

Abstract
Arylalkylamine N-acetyltransferase like 7 (AANATL7) catalyzes the formation of N-acetylarylalkylamides and N-acetylhistamine from acetyl-CoA and the corresponding amine substrate. AANATL7 is a member of the GNAT superfamily of >10000 GCN5-related N-acetyltransferases, many members being linked to important roles in both human metabolism and disease. Drosophila melanogaster utilizes the N-acetylation of biogenic amines for the inactivation of neurotransmitters, the biosynthesis of melatonin, and the sclerotization of the cuticle. We have expressed and purified D. melanogaster AANATL7 in Escherichia coli and used the purified enzyme to define the substrate specificity for acyl-CoA and amine substrates. Information about the substrate specificity provides insight into the potential contribution made by AANATL7 to fatty acid amide biosynthesis because D. melanogaster has emerged as an important model system contributing to our understanding of fatty acid amide metabolism. Characterization of the kinetic mechanism of AANATL7 identified an ordered sequential mechanism, with acetyl-CoA binding first followed by histamine to generate an AANATL7·acetyl-CoA·histamine ternary complex prior to catalysis. Successive pH–activity profiling and site-directed mutagenesis experiments identified two ionizable groups: one with a pKa of 7.1 that is assigned to Glu-26 as a general base and a second pKa of 9.5 that is assigned to the protonation of the thiolate of the coenzyme A product. Using the data generated herein, we propose a chemical mechanism for AANATL7 and define functions for other important amino acid residues involved in substrate binding and regulation of catalysis.
Graphical abstract
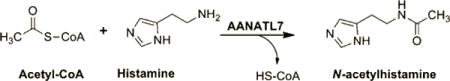
Fatty acid amides, R-CO-NH-R′, represent a diverse family of bioactive lipids produced in both vertebrates and invertebrates.1–4 We have proposed that one route for the biosynthesis of the fatty acid amides involves the reaction of an amine with a fatty acyl-CoA in a reaction catalyzed by an N-acyltransferase: R-CO-S-CoA + H2N-R′ → R-CO-NH-R′ + CoA-SH. Drosophila melanogaster is an excellent model system for the study of fatty acid amide metabolism because these insects (a) produce fatty acid amides,2–4 (b) are inexpensive to grow and maintain, (c) possess a genome that has been sequenced, and (d) can be manipulated genetically with relative ease. Intriguingly, we and others have identified the genes of seven putative N-acyltransferases in D. melanogaster by “genome mining” using the DNA sequences of known N-acetyltransferases.5,6 These seven putative N-acyltransferase enzymes seem to be related to two arylalkylamine N-acetyltransferases (AANATs) of D. melanogaster, AANAT variant A (AANATA) and variant B (AANATB).5–7 As a consequence of their resemblance to AANATA and AANATB, the putative D. melanogaster N-acyltransferases are called arylalkylamine N-acetyltransferase like enzymes, AANATL2–8. Our goal is to clone and overexpress each of these AANATLs, AANATA, and AANATB in Escherichia coli, characterize the recombinant enzymes in vitro, and, ultimately, determine if any of these enzymes do participate in fatty acid amide metabolism in the fly.
Herein, we describe the expression and overproduction of D. melanogaster AANATL7 in E. coli and the subsequent characterization of the recombinant enzyme in vitro. In contrast to AANATL2,4 AANATL7 will not accept long-chain acyl-CoAs as substrates. The specificity of AANATL7 for its acyl-CoA and amine does show some overlap with that of AANATA,6 but there are some differences in the substrate specificities between the two enzymes. While the in vitro substrate specificity data for AANATL7, AANATA, and AANATB suggest that these enzymes are not involved in long-chain fatty acid amide metabolism, Saghatelian et al.8 caution that in vitro specificity data may not accurately predict which substrates are processed by that enzyme in vivo.
Relative to AANATA, AANATL7 exhibits a more narrow preference for the acyl-CoA substrates, but a broader preference for the amine substrates.6 In particular, non-arylalkylamines are not substrates for AANATA and AANATB but are substrates for AANATL7. Histamine, generated by the decarboxylation of histidine, is acetylated by AANATL7.9–11 In the fly, histamine is a neurotransmitter and is found predominately in the nervous system,12–15 with the highest levels being identified in the eye.9,16 The N-acetylation of histamine is likely involved in its inactivation as a neurotransmitter, and our identification and characterization of AANATL7 is the first report of an enzyme in D. melanogaster that catalyzes histamine acetylation.
AANATL7 is a member of the GCN5-related N-acetyltransferase (GNAT) family of enzymes.17,18 Herein, we report work to define the kinetic and chemical mechanism of AANATL7 coupled to targeted site-directed mutagenesis experiments to define active site amino acids involved in catalysis. Substrate binding is ordered, with acetyl-CoA binding first to form the AANATL7·acetyl-CoA complex, followed by amine binding to generate the AANATL7·acetyl-CoA·amine ternary complex. Catalysis occurs after ternary complex formation to generate the N-acetylamide and CoA-SH. Our data point to Glu-26 serving as a general base in catalysis and show that Pro-27 and Arg-138 are also important in the optimal architecture of the active site for substrate binding and efficient catalysis. Lastly, we discuss the structure–function relationships identified for AANATL7 with respect to the different substrates and amino acid residues with respect to structural information available for related N-acyltransferases.
EXPERIMENTAL PROCEDURES
Materials
ProBond nickel-chelating resin was purchased from Invitrogen. Polymerase chain reaction (PCR) primers were purchased from Eurofins MWG Operon. PfuUltra High-Fidelity DNA polymerase was purchased from Agilent. BL21(DE3) E. coli cells, E. coli XL10 competent cells, and the pET-28a(+) vector were purchased from Novagen. NdeI, XhoI, Antarctic Phosphatase, and T4 DNA ligase were purchased from New England Biolabs. Kanamycin monosulfate and isopropyl β-D-1-thiogalactopyranoside (IPTG) were purchased from Gold Biotechnology. Acetyl-CoA, butyryl-CoA, hexanoyl-CoA, octanoyl-CoA, oleoyl-CoA, coenzyme A, and Nω-acetylhistamine were purchased from Sigma-Aldrich. All other reagents were of the highest quality available from either Sigma-Aldrich or Fisher Scientific.
AANATL7 Transcript Localization
We used the D. melanogaster cDNA library generated for the head and thorax-abdomen that was discussed by Dempsey et al.4 Amplification of AANATL7 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) transcripts was achieved by PCR under the following conditions: initial denaturing step of 95 °C for 2 min, and then 45 cycles of 95 °C for 30 s; varying annealing temperatures (45, 50, 55, and 60 °C) for 30 s; 72 °C for 1 min; then a final extension step of 72 °C for 10 min. The primers used to amplify a 268 bp (AANATL7) and 449 bp (GAPDH) product [AANATL7, TTTCCGCAAGATTTTCGACT (forward) and TGTAGGGTTGCTCCGAGAAG (reverse); GAPDH, ATCGTCGAGGGTCTGATGAC (forward) and ACCGAACTCGTTGTCGTACC (reverse)] were synthesized by Eurofins MWG Operon. The amplified products were analyzed by a 1.3% agarose gel, and the band was visualized with 0.5 μg/mL ethidium bromide under ultraviolet light. Each positive band was cut out of the agarose gel, purified by the Promega Wizard SV Gel and PCR Clean-up system, and sequenced by Eurofins MWG Operon.
Cloning of D. melanogaster AANATL7
The following primers were used to amplify the AANATL7 (NCBI reference sequence NM_130653.3) gene from the D. melanogaster head cDNA library: 5′ TCAGCATATGATGGAGTACAAGATGATTGCACCC 3′ (forward) and 5′ TCAGCTCGAGTTAAAGCGACTGCTTCTTCTCAT 3′ (reverse). PfuUltra High-Fidelity DNA polymerase was used with the following PCR conditions: initial denaturing step of 95 °C for 2 min, then 30 cycles (95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min), and then a final extension step of 72 °C for 10 min. The AANATL7 PCR product was then inserted into the NdeI and XhoI restriction sites of pET-28a(+), yielding expression vector AANATL7-pET-28a. The AANATL7-pET-28a vector was then transformed into E. coli XL10 competent cells and spread on a Luria Broth (LB) agar plate supplemented with 40 μg/mL kanamycin and incubated overnight at 37 °C. A single colony was then expressed overnight in LB medium supplemented with 40 μg/mL kanamycin at 37 °C, and the plasmid was purified from the final culture using the Promega Wizard Plus SV Minipreps DNA purification kit. The resulting pure plasmid was sequenced by Eurofins MWG Operon and then transformed into E. coli BL21(DE3) cells for the expression of AANATL7.
Expression and Purification of AANATL7
The E. coli BL21(DE3) cells containing the AANATL7 pET-28a vector were cultured in LB medium supplemented with 40 μg/mL kanamycin at 37 °C. IPTG was used at a final concentration of 1.0 mM to induce the cell culture at an OD600 of 0.6, and the resulting culture was continued for 4 h at 37 °C before being harvested. The final culture was then centrifuged at 5000g for 10 min at 4 °C, and the pellet was collected.
The pellet was then resuspended in the nickel affinity binding buffer [20 mM Tris (pH 7.9), 500 mM NaCl, and 5 mM imidazole], lysed by sonication, and centrifuged at 10000g for 15 min at 4 °C, and the resulting supernatant was collected for subsequent purification steps. The supernatant was then loaded onto a ProBond nickel-chelating resin containing 6 mL of resin. After AANATL7 had been loaded, then the column was washed with 10 column volumes of binding buffer [20 mM Tris-HCl (pH 7.9), 500 mM NaCl, and 5 mM imidazole], followed by a second washing step that utilized 10 column volumes of 20 mM Tris-HCl (pH 7.9), 500 mM NaCl, and 60 mM imidazole. The pure AANATL7 protein was finally eluted in 1 mL fractions of 20 mM Tris-HCl (pH 7.9), 500 mM NaCl, and 500 mM imidazole. Following the purification procedure, the elution buffer containing AANATL7 was exchanged for 50 mM HEPES (pH 8.2), 500 mM NaCl, and 1 mM EDTA, by dialysis for 16 h at 4 °C. The concentration of the final AANATL7 was determined using the Bradford dye binding assay, and the purity was assessed by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) that was visualized using Coomassie stain.
Production of Site-Directed Mutants
The site-directed AANATL7 mutants were generated using the overlap extension method.19 The AANATL7 mutants were amplified using PfuUltra High-Fidelity DNA polymerase with the following set of PCR conditions: initial denaturing step of 95 °C for 2 min, then 30 cycles (95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min), and then a final extension step of 72 °C for 10 min. Primers were designed by using Agilent QuickChange Primer Design tool (Table S1 of the Supporting Information) and synthesized by Eurofins MWG Operon. Using NdeI and XhoI restriction enzymes, the resulting AANATL7 mutant PCR products were then inserted into a pet28a(+) vector. The AANATL7 mutant proteins were expressed and purified using the same protocol that was used for the wild-type enzyme.
