

Abstract
Due to its favorable spectroscopic properties, Cd2+ is frequently used as a probe of Ca2+ sites in proteins. We investigate the ability of Cd2+ to act as a structural and functional surrogate of Ca2+ in protein–membrane interactions. C2 domain from protein kinase Cα (C2α) was chosen as a paradigm for the Ca2+-dependent phosphatidylserine-binding peripheral membrane domains. We identified the Cd2+-binding sites of C2α using NMR spectroscopy, determined the 1.6 Å crystal structure of Cd2+-bound C2α, and characterized metal-ion-dependent interactions between C2α and phospholipid membranes using fluorescence spectroscopy and ultracentrifugation experiments. We show that Cd2+ forms a tight complex with the membrane-binding loops of C2α but is unable to support its membrane-binding function. This is in sharp contrast with Pb2+, which is almost as effective as Ca2+ in driving the C2α-membrane association process. Our results provide the first direct evidence for the specific role of divalent metal ions in mediating protein–membrane interactions, have important implications for metal substitution studies in proteins, and illustrate the potential diversity of functional responses caused by toxic metal ions.
Ca2+-dependent signaling is involved in the regulation of many cellular processes, including those that occur at the membrane surfaces.1–3 The changes in intracellular Ca2+ levels are transduced to the membranes by two families of Ca2+ effector proteins: annexins4,5 and conserved homology-2 (C2) domains.6–8 Both are peripheral membrane proteins that undergo membrane association in Ca2+-dependent manner, with specificity toward negatively charged phospholipids, such as phosphatidylserine (PtdSer).9–11
There are two views on the role of Ca2+ in mediating protein–membrane interactions. One view is that Ca2+ increases the electrostatic potential of the membrane-binding regions of the protein and thereby acts as a nonspecific “electrostatic switch”.12 The other view is that Ca2+ specifically recognizes PtdSer headgroup through the formation of coordination bonds with the carboxyl and/or phosphoryl oxygens.13,14 The latter mechanism implies that the coordination sphere of protein-bound Ca2+ is dynamic: the labile water molecules coordinated by protein-bound Ca2+ are replaced by the lipid headgroup upon membrane association.
Due to its favorable spectroscopic properties (spin I = 1/2), 113Cd2+ can potentially be used in lieu of Ca2+ to directly probe the changes in the metal coordination environment by NMR spectroscopy.15 In this work, we investigated the effect of Cd2+ on the structure and membrane-binding properties of the C2 domain from protein kinase Cα (C2α). Unexpectedly, we found that despite high affinity of Cd2+ to C2α and the similar coordination geometry of protein-bound Cd2+ and Ca2+, Cd2+ is unable to support the membrane-binding function of the protein. This is in sharp contrast with Pb2+ ions, whose coordination geometry in protein-bound state is different from that of Ca2+, yet their ability to drive the C2α-membrane association is comparable.16
Our findings shed light on the mechanism of metal-ion-dependent association of proteins with phospholipid membranes, illustrate how toxic metal ions can potentially interfere with Ca2+ signaling, and have important implications for designing metal substitution studies in proteins.
We used solution NMR to identify the Cd2+ binding sites in C2α. The response of individual cross-peaks to increasing Cd2+ concentration was monitored in a series of 15N-1H HSQC spectra. There are two groups of residues that respond differently to Cd2+ binding, as demonstrated for a selected subset in Figure 1. Group 1 falls into the slow-to-intermediate exchange regime on the NMR chemical-shift time scale, where the cross-peaks of the metal-complexed protein species appear at their final positions and gradually build up upon Cd2+ saturation. Group 2 shows fast-exchange behavior, where the cross-peak positions change smoothly in response to increasing Cd2+ concentration. These data indicate that there are two distinguishable types of Cd2+ binding sites in C2α, with different binding kinetics of Cd2+ ions. The apparent Cd2+ affinities are also quite different, with groups 1 and 2 residues saturating at ~300 µM and ~4 mM Cd2+, respectively.
Figure 1.
