

Abstract
The spindle assembly checkpoint (SAC) monitors chromosome attachment defects and the assembly of SAC proteins at kinetochores is essential for its activation, but the SAC disassembly process remains unknown. We found that deletion of a 14-3-3 protein, Bmh1, or hyper-activation of FEAR (Cdc14 Early Anaphase Release) allows premature SAC silencing in budding yeast, which depends on a kinetochore protein Fin1 that forms a complex with protein phosphatase PP1. Previous works suggest that FEAR-dependent Fin1 dephosphorylation promotes Bmh1-Fin1 dissociation, which enables kinetochore recruitment of Fin1-PP1. We found persistent kinetochore association of SAC protein Bub1 in fin1Δ mutants after anaphase entry. Therefore, we revealed a mechanism that clears SAC proteins from kinetochores. After anaphase entry, FEAR activation promotes kinetochore enrichment of Fin1-PP1, resulting in SAC disassembly at kinetochores. This mechanism is required for efficient SAC silencing after SAC being challenged, and untimely Fin1-kinetochore association causes premature SAC silencing and chromosome missegregation.
Keywords: The spindle assembly checkpoint, Bmh1, Fin1, PP1, FEAR pathway
INTRODUCTION
The kinetochore is the docking site for the spindle assembly checkpoint (SAC) that monitors the defects in chromosome attachment and blocks anaphase onset. Once all chromosomes have achieved bipolar attachment, this checkpoint is silenced to allow anaphase onset, but the SAC silencing process and its disassembly after silencing are not fully understood. Recent evidence indicates that modulation of the phosphorylation status of various proteins at the kinetochore is critical for SAC silencing. Kinetochore-associated protein phosphatase PP1 has been shown to be essential for SAC silencing in yeast cells (Pinsky et al., 2009; Vanoosthuyse and Hardwick, 2009). Moreover, down-regulation of SAC kinase Mps1 also promotes this silencing process (Aravamudhan et al., 2015; Hiruma et al., 2015; Ji et al., 2015). Bipolar attachment generates tension on chromosomes, while lack of tension delays anaphase onset through a conserved protein kinase Ipl1/Aurora B and a centromere binding protein Sgo1 (Biggins and Murray, 2001; Indjeian et al., 2005; Jin et al., 2012). Our recent work indicates that Ipl1-dependent phosphorylation of kinetochore protein Dam1 is sufficient to block SAC silencing (Jin and Wang, 2013). Because Dam1 phosphorylation is reversed by PP1 only after tension generation (Keating et al., 2009; Pinsky et al., 2006a), tension may trigger SAC silencing by inducing Dam1 dephosphorylation.
In budding yeast, PP1 interacts with two kinetochore proteins, Spc105 and Fin1, and mutation of the PP1 binding motif in Spc105 blocks anaphase onset, indicating the critical role of PP1-Spc105 interaction in SAC silencing (Rosenberg et al., 2011). Fin1 was shown to associate with the spindle and spindle poles during anaphase (Woodbury and Morgan, 2007a), but later work indicates that it is also a kinetochore associated protein (Akiyoshi et al., 2009). In addition, Fin1 is a substrate of S-phase cyclin-dependent kinase (CDK) and this phosphorylation is reversed during early anaphase by the phosphatase Cdc14, the chief component of the FEAR pathway (Cdc14 Early Anaphase Release) (Jin et al., 2008; Loog and Morgan, 2005). However, the physiological significance of Fin1 dephosphorylation remains ambiguous.
Prior to anaphase entry, Cdc14 is sequestered within the nucleolus through its association with a nucleolar protein Net1/Cfi1 (Shou et al., 1999; Visintin et al., 1999), which prevents Cdc14 from accessing its nuclear substrates, including Fin1. The release of Cdc14 from the nucleolus is regulated by the phosphorylation of Net1, a substrate of both Clb2/Cdk1 kinase and protein phosphatase PP2ACdc55 (Azzam et al., 2004; Queralt et al., 2006; Wang and Ng, 2006). Before anaphase onset, PP2ACdc55-mediated Net1 dephosphorylation enables Cdc14 sequestration within the nucleolus. Upon anaphase entry, FEAR activation triggers Net1 phosphorylation and Cdc14 release into the nucleus (Queralt et al., 2006). Deletion of PP2A regulatory gene CDC55 leads to premature Cdc14 release (Liang et al., 2009; Wang and Ng, 2006). FEAR-dependent Cdc14 release specifically reverses the phosphorylation imposed by S-phase CDK to facilitate anaphase progression (Jin et al., 2008; Liang et al., 2013). Fin1 is a verified S-phase CDK substrate (Loog and Morgan, 2005), and its phosphorylation promotes its interaction with 14-3-3 proteins, Bmh1 and Bmh2, which prevents the kinetochore association of Fin1 (Akiyoshi et al., 2009; Mayordomo and Sanz, 2002). Since Fin1 binds to PP1, Fin1 dephosphorylation during anaphase could promotes the kinetochore recruitment of Fin1-PP1.
Chromosomes lack tension when sister-chromatid cohesion is eliminated or sister kinetochores are attached by microtubules from the same spindle pole (syntelic attachments). We found that inactivation of the Cik1/Kar3 motor complex increases the frequency of syntelic attachments. Additionally, overexpression of the coiled-coil domain of Cik1 (Cik1-CC) disrupts Cik1-Kar3 interaction, and cells overexpressing Cik1-CC require Ipl1 and Sgo1 to prevent anaphase entry for survival (Jin et al., 2012; Jin and Wang, 2013). We performed a screen for yeast mutants that are sensitive to CIK1-CC overexpression in order to identify more SAC regulators. We found that bmh1Δ mutants and cells with hyperactive FEAR (cdc55Δ swe1Δ) show obvious viability loss after CIK-CC overexpression. These mutant cells exhibit chromosome missegregation and early dephosphorylation of SAC proteins in the presence of tension defects, indicating premature anaphase entry. Interestingly, bmh1Δ and cdc55Δ swe1Δ mutants show untimely Fin1 kinetochore localization, and the premature anaphase entry in these mutants partially depends on Fin1. Moreover, cells with phospho-deficient Fin1-5A protein also show early Fin1 kinetochore localization and sensitivity to syntelic attachments. Importantly, we found that Fin1 is required for the removal of a SAC protein Bub1 from kinetochores. Therefore, our data suggest that the recruitment of the Fin1-PP1 complex onto kinetochores promotes SAC disassembly during anaphase.
RESULTS
Bmh1 is required for the survival of yeast cells with induced syntelic attachments
We showed that overexpression of the coiled-coil domain of CIK1 (CIK1-CC) disrupts Cik1-Kar3 interaction and induces syntelic attachments (Jin et al 2012). Bmh1 and Bmh2 are the two 14-3-3 homologues in budding yeast and the kar3Δ mutant is synthetically lethal with bmh1Δ, but not with bmh2Δ mutants (Tong et al., 2004). To investigate the role of Bmh1 and Bmh2 in response to syntelic attachment, we introduced a plasmid with CIK1-CC under the control of a galactose-inducible promoter (PGALCIK1-CC) into bmh1Δ and bmh2Δ deletion mutants. Their growth was examined on plates containing galactose, which induces CIK1-CC overexpression. Compared to wild-type (WT) cells, bmh1Δ mutants with PGALCIK1-CC plasmid showed more severe sick growth on a galactose plate, although the phenotype was not as dramatic as sgo1Δ mutant. bmh2Δ cells overexpressing CIK1-CC did not show more severe growth defect than WT cells (Fig. 1A), indicating different roles for Bmh1 and Bmh2 in response to syntelic attachments.
Figure 1.
