHD2 and the extruded CPD lesion of the crystal structure (PDB entry 2QSG), yielding a completed productive open complex (Figure 1B).
It has been observed that the wide array of DNA lesions are repaired by NER at very different rates,21,24,28 and the origin of this diversity remains poorly understood. It is hypothesized that differences in lesion-induced local DNA duplex destabilization may play a key role in governing the rate of formation of the productive open complex for lesion recognition by XPC,35 which ultimately leads to excision. Of particular interest are certain bulky DNA lesions that stabilize the DNA duplex and are NER-resistant,36,37 because they persist and can cause cancer-initiating mutations.
A kinetic gating mechanism has recently been proposed on the basis of essentially identical crystal structures of Rad4-Rad23 complexed with damaged1 and undamaged DNA,2 and temperature-jump perturbation spectroscopy (T-jump) experiments: the probability of forming the productive open complex for lesion recognition is a result of the kinetic competition between Rad4 residence time and the rate of forming the open complex at the lesion site.2 At a lesion, the free energy barrier to form the productive open complex, compared with the residence time of XPC, determines its recognition for NER; furthermore, it is proposed that distorting and destabilizing lesions decrease the opening time for productive binding and increase the residence time of XPC. To understand how lesions signal their presence in the face of predominantly undamaged DNA, it is necessary to characterize the dynamic molecular pathway and energetics of Rad4/XPC lesion-specific and productive binding to damaged DNA. The present study addresses this vital gap in our knowledge by investigating the mismatched CPD lesion. Our work provides a complement to experimental investigations, such as single molecule38 and T-jump2 studies that are elucidating binding dynamics of the XPC NER factor.
We have hypothesized two types of mechanisms for binding of Rad4-Rad23 suggested by the crystal structures of its apoprotein without substrate, and in a complex with a CPD lesion (PDB entries 2QSF and 2QSG):1 (1) conformational capture39 and (2) correlated motion (they are illustrated in Figure 2). These are representative of current understanding of protein–DNA recognition mechanisms, whose balance is believed to depend on the particular system.39 An example of the conformational capture mechanism has been observed crystallographically for the AlkD DNA glycosylase.40 In this mechanism, the fraction of the population with flipped-out partners is governed by the local thermodynamic stability: a lower flipping free energy barrier in more destabilized cases produces a larger flipped out population (Figure 2A). No such population is required for the correlated motion mechanism, where flipping occurs in concert with other lesion recognition events (Figure 2B).
Figure 2.
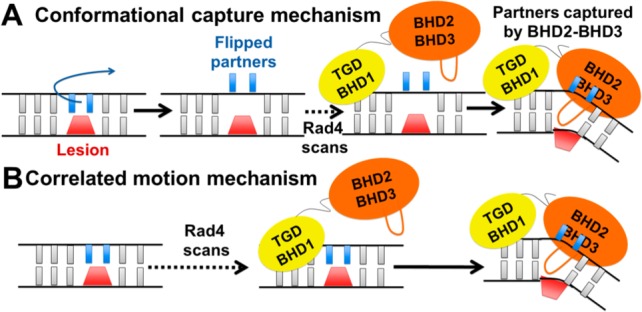
Hypothesized conformational capture and correlated motion mechanisms for lesion recognition by Rad4. (A) Conformational capture mechanism. The ensemble of damaged duplexes contains a population that has one or two bases opposite the lesion flipped out. The Rad4 scans the damaged DNA, flipped out bases in a locally destabilized region are captured by the BHD2 and BHD3 domains, and the BHD3 β-hairpin inserts with eviction of the lesion. (B) Correlated motion mechanism. Scanning is accompanied by probing for a locally destabilized region by BHD2 and BHD3 domains. Once a sufficiently destabilized region is sensed, in correlated motion the BHD3 β-hairpin inserts, the lesion is evicted, and the partner bases flip out to associate with BHD2 and BHD3 domains; the order of these events may be lesion-dependent.