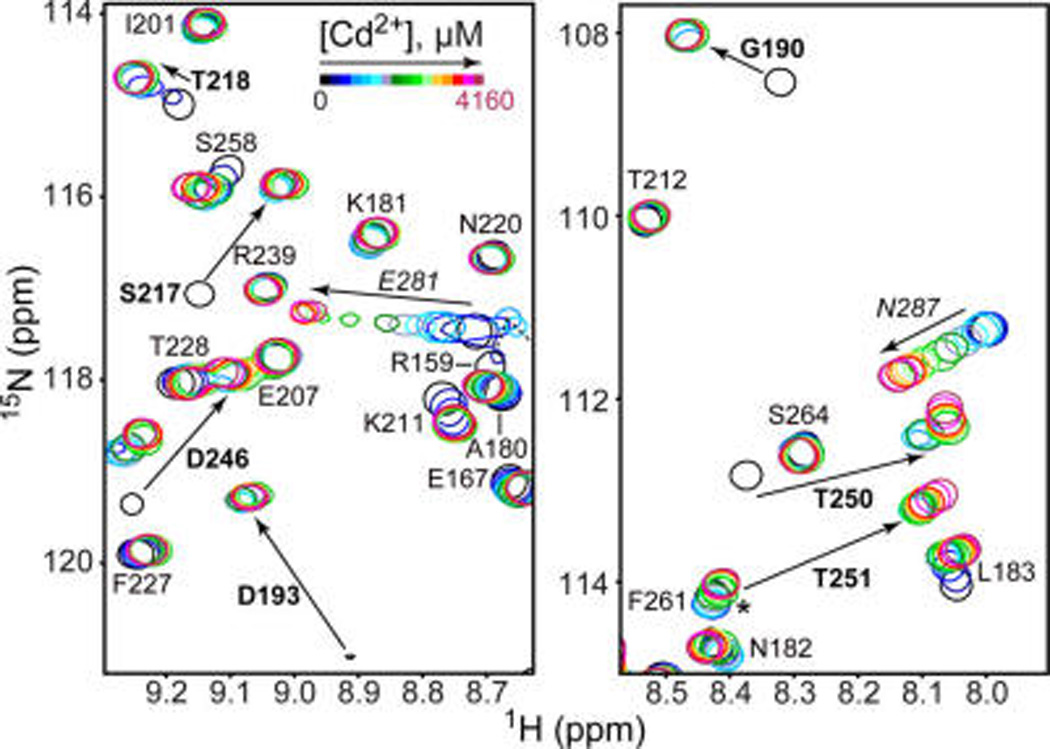
Two types of Cd2+ binding sites in C2α. Expansions of the 15N-1H HSQC spectra showing the chemical shift changes of 110 µM C2α in response to increasing Cd2+ concentration. Residues of groups 1 and 2 are shown in boldface and italics, respectively. Asterisk marks the position of the apo C2α T251 cross peak, which overlaps with F261.
To map the Cd2+ binding sites onto the structural elements of C2α, we exploited their differential Cd2+ affinities and conducted the chemical shift perturbation (CSP) analysis for two pairs of Cd2+ concentration points: 0/240 and 240/4160 µM. Site 1 is mostly formed by the Ca2+- and membrane-binding loops, or CMBLs (Figure 2A, shaded regions). According to the crystal structure of Ca2+-complexed C2α, CMBLs provide all ligands for the binuclear Ca2+ site. The CSP pattern in the low-concentration Cd2+ regime is very similar to that observed previously for the binding of two Ca2+ ions to C2α.16 This suggests that no less than two Cd2+ ions with similar affinities bind to the loop region. We used the fluorescence emission spectra of native tryptophan residues in C2α to estimate the affinity of Cd2+ to the high-affinity CMBL site. The shape of the binding curve indicates a significant degree of cooperativity (Figure 2B). The concentration of Cd2+ required to achieve half-maximal binding is 1.1 ± 0.1 µM. This value can be used as an upper limit for the apparent dissociation constant Kd, given that the total concentration of C2α is comparable (0.5 µM).
Figure 2.