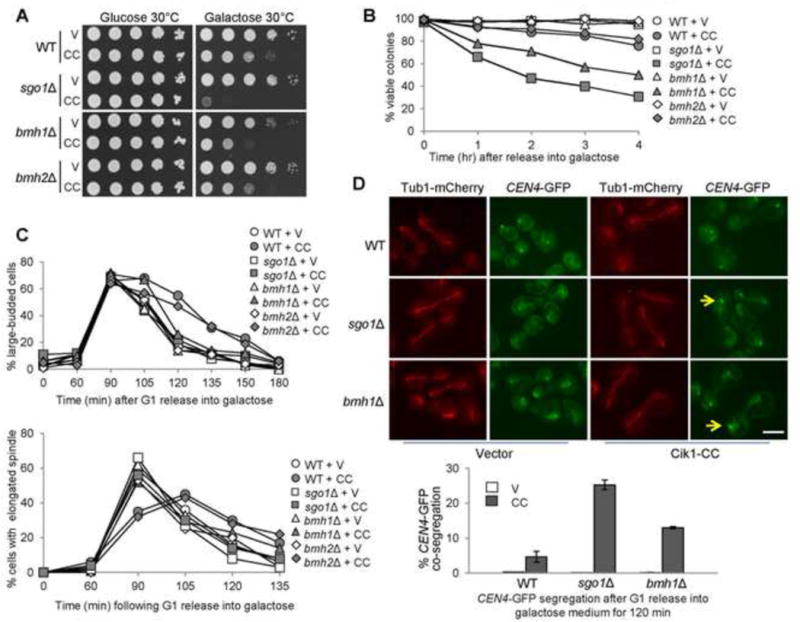
bmh1Δ cells are sensitive to CIK1-CC overexpression. (A) bmh1Δ cells overexpressing CIK1-CC show slow growth. Saturated cells with the indicated genotypes were 10-fold serial diluted, spotted onto glucose and galactose plates, and then incubated at 30°C for 2 days before scanning. V (vector), CC (PGALCIK1-CC). (B) bmh1Δ and sgo1Δ mutants, but not bmh2Δ, exhibit viability loss after CIK1-CC overexpression. Log-phase cells in raffinose were released into 2% galactose medium. Samples were taken and spread onto YPD plates to examine micro-colony formation after overnight incubation at 25°C (n > 300). (C) bmh1Δ and sgo1Δ but not bmh2Δ exhibit premature anaphase entry in the presence of tensionless attachments. PGALCIK1-CC plasmids were introduced to WT, bmh1Δ, sgo1Δ, and bmh2Δ cells with Tub1-mCherry. The transformants were synchronized in G1 phase in raffinose medium and using α factor and then released into galactose medium. Cells were collected and fixed to count the budding index and percentage of cells with elongated spindles (n > 100). (D) bmh1Δ and sgo1Δ cells overexpressing CIK1-CC show chromosome missegregation. PGALCIK1-CC plasmids were introduced to WT, bmh1Δ, and sgo1Δ cells with Tub1-mCherry and GFP-marked centromere of chromosome IV (CEN4-GFP). The transformants were synchronized in G1 in raffinose medium and then released into galactose-containing medium. After release for 120 min, the cells were fixed to examine the spindle morphology and CEN4-GFP segregation. Some representative cells are shown. Arrows indicate the cells with co-segregated CEN4-GFP. Scale bar = 5 μm. The percentage of cells with CEN4-GFP co-segregation was counted (n > 100). The result is the average of three repeats (bottom).
The dramatic slow growth phenotype of bmh1Δ cells overexpressing CIK1-CC could be a consequence of a synthetic defect in kinetochore attachment, or due to a checkpoint defect that leads to premature anaphase entry, resulting in chromosome missegregation and viability loss. We examined the viability of bmh1Δ and bmh2Δ cells overexpressing CIK1-CC. After Cik1-CC induction, both bmh1Δ and sgo1Δ mutants showed obvious viability loss. Only 31% of sgo1Δ cells and 46% of bmh1Δ cells remained viable after Cik1-CC induction for 4 hrs. In contrast, most of the WT and bmh2Δ cells were viable (Fig. 1B), suggesting Bmh1, but not Bmh2, is required for the response to syntelic attachment. Furthermore, we used synchronous cells to analyze cell cycle progression during CIK1-CC overexpression. The budding index and the spindle elongation kinetics indicated similar cell cycle progression in WT and bmh1Δ cells. As shown previously, CIK1-CC overexpression induced a moderate but obvious cell cycle delay in WT cells (Jin et al., 2012). This delay was abolished in sgo1Δ and bmh1Δ mutants, but not in bmh2Δ (Fig. 1C). We also examined Pds1 protein levels to follow the anaphase entry process. WT cells (PDS1-18myc) overexpressing CIK1-CC exhibited a clear delay in Pds1 degradation, but this delay was abolished in sgo1Δ cells (Fig. S1). bmh1Δ PDS1-18myc cells exhibited slow growth, as evidenced by delayed Pds1 turnover, nevertheless CIK1-CC overexpression did not further delay anaphase entry as it did in the WT cells (Fig. S1), indicating the potential role of Bmh1 in the anaphase entry delay induced by syntelic attachments.
In sgo1Δ and ipl1-321 mutants overexpressing CIK1-CC, premature anaphase entry causes a high frequency of chromosome missegregation (Jin et al., 2012). We examined sister chromatid segregation in bmh1Δ cells overexpressing CIK1-CC. G1-arrested WT, sgo1Δ, and bmh1Δ cells with GFP-marked centromere of chromosome IV (CEN4-GFP), Tub1-mCherry, and PGALCIK1-CC plasmids were released into galactose medium. After release for 2 hrs, we examined the GFP signal in cells with an elongated spindle (Tub1-mCherry). For vector control, most of the cells with an elongated spindle showed two separated GFP dots with the spindle poles. When CIK1-CC is overexpressed, almost no CEN4-GFP dots co-segregation was observed in WT cells, whereas sgo1Δ cells showed a co-segregation frequency of 25%. bmh1Δ cells with an elongated spindle also displayed CEN4-GFP co-segregation with a frequency of 14% (Fig. 1D). Our data indicate that some bmh1Δ cells enter anaphase in the presence of syntelic attachment, resulting in chromosome missegregation. The different CEN4-GFP co-segregation rates in bmh1Δ and sgo1Δ mutants indicates that Sgo1 and Bmh1 may play distinct roles in response to syntelic attachments.
bmh1Δ cells show premature SAC silencing in the presence of tension defects
Since premature SAC silencing leads to anaphase entry in sgo1Δ cells with tensionless attachments (Jin and Wang, 2013), we assessed the SAC silencing process in bmh1Δ cells using cohesin mutant mcd1-1. Incubation of mcd1-1 cells at 37°C inactivates cohesin Mcd1 and results in tensionless attachments. The phosphorylation of SAC protein Mad1 indicates checkpoint activation (Hardwick and Murray, 1995; Mirchenko and Uhlmann, 2010), thus we examined Mad1 modification kinetics in synchronized WT, bmh1Δ, mcd1-1, and mcd1-1 bmh1Δ cells growing at 37°C. Both WT and bmh1Δ alone showed weak phosphorylated Mad1 at 60 and 75 min after G1 release, whereas mcd1-1 cells exhibited more persistent Mad1 phosphorylation. In clear contrast, the phospho-variant of Mad1 began to dissipate after 90 min and disappeared at 150 min in mcd1-1 bmh1Δ cells. In line with this result, mcd1-1 bmh1Δ mutants showed less large-budded cells than the mcd1-1 mutants at later time points, indicating the bypass of metaphase arrest (Fig. 2A).
Figure 2.
bmh1Δ cells show premature SAC silencing in response to tension defects. (A) Mad1 phosphorylation kinetics in bmh1Δ cells in the absence of cohesion. MAD1-3HA cells with the indicated genotypes were synchronized in G1 and then released into 37°C YPD medium. The cells were collected over time to examine Mad1 phosphorylation based on the band-shift after western blotting. The budding index and the Mad1 protein levels are shown. (B) The phosphorylation kinetics of Bub1 in bmh1Δ mutant cells in the absence of cohesion. G1-arrested cells with Bub1-13myc were released into YPD at 37°C. Samples were collected periodically and subjected to western blotting with an anti-myc antibody. The budding index and the phosphorylation of Bub1 are shown.
In addition to Mad1, we also examined the phosphorylation kinetics of another SAC protein Bub1 using a similar protocol. WT and bmh1Δ cells showed dramatic Bub1 phosphorylation at the 45 and 60 min, but the top band disappeared by 75 min. mcd1-1 cells maintained Bub1 hyperphosphorylation throughout the time course. The top Bub1 phospho-variant appeared normally in mcd1-1 bmh1Δ cells, but disappeared after 90 min, much earlier than mcd1-1 cells (Fig. 2B). These results support the conclusion that Bmh1 is required for sustained phosphorylation of SAC checkpoint proteins Mad1 and Bub1 in cells with tension defects. Because both Mad1 and Bub1 can be phosphorylated efficiently in bmh1Δ mutants, we speculate that Bmh1 is dispensable for SAC activation. Indeed, bmh1Δ cells exhibited persistent Pds1 stabilization in the presence of microtubule poison nocodazole, indicating competent metaphase arrest (Fig. S2A). Moreover, the viability loss of bmh1Δ mutant in the presence of nocodazole is moderate (Fig. S2B).