Here we have used state-of-the-art simulations to characterize on an atomistic and energetic level the lesion recognition mechanism for forming the productive open complex between Rad4-Rad23 and a CPD-containing DNA duplex with mismatched thymine partners1 (Figure 1). To obtain the molecular pathways and their free energy profiles, we first used umbrella sampling41 with restrained molecular dynamics (MD) simulations to generate structural ensembles along selected reaction coordinates. We used the AMBER1242 or AMBER1443 packages with the ff12SB force field and a customized force field for the CPD lesion (Table S1), explicit water, and counterions. Then, free energy profiles were calculated using the variational free energy profile (vFEP) method44 for the correlated motion mechanism. Full details of modeling, reaction coordinates, and MD simulations are given in the Supporting Information.45−47
Via exploration of many possible combinations of pathways for productive binding to the open complex, we determine that the energetically preferred pathway utilizes a correlated motion mechanism involving three stages (Figures 3 and 4 and Figures S2–S4): (1) initial association of the BHD2 and BHD3 domains induces DNA duplex distortions in bending, unwinding, and stretching, to initiate lesion-partner base flipping; (2) the two partner bases opposite the lesion flip into the protein in a correlated fashion; and (3) the BHD3 β-hairpin inserts into the denatured lesion site to stabilize the open complex.
Figure 3.

Correlated motion pathway. Probing model (PM), intermediate (INT), transition state (TS), and productively bound state (PB) structures are rendered as cartoons with CPD as sticks and F556, F597, and F599 residues as spheres. The BHD2 β-hairpin of PB is transparent to reveal BHD3 β-hairpin (green) insertion. See the Supporting Movie for the dynamic pathway.
Figure 4.
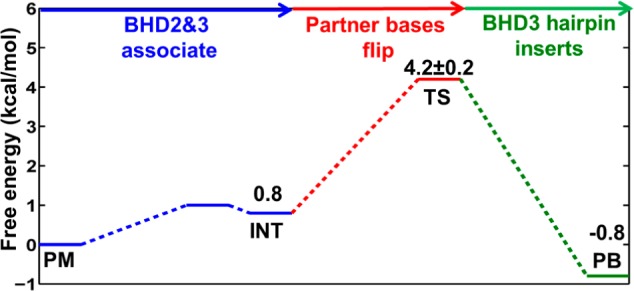
Free energy profile for the correlated motion pathway. The indicated states correspond to those in Figure 3. The statistical error for the transition state (TS) is estimated by averaging the free energy difference between 10–35 and 35–60 ns shown in Figure S4.
For stage (1), we first docked the apo Rad4-Rad23 TGD and BHD1 domains to the DNA duplex positioned as in the crystal structure of the complex,1 but without engagement of the BHD2 and BHD3 domains (docking model, Figure S1); then these domains were guided to approach the lesion site (Figure S2). A structural ensemble we term the probing model (PM) (Figure 3) was obtained, whose energy is ∼1 kcal/mol lower than that of the docking model (Figure S2). In this structural ensemble, the DNA duplex bends slightly in the direction of the productive binding model, the BHD2 β-hairpin tip gains initial interactions with the DNA backbone on the minor groove side, and the CPD and its partner bases maintain their stacked-in conformation as in the docking model (Figure S1). As BHD2 and BHD3 associate further, an intermediate state (INT) was identified with an energy 0.8 kcal/mol higher than that of the PM structure (Figures 3 and 4 and Figure S2). This intermediate state was confirmed by two unrestrained MD simulations [initiated from two different structures (detailed in the Supporting Information), which converged to INT (Figure S2)]. The approach of BHD2 and BHD3 to the lesion site in proceeding from PM to INT (Figure 3) is correlated with further DNA bending and untwisting around the lesion site; these are accompanied by gradual displacement of the CPD from its stacked-in position in the PM (Figure S5). The distortions of the DNA duplex cause the two partner thymines to approach Phe599 and enter a “flipping pathway” comprised of three Phe residues (Phe599, Phe597, and Phe556) that direct the partner bases into their binding pocket at the BHD2-BHD3/DNA interface (Figure 3).