Measurement of the Enzyme Assay
Ellman’s reagent20 was used to assay the AANATL7 activity by measuring the release of coenzyme A at 412 nm with a molar extinction coefficient of 13600 M−1 cm−1 in a buffered solution containing 300 mM Tris (pH 8.0), 150 μM DTNB, and the desired concentrations of substrates and/or inhibitors. Initial velocities were measured by using a Cary 300 Bio UV–visible spectrophotometer at 22 °C. The apparent kinetic constants were generated for each amine substrate by holding acetyl-CoA at a fixed saturating concentration. Apparent kinetic constants for each acyl-CoA substrate were determined by holding histamine at a fixed saturating concentration. The resulting data were fit to eq 1 using Sigma Plot 12.0 to generate the steady-state kinetic constants
| (1) |
where vo is the initial velocity, Vmax,app is the apparent maximal velocity, Km,app is the apparent Michaelis constant, and [S] is the substrate concentration. Each assay was performed in triplicate. Uncertainty calculations for kcat,app, (kcat/Km)app, and relative (kcat/Km)app were calculated using eq 2
| (2) |
where σ is the standard error.21
Kinetic Mechanism and Inhibitor Analysis
Initial velocity patterns for acetyl-CoA and histamine were assessed by varying the concentration of one substrate while holding the concentration of the other substrate at a fixed concentration. The resulting data were then fit to eq 3 for an ordered bi-bi mechanism and plotted as a double-reciprocal plot using IGOR Pro 6.34A. The two plots were generated by holding the histamine concentration constant (100 μM, 200 μM, 500 μM, 1.0 mM, and 3.0 mM) and varying the concentration of acetyl-CoA. The second plot held the concentration of acetyl-CoA constant (5 μM, 10 μM, 25 μM, 50 μM, and 150 μM) and varied the concentration of histamine.
| (3) |
In combination with the initial velocity analysis, we used dead-end inhibition to solve the kinetic mechanism. Oleoyl-CoA and tyrosol were evaluated as dead-end inhibitors for AANATL7. The initial velocities were generated by holding one substrate (acetyl-CoA or histamine) at a fixed concentration and varying the concentration of the other substrate, with each data set evaluated at different fixed concentrations of the inhibitor. Using SigmaPlot 12.0, the resulting data were fit to eqs 4–6 for competitive, noncompetitive, and uncompetitive inhibition, respectively
| (4) |
| (5) |
| (6) |
where vo is the initial velocity, Vmax,app is the apparent maximal velocity, Km,app is the apparent Michaelis constant, [S] is the substrate concentration, [I] is the inhibitor concentration, and Ki is the inhibition constant. Each assay was performed in triplicate.
Rate versus pH Dependence
The pH dependence on the apparent kinetic constants was determined for both acetyl-CoA and histamine. The kinetic constants for each substrate were evaluated at intervals of 0.5 pH unit from pH 6.0 to 9.5. The following buffers were used for the pH dependence experiments: MES (pH 6.0–7.0), Tris (pH 7.0–9.0), and AMeP (pH 9.0–9.5). Using Igor Pro 6.34A, the resulting data were fit to eq 7 (log kcat/Km-acetyl-CoA and log kcat/Km-histamine) and eq 8 (log kcat), where c is the pH-independent plateau, to determine the apparent pKa of measured ionizable groups. The wild-type enzyme is reported from triplicate experiments, whereas the E26A mutant was performed in duplicate.
| (7) |
| (8) |
Intrinsic Fluorescence Measurements for the Determination of the Coenzyme A Dissociation Constant
A JASCO FP-8300 spectrofluorometer was used to generate fluorescence spectra for the AANATL7 wild-type and R138A mutant enzymes. Fluorescence emission spectra (excitation at 280 nm, emission at 290–300 nm) were generated for both wild-type AANATL7 and the R138A mutant in a 0.4 cm path length cell containing 400 μL of 300 mM Tris-HCl (pH 8.0), a fixed protein concentration of 0.1 mg/mL, and varying concentrations of coenzyme A at 22 °C. Emission spectra were collected with a 50 nm/min scan speed, an excitation bandwidth of − 5 nm, and an emission bandwidth of − 2.5 nm in triplicate. The coenzyme A dissociation constant for wild-type AANATL7 and the R138A mutant were determined by fitting the data to eq 9 using SigmaPlot 12.0
| (9) |
where ΔFmax is the maximal change in fluorescence at infinite ligand concentration, ΔF is the change in intrinsic fluorescence, [L] is the ligand concentration, and Kd is the ligand dissociation constant.
Product Characterization
A Phenomenex Kinetex 2.6 μm C18 100 Å (50 mm × 2.1 mm) reverse phase column coupled to an Agilent 6540 instrument using a liquid chromatography/quadrupole time-of-flight mass spectrometer (LC/QTOF-MS) in positive ion mode was used to characterize the AANATL7-catalyzed product from the substrates acetyl-CoA and histamine. An enzyme reaction mixture containing 300 mM Tris-HCl (pH 8.0), 500 μM acetyl-CoA, 50 mM histamine, and 9.3 μg of AANATL7 in a final volume of 750 μL was incubated for 30 min at 22 °C. Following the 30 min incubation period, AANATL7 was removed from the reaction mixture using a Millipore 10 kDa ultrafilter in which the reaction product flowed through the filter while AANATL7 was retained in the hold-up volume on the filter. The final reaction product was then injected onto the LC/QTOF-MS instrument to analyze the retention time and high-resolution mass. The resulting retention time and high-resolution mass for the reaction product were compared to those of a commercial standard of Nω-acetylhistamine. The experimental conditions for LC/QTOF-MS analysis are reported in ref 4.
RESULTS AND DISCUSSION
Overexpression, Purification, and Transcript Localization of AANATL7
The gene for AANATL7 was amplified from a cDNA library generated from D. melanogaster heads and inserted into a pet28a(+) vector that encodes an N-terminal His6 tag. AANATL7 was purified using ProBond nickel-chelating resin to homogeneity yielding a 27 mg/L culture. The purity was assayed to be ≥95% pure by 10% SDS–PAGE (Figure S1 of the Supporting Information).
The localization of the D. melanogaster AANATL7 transcript was evaluated using reverse transcription polymerase chain reaction (RT-PCR). The agarose gel for the amplification of the AANATL7 transcript (Figure 1) showed a similar trend in both the fly head and thorax-abdomen compared to that of the GAPDH control. These data suggest that the AANATL7 transcript is found at similar levels in both anatomical regions, indicating this enzyme will catalyze the acyl-CoA-dependent N-acylation of biogenic amines in both the head and thorax-abdomen of the fly. Further evaluation of the respective substrates and final products of the AANATL7-catalyzed reaction will be discussed in subsequent sections.
Figure 1.
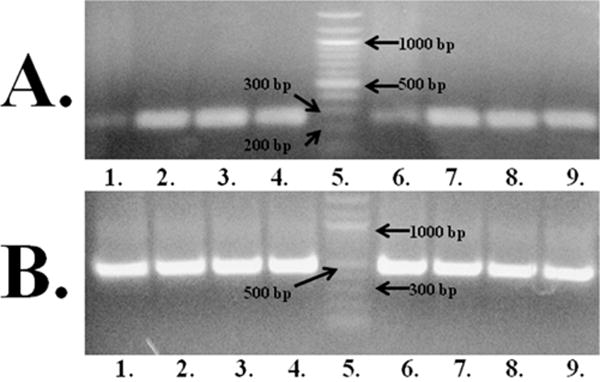
AANATL7 RT-PCR. (A) AANATL7, 268 bp product: lanes 1–4, RT-PCR product from the D. melanogaster cDNA library generated from the heads with different PCR annealing temperatures of 45, 50, 55, and 60 °C, respectively; lane 5, New England Biolabs Quick-Load 100 bp DNA Ladder; lanes 6–9, RT-PCR product from the D. melanogaster cDNA library generated from the thorax-abdomen with different PCR annealing temperatures of 45, 50, 55, and 60 °C, respectively. (B) GAPDH control (NM_001038847.1), 449 bp product: lanes 1–4, RT-PCR product from the D. melanogaster cDNA library generated from the heads with different PCR annealing temperatures of 45, 50, 55, and 60 °C, respectively; lane 5, New England Biolabs Quick-Load 100 bp DNA Ladder; lanes 6–9, RT-PCR product from the D. melanogaster cDNA library generated from the thorax-abdomen with different PCR annealing temperatures of 45, 50, 55, and 60 °C, respectively.
Steady-State Kinetic Characterization of AANATL7 Acyl-CoA Substrates
Activity was assayed using Ellman’s reagent20 by monitoring the rate of coenzyme A release at 412 nm. Different acyl-CoA substrates with increasing acyl chain lengths were evaluated with AANATL7 to determine their respective substrate specificities at saturating histamine concentrations. The relative (kcat/Km)app for the acyl-CoA substrates decreased as the acyl chain length was increased (Table 1). Butyryl-CoA and hexanoyl-CoA were found to have only 10% and 0.082% of the (kcat/Km)app of acetyl-CoA, respectively. The significant decrease in the second-order rate constant resulted mainly from the kcat,app value, while the Km,app value was similar for each acyl-CoA substrate. Octanoyl-CoA and oleoyl-CoA were also evaluated as substrates with histamine as the amine substrate, and activity was not observed at a substrate of 500 μM each. The acyl-CoA substrate specificity of AANATL7 differed from that of AANATA: the (kcat/Km)app values for butyryl-CoA, hexanoyl-CoA, and octanoyl-CoA as AANATA substrates did not decrease as dramatically relative to that of acetyl-CoA as we measured for these same substrates for AANATL7 (Table 1). The differences observed between respective acyl-CoA specificities for AANATA and AANATL7 could result from either the amine substrate used in the evaluation of the acyl-CoA substrates or a structural difference in the AANATL7 active site such that the AANATL7 is less accommodating with respect to the longer-chain (>C6) acyl-CoA substrates. To assess if the significant decrease in (kcat/Km)app-acetyl-CoA/(kcat/Km)app-acyl-CoA observed for AANATL7 is derived from the amine substrate, we reevaluated the kinetic constants for acetyl-CoA and butyryl-CoA at saturating tyramine concentrations. A similar trend was observed for the acyl-CoA kinetic constants when tyramine was the saturating amine substrate: the (kcat/Km)app values for acetyl-CoA and butyryl-CoA are (1.1 ± 0.1) × 106 M−1 s−1 and (2.9 ± 0.2) × 105 M−1 s−1, respectively (kcat,app-acetyl-CoA = 21 ± 1 s−1, and Km,app-acetyl-CoA = 20 ± 1 μM; kcat,app-butyryl-CoA = 1.87 ± 0.04 s−1, and Km,app-butyryl-CoA = 7 ± 1 μM). A structural rationale for these data is difficult without an AANATL7 crystal structure. Most likely, the longer-chain acyl-CoA substrate extends into the amine binding pocket within the AANATL7 active site to either block amine substrate binding altogether or perturb amine binding to prevent effective nucleophilic attack by the amine at the carbonyl of the acetyl-CoA thioester moiety. We found that the Km,app-histamine did increase as the acyl chain length increased for the acyl-CoA substrates (Table 2), consistent with our hypothesis that the longer acyl chains either block or perturb amine substrate binding.