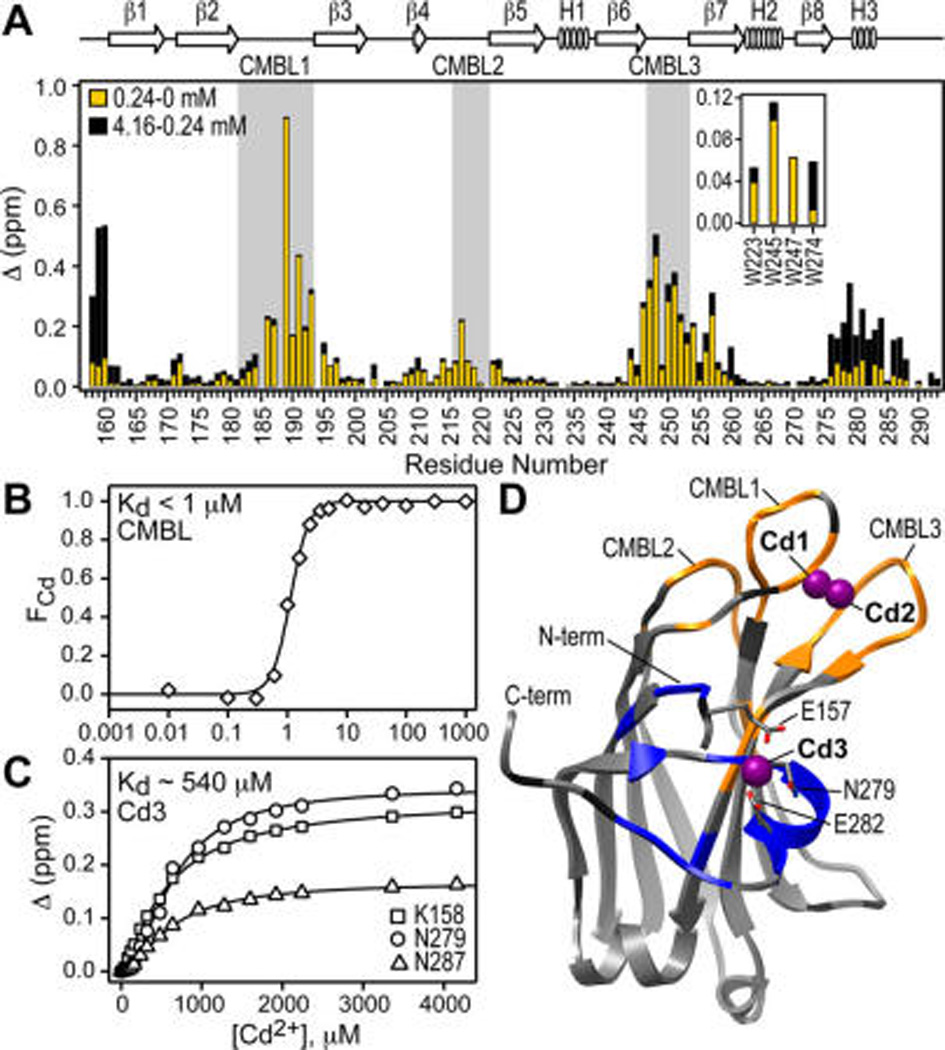
Cd2+ binds to two distinct C2α regions: CMBLs and N-terminal/Helix3 region. (A) Chemical shift perturbation Δ for low-(yellow) and high-concentration (black) Cd2+ regimes, demonstrating the presence of two types of sites. Inset shows Δ values for the indole N–H groups of Trp side chains. (B) Fraction of the Cd2+-complexed C2α, FCd, calculated using the change in the spectral center of mass of the Trp fluorescence emission spectra, plotted against Cd2+ concentration. (C) Representative NMR-detected Cd2+ binding curves for N-terminal/Helix3 region. (D) Crystal structure of the Cd2+-complexed C2α (PDB ID 4L1L). Residues showing intermediate-to-slow and fast exchange regimes upon Cd2+ binding are highlighted in orange and blue, respectively. Pro and residues with no data are dark gray.
The N-terminal/Helix3 region of C2α forms the low-affinity Cd2+ site (Figure 2A). This behavior is unique for Cd2+, as the binding of Ca2+ to this region of C2α has not been observed. The Cd2+ dissociation constant for this site can be estimated from the NMR data and is ~540 µM (Figure 2C). Note that the separation of the Cd2+ binding sites was possible due to the different total protein concentration regimes used for the fluorescence (0.5 µM) and NMR (110 µM) experiments.