For stage (2), the 5′ partner thymine is guided to approach its position in the binding pocket (Figure S3). This process is the most energetically demanding one in the preferred pathway of the correlated motion mechanism. As the 5′ thymine flips into its binding pocket, the free energy gradually increases to 4.2 kcal/mol, which is denoted as a transition state (TS) in the free energy profile (Figure 4). In the TS ensemble, the 5′ thymine is in its binding pocket positioned further along the Phe flipping path, stacked with Phe597; this is accompanied by unguided flipping of the 3′ thymine to stack on the edge of Phe597, but it is not totally inserted into its binding pocket in BHD3 (Figure S3). The lesion site is now denatured, and Phe599 in the BHD3 β-hairpin has reoriented for insertion of the hairpin into the duplex to fill the cavity (Figure 3 and Figure S5). These structural changes result in further unwinding and stretching around the lesion site (Figure S5).
For stage (3), after both 5′ and 3′ partner thymines are flipped out with the 5′ thymine in its binding pocket, the insertion of the BHD3 β-hairpin is achieved by guiding the hairpin to approach the center of the denatured lesion site (Figure S4). From the TS, the energy drops to −0.8 kcal/mol as the BHD3 β-hairpin inserts, providing an energy gain of ∼5 kcal/mol (Figure 4). The ensemble at −0.8 kcal/mol exhibits a stably inserted BHD3 hairpin and converges with an unrestrained MD initiated from our productive binding (PB) model (Figure 3 and Figure S4). Hence, this structural ensemble has achieved the productive binding state. The hairpin insertion is a spontaneous process once the local denaturation at the lesion site has crossed the maximal energy barrier at the TS, where both partner bases are flipped. Along the insertion path of the BHD3 β-hairpin, in correlated motion, the CPD lesion extrudes out of the major groove (Figure S5), the lesion site further unwinds (Figure S5), the 3′ thymine follows the PHE flipping path to insert completely into its binding pocket in BHD3, stacking with Phe556 (Figure 3), and the bend angle of the DNA duplex adjusts toward the value of 47° (Figure S5) as in our productive binding model (Figure 2A).
The complete free energy profile of our preferred correlated motion mechanism is shown in Figure 4. The free energy barrier is 4.2 ± 0.2 kcal/mol, where flipping of partner bases accounts for most of this energetic cost. We also explored other possibilities for the order of events, which showed higher energy barriers and/or led to nonproductive binding states (Figures S6 and S7). Compared with the free energy barrier of our conformational capture mechanism of 9.6 kcal/mol (detailed in Figures S8–S11 of the Supporting Information), the correlated motion mechanism presents a much lower free energy barrier for the Rad4-CPD-containing DNA duplex to achieve the productive binding state. We hypothesize that the energetic relationship between the two mechanisms may depend on the nature of the lesion; e.g., some lesions, such as the benzo[a]pyrene (B[a]P)-derived cis-B[a]P-dG adduct, whose normal partner C is preflipped in a base-displaced intercalated conformation,48 may reveal lower barriers for the conformational capture mechanism.
Our mechanism sheds light on the function of BHD2 in lesion sensing. It has been shown experimentally with human XPC that deletion of the BHD3 domain does not entirely destroy the XPC DNA-damage sensor function;49 this is consistent with the initial stage of our correlated motion mechanism, in which the PM structure shows engagement first of BHD2 with the minor groove of the DNA damage site, which leads to further DNA bending at INT (Figure 3) that facilitates partner base flipping.
Chen et al. have used temperature-jump perturbation spectroscopy to obtain an estimate of the activation enthalpy for forming the open complex when Rad4 is bound to mismatched duplexes, and obtained a value of 7.9 ± 4.7 kcal/mol2 for the most relevant mismatch sequence investigated. To the best of our knowledge, this is the only experimental estimate available to compare with our free energy barrier of 4.2 kcal/mol, although the systems are not the same and our work provides the free energy; nonetheless, it is interesting that our computed barrier is of the same order as this experimental quantity.