The function of Bmh1 in checkpoint regulation depends on Fin1
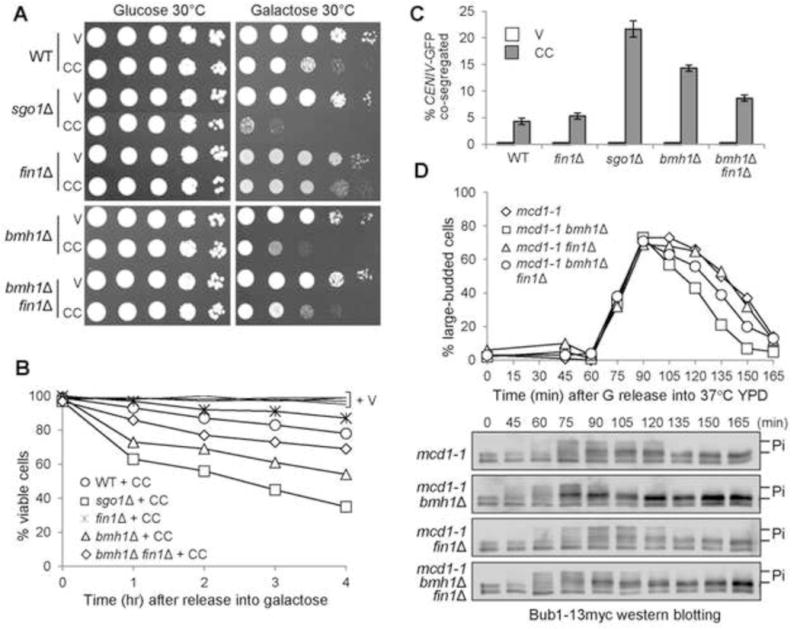
The kinetochore protein Fin1 binds to both Bmh1 and protein phosphatase PP1, but only CDK-phosphorylated Fin1 binds to Bmh1, which prevents kinetochore association of Fin1-PP1 (Akiyoshi et al., 2009). Thus, the premature SAC silencing observed in bmh1Δ could be attributed to precocious kinetochore association of Fin1-PP1. To test this idea, we first compared the growth of bmh1Δ single and bmh1Δ fin1Δ double mutant cells overexpressing CIK1-CC. The double mutant cells harboring a PGALCIK1-CC plasmid showed better growth than bmh1Δ single mutants on galactose plates, but not as well as WT cells, indicating a partial rescue (Fig. 3A). We further assessed their viability after CIK1-CC overexpression. In agreement with the growth on galactose plates, fin1Δ partially suppressed the viability loss of bmh1Δ (Fig. 3B). After incubation in galactose medium for 4 hrs, 54% of bmh1Δ cells were viable, but this number was 69% for bmh1Δ fin1Δ cells. Deleting FIN1 also partially rescued the chromosome segregation defect in bmh1Δ cells when syntelic attachment is induced by CIK1-CC overexpression (Fig. 3C). bmh1Δ fin1Δ mutants overexpressing CIK1-CC showed less chromosome missegregation after release from G1 arrest for 120 min, at 9%, compared with bmh1Δ cells at 14%.
Figure 3.
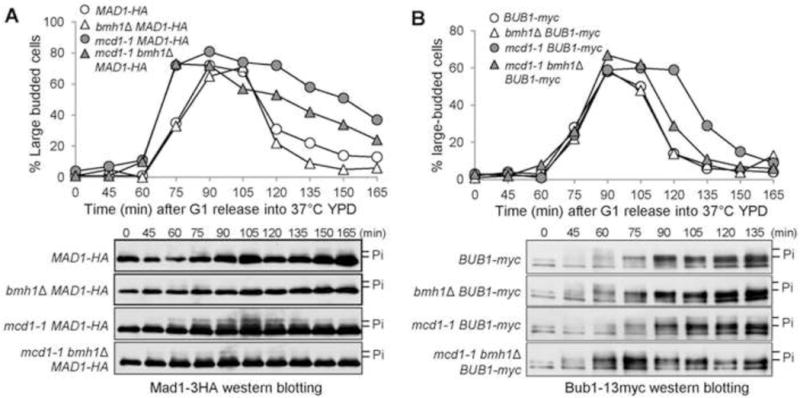
The premature SAC silencing in bmh1Δ mutants is partially Fin1-dependent. (A) Cells were 10-fold diluted and spotted onto glucose and galactose plates. The plates were incubated at 30°C for 2 days before scanning. (B) fin1Δ deletion partially suppresses the viability loss of bmh1Δ cells overexpressing CIK1-CC. Asynchronous cells with either a vector or a PGALCIK1- CC plasmid were grown to log-phase in raffinose medium and then galactose was added to a final concentration of 2%. The cells were collected over time and spread onto YPD plates to determine the plating efficiency after overnight incubation. (C) fin1Δ deletion partially suppresses chromosome mis-segregation in bmh1Δ mutant cells. G1-arrested CEN4-GFP TUB1-mCherry cells with indicated genotypes in raffinose medium were released into galactose medium for 120 min at 30°C. The cells were collected to visualize the spindle morphology and CEN4-GFP distribution. The percentage of cells with co-segregated CEN4-GFP was counted (n > 100). The percentage is the average from three independent experiments. (D) fin1Δ deletion partially suppresses the premature Bub1 dephosphorylation in bmh1Δ cells lacking tension. BUB1-13myc cells with the indicated genotypes were synchronized in G1 phase and then released into YPD at 37°C to inactivate cohesin Mcd1. The cells were collected every 15 min for the budding index and the examination of Bub1 phosphorylation.
Because mcd1-1 bmh1Δ mutant cells exhibit earlier Bub1 dephosphorylation than mcd1-1 cells (Fig. 2B), we also assessed whether this phenotype depends on the presence of Fin1. We analyzed the kinetics of Bub1 phosphorylation in synchronous mcd1-1 bmh1Δ and mcd1-1 bmh1Δ fin1Δ mutant cells incubated at 37°C. As shown previously, mcd1-1 displayed postponed Bub1 dephosphorylation, and complete disappearance of the top band occurred 30 min earlier in mcd1-1 bmh1Δ mutant cells. However, mcd1-1 bmh1Δ fin1Δ mutants exhibited delayed Bub1 dephosphorylation compared to mcd1-1 bmh1Δ mutants (Fig. 3D). Together, these results support the conclusion that the premature anaphase entry in bmh1Δ mutant cells at least partially depends on Fin1.
Hyperactive FEAR results in sensitivity to induced syntelic attachment
Bmh1 interacts with Fin1 and the phosphorylation of Fin1 by S-phase CDK promotes this interaction (Akiyoshi et al., 2009; Mayordomo and Sanz, 2002). FEAR-induced Cdc14 release during early anaphase reverses the phosphorylation imposed by S-phase CDK (Jin et al., 2008). Thus hyperactive FEAR will compromise Fin1-Bmh1 interaction and promote SAC silencing. PP2ACdc55 dephosphorylates Net1 to facilitate Net1-Cdc14 interaction and the nucleolar localization of Cdc14, whereas the phosphorylation of Net1 by Clb2/Cdk1 promotes Cdc14 release (Queralt et al., 2006; Wang and Ng, 2006). cdc55Δ swe1Δ mutants, which lacks both of the PP2A regulatory subunit Cdc55 and the negative regulator of Clb2/Cdk1 Swe1, show hyperactive FEAR (Liang et al., 2009; Liu and Wang, 2006). To determine the role of FEAR in SAC regulation, we first examined the sensitivity of cdc55Δ swe1Δ mutants to syntelic attachment induced by CIK1-CC overexpression. WT cells showed slow growth after CIK1-CC overexpression as described, but cdc55Δ swe1Δ mutants with PGALCIK1-CC failed to grow on galactose plates (Fig. S3A). cdc55Δ single mutant cells also exhibited sensitivity to CIK1-CC overexpression, but were not as dramatic as cdc55Δ swe1Δ mutants, presumably due to the more active FEAR in the double mutants.