Table 1.
Steady-State Kinetic Constants for Different Acyl-CoA Substratesa
| substrate | Km,app (μM) | kcat,app (s−1) | (kcat/Km)app (M−1 s−1) | relative (kcat/Km)appd |
|---|---|---|---|---|
| acetyl-CoAb | 29 ± 2 | 32 ± 1 | (1.1 ± 0.1) × 106 | 470 ± 750 |
| butyryl-CoAb | 6 ± 1 | 0.66 ± 0.02 | (1.1 ± 0.2) × 105 | 49 ± 13 |
| hexanoyl-CoAc | 40 ± 5 | 0.093 ± 0.004 | (2.3 ± 0.3) × 102 | 1.0 ± 0.2 |
Kinetic constants are reported as ± the standard error (n = 3).
Reaction conditions: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, 7.5 mM histamine, and varying concentrations of acyl-CoA.
Reaction conditions: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, 50 mM histamine, and varying concentrations of hexanoyl-CoA.
The relative (kcat/Km)app-acyl-CoA is indexed relative to hexanoyl-CoA.
Table 2.
Steady-State Kinetic Constants for Histamine Using Different Acyl-CoA Substratesa
| varied amine substrate | saturated acyl-CoA substrate | Km,app (mM) | kcat,app (s−1) | (kcat/Km)app (M−1 s−1) |
|---|---|---|---|---|
| histamine | acetyl-CoA | 0.52 ± 0.05 | 30 ± 1 | (5.8 ± 0.6) × 104 |
| butyryl-CoA | 2.9 ± 0.3 | 0.87 ± 0.03 | (3.0 ± 0.4) × 102 | |
| hexanoyl-CoA | 15 ± 2 | 0.10 ± 0.01 | 7 ± 1 |
Reaction conditions: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, 500 μM acyl-CoA, and varying concentrations of histamine. Kinetic constants are reported as ± the standard error (n = 3).
Steady-State Kinetic Characterization of AANATL7 Amine Substrates
Different amine substrates were characterized to determine the substrate specificity for AANATL7. An array of arylalkylamines, polyamines, histamine, and ethanolamine were evaluated as substrates at saturating concentrations of acetyl-CoA. The steady-state kinetic constants for the amines that are AANATL7 substrates are listed in Table 3. This set of substrates provides an opportunity to define how the structural features of the amines affect binding and/or catalysis for AANATL7. These structural parameters of the amine substrates were divided into five general classes: (a) indole ring and phenyl ring modifications, (b) length of the spacer group between the phenyl ring and the nucleophilic primary amine, (c) modification of the α-position of the ethylamine spacer group, (d) modification of the β-position of the ethylamine spacer group, and (e) evaluation of other non-arylalkylamine substrates.
Table 3.
Steady-State Kinetic Constants for the Different Amine Substratesa
| Substrate | Structure | Km, app | kcat,app | (kcat/Km)app |
|---|---|---|---|---|
| μM | s−1 | M−1s−1 | ||
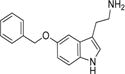
| 5-Benzyloxytryptamine |
|
9 ± 1 | 5.7 ± 0.1 | (6.3 ± 0.4) × 105 |
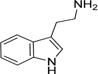
| Tryptamine |
|
26 ± 2 | 16 ± 0.3 | (6.0 ± 0.4) × 105 |
| Tyramine |
|
42 ± 2 | 238 ± 0.4 | (5.5 ± 0.3) × 105 |
| 3-Methoxyphenethylamine |
|
61 ± 4 | 26 ± 1 | (4.3 ± 0.3) × 105 |
| 5-Methoxytryptamine |
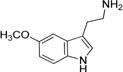
|
42 ± 2 | 13.1 ± 0.2 | (3.1 ± 0.2) × 105 |
| 3-(Trifluoromethyl)phenethylamine |
|
180 ± 10 | 40 ± 1 | (2.2 ± 0.1) × 105 |
| Phenethylamine |
|
320 ± 40 | 42 ± 2 | (1.3 ± 0.2) × 105 |
| Octopamine |
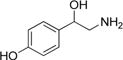
|
120 ± 10 | 16 ± 0.3 | (1.3 ± 0.1) × 105 |
| Dopamine |
|
170 ± 20 | 21 ± 1 | (1.2 ± 0.1) × 105 |
| β-Methylphenethylamine |
|
260 ± 30 | 29 ± 1 | (1.1 ± 0.1) × 105 |
| Serotonin |
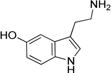
|
160 ± 20 | 17 ± 1 | (1.1 ± 0.1) × 105 |
| 4-Methoxyphenethylamine |
|
190 ± 20 | 15 ± 0.4 | (8.0 ± 0.7) × 104 |
| 3,4-Methylenedioxyphenethylamine |
|
320 ± 30 | 23 ± 1 | (7.4 ± 0.7) × 104 |
| Histamine |
|
520 ± 50 | 30 ± 1 | (5.8 ± 0.6) × 104 |
| Norepinephrine |
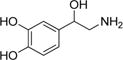
|
230 ± 50 | 13 ± 1 | (6 ± 1) × 104 |
| 4-Phenylbutylamine |
|
610 ± 30 | 8.5 ± 0.2 | (1.4 ± 0.1) × 104 |
| Putrescine |
|
81 ± 11 | 0.25 ± 0.01 | (3.0 ± 0.4) × 103 |
| Benzylamine |
|
890 ± 80 | 0.87 ± 0.02 | 980 ± 100 |
| Agmatine |
|
790 ± 100 | 0.68 ± 0.03 | 850 ± 110 |
| Ethanolamine |
|
(1.00 ± 0.01) × 105 | 2.3 ± 0.1 | 23 ± 1.0 |
Reaction condition −300 mM Tris-HCl pH 8.0, 150 μM DTNB, 500 μM acetyl-CoA, and varying concentration of the amine substrate. Kinetic constants are reported as ± standard error (n = 3).
Arylalkylamine substrates with different functional groups (-H, -OH, -OMe, and -OCH2C6H5) at position 5 of the indole ring yielded (kcat/Km)app values that decreased in the following order: 5-benzyloxytryptamine (-OCH2C6H5) > tryptamine (-H) > 5-methoxytryptamine (-OMe) > serotonin (5-hydroxytryptamine, -OH) (Table 3). The (kcat/Km)app order predominantly resulted from the Km,app value, suggesting that amine substrate binding is important for its specificity. Serotonin is the most hydrophilic modification we evaluated, and this substrate yielded the highest Km,app value (160 ± 20 μM). 5-Benzyloxytryptamine is the most hydrophobic modification we evaluated, and this substrate yielded the lowest Km,app value (9 ± 1 μM). These data suggest that one parameter important in amine substrate binding affinity is the hydrophobicity of the group at position 5 of the indole ring; greater hydrophobicity at this position leads to higher AANATL7 affinity (a lower Km,app value). There was not a significant difference in the observed kcat,app values within our panel of indole-containing amine substrates, except for 5-benzyloxy-tryptamine, which exhibited a 2.3–3.0-fold lower turnover number compared to those of tryptamine, serotonin, and 5-methoxytryptamine.
The biosynthesis of melatonin (N-acetyl-5-methoxytryptamine) from L-tryptophan is reported to occur in four sequential reactions: hydroxylation of L-tryptophan to generate 5-hydroxytryptophan, decarboxylation to serotonin, N-acetylation to N-acetylserotonin, and, finally, methylation to generate melatonin.22,23 Our amine specificity data hint at an alternative pathway for the in vivo production of melatonin by the direct acetylation of 5-methoxytryptamine. We found that the (kcat/Km)app value for 5-methoxytryptamine is 2.8-fold higher than the value for serotonin, consistent with what was found for D. melanogaster AANATA,6 yeast AANAT,24 human AANAT,24 ovine AANAT,24 and other vertebrate and nonvertebrate AANAT enzymes.25 These data suggest that AANATL7 could catalyze the direct formation of melatonin in D. melanogaster and other organisms; however, further work is necessary to evaluate if this chemistry is biologically relevant.
Next, we evaluated a set of phenethylamine substrates that are analogues of phenylalanine or tyrosine. These data showed that tyramine has the highest (kcat/Km)app value and norepinephrine the lowest (kcat/Km)app value for this family of amine substrates with a relatively narrow 9.2-fold range in the (kcat/Km)app values. Overall, we found relatively small differences in Km,app, kcat,app, and (kcat/Km)app for the phenethylamine-based substrates, indicating the AANATL7 active site is relatively tolerant of decorations to the phenyl ring and to the β-position of the -CH2CH2- spacer between the phenyl ring and the nucleophilic amine (Table 3). While the differences within this data set are small, we note that the greatest difference were observed in the Km,app values, the greatest difference being the change from 42 ± 2 μM to 320 ± 40 μM (tyramine to phenethylamine). One trend observed for the Km,app values was an increase for the substrates without a modified phenyl ring, except for 3,4-methylenedioxyphenethylamine.
Another structural feature that has an effect on enzyme activity is the length of the spacer group between the amino group and the phenyl ring. Increasing the spacer length from two methylene groups to four methylene groups (4-phenyl-butylamine) yields a 9-fold decrease in the (kcat/Km)app value when compared to that of phenethylamine. The decrease in (kcat/Km)app resulted from an 2-fold increase in the Km,app and a 5-fold decrease in the kcat,app. Decreasing the spacer length by one methylene group, as found in benzylamine, resulted in a 133-fold decrease in the (kcat/Km)app value relative to that of phenethylamine. The decrease in (kcat/Km)app resulted from a 3-fold increase in Km,app and a 48-fold decrease in kcat,app. These data suggest that the positioning of the primary amine in an optimal region for nucleophilic attack is critical; therefore, changes to the length will impact substrate binding and enzyme catalysis. Our data indicate the optimal length of the spacer would be two methylene groups (phenethylamine).