Our results have identified a PHE “flipping path” in BHD3 (Figure 3), which can direct destabilized partner bases into their binding pocket for productive binding. The loss of this aromatic pathway would impede the flipping of the partner bases into the protein for productive binding, which is subject to experimental testing.
While the NER mechanism recognizes a wide array of DNA lesions, they are repaired at very different rates, but the origin of this diversity is not understood. It will be interesting to determine if lesions with different local thermodynamic stabilities present different free energy barriers for forming the productive open complex, to help explain this diversity. Furthermore, stabilizing, repair-resistant bulky lesions36,37 might manifest very high barriers that inhibit formation of the productive open complex, which would better explain the origin of their NER resistance on an energetic and molecular level.
Acknowledgments
This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation Grant MCB060037 (to S.B.), and the high-performance computing resources of New York University (NYU-ITS).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00840.
This research was supported by National Institutes of Health Grants R01-CA28038 and R01-CA75449 (to S.B.), R01-GM079223 (to Y.Z.), and 1R01ES024050 (to N.E.G.).
The authors declare no competing financial interest.
Supplementary Material
References
- Min J. H.; Pavletich N. P. (2007) Nature 449, 570–575. 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- Chen X.; Velmurugu Y.; Zheng G.; Park B.; Shim Y.; Kim Y.; Liu L.; Van Houten B.; He C.; Ansari A.; Min J. H. (2015) Nat. Commun. 6, 5849. 10.1038/ncomms6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Clauson C. L.; Robbins P. D.; Niedernhofer L. J.; Wang Y. (2012) Aging Cell 11, 714–716. 10.1111/j.1474-9726.2012.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharer O. D. (2013) Cold Spring Harbor Perspect. Biol. 5, a012609. 10.1101/cshperspect.a012609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedberg E. C., Walker G. C., Siede W., Wood R. D., Schultz R. A., and Ellenberger T. (2005) DNA Repair and Mutagenesis, 2nd ed., American Society for Microbiology Press, Washington, DC. [Google Scholar]
- DiGiovanna J. J.; Kraemer K. H. (2012) J. Invest. Dermatol. 132, 785–796. 10.1038/jid.2011.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K.; Ng J. M.; Masutani C.; Iwai S.; van der Spek P. J.; Eker A. P.; Hanaoka F.; Bootsma D.; Hoeijmakers J. H. (1998) Mol. Cell 2, 223–232. 10.1016/S1097-2765(00)80132-X. [DOI] [PubMed] [Google Scholar]
- Volker M.; Mone M. J.; Karmakar P.; van Hoffen A.; Schul W.; Vermeulen W.; Hoeijmakers J. H.; van Driel R.; van Zeeland A. A.; Mullenders L. H. (2001) Mol. Cell 8, 213–224. 10.1016/S1097-2765(01)00281-7. [DOI] [PubMed] [Google Scholar]
- Riedl T.; Hanaoka F.; Egly J. M. (2003) EMBO J. 22, 5293–5303. 10.1093/emboj/cdg489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi M.; Masutani C.; Maekawa T.; Sugasawa K.; Ohkuma Y.; Hanaoka F. (2000) J. Biol. Chem. 275, 9870–9875. 10.1074/jbc.275.13.9870. [DOI] [PubMed] [Google Scholar]