To gain insight into the coordination geometry of protein-bound Cd2+, we determined the crystal structure of the complex. The structure was refined to 1.60 Å, with an R-free of 19.5%. Out of 6 Cd2+ ions bound to C2α, only 3 have 4 or more protein ligands: the binuclear Cd2+ cluster in the CMBL region, with occupancies of 1.0 (Cd1) and 0.72 (Cd2); and Cd3 located between the N-terminal region and Helix3, with the occupancy of 0.4 (Figure 2D). The crystal structure of the complex is in good agreement with the NMR and fluorescence data: there are at least two CMBL Cd2+ sites that are high-affinity (Figure 2B) and show intermediate-to-slow exchange regime with respect to Cd2+ binding (Figure 2D, orange); and there is one Cd2+ site that is low-affinity (Figure 2C) and shows fast exchange with respect to Cd2+ binding (Figure 2D, blue). All details of fluorescence, NMR, and X-ray crystallography experiments are given in the SI, Sections 1–3.
The coordination geometry of CMBL-bound Cd2+ is pentagonal bipyramidal and is very similar to that of Ca2+ (Figure 3A and Table S3). The distance between the two metal ions is 3.7 and 3.8 Å for Cd2+ and Ca2+, respectively. The protein ligands are: the carbonyl oxygens of W247 and M186 and the carboxyl oxygens of aspartate side chains in either bidentate (D187 and D248) or monodentate (D193, D246, and D254) coordination modes. The axial ligands of Cd1 and Cd2 sites are identical and include water molecules above and the carboxyl oxygens of the D246 side chain below the equatorial plane. Note that the Ca2+-complexed C2α structure contains a short-chain PtdSer analog, whose phosphoryl oxygens serve as axial ligands of Ca1.
Figure 3.
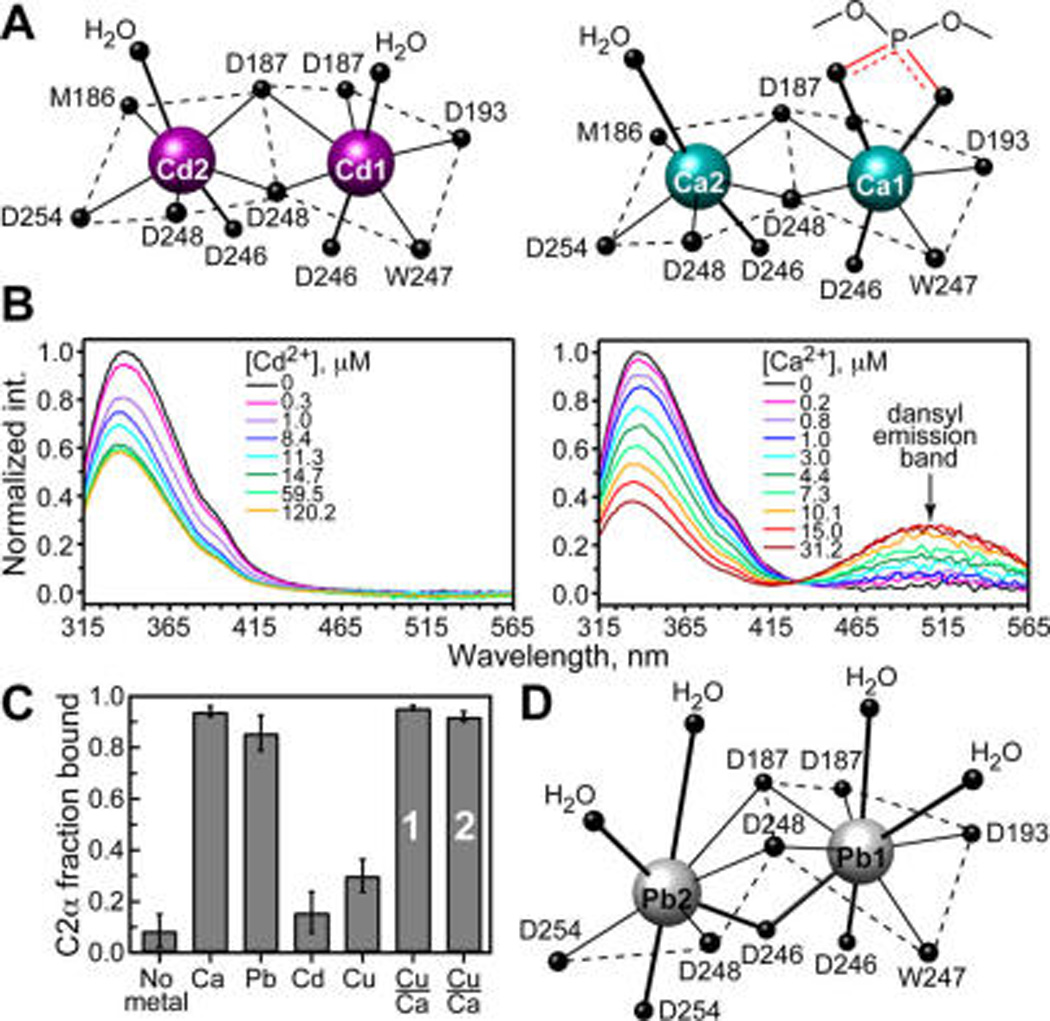
Coordination geometry of metal ions does not correlate with their ability to mediate C2α-membrane association. (A) Coordination geometry of CMBL-bound Cd2+ (4L1L) and Ca2+ (1DSY).13 (B) Fluorescence emission spectra, obtained as the difference between the spectra of C2α-containing and C2α-free dansyl-doped LUVs. Build-up of the dansyl band in Ca2+ experiments is due to protein–membrane FRET. (C) Fractional population of membrane-bound C2α obtained in ultracentrifugation binding assays with LUVs having 30% PtdSer component. Total concentration of metal ions was 175 µM, except for the Cu2+/Ca2+ displacement experiments that were carried out at 12.5/175 µM (bar 1) and 175/2450 µM (bar 2). (D) Coordination geometry of CMBL-bound Pb2+ (3TWY).16