We next evaluated other non-arylalkylamines as AANATL7 substrates: histamine, ethanolamine, putrescine, agmatine, histidine, Nω-acetylhistamine, and isoniazid. In decreasing order of their (kcat/Km)app value, these non-arylalkylamine substrates rank as follows: histamine > putrescine > agmatine ≫ ethanolamine. The ability of AANATL7 to utilize non-arylalkylamines as substrates highlights a promiscuity for the amine substrates greater than that observed for D. melanogaster AANATA. Histamine is the non-arylalkylamine substrate with the highest (kcat/Km)app, which is only 9% of that of the “best” arylalkylamine substrate, 5-benzyloxytryptamine. This largely results from a 58-fold increase in Km,app, relative to that of 5-benzyloxytryptamine, because the kcat,app value for histamine is actually 5.3-fold higher than the kcat,app of 5-benzyloxytryptamine (Table 3). In fact, histamine possesses the third highest kcat,app value of all the amine substrates evaluated in this study; only phenethylamine and 3-(trifluoromethyl)phenethylamine exhibited higher kcat,app values. Because we measure CoA-SH release to monitor AANATL7 activity, it is possible that AANATL7 catalyzes the formation of Nω-acetylhistamine, Nτ-acetylhistamine, or both. Characterization of the product formed by the AANATL7-catalyzed reaction of acetyl-CoA with histamine by LC-QTOF/MS shows only Nω-acetylhistamine (Table S2 of the Supporting Information), indicating that acetylation occurs at the ω-amine of histamine. Also, we evaluated Nω-acetylhistamine as a substrate and found no activity at 1.0 mM, providing additional evidence suggesting that AANATL7 will not catalyze the acetylation of the imidazole nitrogen atoms of histamine.
The in vivo role of AANATL7 in catalyzing the formation of Nω-acetylhistamine is unknown; however, there is evidence suggesting it could function in this chemistry. First, the total amount of histamine found in D. melanogaster head is 1.98 ± 0.15 ng, yielding concentrations of 540 μM and 574 μM in the heads and eyes, respectively.15,16 The cellular concentrations of histamine correlate well with the Km,app value (520 ± 50 μM). Second, Nω-acetylhistamine and the AANATL7 transcript are found in the fly head.9 In summary, these data point toward a role for AANATL7 in the N-acetylation of histamine in vivo. There are reports of other N-acetyltransferases26–28 catalyzing the acetyl-CoA-dependent formation of Nω-acetylhistamine, a polyamine N-acetyltransferase in the parasitic flatworm Fasciola hepatica,26 and two arylalkylamine N-acetyltransferases (aaNAT2 and aaNAT5b) in Aedes aegypti.28 The Km,app values for histamine for these enzymes (760 and 740 μM, respectively) are similar to the AANATL7 value (660 μM), suggesting that Nω-acetylation of histamine could occur across a broad range of different species.26,28
Putrescine, agmatine, and ethanolamine are poor substrates for AANATL7, with (kcat/Km)app values that are 0.48, 0.13, and 0.0037% of the (kcat/Km)app value for 5-benzyloxytryptamine, respectively. This is the second report of an enzyme that will catalyze the formation of N-acetylagmatine, albeit with a specificity that is low compared to those of the other arylalkylamine substrates.29 In summary, our data addressing the amine substrate specificity of AANATL7 indicate that this enzyme will catalyze the acetyl-CoA-dependent acetylation of most primary amines that are separated from a diversity of chemical moieties, a -(CH2)n- spacer with n = 2 representing the preferred length. Then we showed that isoniazid, which is a first-line drug in the treatment of tuberculosis,30–32 will not serve as a substrate for AANATL7.
Finally, we evaluated the cognate amino acids related to a number of the amine substrates as substrates for AANATL7. No CoA-SH release was observed at 1.0 mM for any of the four amino acids (tyrosine, tryptophan, phenylalanine, and histidine), indicating that AANATL7 will not catalyze the formation of N-acetyl amino acids. The presence of the α-carboxylate prevents the amino acid from serving as a substrate because of either an electrostatic effect or a steric effect that perturbs binding of substrate to the AANATL7 active site. To distinguish between these possibilities, we assessed tyrosine methyl ester, which would remove the electrostatic effect induced by the negatively charged carboxylate. We found that 1.0 mM tyrosine methyl ester was not a substrate, which suggests that modification at the α-position results in a steric effect that interferes with binding of the amine substrate to the AANATL7 active site. To further test this hypothesis, each amino acid and tyrosine methyl ester were tested as inhibitors for the AANATL7-catalyzed formation of N-acetylhistamine. Tyrosine, tryptophan, phenylalanine, histidine, and tyrosine methyl ester were evaluated individually at 1.0 mM as inhibitors, while acetyl-CoA and histamine were held at an initial concentration equal to their respective Km,app value. No inhibition was observed, indicating that tyrosine, tryptophan, phenylalanine, histidine, and tyrosine methyl ester either do not bind to AANATL7 or bind with low affinity (Kd > 10 mM). Clearly, modification of the spacer moiety of the amine substrates to an α-carboxylate or α-carboxylate ester decreases binding affinity for AANATL7 due to steric hindrance. Interestingly, these data are in direct contrast to what was observed for modification of the β-position of the spacer group (β-methylphenethylamine, octopamine, and norepinephrine), where no significant differences were found in comparing the kinetic constants between the cognate unmodified substrate (phenethylamine, tyramine, and dopamine, respectively).
A summary of the structure–activity relationships of amine substrates from Table 3 follows. (a) Arylalkylamine substrates exhibited tighter binding when a more hydrophobic group was positioned at position 5 of the indole ring, while the kcat,app value is relatively unchanged (except for that of 5-benzyloxtryptamine). (b) The length of the spacer group affects both binding and catalysis, with the optimal length being two methylene groups (-CH2-CH2-). (c) The Km,app values for phenyl ring-substituted analogues were generally lower than those for phenethylamines. (d) Substitution at the α-position of the spacer group induces steric interference for binding to the active site of AANATL7, while substitution at the β-position had a minimal impact on the kinetic constants. (e) AANATL7 catalyzes the formation of some non-arylalkylamine substrates consisting of histamine, putrescine, agmatine, and ethanolamine. Overall, AANATL7 exhibits promiscuity for amine substrates greater than that of D. melanogaster AANATA.6
Kinetic Mechanism
Substrate binding order is often one aspect of the detailed characterization of a multisubstrate enzyme. Such data are valuable in comparing related enzymes, in understanding the chemical mechanism, and in inhibitor design. We have additional interest in substrate binding order because one of our long-term goals is to develop activity-based profiling probes to identify other novel acyltransferases and/or other novel acyl-CoA-metabolizing enzymes. Our design of profiling probes as analogues of the acyl-CoAs, amines, or bisubstrate mimics will be guided by the substrate binding order.
Initial velocities for AANATL7 were measured by varying the initial concentration of either acetyl-CoA or histamine, while holding the fixed initial concentration of the other substrate constant. The resulting double-reciprocal plots (Figure 2) show a pattern of intersecting lines for both acetyl-CoA and histamine. The intersecting lines indicate that AANATL7 catalyzes the formation of N-acetylhistamine by a sequential mechanism, in which both substrates must be bound to generate a ternary complex before catalysis can occur. The next question is whether the kinetic mechanism is an ordered or random sequential mechanism. Dead-end inhibition studies were used to distinguish between these respective sequential mechanisms, using oleoyl-CoA and tyrosol as the inhibitors. Oleoyl-CoA is an analogue of acetyl-CoA and did not show a rate of reaction above the baseline rate of hydrolysis. Tyrosol was used as a structural analogue of the amine substrate, in which the primary amine is replaced with a hydroxyl group. Tyrosol did not show a rate of coenzyme A release above the background rate of hydrolysis at a concentration of 25 mM, which also suggests that AANATL7 will not catalyze the O-acetylation (at least for this substrate). These two inhibitors were previously used as dead-end inhibitors for the determination of the kinetic mechanism for D. melanogaster AANATA,6 while oleoyl-CoA was also known to inhibit mammalian serotonin N-acetyltransferases.33,34 Oleoyl-CoA was competitive versus acetyl-CoA and pure noncompetitive versus histamine, with inhibition constants of 200 ± 10 nM and 410 ± 10 nM, respectively (Figure 3). Tyrosol was uncompetitive versus acetyl-CoA and competitive versus histamine, with inhibition constants of 620 ± 20 μM and 430 ± 20 μM, respectively. These inhibition patterns demonstrate that AANATL7 will catalyze the formation of N-acetylhistamine as an ordered sequential mechanism, with acetyl-CoA binding first followed by histamine to form a precatalytic ternary AANATL7·acetyl-CoA·histamine complex. An ordered sequential kinetic mechanism has been reported for both D. melanogaster AANATA6 and sheep serotonin N-acetyltransferase.35
Figure 2.
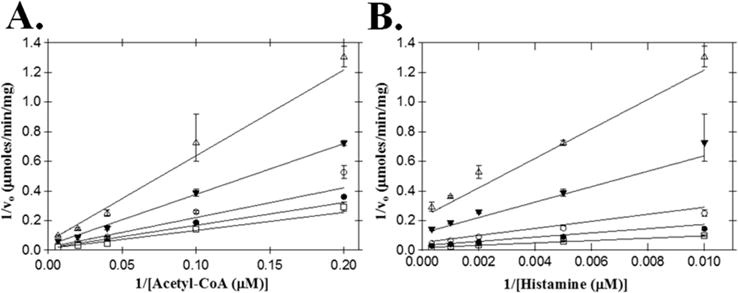
Double-reciprocal plot of initial velocities for acetyl-CoA and histamine. (A) Velocities measured at a fixed concentration of histamine: 3.0 mM (□), 1.0 mM (●), 500 μM (○), 200 μM (▼), and 100 μM (△). (B) Velocities measured at a fixed concentration of acetyl-CoA: 150 μM (□), 50 μM (●), 25 μM (○), 10 μM (▼), and 5 μM (△).
Figure 3.
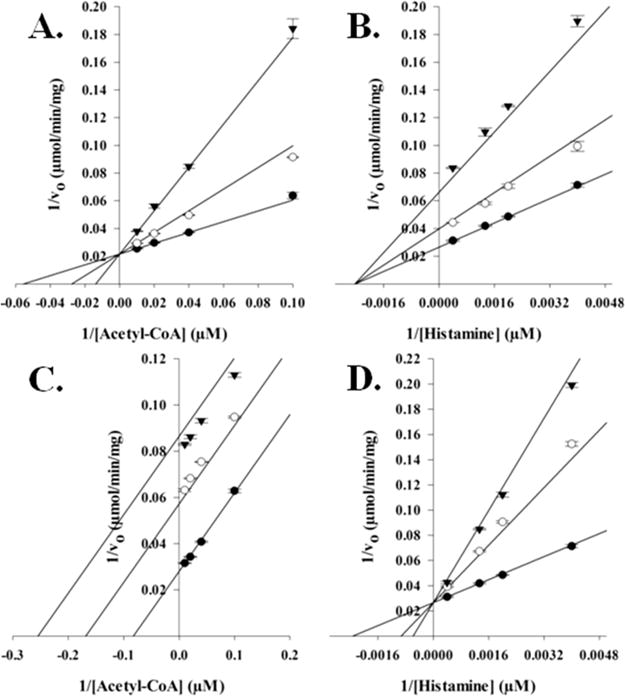
Dead-end inhibition analysis of AANATL7. (A) Velocities measured at a fixed concentration of histamine (520 μM), varying the concentration of acetyl-CoA, and varying the concentration of the inhibitor, oleoyl-CoA: 0 nM (●), 200 nM (○), and 600 nM (▼) (Ki = 200 ± 10 nM). (B) Velocities were measured at a fixed concentration of acetyl-CoA (29 μM), varying the concentration of histamine, and varying the concentration of the inhibitor, oleoyl-CoA: 0 nM (●), 200 nM (○), and 600 nM (▼) (Ki = 410 ± 10 nM). (C) Velocities measured at a fixed concentration of histamine (520 μM), varying the concentration of acetyl-CoA, and varying the concentration of the inhibitor, tyrosol: 0 μM (●), 650 μM (○), and 1.3 mM (▼) (Ki = 620 ± 20 μM). (D) Velocities measured at a fixed concentration of acetyl-CoA (29 μM), varying the concentration of histamine, and varying the concentration of the inhibitor, tyrosol: 0 μM (●), 650 μM (○), and 1.3 mM (▼) (Ki = 430 ± 20 μM).