- Egly J. M.; Coin F. (2011) DNA Repair 10, 714–721. 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- Kuper J.; Braun C.; Elias A.; Michels G.; Sauer F.; Schmitt D. R.; Poterszman A.; Egly J. M.; Kisker C. (2014) PLoS Biol. 12, e1001954. 10.1371/journal.pbio.1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans E.; Moggs J. G.; Hwang J. R.; Egly J. M.; Wood R. D. (1997) EMBO J. 16, 6559–6573. 10.1093/emboj/16.21.6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapias A.; Auriol J.; Forget D.; Enzlin J. H.; Scharer O. D.; Coin F.; Coulombe B.; Egly J. M. (2004) J. Biol. Chem. 279, 19074–19083. 10.1074/jbc.M312611200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugasawa K.; Okamoto T.; Shimizu Y.; Masutani C.; Iwai S.; Hanaoka F. (2001) Genes Dev. 15, 507–521. 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scrima A.; Konickova R.; Czyzewski B. K.; Kawasaki Y.; Jeffrey P. D.; Groisman R.; Nakatani Y.; Iwai S.; Pavletich N. P.; Thoma N. H. (2008) Cell 135, 1213–1223. 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D.; Tursun M.; Duckett D. R.; Drummond J. T.; Modrich P.; Sancar A. (1997) Mol. Cell. Biol. 17, 760–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. (2000) Nucleic Acids Res. 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schärer O. D. (2007) Mol. Cell 28, 184–186. 10.1016/j.molcel.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Geacintov N. E.; Broyde S.; Buterin T.; Naegeli H.; Wu M.; Yan S.; Patel D. J. (2002) Biopolymers 65, 202–210. 10.1002/bip.10239. [DOI] [PubMed] [Google Scholar]
- Gunz D.; Hess M. T.; Naegeli H. (1996) J. Biol. Chem. 271, 25089–25098. 10.1074/jbc.271.41.25089. [DOI] [PubMed] [Google Scholar]
- Evans E.; Fellows J.; Coffer A.; Wood R. D. (1997) EMBO J. 16, 625–638. 10.1093/emboj/16.3.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess M. T.; Schwitter U.; Petretta M.; Giese B.; Naegeli H. (1997) Proc. Natl. Acad. Sci. U. S. A. 94, 6664–6669. 10.1073/pnas.94.13.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood R. D. (1999) Biochimie 81, 39–44. 10.1016/S0300-9084(99)80036-4. [DOI] [PubMed] [Google Scholar]
- Sugasawa K.; Shimizu Y.; Iwai S.; Hanaoka F. (2002) DNA Repair 1, 95–107. 10.1016/S1568-7864(01)00008-8. [DOI] [PubMed] [Google Scholar]
- Dip R.; Camenisch U.; Naegeli H. (2004) DNA Repair 3, 1409–1423. 10.1016/j.dnarep.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Buterin T.; Meyer C.; Giese B.; Naegeli H. (2005) Chem. Biol. 12, 913–922. 10.1016/j.chembiol.2005.06.011. [DOI] [PubMed] [Google Scholar]
- Gillet L. C.; Schärer O. D. (2006) Chem. Rev. 106, 253–276. 10.1021/cr040483f. [DOI] [PubMed] [Google Scholar]
- Maillard O.; Camenisch U.; Blagoev K. B.; Naegeli H. (2008) Mutat. Res., Rev. Mutat. Res. 658, 271–286. 10.1016/j.mrrev.2008.01.007. [DOI] [PubMed] [Google Scholar]
- Maillard O.; Camenisch U.; Clement F. C.; Blagoev K. B.; Naegeli H. (2007) Trends Biochem. Sci. 32, 494–499. 10.1016/j.tibs.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y.; Masutani C.; Mizukoshi T.; Kondo J.; Hanaoka F.; Iwai S. (1999) J. Biol. Chem. 274, 20027–20033. 10.1074/jbc.274.28.20027. [DOI] [PubMed] [Google Scholar]
- Sugasawa K.; Akagi J.; Nishi R.; Iwai S.; Hanaoka F. (2009) Mol. Cell 36, 642–653. 10.1016/j.molcel.2009.09.035. [DOI] [PubMed] [Google Scholar]