Previous work showed that Net1 phosphorylation by Clb2/Cdk1 triggers Cdc14 release, and mutation of the six phosphorylation sites (net1-6Cdk) blocks FEAR activation and the premature Cdc14 release in cdc55Δ mutants (Azzam et al., 2004; Liang et al., 2013). To further assess if the sensitivity of cdc55Δ swe1Δ cells to syntelic attachment is due to hyperactive FEAR, we compared the growth of cdc55Δ swe1Δ and cdc55Δ swe1Δ net1-6Cdk cells overexpressing CIK1-CC. Obviously, net1-6Cdk was able to rescue the growth of cdc55Δ swe1Δ on galactose plates (Fig. S3A). In addition, the viability loss of cdc55Δ swe1Δ induced by CIK1-CC overexpression was also partially restored by net1-6Cdk (Fig. S3B). cdc55Δ mutants are sensitive to microtubule-depolymerizing agents benomyl and nocodazole (Wang and Burke, 1995). net1-6Cdk mutant also suppressed the growth defect of cdc55Δ swe1Δ cells on benomyl plates and their viability loss after nocodazole treatment (Fig. S3C and D). We further examined if CIK1-CC overexpression delays FEAR activation in cdc15-2 cells, in which the Cdc14 release is dependent exclusively on the FEAR (Stegmeier et al., 2002), and we observed a clear delay of Cdc14 release from the nucleolus (Fig. S4). Taken together, these results suggest that syntelic attachment delays FEAR activation, but cells with hyperactive FEAR are sensitive to the syntelic attachments.
Cells with hyperactive FEAR show premature SAC silencing
Next, we asked if CIK1-CC-induced viability loss in cdc55Δ swe1Δ mutants is due to chromosome missegregation. After G1 release for 120 minutes, the localization of CEN4-GFP in cells with an elongated spindle was examined. In the absence of CIK1-CC overexpression, almost no missegregation was observed. When CIK1-CC is overexpressed, however, cdc55Δ swe1Δ cells showed a missegregation frequency of 16%, nevertheless most of WT cells showed normal CEN4-GFP segregation (Fig. 4A). Consistent with premature SAC silencing, cdc55Δ swe1Δ cells overexpressing CIK1-CC exhibited less large-budded cells at later time points compared to WT cells. Therefore, in the presence of syntelic attachment, hyperactive FEAR causes chromosome missegregation, phenotypically similar to bmh1Δ. To examine SAC silencing process in cells with hyperactive FEAR, we also compared Bub1 phosphorylation kinetics in synchronized mcd1-1 and mcd1-1 cdc55Δ swe1Δ cells incubated at 37°C. mcd1-1 cells exhibited delayed Bub1 dephosphorylation, but the phospho-variant of Bub1 was reduced significantly after 90 min in mcd1-1 cdc55Δ swe1Δ cells following G1 release. Consistently, mcd1-1 cdc55Δ swe1Δ mutants showed less large-budded cells than mcd1-1 mutants at later time points (Fig. 4B). Therefore, cells with hyperactive FEAR are unable to maintain Bub1 hyperphosphorylation in the presence of tensionless attachments.
Figure 4.
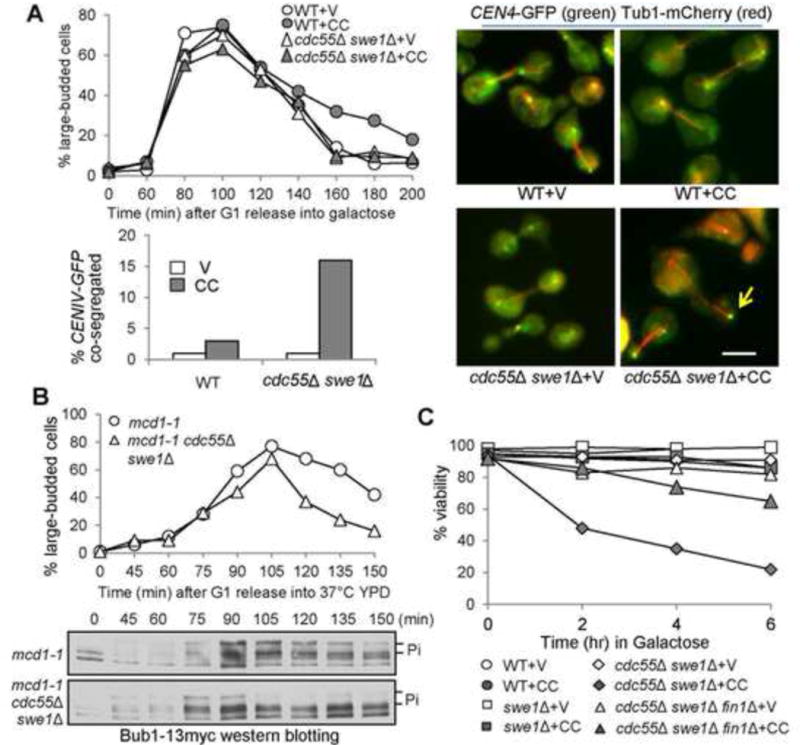
Cells with hyperactive FEAR are sensitive to syntelic attachment and show chromosome mis-segregation. (A) CIK1-CC overexpression causes chromosome mis-segregation in cdc55Δ swe1Δ cells. PGALCIK1-CC plasmids were introduced to WT and cdc55Δ swe1Δ cells with Tub1-mCherry and CEN4-GFP. The transformants were synchronized in G1 in raffinose medium and then released into galactose medium. After release for 120 min, the cells were fixed to examine the spindle morphology and CEN4-GFP segregation. The percentage of cells with CEN4-GFP co-segregation was counted (n > 100). Some representative cells are shown in the right panel. The arrow indicates a cell with co-segregated CEN4-GFP. Scale bar = 5 μm. (B) cdc55Δ swe1Δ cells exhibit premature Bub1 dephosphorylation in the presence of tensionless attachments. G1-arrested mcd1-1 and mcd1-1 cdc55Δ swe1Δ cells with Bub1-13myc were released into YPD at 37°C. Samples were collected periodically and subjected to western blotting. Here shows the budding index as well as the phosphorylation of Bub1. (C) The viability loss of cdc55Δ swe1Δ mutants induced by CIK1-CC overexpression is partially Fin1-dependent. Log-phase cells in raffinose were released into 2% galactose medium. Samples were taken every 2 hr and spread onto YPD plates. After incubation at 25°C overnight, the percentage of viable cells was counted (n > 300).
If the FEAR regulates Fin1 sequestration through Bmh1, then deletion of FIN1 is expected to rescue the sensitivity of cdc55Δ swe1Δ as well. We found that fin1Δ suppressed the viability loss in cdc55Δ swe1Δ cells overexpressing CIK1-CC (Fig. 4C). After 6 hr induction of Cik1-CC in galactose, only 22% of cdc55Δ swe1Δ cells were viable, but the viability rate increased to 65% for cdc55Δ swe1Δ fin1Δ cells. Deletion of FIN1 also partially suppressed the growth defect of cdc55Δ swe1Δ cells with PGALCIK1-CC on galactose plates (Fig. S5). Thus, we conclude that the FEAR promotes SAC silencing partially through Fin1.
The FEAR and Bmh1 regulate the kinetochore localization of Fin1
Because phosphorylation-dependent Fin1-Bmh1 interaction prevents the kinetochore association of Fin1-PP1 (Akiyoshi et al., 2009), hyperactive FEAR or the absence of Bmh1 may lead to premature Fin1 kinetochore localization. To test this idea, we first confirmed the colocalization of Fin1-GFP with a kinetochore protein Nuf2-mCherry in asynchronous cells. We further used live-cell imaging to follow Fin1 localization during cell cycle. Before anaphase entry, yeast cells exhibited nuclear Fin1 localization. Fin1-GFP co-localized with Nuf2-mCherry as anaphase initiated, and increased kinetochore intensity of Fin1-GFP was observed as the two kinetochore clusters separate (Fig. 5A). Therefore, Fin1 protein is enriched at kinetochores after anaphase onset.
Figure 5.