Evaluation of the pH Dependence on the Kinetic Constants and the Use of Site-Directed Mutagenesis To Identify Catalytic Amino Acids
We used a combination of pH–activity and site-directed mutagenesis studies to aid in the identification of the AANATL7 catalytic residues and, subsequently, propose a chemical mechanism for AANATL7. In the absence of a crystal structure, we indexed all the data generated herein, to D. melanogaster AANATA,6,7 sheep serotonin N-acetyltransferase,36–41 and other GNAT enzymes, to define a function for the amino acid residues identified in our work. We first examined the pH dependence of the steady-state kinetic parameters to help delineate the amino acids involved in catalysis. A rising (kcat/Km)app pH rate profile was observed for both acetyl-CoA (Figure 4A) and histamine (Figure 4B), with pKa,app values of 6.8 ± 0.1 and 7.6 ± 0.1, respectively. The (kcat/Km)app pH rate profile represents the ionizable groups that are important for binding and catalysis for free enzyme and substrate, up until the first irreversible step in catalysis (E + S ⇋ ES); therefore, it is likely that these two apparent pKa values both represent the general base in catalysis. The kcat,app pH profiles for both acetyl-CoA and histamine were bell-shaped (Figure 4C,D) with two pKa,app values (pKa,app-acetyl-CoA = 7.1 ± 0.1 and 9.5 ± 0.1; pKa,app-histamine = 7.2 ± 0.1 and 9.6 ± 0.1). The apparent pKa value of 7.1–7.2 is attributed to the general base identified in both (kcat/Km)app profiles, while the apparent pKa value of 9.5–9.6 could represent either a general acid in catalysis or the thiol group of the departing coenzyme A product. The pKa,app value of 9.5–9.6 was not observed in either (kcat/Km)app pH rate profile, suggesting that this ionizable group contributes to catalysis after the first irreversible step (ES → E + P). A lack of a pKa,app value of ≥9.5 in the (kcat/Km)app profiles can also result from an inability to evaluate AANATL7 at higher pH values, because of the instability of Ellman’s reagent and/or base-catalyzed hydrolysis of the acetyl-CoA substrate. Note that the pKa,app value identified in the kcat,app profiles is 9.5–9.6, which is greater than or equal to the highest experimental pH we employed in our studies. The pKa,app value of 9.5–9.6 comes from the best fit of our data to eq 8. Therefore, the lack of a pKa,app value of ≥9.5 in the (kcat/Km)app profiles may reflect our inability to assess AANATL7 at pH >9.5. The observed difference in the pKa,app values between the (kcat/Km)app and kcat,app pH rate profiles for the general base likely results from the kinetic stickiness of the substrates evaluated in this study.
Figure 4.
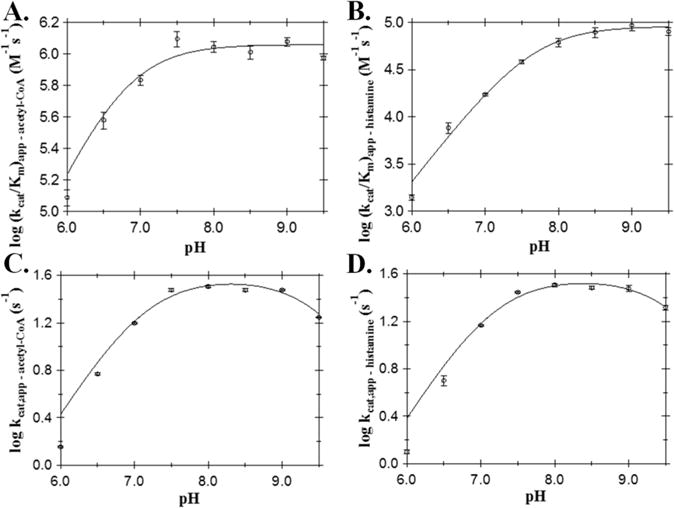
AANATL7 pH rate profile: (A) (kcat/Km)app for acetyl-CoA, (B) (kcat/Km)app for histamine, (C) kcat,app for acetyl-CoA, and (D) kcat,app for histamine.
The pH–activity profiles do not provide definitive information about the specific residues involved in catalysis. Therefore, we further evaluated amino acid residues that can function as either a general base or acid by site-directed mutagenesis. With the lack of a crystal structure, we utilized a primary sequence alignment of AANATL7 with D. melanogaster AANATA6 to identify possible amino acid residues that can function in substrate binding and/or catalysis. AANATA has been characterized with the function of specific residues clearly defined, including a general base in catalysis and other residues critical in substrate binding and/or regulation of catalysis. From the primary sequence alignment, the proposed general base of AANATA (Glu-47) is conserved, corresponding to Glu-26 in AANATL7.6 Two serine residues in D. melanogaster (Ser-182 and Ser-186) have been proposed to function in catalysis, which correspond to Thr-167 and Ser-171, respectively, in AANATL7.6 Other amino acid residues with defined functions in AANATA are Pro-48, Arg-153, and His-220.6 These particular amino acids are conserved in AANATL7,6 being Pro-27, Arg-138, and His-206, respectively.6 We mutated each of these residues to an alanine to define their respective function in AANATL7 and referenced the resulting data to that reported for AANATA.
Mutation of Glu-26 to alanine resulted in a 22–32-fold decrease in the kcat,app relative to that of the wild type (Table 4). The (kcat/Km)app-acetyl-CoA and (kcat/Km)app-histamine values were only 7.0 and 0.15% of that of the wild type, respectively. In addition, the E26A mutant has a Km,app-acetyl-CoA similar to that of the wild type; however, the Km,app-histamine was increased ~30-fold. On the basis of the catalytic deficiency of the E26A mutant, we propose that Glu-26 is the general base in AANATL7 catalysis with a pKa,app value of ~7.2 (Figure 4). The pH–rate profiles generated for the E26A mutant are consistent with this hypothesis. The kcat,app-acetyl-CoA and (kcat/Km)app-acetyl-CoA pH rate profiles for the E26A mutant resulted in a linear increase as the pH was increased from 7.5 to 9.5, with slopes of 0.6 and 0.2, respectively (Figure 5). We were not able to accurately measure a velocity for E26A above the background rate of acetyl-CoA hydrolysis at pH ≤7.0, suggesting the mutant enzyme is catalytically dead at pH <7.0. The linear pH–rate profiles we obtain likely result from hydroxide chemically rescuing the function of Glu-26 in deprotonation of the positively charged primary amino group of histamine. These data for the E26A mutant are consistent with what has been published for D. melanogaster AANATA.6 Furthermore, other enzymes of the GNAT family are proposed to employ an active site Glu residue as the general base, including human spermidine/spermine N1-acetyltransferase42,43 and the Gcn5/PCAF family of histone N-acetyltransferases.44,45
Table 4.
Steady-State Kinetic Constants for AANATL7 Site-Directed Mutantsa
| variantb | Km,app (μM) | kcat,app (s−1) | Acetyl-CoA (kcat/Km)app (M−1 s−1) |
{[(kcat/Km)app-mutant]/[(kcat/Km)app-wild-type]} × 100 (%) |
|---|---|---|---|---|
| wild-type | 29 ± 2 | 32 ± 1 | (1.1 ± 0.1) × 106 | 100 |
| E26A | 13 ± 1 | 1.01 ± 0.02 | (7.7 ± 0.5) × 104 | 7.0 |
| R138A | 550 ± 60 | 88 ± 3 | (1.6 ± 0.2) × 105 | 15 |
| T167A | 140 ± 4 | 1.84 ± 0.02 | (1.36 ± 0.05) × 104 | 1.2 |
| S171A | 19 ± 1 | 8.6 ± 0.1 | (4.6 ± 0.1) × 105 | 42 |
| T167A/S171A | 29 ± 2 | 1.61 ± 0.03 | (5.6 ± 0.3) × 104 | 5.1 |
| H206A | 210 ± 5 | 9.6 ± 0.1 | (4.6 ± 0.1) × 104 | 4.2 |
| variantc | Km,app (μM) | kcat,app (s−1) | Histamine (kcat/Km)app (M−1 s−1) |
{[(kcat/Km)app-mutant]/[(kcat/Km)app-wild-type]} × 100 (%) |
|---|---|---|---|---|
| wild-type | 0.52 ± 0.05 | 30 ± 1 | (5.8 ± 0.6) × 104 | 100 |
| E26A | 16 ± 1 | 1.39 ± 0.04 | 86 ± 32 | 0.15 |
| R138A | 4.9 ± 0.4 | 71 ± 2 | (1.5 ± 0.1) × 104 | 26 |
| T167A | 6.7 ± 0.7 | 1.9 ± 0.1 | 280 ± 30 | 0.48 |
| S171A | 3.4 ± 0.2 | 10.4 ± 0.2 | (3.0 ± 0.2) × 103 | 5.2 |
| T167A/S171A | 13 ± 1 | 2.2 ± 0.1 | 160 ± 20 | 0.28 |
| H206A | 2.3 ± 0.1 | 7.7 ± 0.1 | (3.3 ± 0.2) × 103 | 5.7 |
Kinetic constants are reported as ± the standard error (n = 3).
Reaction conditions: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, a constant saturating concentration of histamine (>3Km,app), and varying concentrations of acetyl-CoA.
Reaction conditions: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, a constant saturating concentration of acetyl-CoA (>3Km,app), and varying concentrations of histamine.
Figure 5.