Two independent methods were used to quantify the metal-ion-dependent association of C2α with membranes: protein-to-membrane FRET and lipid ultracentrifugation binding assays. FRET experiments relied on the native Trp residues and membrane-incorporated 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl (dansyl-PE) as donor and acceptor fluorophores, respectively.16,17 Titration of Ca2+ into solution containing C2α and large unilamellar vesicles (LUVs) comprising 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-l-serine/dansyl-PE at a molar ratio of 73:20:7 resulted in the increase of dansyl fluorescence at 505 nm due to the protein-to-membrane FRET. In contrast, no changes in dansyl fluorescence were observed in the Cd2+ titration experiments (Figure 3B). The absence of FRET cannot be attributed to the nonoptimal orientation of the transition dipoles because dansyl-PE is distributed randomly in the membrane. Cd2+ alone neither quenches dansyl fluorescence nor associates appreciably with the PtdSer component of the LUVs under the conditions of our experiments (Figures S6 and S7). We conclude that Cd2+ is unable to mediate the association of C2α with membranes. This was further demonstrated in the ultracentrifugation lipid-binding experiments, where the sucrose-loaded LUVs were incubated with C2α and a solution of the corresponding metal ion and then pelleted by ultracentrifugation. The amount of protein left in the supernatant was quantified and used to calculate the fraction bound to the membranes. Addition of Ca2+ results in full membrane binding, while addition of Cd2+ does not (Figure 3C).
These findings are surprising in the context of our previous study on Pb2+ interactions with C2α. In contrast to the structure of Ca2+-bound C2α, M186 does not coordinate to Pb1 via the carbonyl oxygen; D246 and D254 serve as bidentate ligands to Pb1 and Pb2, respectively; and instead of pentagonal bipyramidal, Pb2 adopts hemidirected coordination geometry, with all eight ligands accommodated in one hemisphere (Figure 3D). Despite these differences, Pb2+ is almost as effective as Ca2+ in driving the protein–membrane interactions (Figure 3C and our previous work).16
We attribute this behavior to the coordination preferences of metal ions. Cd2+ is a soft Lewis acid,18 with preference toward soft ligands, such as thiol groups of cysteine residues.19 Its most frequently encountered coordination number (CN) in proteins is either 4 or 6.20 The crystal structures of Ca2+-bound C2α13 and annexin V14 in complex with PtdSer analogs suggest that a Ca2+-coordinated water molecule is replaced by either phosphoryl and/or carboxyl oxygen(s) of PtdSer upon membrane association. This may be accompanied by an expansion of the metal ion coordination sphere, as shown for Ca2+-bound C2α in Figure 3A. While Cd2+ can adopt a hepta-coordinated state when bound to C2α, the rearrangement and possible expansion of its all-oxygen coordination sphere required for PtdSer interactions may be unfavorable. In contrast, Ca2+ is a hard Lewis acid that favors all-oxygen coordination environment with CNs of 6–8.21 Pb2+ is a borderline Lewis acid that interacts readily with oxygen-containing ligands and can adopt CNs up to 9 in proteins.22