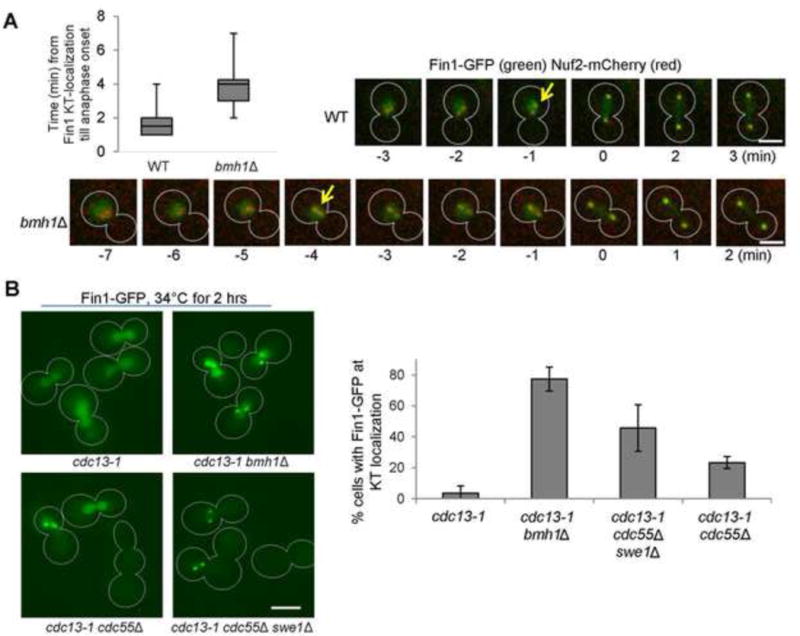
bmh1Δ mutant cells show earlier Fin1 kinetochore localization. (A) Live-cell imaging of Fin1 localization in a WT and bmh1Δ cell. Log-phase WT and bmh1Δ cells with Nuf2-mCherry and Fin1-GFP were spotted onto the surface of a slide covered with agarose medium and subjected to live-cell microscopy. At each time point, a Z-stack with 13 planes, separated by 0.4 μm, was acquired and subsequently projected. We set time 0 as the point when the distance between two Nuf2-mCherry foci is more than 3μm. Time from the clear co-localization of GFP dots with Nuf2-mCherry to time 0 was measured and the average is shown at top-left (n = 20). Arrows indicate the starting point for Fin1 and Nuf2 co-localization. Scale bar = 5 μm. (B) Premature Fin1 kinetochore localization in bmh1Δ, cdc55Δ, and cdc55Δ swe1Δ mutants arrested with DNA damage (cdc13-1). Asynchronous cells were grown at 25°C to log-phase, and then shifted to 34°C for 2 hrs. Cells were collected, and Fin1-GFP localization was counted without fixation (n >100). The Fin1-GFP kinetochore localization in representative cells is shown in the left panel. Scale bar = 5 μm. The average percentage of cells with kinetochore-localized Fin1-GFP from three experiments is shown in the right panel.
To test if Bmh1 regulates Fin1 kinetochore localization, we compared the co-localization kinetics of Fin1-GFP and Nuf2-mCherry in WT and bmh1Δ cells. To facilitate the comparison, we set up the time 0 as the point when the two Nuf2-mCherry dots have initiated separation (> 3μm). We measured the difference between the first time point when Fin1-GFP dots co-localized with Nuf2-mCherry and time 0 in WT and bmh1Δ cells. The average time for WT cells is 1.65 min, indicating the coordinated anaphase onset and Fin1 kinetochore localization. The average time for bmh1Δ mutant cells is, however, 3.75 min, obviously longer than that of WT cells (Fig. 5A). Because WT and bmh1Δ cells show similar cell cycle progression (Fig. 1C), this result indicates premature Fin1 kinetochore localization in the mutant cells. To our surprise, we did not observe obvious premature Fin1-GFP foci formation in cdc55Δ swe1Δ cells, suggesting that hyperactive FEAR is not sufficient to induce premature kinetochore localization of Fin1 in an undisturbed cell cycle.
We also examined Fin1 kinetochore localization in cdc13-1 mutant cells, which arrest prior to anaphase when grown at the restrictive temperature because uncapped telomeres activate the DNA damage checkpoint. When arrested, the SAC is silenced but the FEAR pathway is inactive (Liang and Wang, 2007; Wang et al., 2001). After incubation at 34°C for 2 hrs, almost all cdc13-1 cells exhibited nuclear Fin1 localization and no Fin1-GFP foci were observed (Fig. 5B). Strikingly, two clear Fin1-GFP foci were observed in 77% of cdc13-1 bmh1Δ cells and in 45% of cdc13-1 cdc55Δ swe1Δ cells, indicating kinetochore enrichment of Fin1. In cdc13-1 cdc55Δ cells, 23% of them showed Fin1-GFP foci. These results indicate that deletion of BMH1 or FEAR hyperactivity leads to precocious Fin1 kinetochore localization in cdc13-1 arrested cells.
Next, we asked if Fin1 dephosphorylation is sufficient for its kinetochore localization. For this purpose, we examined the localization of nonphosphorylatable Fin1-5A-GFP, in which the CDK phosphorylation sites are mutated to alanine (Woodbury and Morgan, 2007a). In cdc13-1 arrested cells, no kinetochore localization of Fin1-GFP was observed, but 91% of cdc13-1 fin1-5A-GFP cells showed two clear GFP foci, indicating kinetochore localization of Fin1-5A (Fig. 6A). Interestingly, fin1-5A mutants are also sensitive to syntelic attachment induced by CIK1-CC, as seen by the slow growth and the viability loss in galactose medium (Fig. 6B and C). After 6 hr incubation in galactose, 45% of fin1-5A cells maintained viability, compared to 83% for cells with WT Fin1. To test if the sensitivity of fin1-5A to syntelic attachments requires Fin1-PP1 interaction, we mutated the PP1 binding motifs in Fin1-5A (fin1-5A-AA) (Akiyoshi et al., 2009). Interestingly, fin1-5A-AA did not show any sensitivity to CIK1-CC overexpression (Fig. 6B and C), suggesting that the premature Fin1 kinetochore localization likely silences the SAC through Fin1-associated PP1.
Figure 6.
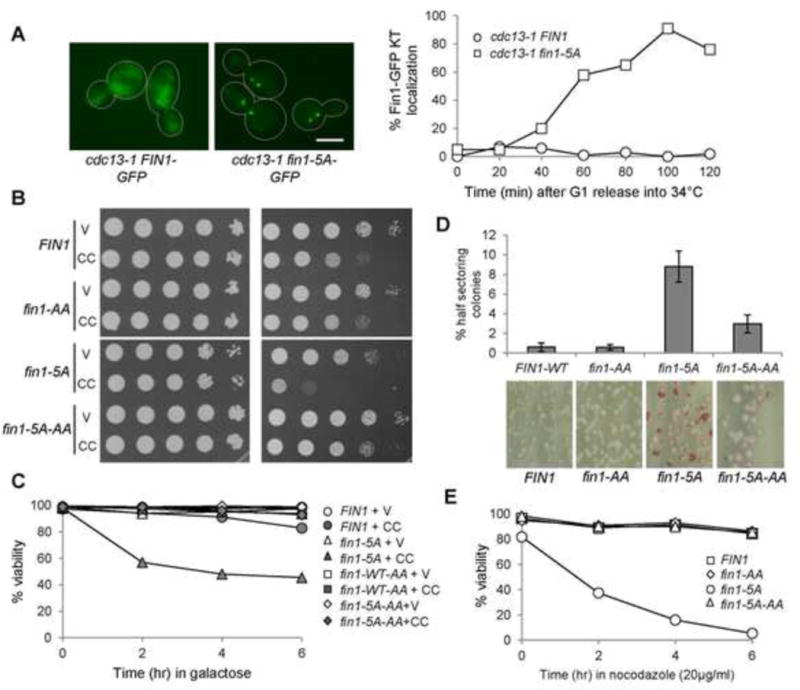
Nonphosphorylatable Fin1-5A show premature kinetochore localization and fin1-5A mutants are sensitivity to CIK1-CC overexpression. (A) Fin1-5A-GFP prematurely localizes at the kinetochore in cdc13-1 arrested cells. cdc13-1 fin1Δ cells with Fin1-GFP or Fin1-5A-GFP were synchronized in G1 at 25°C and then released into 34° YPD medium. Cells were collected over time and fixed. The percentage of cells with two clear GFP dots was counted (n >100). The localization of Fin1-GFP and Fin1-5A-GFP in some cdc13-1-arrested cells is shown in the left. Scale bar, 5μm. (B) fin1-5A mutants show PP1-dependent sensitivity to CIK1-CC overexpression. Saturated cells with indicated genotypes were 10-fold serial diluted, spotted onto glucose and galactose plates, and then incubated at 30°C for 2 days before scanning. fin1-5A: 5 CDK phosphorylation sites are mutated. fin1-5A-AA: both CDK phosphorylation sites and PP1 binding motif are mutated. V (vector), CC (PGALCIK1-CC). (C) fin1-5A cells overexpressing CIK1-CC show viability loss, which depends on Fin1’s PP1 binding. Log-phase cells in raffinose were released into 2% galactose medium. Samples were taken every 2 hr and spread onto YPD plates to determine plating efficiency (n > 300). (D) fin1-5A mutant cells show elevated chromosome loss, which depends on its PP1 binding. Cells harboring chromosome fragment III (CFIII) with indicated genotypes were incubated in URA dropout medium and then spread onto URA+ plates lacking adenine. The percentage of half-red colonies was counted after incubation for 3 days at 25°C. Here shows the average of three independent experiments (n > 400). (E) fin1-5A mutant cells show PP1-binding dependent sensitivity to nocodazole exposure. Cells with the indicated genotypes were released into YPD containing 20μg/ml of nocodazole. Cells were collected every 2 hr and spread onto YPD plates to determine the viability.