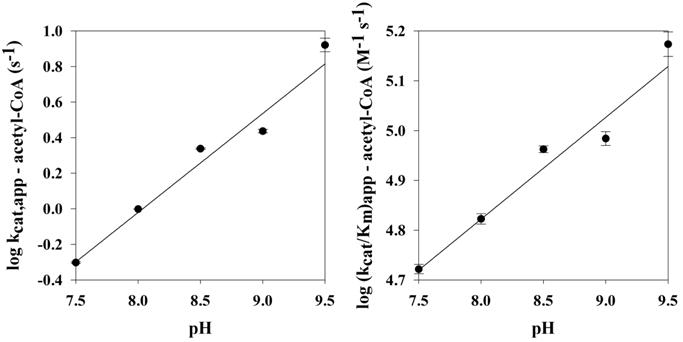
AANATL7 E26A pH–rate profile: (A) kcat,app for acetyl-CoA and (B) (kcat/Km)app for acetyl-CoA.
Next, we evaluated the role of Thr-167 and Ser-171 in binding and/or catalysis. The corresponding residues in AANATA (Ser-182 and Ser-186, respectively)6 were proposed to function in catalysis.7 Therefore, we mutated Thr-167 and Ser-171 each to an alanine to define a specific function in AANATL7 catalysis. The T167A mutant produced a decrease in kcat,app of 16–17-fold relative to that of the wild-type enzyme. (kcat/Km)app-acetyl-CoA and (kcat/Km)app-histamine for T167A were only 1.2% and 0.49% of that of the wild-type enzyme, respectively. In addition, the T167A mutant yielded a Km,app-acetyl-CoA that increases 5-fold compared to that of the wild type and a Km,app-histamine that was increased 13-fold. The S171A mutant produced a kcat,app decrease of 3–4-fold relative to that of the wild-type enzyme. (kcat/Km)app-acetyl-CoA and (kcat/Km)app-histamine for S171A were only 42 and 5.2% of that of the wild-type enzyme, respectively. In addition, the S171A mutant has a Km,app-acetyl-CoA similar to that of the wild type; however, the Km,app-histamine was increased 7-fold. These data suggest that Thr-167 and Ser-171 could function in catalysis and/or substrate binding; however, a direct role as a general acid is unlikely. Because threonine and serine are chemically similar (both possess a hydroxyl group), we evaluated a T167A/S171A double mutant to delineate if these residues function in a redundant role. The T167A/S171A double mutant produced a kcat,app decrease of 14–20-fold relative to that of the wild-type enzyme, similar to what was observed for the T167A mutant. (kcat/Km)app-acetyl-CoA and (kcat/Km)app-histamine for T167A/S171A were only 5.1% and 0.28% of that of the wild-type enzyme, respectively. In addition, the T167A/S171A mutant has the same Km,app-acetyl-CoA as the wild type; however, the Km,app-histamine was increased 25-fold. These data, as well as data generated for the corresponding residues from D. melanogaster AANATA,6,7 allow us to propose a role for both of these residues in AANATL7 catalysis. One proposed role for Ser-186 in AANATA (Ser-171 in AANATL7) was to serve as the general acid protonating the departing thiolate of CoA-S−,7 thus accounting for the pKa of 9.5–9.6 identified by our pH–rate studies. We are not in favor of either Thr-167 or Ser-171 serving as a general acid in AANATL7 catalysis. Such a mechanism would require a significant depression in the pKa of the hydroxyl group of either Thr or Ser (3–5 pH units) and invokes a thermodynamically unfavorable transfer of a proton from a hydroxyl to a thiolate. A redundant role in catalysis for these residues is unlikely because the T167A/S171A double mutant was as catalytically impaired as the T167A mutant (within experimental error) (Table 4). Instead, we propose that Thr-167 and Ser-171 function to stabilize the proper active site architecture, most likely through a network of hydrogen bonds, similar to what was observed for the corresponding serine residues in AANATA.6
A remaining question is the nature of the ionizable group responsible for the pKa,app of 9.5–9.6 we identified in the kcat,app pH rate profiles. We propose that the thiol group of the CoA-SH product is responsible for this pKa,app, which has been reported to range from 9.6 to 10.4.46,47 Protonation of CoA-S− to CoA-SH is a step that occurs during the collapse of the tetrahedral intermediate and is likely necessary for catalysis (Scheme 1). The decrease in kcat,app at pH ≥9.0 results from the deprotonation of CoA-SH to yield CoA-S−, inhibiting its release from the active site. This proposal eliminates the need to invoke an active site amino acid as the general acid in catalysis.
Scheme 1. Proposed Chemical Mechanism for D. melanogaster AANATL7a.
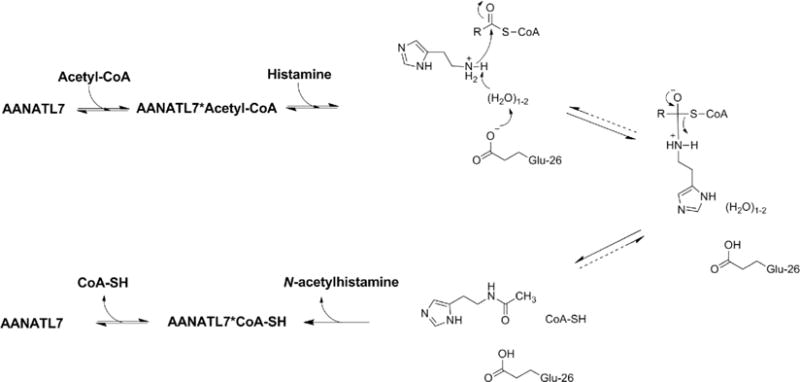
aThe dashed arrow represents the potential positions of the irreversible step based on data generated herein and for other D. melanogaster AANATL enzymes.6,7
Use of Site-Directed Mutagenesis To Define the Function of AANATL7 Amino Acids in Binding and/or Regulation of Catalysis
From the primary sequence alignment, AANATL7 Pro-27, Arg-138, and His-206 are conserved in AANATA (Pro-48, Arg-153, and His-220, respectively).6 Therefore, we mutated each of these residues to an alanine and evaluated the kinetic constants of each mutant (Tables 4 and 5) in an effort to define their function. The P27A mutant produced a 38–42-fold decrease in the kcat,app value when histamine was used as the amine substrate. Additionally, Km,app-acetyl-CoA and Km,app-histamine increased 3-fold for both substrates, yielding (kcat/Km)app-acetyl-CoA and (kcat/Km)app-histamine values that were only 0.67 and 0.76% of the value for the wild-type enzyme, respectively. The significant decrease in kcat,app and an increase in Km,app for the P27A mutant suggest that Pro-27 functions to organize the active site and regulate catalysis. The corresponding residues from sheep serotonin N-acetyltransferase (Pro-64)37,48,49 and D. melanogaster AANATA (Pro-48)6 are located on a flexible loop that undergoes a major conformational change to form the amine binding pocket. This conformational change converts the substrate binding pocket from a low-affinity state to a high-affinity state.6,49 For sheep serotonin N-acetyltransferase, two crystal structures have been published, one of the apoenzyme (PDB entry 1B6B)48 and the other of the enzyme cocrystallized with the bisubstate inhibitor, tryptamine-acetyl-CoA (PDB entry 1CJW).37 These crystal structures show that Pro-64 is located in the acetyl-CoA binding pocket in the low-affinity state (apo form), whereas the conformational change yields a high-affinity state where the proline residue migrates ~8 Å to form a direct π-stacking interaction with the tryptamine moiety of the bisubstrate inhibitor.37,48,49 The 3-fold increase in Km,app-histamine for the P27A mutant suggests that a similar π-stacking interaction does take place between Pro-27 and the imidazole ring of histamine in the wild-type enzyme.
Table 5.
Steady-State Kinetic Constants for the Pro-27 Site-Directed Mutanta
| variant | varied substrate | Km,app (μM) | kcat,app (s−1) | (kcat/Km)app (M−1 s−1) | {[(kcat/Km)app-mutant]/[(kcat/Km)app-wild-type]} × 100 (%) |
|---|---|---|---|---|---|
| wild-typeb | acetyl-CoA | 29 ± 2 | 32 ± 1 | (1.1 ± 0.1) × 106 | 100 |
| P27Ab | 105 ± 4 | 0.77 ± 0.02 | (7.4 ± 0.3) × 103 | 0.67 | |
| wild-typec | histamine | 520 ± 54 | 30 ± 1 | (5.8 ± 0.6) × 104 | 100 |
| P27Ac | 1800 ± 100 | 0.80 ± 0.02 | 440 ± 30 | 0.76 | |
| wild-typec | agmatine | 790 ± 100 | 0.68 ± 0.03 | 850 ± 110 | 100 |
| P27Ac | 1300 ± 100 | 0.012 ± 0.001 | 9.7 ± 0.7 | 1.1 |
Kinetic constants are reported as ± the standard error (n = 3).
Reaction conditions: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, a constant saturating concentration of histamine (>3Km,app), and varying concentrations of acetyl-CoA.
Reaction conditions: 300 mM Tris-HCl (pH 8.0), 150 μM DTNB, a constant saturating concentration of acetyl-CoA (>3Km,app), and varying concentrations of amine substrate.
The amine promiscuity of AANTL7 allows us to further test for a π-stacking interaction between Pro-27 and histamine by comparing the kinetic constants of histamine to those measured for agmatine. If a π-stacking interaction between Pro-27 and histamine contributes significantly to the binding of histamine to AANATL7, we would expect a higher Km,app-P27A/Km,app-wild-type ratio for histamine than for agmatine, an amine substrate with a positively charged guanidinium group that is not an effective π-acceptor. The P27A mutant produced a kcat,app-agmatine that decreased 57-fold and a Km,app-agmatine that increased only 1.6-fold relative to those of the wild-type enzyme (Table 5). The P27A data for histamine and agmatine yielded a (Km,app-P27A/Km,app-wild-type)histamine/(Km,app-P27A/Km,app-wild-type)agmatine ratio of only 2.2, suggesting that, at most, a π-stacking interaction between Pro-27 and histamine contributes modestly to histamine binding and that the most important function of Pro-27 is in the optimal organization of the AANATL7 active site for catalysis.