To test if the ability of a metal ion to expand the coordination sphere is important for membrane binding, we characterized the interactions of C2α with Cu2+. Similar to Pb2+, Cu2+ is a borderline Lewis acid, albeit with a well-documented preference for CN = 4 in proteins.20 In addition, Cd2+ and Cu2+ have been used with success as a diamagnetic–paramagnetic pair in structural studies.23 In the presence of 50 µM Cu2+, cross-peaks of C2α residues in the N-terminal/Helix3 region broaden beyond detection due to the paramagnetic relaxation enhancement (PRE) caused by bound Cu2+ (Figure 4A). Increasing the concentration of Cu2+ to 150 µM results in efficient relaxation of all CMBL regions. These data suggest that Cu2+ populates the same set of C2α sites as Cd2+, albeit with the opposite pattern of affinities (Figures S8 and S9). The lipid-binding assays show that like Cd2+, Cu2+ is not an effective mediator of protein–membrane interactions: only ~30% of Cu2+-complexed C2α is membrane bound (Figure 3C). Reducing the concentration of PtdSer from 30% to 20% eliminates the membrane binding of C2α in the presence of Cu2+ (Figure S10).
Figure 4.
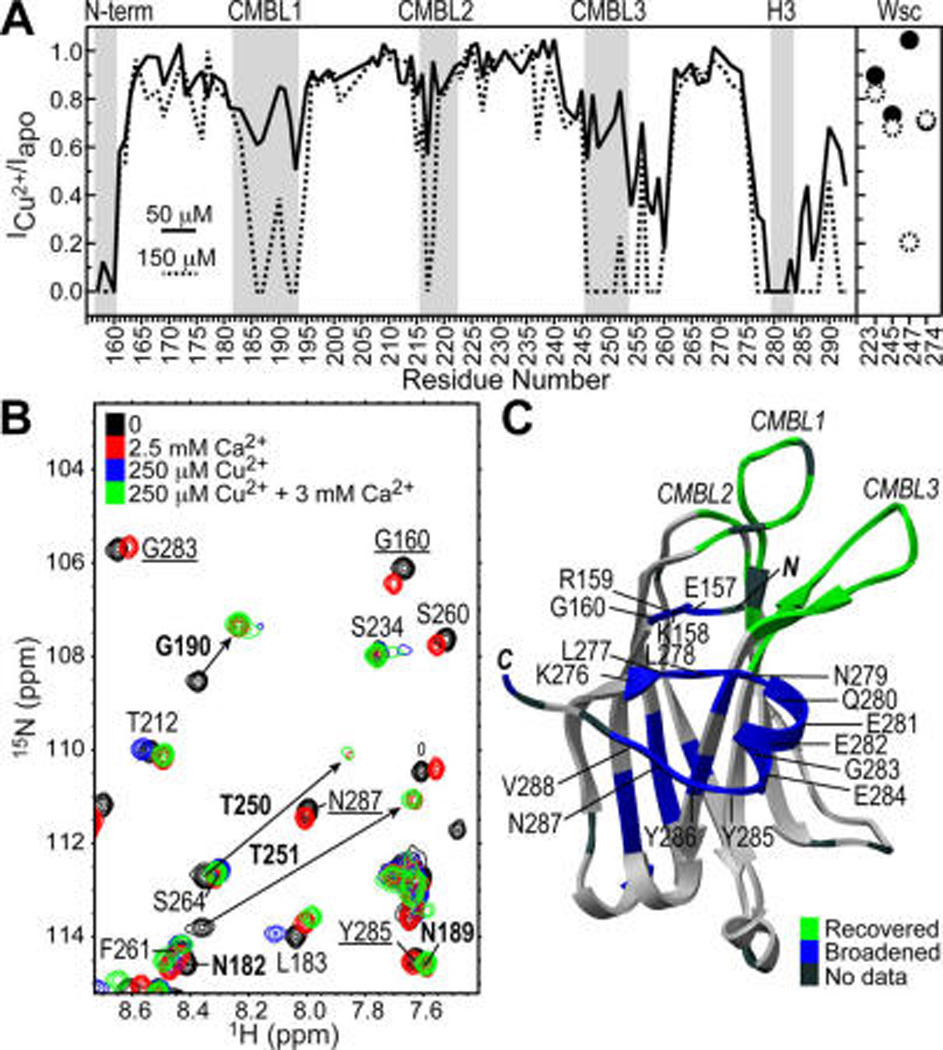
Ca2+ displaces Cu2+ from the CMBL region of C2α, with formation of mixed Ca2+/Cu2+-bound species. (A) PRE, calculated as ratio of NMR cross-peak intensities in Cu2+-bound C2α to those in apo C2α, for two Cu2+ concentrations: 50 and 150 µM. Data were normalized to residue K236. (B) Overlay of 15N-1H HSQC spectra of 100 µM C2α at several concentrations of Ca2+ and Cu2+. Residues that remain broadened upon addition of 12-fold Ca2+ excess over Cu2+ are underlined. Residues whose intensity fully recovers upon addition of Ca2+ are shown in boldface. Arrows indicate direction of cross-peaks’ shift due to Ca2+ binding. (C) Mapping of broadened residues (blue) and residues recovered upon Ca2+ addition (green) onto the structure of apo C2α (3RDJ).16
We carried out Ca2+/Cu2+ displacement experiments to assess the role of the N-terminal/Helix3 metal ion-binding site in protein–membrane interactions. In the membrane-free environment, the cross-peaks of CMBL residues that are broadened beyond detection in the presence of the 250 µM Cu2+ are recovered upon addition of 3.0 mM Ca2+ (Figure 4B,C). In contrast, the resonances in the vicinity of the N-terminal/Helix3 site remain broadened due to the presence of bound