The hypersensitivity of fin1-5A to syntelic attachment indicates the critical role of Fin1 in accurate chromosome segregation. We measured the rate of chromosome loss in the mutants using the color sectoring assay (Spencer et al., 1990). ade2 cells that have lost the chromosome III fragment carrying the SUP11 gene turn red on plates lacking adenine. We counted half-sectored colonies, which suggests fragment loss during the first cell division. WT cells showed low frequency of chromosome loss, as 0.6% of cells displayed half sectoring colonies. In contrast, the rate of chromosome loss in fin1-5A mutants was as high as 9% (Fig.6D). Additionally, 76% of colonies of fin1-5A showed a mixture of red and white color, indicating high rate missegregation during following cell divisions. However, the chromosome loss phenotype of fin1-5A was partially rescued by mutating the PP1 binding motifs, as fin1-5A-AA mutant showed 3% half-sectoring colonies and 37% mixed colonies, indicating that PP1 activity is required for chromosome loss (Fig. 6D). Consistently, fin1-5A mutant cells are very sensitive to nocodazole exposure, as evidenced by dramatic viability loss, which is also dependent on PP1 binding (Fig. 6E). Therefore, premature association of Fin1-PP1 with the kinetochore causes dramatic chromosome missegregation.
Fin1 promotes the removal of SAC protein Bub1 from the kinetochore in anaphase
Although precocious Fin1-PP1 kinetochore localization induces premature SAC silencing, Fin1-PP1 is unlikely essential for SAC silencing because their kinetochore localization occurs in anaphase when the SAC has been already satisfied. Since some anaphase cells show Bub1 kinetochore localization (Gillett et al., 2004; Shimogawa, 2010), Bub1 delocalization from kinetochores may occur after SAC silencing. To test if Fin1 promotes Bub1 delocalization, we analyzed Bub1-GFP localization in synchronous WT and fin1Δ cells with Nuf2-mCherry. Most of the WT and fin1Δ cells in metaphase exhibited co-localization of Bub1 with kinetochore protein Nuf2 (75 min, Fig. 7A). Very few WT cells in anaphase, however, showed kinetochore-localized Bub1 (90 and 105 min). In clear contrast, much more fin1Δ cells in anaphase exhibited kinetochore localization of Bub1 (Fig. 7A). We further examined the role of Fin1 in Bub1 localization using synchronous cdc15-2 cells, which arrest in telophase at high temperature. After G1 release into 36°C medium for 60 min, most of cdc15-2 and cdc15-2 fin1Δ cells were in metaphase and these metaphase cells showed kinetochore-localized Bub1. After anaphase entry, about 20% cdc15-2 cells still showed Bub1 kinetochore localization (75, 90, and 105 min in Fig. 7B). However, more than 70% cdc15-2 fin1Δ cells showed Bub1 kinetochore localization after anaphase entry (Fig. 7B). As ase1Δ is synthetically lethal with fin1Δ (Tong et al., 2004; Woodbury and Morgan, 2007b), we also examined Fin1 localization in cdc15-2 ase1Δ mutant, which was the same as cdc15-2 single mutants cells (Fig. 7B). Thus, the kinetochore localization of Fin1-PP1 during anaphase clears the SAC protein Bub1 from kinetochores.
Figure 7.
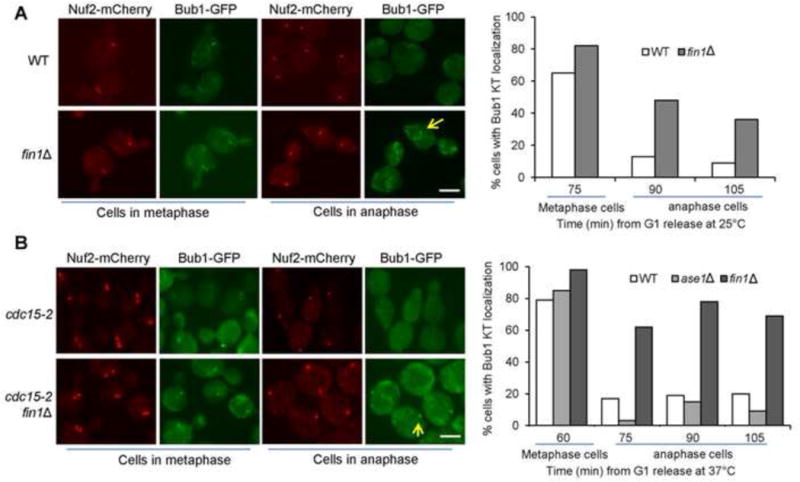
Fin1 promotes the dissociation of Bub1 from kinetochores in anaphase. (A) The kinetochore localization of Bub1 in WT and fin1Δ mutant cells. Cells were synchronized in G1 and then released into cell cycle at 25°C. The localization of Bub1-GFP and a kinetochore protein Nuf2-mCherry was examined over time. At 75 min after release, the majority of cells are in metaphase, and the percentage of metaphase cells with kinetochore-localized Bub1 is shown. The percentage of anaphase cells that show kinetochore-localized Bub1 was counted for the 90 and 105 min samples. Some representative cells are shown in the left panel. Scale bar, 5μm. (B) The kinetochore localization of Bub1 in cdc15-2 mutants. G1-arrested cdc15-2, cdc15-2 fin1Δ and cdc15-2 ase1Δ cells with Nuf2-mCherry Bub1-GFP were released into 37°C YPD. The percentage of metaphase cells with kinetochore-localized Bub1 was counted at 60 min as majority of the cells are in metaphase. The percentage of anaphase cells with kinetochore-localized Bub1 was assessed at 75, 90 and 105 min. Some representative cells are shown in the left panel. Scale bar, 5μm.
Our data support a working model that the kinetochore association of Fin1-PP1 promotes the removal of SAC components from kinetochores. What is the physiological significance of this regulation following SAC silencing? We compared the cell cycle progression of WT and fin1Δ cells, but no obvious cell cycle delay was noticed for fin1Δ cells (Fig. S6A). However, fin1Δ cells exhibited a moderate but reproducible cell cycle delay in the presence of low concentration of nocodazole (3μg/ml). We further compared cell cycle progression of WT and fin1Δ mutants after exposure with 20μg/ml of nocodazole that arrests cell in metaphase. fin1Δ cells showed delayed transition from large budded to single cells after nocodazole exposure (Fig. S6B). We also found that fin1Δ cells grew better than WT cells on benomyl plates. In contrast, ipl1-321 cells were sick on benomyl plates, presumably due to its role in the correction of erroneous kinetochore attachments and the prevention of SAC silencing (Jin and Wang, 2013; Pinsky et al., 2006b; Tanaka et al., 2002). fin1Δ clearly suppressed the benomyl sensitivity of ipl1-321 (Fig. S6C). Therefore, Fin1-mediated enrichment of PP1 at the kinetochore might be required for efficient SAC silencing only when the SAC is challenged with microtubule poisons.
DISCUSSION
Several mechanisms have been proposed for SAC silencing. Recent studies suggest that end-on microtubule attachment to the kinetochore may physically separate the Mps1 kinase from its substrates or remove Mps1 from the kinetochore for SAC silencing (Aravamudhan et al., 2015; Hiruma et al., 2015). PP1 has been shown to be essential for SAC silencing (London et al., 2012; Rosenberg et al., 2011; Vanoosthuyse and Hardwick, 2009). We previously found that tension-dependent dephosphorylation of kinetochore protein Dam1 in budding yeast is necessary for SAC silencing most likely through PP1 (Jin and Wang, 2013). Here, we identified an additional layer of SAC regulation that occurs after its silencing. Prior to anaphase onset, Fin1 phosphorylation prevents its kinetochore localization. After anaphase onset, however, Fin1 dephosphorylation triggers the kinetochore association of Fin1-PP1, which helps remove SAC protein Bub1 from kinetochores for SAC disassembly.