Next, we mutated Arg-138 to an alanine to interrogate its respective function. The R138A mutant resulted in increases in kcat,app (2.4–2.8-fold) and both Km,app-acetyl-CoA (19-fold) and Km,app-acetyl-CoA (9-fold) values. The increase in the kcat,app value for R138A was balanced by increases in the Km,app values to yield a mutant enzyme with (kcat/Km)app-acetyl-CoA and (kcat/Km)app-histamine values that were only 15% and 26% of that of the wild type, respectively (Table 4). These data suggest that Arg-138 functions in both catalysis and substrate binding. Most likely, Arg-138 does not have a direct role in catalysis as a general acid or base. However, the increase in the kcat,app value is consistent with a conformation, dependent upon Arg-138, which binds product relatively tightly so that product release is, at least, partially rate-determining in the wild-type enzyme. Mutation of Arg-138 to alanine results in a loss of this conformation, inducing domain mobility to alleviate the partially rate-limiting product release step. The corresponding residue in D. melanogaster AANATA (Arg-153) produced a similar increase in the kcat,app value upon mutation to alanine, which was attributed to the elimination of a salt bridge that was key to a conformation that decreased the rate of CoA-SH release.6 The AANATL7 R138A mutant likely eliminates a salt bridge, which would increase the rate of CoA-SH release and increase kcat,app (similar to what was observed for AANATA).6 We would expect that if the R138A mutant is increasing the koff for the CoA-SH product, then the dissociation constant for CoA-SH should increase because the Kd is the koff-CoA-SH/kon-CoA-SH ratio. We determined the Kd for binding of CoA-SH for the wild-type enzyme and R138A mutant by measuring the quenching of intrinsic AANATL7 fluorescence by the increasing CoA-SH concentration. These experiments yielded a Kd-wild-type of 530 ± 110 μM and a Kd-R138A of 1.1 ± 0.2 mM (Figure 6 and Figures S4 and S5 of the Supporting Information), a 2.1-fold decrease in the binding affinity for the R153A mutant relative to that for the wild-type enzyme. The Kd-R153/Kd-wild-type ratio (2.1-fold) is in reasonable agreement with the kcat-R153A/kcat-wild-type ratio (2.4–2.8-fold) (Table 4). These data suggest, but do not prove, that a salt bridge involving Arg-138 could be one important component in the regulation of dynamics in AANATL7, contributing to a partial rate-limiting release of CoA-SH. These data are consistent with what is observed with D. melanogaster AANATA6 and other GNAT enzymes that have product release as a partially rate-limiting step.18,36,50,51
Figure 6.
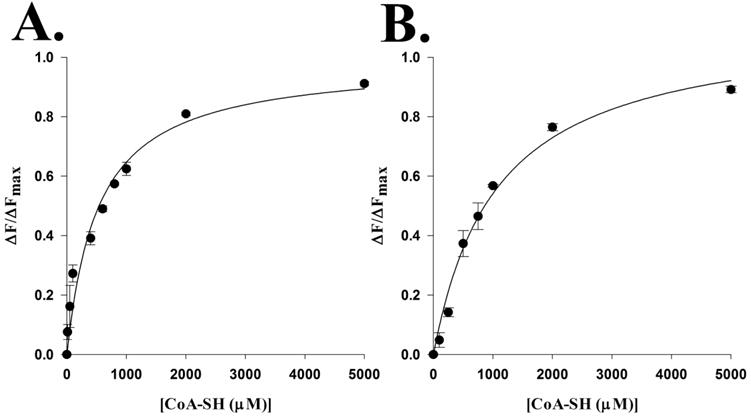
Determination of the coenzyme A dissociation constant by measuring the intrinsic fluorescence quenching of AANATL7: (A) wild type and (B) R153A mutant.
Similar to Pro-27 and Arg-138, His-206 is conserved relative to D. melanogaster AANATA (His-220).6 The H206A mutant produced a 3–4-fold decrease in the kcat,app value (Table 5) and an increase in the Km,app for both acetyl-CoA (7-fold) and histamine (4-fold). This suggests that His-206 functions to organize the binding pocket for both substrates, similar to what is proposed for the corresponding residue in AANATA (His-220).6
Proposed Chemical Mechanism for AANATL7 Catalysis
A chemical mechanism consistent with our data is shown in Scheme 1. The details of our proposed mechanism are as follows: (a) ordered substrate binding, with acetyl-CoA binding first followed by histamine to generate an AANATL7·acetyl-CoA·histamine ternary complex prior to catalysis, (b) Glu-26 serving as the general base to deprotonate the positively charged primary amino group of histamine through ordered water molecules (consistent with what was observed for AANATA and other GNAT enzymes), (c) nucleophilic attack of histamine on the carbonyl of the acetyl-CoA thioester to generate the zwitterionic tetrahedral intermediate, (d) collapse of the zwitterionic tetrahedral intermediate followed by protonation the coenzyme A thiolate, and (e) and ordered product release, with N-acetylhistamine leaving first, followed by CoA-SH. Evidence supporting ordered product release is produced by our measurement of the direct binding of CoA-SH to AANATL2 in the absence of the N-acetylated product, which is also consistent with a synergistic effect caused by ordered substrate binding. This chemical mechanism is consistent with the data generated herein and with the mechanism proposed for D. melanogaster AANATA6 and other GNAT enzymes.52,53
CONCLUSIONS
We showed that AANATL7 will catalyze the formation of N-acylamides from the corresponding acyl-CoA and amine substrates. AANATL7 is relatively discriminative for the acyl-CoA substrates, showing that the (kcat/Km)app for acetyl-CoA is ≥10-fold higher than that for any other acyl-CoA substrate analyzed in this study. In contrast, AANATL7 is less discriminatory with regard to the amine substrates with arylalkylamines, histamine, putrescine, agmatine, and ethanolamine all serving as substrates. We proposed a chemical mechanism for the AANATL7-catalyzed formation of N-acetylhistamine as illustrated in Scheme 1. First, substrate binding is ordered sequential with acetyl-CoA binding first, followed by histamine to generate the ternary complex (AANATL7·acetyl-CoA·histamine) prior to catalysis. Next, we invoked Glu-26 as the general base in catalysis with a pKa,app of ~7.2, which will deprotonate the positively charged primary amino group of histamine through ordered water molecules that make up an active site “proton wire”. Finally, we suggest that protein dynamics has a significant regulatory effect on AANATL7 catalysis, where noncatalytic amino acid residues (Arg-138 and Pro-27) have a significant impact on the rate of catalysis.
Supplementary Material
Acknowledgments
Support for this work has been provided by Mass Spectrometry and Peptide Facility, Department of Chemistry, University of South Florida.
Funding
This work has been supported, in part, by grants from the Florida Center for Excellence for Biomolecular and Targeted Therapeutics (FCoE-BITT Grant GALS020), the University of South Florida (a Research Scholarship grant from the College of Arts and Sciences), the Shirley W. and William L. Griffin Foundation, the National Institute of Drug Abuse of the National Institutes of Health (R03-DA034323), and the National Institute of General Medical Science of the National Institutes of Health (R15-GM107864) (all to D.J.M.) and a Dissertation Completion Fellowship from the Office of Graduate Studies at the University of South Florida to D.R.D.
ABBREVIATIONS
- AANATA
arylalkylamine N-acetyltransferase variant A
- AANATB
arylalkylamine N-acetyltransferase variant B
- AANATL
arylalkylamine N-acetyltransferase like
- AANATL2
arylalkylamine N-acetyltransferase like 2
- AANATL7
arylalkylamine N-acetyltransferase like 7
- AMeP
2-amino-2-methyl-1-propanol hydrochloride
- DTNB
5,5′-dithiobis(2-nitrobenzoic acid)
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GNAT
GCN5-related N-acetyltransferase
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- LC/QTOF-MS
liquid chromatography/quadrupole time-of-flight mass spectroscopy
- MES
2-(N-morpholino)ethanesulfonic acid
- Tris
tris(hydroxymethyl)-aminomethane
Footnotes
Supporting Information
Two tables consisting of PCR primers for the site-directed mutants and the LC/QTOF-MS data for product characterization and three figures consisting of an agarose gel and SDS–PAGE gel for the cloning and purification of AANATL7, two AANATL7 site-directed mutant gels, and the raw data for the intrinsic protein fluorescence measurements of binding of CoA-H to wild-type and R138A mutant enzymes. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.Farrell EK, Merkler DJ. Biosynthesis, degradation and pharmacological importance of the fatty acid amides. Drug Discovery Today. 2008;13:558–568. doi: 10.1016/j.drudis.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tortoriello G, Rhodes BP, Takacs SM, Stuart JM, Basnet A, Raboune S, Widlanski TS, Doherty P, Harkany T, Bradshaw HB. Targeted lipidomics in Drosophila melanogaster identifies novel 2-monoacylglycerols and N-acyl amides. PLoS One. 2013;8:e67865. doi: 10.1371/journal.pone.0067865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeffries KA, Dempsey DR, Behari AL, Anderson RL, Merkler DJ. Drosophila melanogaster as a model system to study long-chain fatty acid amide metabolism. FEBS Lett. 2014;588:1596–1602. doi: 10.1016/j.febslet.2014.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dempsey DR, Jeffries KA, Anderson RL, Carpenter AM, Rodriquez Opsina S, Merkler DJ. Identification of an arylalkylamine N-acyltransferase from Drosophila melanogaster that catalyzes the formation of long-chain N-acylserotonins. FEBS Lett. 2014;588:594–599. doi: 10.1016/j.febslet.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amherd R, Hintermann E, Walz D, Affolter M, Meyer UA. Purification, cloning, and characterization of a second arylalkylamine N-acetyltransferase from Drosophila melanogaster. DNA Cell Biol. 2000;19:697–705. doi: 10.1089/10445490050199081. [DOI] [PubMed] [Google Scholar]
- 6.Dempsey DR, Jeffries KA, Bond JD, Carpenter AM, Rodriguez-Ospina S, Breydo L, Caswell KK, Merkler DJ. Mechanistic and structural analysis of Drosophila melanogaster arylalkylamine N-acetyltransferases. Biochemistry. 2014;53:7777–7793. doi: 10.1021/bi5006078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng KC, Liao JN, Lyu PC. Crystal structure of the dopamine N-acetyltransferase-acetyl-CoA complex provides insights into the catalytic mechanism. Biochem J. 2012;446:395–404. doi: 10.1042/BJ20120520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saghatelian A, Cravatt BF. Discovery metabolite profiling-forging functional connections between the proteome and metabolome. Life Sci. 2005;77:1759–1766. doi: 10.1016/j.lfs.2005.05.019. [DOI] [PubMed] [Google Scholar]
- 9.Sarthy PV. Histamine: A neurotransmitter candidate for Drosophila photoreceptors. J Neurochem. 1991;57:1757–1768. doi: 10.1111/j.1471-4159.1991.tb06378.x. [DOI] [PubMed] [Google Scholar]