Cu2+. These data indicate the formation of mixed metal-ion species, with Ca2+ and Cu2+ bound to the CMBL and N-terminal/Helix3 regions of C2α, respectively. The ability of this species to associate with PtdSer-containing membranes was tested in ultracentrifugation lipid-binding experiments for two concentration regimes, with full membrane binding observed in both cases (Figure 3C, bars 1 and 2). Thus, having a divalent metal ion bound to the N-terminal/Helix3 site does not negatively affect the C2α membrane-binding properties.
Taken together, our findings support the view that a divalent metal ion plays a specific role in mediating protein–membrane interactions, rather than the nonspecific “electrostatic switch” model. All three non-native metal ions: Cd2+, Cu2+, and Pb2+ can increase the electrostatic potential of C2α through direct binding to the aspartate-rich CMBL region. However, the formation of metal ion-protein complex is not sufficient to promote protein–membrane association, as demonstrated for Cd2+ and Cu2+. We conclude that specific interactions of a divalent metal ion with PtdSer group(s) are required for productive protein–membrane interactions.
Favorable spectroscopic properties and the nearly identical ionic radius have made Cd2+ a popular substitute for Ca2+.15 Our results show that the preferred coordination geometry of metal ions, their ability to expand the coordination sphere, and the chemical identity of protein ligands need to be taken into account when designing metal substitution studies. This is particularly important for proteins that rely on metal ions to carry out a specific function. A case in point is the C2 domains, which are the second most abundant lipid-binding domain behind the PH:24 a UniProt25 search produced >140 different human C2-containing proteins whose function includes signal transduction and membrane trafficking.
Cd2+ is a toxic metal ion with no safe limit of exposure.26 In our study, the affinity of Cd2+ to the CMBL region exceeds the previously reported Ca2+ value27 >30-fold. It is plausible that bioavailable Cd2+ can influence the function of C2-domain containing proteins through a direct competition with Ca2+ for the CMBL regions. Our results demonstrate that toxic metal ions, such as Cd2+ and Pb2+, can elicit very different functional responses with respect to the in vitro membrane binding: Pb2+ promotes membrane association of C2α, whereas Cd2+ does not. Either scenario, if occurring in the cell, would lead to the aberration in the Ca2+ signaling response.
Supplementary Material
Acknowledgments
This work was supported in part by the NSF CAREER (CHE-1151435) and Welch Research Foundation (A-1784) awards to T.I.I.