We found that syntelic attachments delay anaphase onset by preventing SAC silencing through tension sensing proteins Ipl1 and Sgo1 (Jin and Wang, 2013). Here we show that bmh1Δ and cells with hyperactive FEAR also allow premature anaphase entry in the presence of syntelic attachments. Interestingly, FIN1 deletion suppresses the premature anaphase entry in these cells. FEAR activation in early anaphase promotes Fin1 dephosphorylation, which triggers Bmh1-Fin1 dissociation (Akiyoshi et al., 2009; Jin et al., 2008). Thus, FEAR and Bmh1 regulate the SAC through Fin1. Indeed, we found that bmh1Δ and cells with hyperactive FEAR exhibit premature Fin1 kinetochore localization. Cells expressing a phospho-deficient Fin1 also show premature kinetochore localization and display sensitivity to CIK1-CC overexpression. Since we observed that the sensitivity depends on Fin1-PP1 interaction, the recruitment of PP1 to the kinetochore by Fin1 likely promotes premature anaphase entry. Importantly, we show that fin1Δ mutant cells retain Bub1 kinetochore localization even after anaphase entry. Therefore, we identified a mechanism that regulates SAC assembly at the kinetochore. Our results support a model that S-phase CDK-dependent Fin1 phosphorylation promotes Bmh1-Fin1 interaction, which prevents kinetochore association of Fin1-PP1 and allows SAC assembly. After anaphase entry, however, FEAR-dependent Fin1 dephosphorylation triggers the recruitment of Fin1-PP1 onto kinetochores, which triggers the removal of SAC proteins from kinetochores (Fig. S7).
This Fin1-dependent SAC regulation is unlikely required for SAC silencing during normal cell cycle. We found that fin1Δ mutant cells exhibited delayed cell cycle in the presence of low concentration of nocodazole or after nocodazole exposure, indicating that this mechanism might be required for efficient SAC silencing only when SAC is challenged by spindle poison. One untested possibility is that the removal of Bub1 from kinetochore after anaphase entry prevents SAC reactivation, creating a point of no return for anaphase entry. Because CDK-dependent Fin1 phosphorylation prevents the kinetochore association of Fin1-PP1, this mechanism limits SAC activation within a window during the cell cycle.
Although bmh1Δ and cdc55Δ swe1Δ cells are sensitive to Cik1-CC induced syntelic attachment, the viability loss and the frequency of chromosome missegregation are less pronounced than the tension sensing mutant sgo1. Thus, premature Fin1 kinetochore localization cannot induce an efficient anaphase onset. Currently, only two kinetochore proteins, Spc105 and Fin1, have been shown to interact with PP1 (Akiyoshi et al., 2009; Rosenberg et al., 2011). Kinetochore-associated PP1 through Spc105 or Fin1 may exhibit different substrate specificity due to their distinct spacial and temporal kinetochore location. Spc105-associated PP1 is essential for SAC silencing (Rosenberg et al., 2011), but Fin1-PP1 helps clear SAC proteins from kinetochores after its silencing. It is our future interest to define the substrate specificity of Spc105 and Fin1-associated PP1 as well as their unique roles is SAC regulation.
We noticed that the suppression of viability loss in bmh1Δ by fin1Δ in response to CIK1-CC overexpression is incomplete. Bmh1 may play Fin1-independent roles, as Bmh1 binds to many proteins (Kakiuchi et al., 2007; van Heusden, 2009). For instance, Bmh1 regulates mitotic exit through its interaction with Bfa1, a negative regulator of mitotic exit network (Caydasi et al., 2014). The FEAR pathway regulates anaphase onset through multiple mechanisms in addition to Fin1. As PP2ACdc55 delays anaphase onset by dephosphorylating and inhibiting the anaphase promoting complex (APC) (Lianga et al., 2013; Rossio et al., 2013; Vernieri et al., 2013), hyperactive APC in cdc55Δ may also contribute to premature anaphase onset in response to syntelic attachment. Moreover, FEAR-dependent Cdc14 release might dephosphorylate additional substrates to promote mitotic progression. For example, Cdc14-dependent dephosphorylation of Sli15, a subunit of Ipl1 kinase complex, promotes Ipl1 spindle localization (Mirchenko and Uhlmann, 2010; Pereira and Schiebel, 2003). Therefore, the hyperactive FEAR is expected to promote mitotic progression by dephosphorylating multiple proteins.
Our data demonstrate the premature anaphase entry in bmh1Δ and cdc55Δ swe1Δ mutant cells in the presence of syntelic attachments. Both bmh1Δ and cdc55Δ mutant are also sensitive to microtubule-depolymerizing agents (Grandin and Charbonneau, 2008; Minshull et al., 1996; Wang and Burke, 1997). It is unclear whether the premature anaphase entry contributes to the sensitivity of these mutants to spindle poisons. We examined the cell cycle progression in bmh1Δ cells treated with nocodazole, but the persistent Pds1 level indicates no premature anaphase onset. In nocodazole-treated bmh1Δ cells, the premature kinetochore recruitment of Fin1-PP1 may not be sufficient to drive anaphase onset due to the hyperactive checkpoint kinase. However, cells with premature kinetochore association of Fin1-PP1 may fail to maintain a prolonged metaphase arrest. In summary, our results demonstrate an extra layer of SAC regulation that removes SAC proteins from kinetochores, which resets the SAC at kinetochores in anaphase. Untimely activation of this pathway leads to premature SAC silencing and chromosome missegregation.
MATERIALS AND METHODS
Yeast strains, growth and media
The relevant genotypes and sources of the yeast strains used in this study are listed in Table S1. All the strains listed are isogenic to Y300, a W303 derivative, and they were constructed by tetrad dissection. The bmh1Δ and bmh2Δ strains were originally from the ATCC yeast deletion collection, and were crossed with Y300 three times. We deleted the FIN1 and BMH1 genes with a Sphis5+ marker as described previously (Longtine et al., 1998), and the resulting deletion mutants were confirmed using PCR. Yeast cell growth, synchronization, and CIK1-CC overexpression were performed as described (Jin et al., 2012).
Cytological Techniques
For fluorescence microscopy, collected yeast cells were fixed with 3.7% formaldehyde for 20 min and then washed once with 1×PBS (pH7.2). The cells were resuspended in 1×PBS for the examination of fluorescence signals using a microscope with a 60× objective lens (EVOS from Lifetechnologies). Live-cell microscopy was carried out with the Andor Revolution SD imaging system. Cells were spotted onto an agarose pad filled with synthetic complete medium. All live-cell images were acquired at 25°C with a 100× objective lens. A z-stack with 13 planes separated by 0.4μm was acquired every 1 min and converted to maximum projection using Andor IQ2 software.
Western blotting
Yeast cells (1.5 ml) were collected by centrifugation and the cell pellets were resuspended in 200μl 0.1 M NaOH. After incubation at room temperature for 5 min, the samples were centrifuged and the pellets were resuspended in 100μl 1×SDS protein loading buffer. The protein samples were boiled for 5 min and resolved by 8% SDS-PAGE. After probing with antimyc or anti-HA primary antibodies (Covance Research Products, Inc.) followed by HRP-conjugated secondary antibody (Jackson ImmunoResearch, Inc.), the proteins were detected with ECL (Perkin Elmer LAS, Inc.). We used anti-Pgk1 antibody (Molecular Probes) to detect Pgk1 protein levels, which are used as a loading control.
Supplementary Material
Acknowledgments
We are grateful for the yeast community at Florida State University for reagents and suggestions. We thank Dr. Sue Biggins for yeast strains and plasmids. We thank Ruth Didier for her assistance in live-cell imaging, and Jodi Slade for her assistance of the graphical abstract. This work was supported by R15GM097326 and RO1GM102115 from NIH/NIGMS to Y.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, Y.W. and M.B.; Investigation, M.B., C.G., F.J., D.R., and Y.W.; Writing – original draft, C.G. and M.B.; Writing – Review & Editing, Y.W. and M.B.; Funding Acquisition, Y.W.; Supervision, Y.W.