- 10.Melzig J, Burg M, Gruhn M, Pak WL, Buchner E. Selective histamine uptake rescues photo- and mechanoreceptor function of histidine decarboxylase-deficient Drosophila mutant. J Neurosci. 1998;18:7160–7166. doi: 10.1523/JNEUROSCI.18-18-07160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burg MG, Sarthy PV, Koliantz G, Pak WL. Genetic and molecular identification of a Drosophila histidine decarboxylase gene required in photoreceptor transmitter synthesis. EMBO J. 1993;12:911–919. doi: 10.1002/j.1460-2075.1993.tb05732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elias MS, Evans PD. Histamine in the insect nervous system: Distribution, synthesis and metabolism. J Neurochem. 1983;41:562–568. doi: 10.1111/j.1471-4159.1983.tb04776.x. [DOI] [PubMed] [Google Scholar]
- 13.Monastirioti M. Biogenic amine systems in the fruit fly Drosophila melanogaster. Microsc Res Tech. 1999;45:106–121. doi: 10.1002/(SICI)1097-0029(19990415)45:2<106::AID-JEMT5>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Nassel DR. Histamine in the brain of insects: A review. Microsc Res Tech. 1999;44:121–136. doi: 10.1002/(SICI)1097-0029(19990115/01)44:2/3<121::AID-JEMT6>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 15.Borycz J, Vohra M, Tokarczyk G, Meinertzhagen IA. The determination of histamine in the Drosophila head. J Neurosci Methods. 2000;101:141–148. doi: 10.1016/s0165-0270(00)00259-4. [DOI] [PubMed] [Google Scholar]
- 16.Borycz JA, Borycz J, Kubow A, Kostyleva R, Meinertzhagen IA. Histamine compartments of the Drosophila brain with an estimate of the quantum content at the photoreceptor synapse. J Neurophysiol. 2005;93:1611–1619. doi: 10.1152/jn.00894.2004. [DOI] [PubMed] [Google Scholar]
- 17.Dyda F, Klein DC, Hickman AB. GCN5-related N-acetyltransferases: A structural overview. Annu Rev Biophys Biomol Struct. 2000;29:81–103. doi: 10.1146/annurev.biophys.29.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vetting MW, de Carvalho LPS, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys. 2005;433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 19.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 20.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 21.Morrison JF, Uhr ML. The function of bivalent metal ions in the reaction catalysed by ATP:creatine phosphotransferase. Biochim Biophys Acta. 1966;122:57–74. doi: 10.1016/0926-6593(66)90091-9. [DOI] [PubMed] [Google Scholar]
- 22.Finocchiaro L, Callebert J, Launay JM, Jallon JM. Melatonin biosynthesis in Drosophila: Its nature and its effects. J Neurochem. 1988;50:382–387. doi: 10.1111/j.1471-4159.1988.tb02923.x. [DOI] [PubMed] [Google Scholar]
- 23.Gilvarg C. Metabolism of amino acids. Annu Rev Biochem. 1961;30:239–268. [Google Scholar]
- 24.Ganguly S, Mummaneni P, Steinbach PJ, Klein DC, Coon SL. Characterization of the Saccharomyces cerevisiae homolog of the melatonin rhythm enzyme arylalkylamine N-acetyltransferase (EC 2.3.1.87) J Biol Chem. 2001;276:47239–47247. doi: 10.1074/jbc.M107222200. [DOI] [PubMed] [Google Scholar]
- 25.Falcon J, Coon SL, Besseau L, Cazamea-Catalan D, Fuentes M, Magnanou E, Paulin CH, Boeuf G, Sauzet S, Jorgensen EH, Mazan S, Wolf YI, Koonin EV, Steinbach PJ, Hyodo S, Klein DC. Drastic neofunctionalization associated with evolution of the timezyme AANAT 500 Mya. Proc Natl Acad Sci USA. 2014;111:314–319. doi: 10.1073/pnas.1312634110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aisien SO, Walter RD. Biogenic-amine acetylation: An additional function of the N-acetyltransferase from Fasciola hepatica. Biochem J. 1993;291(Part 3):733–737. doi: 10.1042/bj2910733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wittich RM, Walter RD. Putrescine N-acetyltransferase in Onchocerca volvulus and Ascaris suum, an enzyme which is involved in polyamine degradation and release of N-acetylputrescine. Mol Biochem Parasitol. 1990;38:13–17. doi: 10.1016/0166-6851(90)90199-v. [DOI] [PubMed] [Google Scholar]
- 28.Han Q, Robinson H, Ding H, Christensen BM, Li J. Evolution of insect arylalkylamine N-acetyltransferases: Structural evidence from the yellow fever mosquito, Aedes aegypti. Proc Natl Acad Sci USA. 2012;109:11669–11674. doi: 10.1073/pnas.1206828109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forouhar F, Lee IS, Vujcic J, Vujcic S, Shen J, Vorobiev SM, Xiao R, Acton TB, Montelione GT, Porter CW, Tong L. Structural and functional evidence for Bacillus subtilis PaiA as a novel N1-spermidine/spermine acetyltransferase. J Biol Chem. 2005;280:40328–40336. doi: 10.1074/jbc.M505332200. [DOI] [PubMed] [Google Scholar]
- 30.Chang KC, Yew WW, Tam CM, Leung CC. WHO group 5 drugs and difficult multidrug-resistant tuberculosis: A systematic review with cohort analysis and meta-analysis. Antimicrob Agents Chemother. 2013;57:4097–4104. doi: 10.1128/AAC.00120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vernon A. Treatment of latent tuberculosis infection. Seminars in Respiratory Critical Care Medicine. 2013;34:67–86. doi: 10.1055/s-0032-1333544. [DOI] [PubMed] [Google Scholar]
- 32.Zumla A, Raviglione M, Hafner R, von Reyn CF. Tuberculosis. N Engl J Med. 2013;368:745–755. doi: 10.1056/NEJMra1200894. [DOI] [PubMed] [Google Scholar]
- 33.Khalil EM, Cole PA. A potent inhibitor of the melatonin rhythm enzyme. J Am Chem Soc. 1998;120:6195–6196. [Google Scholar]
- 34.Ferry G, Loynel A, Kucharczyk N, Bertin S, Rodriguez M, Delagrange P, Galizzi JP, Jacoby E, Volland JP, Lesieur D, Renard P, Canet E, Fauchere JL, Boutin JA. Substrate specificity and inhibition studies of human serotonin N-acetyltransferase. J Biol Chem. 2000;275:8794–8805. doi: 10.1074/jbc.275.12.8794. [DOI] [PubMed] [Google Scholar]
- 35.De Angelis J, Gastel J, Klein DC, Cole PA. Kinetic analysis of the catalytic mechanism of serotonin N-acetyltransferase (EC 2.3.1.87) J Biol Chem. 1998;273:3045–3050. doi: 10.1074/jbc.273.5.3045. [DOI] [PubMed] [Google Scholar]
- 36.Khalil EM, De Angelis J, Cole PA. Indoleamine analogs as probes of the substrate selectivity and catalytic mechanism of serotonin N-acetyltransferase. J Biol Chem. 1998;273:30321–30327. doi: 10.1074/jbc.273.46.30321. [DOI] [PubMed] [Google Scholar]
- 37.Hickman AB, Namboodiri MA, Klein DC, Dyda F. The structural basis of ordered substrate binding by serotonin N-acetyltransferase: Enzyme complex at 1.8 Å resolution with a bisubstrate analog. Cell. 1999;97:361–369. doi: 10.1016/s0092-8674(00)80745-x. [DOI] [PubMed] [Google Scholar]
- 38.Scheibner KA, De Angelis J, Burley SK, Cole PA. Investigation of the roles of catalytic residues in serotonin N-acetyltransferase. J Biol Chem. 2002;277:18118–18126. doi: 10.1074/jbc.M200595200. [DOI] [PubMed] [Google Scholar]
- 39.Zheng WP, Cole PA. Serotonin N-acetyltransferase: Mechanism and inhibition. Curr Med Chem. 2002;9:1187–1199. doi: 10.2174/0929867023370013. [DOI] [PubMed] [Google Scholar]
- 40.Wolf E, De Angelis J, Khalil EM, Cole PA, Burley SK. X-ray crystallographic studies of serotonin N-acetyltransferase catalysis and inhibition. J Mol Biol. 2002;317:215–224. doi: 10.1006/jmbi.2001.5371. [DOI] [PubMed] [Google Scholar]
- 41.Klein DC. Arylalkylamine N-acetyltransferase: “The timezyme”. J Biol Chem. 2007;282:4233–4237. doi: 10.1074/jbc.R600036200. [DOI] [PubMed] [Google Scholar]
- 42.Bewley MC, Graziano V, Jiang J, Matz E, Studier FW, Pegg AE, Coleman CS, Flanagan JM. Structures of wild-type and mutant human spermidine/spermine N1-acetyltransferase. A potential therapeutic drug target. Proc Natl Acad Sci USA. 2006;103:2063–2068. doi: 10.1073/pnas.0511008103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hegde SS, Chandler J, Vetting MW, Yu M, Blanchard JS. Mechanistic and structural analysis of human spermidine/spermine N1-acetyltransferase. Biochemistry. 2007;46:7187–7195. doi: 10.1021/bi700256z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanner KG, Trievel RC, Kuo MH, Howard RM, Berger SL, Allis CD, Marmorstein R, Denu JM. Catalytic mechanism and function of invariant glutamic acid 173 from the histone acetyltransferase GCN5 transcriptional coactivator. J Biol Chem. 1999;274:18157–18160. doi: 10.1074/jbc.274.26.18157. [DOI] [PubMed] [Google Scholar]
- 45.Yuan H, Marmorstein R. Histone acetyltransferases: Rising ancient counterparts to protein kinases. Biopolymers. 2013;99:98–111. doi: 10.1002/bip.22128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.The Merck Index. 12th. Merck and Co.; White House Station, NJ: 1996. pp. 417–418. [Google Scholar]
- 47.Pitman IH, Morris IJ. Coenzyme A: pKa and gamma values. Aust J Chem. 1980;33:1625–1630. [Google Scholar]
- 48.Hickman AB, Klein DC, Dyda F. Melatonin biosynthesis: The structure of serotonin N-acetyltransferase at 2.5 Å resolution suggests a catalytic mechanism. Mol Cell. 1999;3:23–32. doi: 10.1016/s1097-2765(00)80171-9. [DOI] [PubMed] [Google Scholar]
- 49.Pavlicek J, Coon SL, Ganguly S, Weller JL, Hassan SA, Sackett DL, Klein DC. Evidence that proline focuses movement of the floppy loop of arylalkylamine N-acetyltransferase (EC 2.3.1.87) J Biol Chem. 2008;283:14552–14558. doi: 10.1074/jbc.M800593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farazi TA, Manchester JK, Gordon JI. Transient-state kinetic analysis of Saccharomyces cerevisiae myristoyl-CoA:protein N-myristoyltransferase reveals that a step after chemical transformation is rate limiting. Biochemistry. 2000;39:15807–15816. doi: 10.1021/bi002074t. [DOI] [PubMed] [Google Scholar]
- 51.Draker KA, Northrop DB, Wright GD. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC(6′)-Ii from Enterococcus faecium: Evidence of dimer subunit cooperativity. Biochemistry. 2003;42:6565–6574. doi: 10.1021/bi034148h. [DOI] [PubMed] [Google Scholar]
- 52.Thoden JB, Cook PD, Schaffer C, Messner P, Holden HM. Structural and functional studies of QdtC: An N-acetyltransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-D-glucose. Biochemistry. 2009;48:2699–2709. doi: 10.1021/bi802313n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Thoden JB, Holden HM. Molecular structure of WlbB, a bacterial N-acetyltransferase involved in the biosynthesis of 2,3-diacetamido-2,3-dideoxy-D-mannuronic acid. Biochemistry. 2010;49:4644–4653. doi: 10.1021/bi1005738. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.