- Gold B.; Stone M. P.; Marky L. A. (2014) Acc. Chem. Res. 47, 1446–1454. 10.1021/ar500061p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.; Zhang K.; Ren Y.; Nadji S.; Sinha N.; Taylor J. S.; Kang C. (2002) Proc. Natl. Acad. Sci. U. S. A. 99, 15965–15970. 10.1073/pnas.242422699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Zheng H.; Ding S.; Kropachev K.; Schwaid A. G.; Tang Y.; Mu H.; Wang S.; Geacintov N. E.; Zhang Y.; Broyde S. (2013) Chem. Res. Toxicol. 26, 1115–1125. 10.1021/tx400156a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y.; Geacintov N. E.; Broyde S. (2012) Biochemistry 51, 1486–1499. 10.1021/bi201794x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Reeves D.; Kropachev K.; Cai Y.; Ding S.; Kolbanovskiy M.; Kolbanovskiy A.; Bolton J. L.; Broyde S.; Van Houten B.; Geacintov N. E. (2011) DNA Repair 10, 684–696. 10.1016/j.dnarep.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C. D.; Simons M.; Mackenzie C. E.; Van Houten B.; Kad N. M. (2014) DNA Repair 20, 2–13. 10.1016/j.dnarep.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; Nussinov R.; Wright P. E. (2009) Nat. Chem. Biol. 5, 789–796. 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinson E. H.; Gowda A. S.; Spratt T. E.; Gold B.; Eichman B. F. (2010) Nature 468, 406–411. 10.1038/nature09428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B. (1995) Comput. Phys. Commun. 91, 275–282. 10.1016/0010-4655(95)00053-I. [DOI] [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E. III, Simmerling C. L., Wang J., Duke R. E., Luo R., Walker R. C., Zhang W., Merz K. M., Roberts B., Hayik S., Roitberg A., Seabra G., Swails J., Götz A. W., Kolossváry I., Wong K. F., Paesani F., Vanicek J., Wolf R. M., Liu J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Cai Q., Ye X., Wang J., Hsieh M.-J., Cui G., Roe D. R., Mathews D. H., Seetin M. G., Salomon-Ferrer R., Sagui C., Babin V., Luchko T., Gusarov S., Kovalenko A., and Kollman P. A. (2012) AMBER12, University of California, San Francisco, San Francisco. [Google Scholar]
- Case D. A., Babin V., Berryman J. T., Betz R. M., Cai Q., Cerutti D. S., Cheatham T. E. III, Darden T. A., Duke R. E., Gohlke H., Goetz A. W., Gusarov S., Homeyer N., Janowski P., Kaus J., Kolossváry I., Kovalenko A., Lee T. S., LeGrand S., Luchko T., Luo R., Madej B., Merz K. M., Paesani F., Roe D. R., Roitberg A., Sagui C., Salomon-Ferrer R., Seabra G., Simmerling C. L., Smith W., Swails J., Walker R. C., Wang J., Wolf R. M., Wu X., and Kollman P. A. (2014) AMBER14, University of California, San Francisco, San Francisco. [Google Scholar]
- Lee T. S.; Radak B. K.; Huang M.; Wong K. Y.; York D. M. (2014) J. Chem. Theory Comput. 10, 24–34. 10.1021/ct400691f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuznetsov N. A.; Bergonzo C.; Campbell A. J.; Li H.; Mechetin G. V.; de los Santos C.; Grollman A. P.; Fedorova O. S.; Zharkov D. O.; Simmerling C. (2015) Nucleic Acids Res. 43, 272–281. 10.1093/nar/gku1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song K.; Campbell A. J.; Bergonzo C.; de los Santos C.; Grollman A. P.; Simmerling C. (2009) J. Chem. Theory Comput. 5, 3105–3113. 10.1021/ct9001575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X. J.; Olson W. K. (2008) Nat. Protoc. 3, 1213–1227. 10.1038/nprot.2008.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosman M.; de los Santos C.; Fiala R.; Hingerty B. E.; Ibanez V.; Luna E.; Harvey R.; Geacintov N. E.; Broyde S.; Patel D. J. (1993) Biochemistry 32, 4145–4155. 10.1021/bi00067a001. [DOI] [PubMed] [Google Scholar]
- Camenisch U.; Trautlein D.; Clement F. C.; Fei J.; Leitenstorfer A.; Ferrando-May E.; Naegeli H. (2009) EMBO J. 28, 2387–2399. 10.1038/emboj.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.