Footnotes
ASSOCIATED CONTENT
Experimental details and crystallization data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Berridge MJ, Lipp P, Bootman MD. Nat. Rev. Mol. Cell Biol. 2000;1:11. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 2.Clapham DE. Cell. 2007;131:1047. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- 3.Patergnani S, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Giorgi C, Marchi S, Missiroli S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Cell Commun. Signaling. 2011;9:19. doi: 10.1186/1478-811X-9-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lizarbe MA, Barrasa JI, Olmo N, Gavilanes F, Turnay J. Int. J. Mol. Sci. 2013;14:2652. doi: 10.3390/ijms14022652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerke V, Creutz CE, Moss SE. Nat. Rev. Mol. Cell Biol. 2005;6:449. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 6.Nalefski EA, Falke JJ. Protein Sci. 1996;5:2375. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rizo J, Sudhof TC. J. Biol. Chem. 1998;273:15879. doi: 10.1074/jbc.273.26.15879. [DOI] [PubMed] [Google Scholar]
- 8.Corbalán-García S, Gómez-Fernández JC. BioFactors. 2010;36:1. doi: 10.1002/biof.68. [DOI] [PubMed] [Google Scholar]
- 9.Richter RP, Him JL, Tessier B, Tessier C, Brisson AR. Biophys. J. 2005;89:3372. doi: 10.1529/biophysj.105.064337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vats K, Knutson K, Hinderliter A, Sheets ED. ACS Chem. Biol. 2010;5:393. doi: 10.1021/cb900303s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stahelin RV, Rafter JD, Das S, Cho W. J. Biol. Chem. 2003;278:12452. doi: 10.1074/jbc.M212864200. [DOI] [PubMed] [Google Scholar]
- 12.Murray D, Honig B. Mol. Cell. 2002;9:145. doi: 10.1016/s1097-2765(01)00426-9. [DOI] [PubMed] [Google Scholar]
- 13.Verdaguer N, Corbalán-García S, Ochoa WF, Fita I, Gómez-Fernández JC. EMBO J. 1999;18:6329. doi: 10.1093/emboj/18.22.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swairjo MA, Concha NO, Kaetzel MA, Dedman JR, Seaton BA. Nat. Struct. Biol. 1995;2:968. doi: 10.1038/nsb1195-968. [DOI] [PubMed] [Google Scholar]
- 15.Armitage IM, Drakenberg T, Reilly B. Met. Ions Life Sci. 2013;11:117. doi: 10.1007/978-94-007-5179-8_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morales KA, Lasagna M, Gribenko AV, Yoon Y, Reinhart GD, Lee JC, Cho W, Li P, Igumenova TI. J. Am. Chem. Soc. 2011;133:10599. doi: 10.1021/ja2032772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nalefski EA, Falke JJ. Methods Mol. Biol. 2002;172:295. doi: 10.1385/1-59259-183-3:295. [DOI] [PubMed] [Google Scholar]
- 18.Pearson RG. J. Am. Chem. Soc. 1963;85:3533. [Google Scholar]
- 19.Chakraborty S, Kravitz JY, Thulstrup PW, Hemmingsen L, DeGrado WF, Pecoraro VL. Angew. Chem., Int. Ed. Engl. 2011;50:2049. doi: 10.1002/anie.201006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rulíšek L, Vondrášek J. J. Inorg. Biochem. 1998;71:115. doi: 10.1016/s0162-0134(98)10042-9. [DOI] [PubMed] [Google Scholar]
- 21.Kirberger M, Wang X, Deng H, Yang W, Chen G, Yang JJ. J. Biol. Inorg. Chem. 2008;13:1169. doi: 10.1007/s00775-008-0402-7. [DOI] [PubMed] [Google Scholar]
- 22.Kirberger M, Yang JJ. J. Inorg. Biochem. 2008;102:1901. doi: 10.1016/j.jinorgbio.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen MR, Hansen DF, Ayna U, Dagil R, Hass MAS, Christensen HEM, Led JJ. Magn. Reson. Chem. 2006;44:294. doi: 10.1002/mrc.1771. [DOI] [PubMed] [Google Scholar]
- 24.Cho W, Stahelin RV. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2006;1761:838. [Google Scholar]
- 25.UniProt Consortium. Nucleic Acids Res. 2012;40:D71. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thévenod F, Lee WK. Met. Ions Life Sci. 2013;11:415. doi: 10.1007/978-94-007-5179-8_14. [DOI] [PubMed] [Google Scholar]
- 27.Kohout SC, Corbalán-García S, Torrecillas A, Gómez-Fernández JC, Falke JJ. Biochemistry. 2002;41:11411. doi: 10.1021/bi026041k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.