References
- Akiyoshi B, Nelson CR, Ranish JA, Biggins S. Quantitative proteomic analysis of purified yeast kinetochores identifies a PP1 regulatory subunit. Genes Dev. 2009;23:2887–2899. doi: 10.1101/gad.1865909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravamudhan P, Goldfarb AA, Joglekar AP. The kinetochore encodes a mechanical switch to disrupt spindle assembly checkpoint signalling. Nat Cell Biol. 2015;17:868–879. doi: 10.1038/ncb3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam R, Chen SL, Shou W, Mah AS, Alexandru G, Nasmyth K, Annan RS, Carr SA, Deshaies RJ. Phosphorylation by cyclin B-Cdk underlies release of mitotic exit activator Cdc14 from the nucleolus. Science. 2004;305:516–519. doi: 10.1126/science.1099402. [DOI] [PubMed] [Google Scholar]
- Biggins S, Murray AW. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001;15:3118–3129. doi: 10.1101/gad.934801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caydasi AK, Micoogullari Y, Kurtulmus B, Palani S, Pereira G. The 14-3-3 protein Bmh1 functions in the spindle position checkpoint by breaking Bfa1 asymmetry at yeast centrosomes. Mol Biol Cell. 2014;25:2143–2151. doi: 10.1091/mbc.E14-04-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillett ES, Espelin CW, Sorger PK. Spindle checkpoint proteins and chromosome-microtubule attachment in budding yeast. J Cell Biol. 2004;164:535–546. doi: 10.1083/jcb.200308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandin N, Charbonneau M. Budding yeast 14-3-3 proteins contribute to the robustness of the DNA damage and spindle checkpoints. Cell Cycle. 2008;7:2749–2761. doi: 10.4161/cc.7.17.6592. [DOI] [PubMed] [Google Scholar]
- Hardwick KG, Murray AW. Mad1p, a phosphoprotein component of the spindle assembly checkpoint in budding yeast. J Cell Biol. 1995;131:709–720. doi: 10.1083/jcb.131.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiruma Y, Sacristan C, Pachis ST, Adamopoulos A, Kuijt T, Ubbink M, von Castelmur E, Perrakis A, Kops GJ. CELL DIVISION CYCLE. Competition between MPS1 and microtubules at kinetochores regulates spindle checkpoint signaling. Science. 2015;348:1264–1267. doi: 10.1126/science.aaa4055. [DOI] [PubMed] [Google Scholar]
- Indjeian VB, Stern BM, Murray AW. The centromeric protein Sgo1 is required to sense lack of tension on mitotic chromosomes. Science. 2005;307:130–133. doi: 10.1126/science.1101366. [DOI] [PubMed] [Google Scholar]
- Ji Z, Gao H, Yu H. CELL DIVISION CYCLE. Kinetochore attachment sensed by competitive Mps1 and microtubule binding to Ndc80C. Science. 2015;348:1260–1264. doi: 10.1126/science.aaa4029. [DOI] [PubMed] [Google Scholar]
- Jin F, Liu H, Li P, Yu HG, Wang Y. Loss of function of the cik1/kar3 motor complex results in chromosomes with syntelic attachment that are sensed by the tension checkpoint. PLoS Genet. 2012;8:e1002492. doi: 10.1371/journal.pgen.1002492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Liu H, Liang F, Rizkallah R, Hurt MM, Wang Y. Temporal control of the dephosphorylation of Cdk substrates by mitotic exit pathways in budding yeast. Proc Natl Acad Sci U S A. 2008;105:16177–16182. doi: 10.1073/pnas.0808719105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Wang Y. The signaling network that silences the spindle assembly checkpoint upon the establishment of chromosome bipolar attachment. Proc Natl Acad Sci U S A. 2013;110:21036–21041. doi: 10.1073/pnas.1307595111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakiuchi K, Yamauchi Y, Taoka M, Iwago M, Fujita T, Ito T, Song SY, Sakai A, Isobe T, Ichimura T. Proteomic analysis of in vivo 14-3-3 interactions in the yeast Saccharomyces cerevisiae. Biochemistry. 2007;46:7781–7792. doi: 10.1021/bi700501t. [DOI] [PubMed] [Google Scholar]
- Keating P, Rachidi N, Tanaka TU, Stark MJ. Ipl1-dependent phosphorylation of Dam1 is reduced by tension applied on kinetochores. J Cell Sci. 2009;122:4375–4382. doi: 10.1242/jcs.055566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Jin F, Liu H, Wang Y. The molecular function of the yeast polo-like kinase Cdc5 in Cdc14 release during early anaphase. Mol Biol Cell. 2009;20:3671–3679. doi: 10.1091/mbc.E08-10-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Richmond D, Wang Y. Coordination of Chromatid Separation and Spindle Elongation by Antagonistic Activities of Mitotic and S-Phase CDKs. PLoS Genet. 2013;9:e1003319. doi: 10.1371/journal.pgen.1003319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Wang Y. DNA damage checkpoints inhibit mitotic exit by two different mechanisms. Mol Cell Biol. 2007;27:5067–5078. doi: 10.1128/MCB.00095-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lianga N, Williams EC, Kennedy EK, Dore C, Pilon S, Girard SL, Deneault JS, Rudner AD. A Wee1 checkpoint inhibits anaphase onset. J Cell Biol. 2013;201:843–862. doi: 10.1083/jcb.201212038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang Y. The function and regulation of budding yeast Swe1 in response to interrupted DNA synthesis. Mol Biol Cell. 2006;17:2746–2756. doi: 10.1091/mbc.E05-11-1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London N, Ceto S, Ranish JA, Biggins S. Phosphoregulation of Spc105 by Mps1 and PP1 Regulates Bub1 Localization to Kinetochores. Curr Biol. 2012 doi: 10.1016/j.cub.2012.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast. 1998;14:953–961. doi: 10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Loog M, Morgan DO. Cyclin specificity in the phosphorylation of cyclin-dependent kinase substrates. Nature. 2005;434:104–108. doi: 10.1038/nature03329. [DOI] [PubMed] [Google Scholar]
- Mayordomo I, Sanz P. The Saccharomyces cerevisiae 14-3-3 protein Bmh2 is required for regulation of the phosphorylation status of Fin1, a novel intermediate filament protein. The Biochemical journal. 2002;365:51–56. doi: 10.1042/BJ20020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshull J, Straight A, Rudner AD, Dernburg AF, Belmont A, Murray AW. Protein phosphatase 2A regulates MPF activity and sister chromatid cohesion in budding yeast. Curr Biol. 1996;6:1609–1620. doi: 10.1016/s0960-9822(02)70784-7. [DOI] [PubMed] [Google Scholar]
- Mirchenko L, Uhlmann F. Sli15(INCENP) dephosphorylation prevents mitotic checkpoint reengagement due to loss of tension at anaphase onset. Curr Biol. 2010;20:1396–1401. doi: 10.1016/j.cub.2010.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira G, Schiebel E. Separase regulates INCENP-Aurora B anaphase spindle function through Cdc14. Science. 2003;302:2120–2124. doi: 10.1126/science.1091936. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Kotwaliwale CV, Tatsutani SY, Breed CA, Biggins S. Glc7/protein phosphatase 1 regulatory subunits can oppose the Ipl1/aurora protein kinase by redistributing Glc7. Mol Cell Biol. 2006a;26:2648–2660. doi: 10.1128/MCB.26.7.2648-2660.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinsky BA, Kung C, Shokat KM, Biggins S. The Ipl1-Aurora protein kinase activates the spindle checkpoint by creating unattached kinetochores. Nat Cell Biol. 2006b;8:78–83. doi: 10.1038/ncb1341. [DOI] [PubMed] [Google Scholar]
- Pinsky BA, Nelson CR, Biggins S. Protein phosphatase 1 regulates exit from the spindle checkpoint in budding yeast. Curr Biol. 2009;19:1182–1187. doi: 10.1016/j.cub.2009.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queralt E, Lehane C, Novak B, Uhlmann F. Downregulation of PP2A(Cdc55) phosphatase by separase initiates mitotic exit in budding yeast. Cell. 2006;125:719–732. doi: 10.1016/j.cell.2006.03.038. [DOI] [PubMed] [Google Scholar]
- Rosenberg JS, Cross FR, Funabiki H. KNL1/Spc105 recruits PP1 to silence the spindle assembly checkpoint. Curr Biol. 2011;21:942–947. doi: 10.1016/j.cub.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossio V, Michimoto T, Sasaki T, Ohbayashi I, Kikuchi Y, Yoshida S. Nuclear PP2A-Cdc55 prevents APC-Cdc20 activation during the spindle assembly checkpoint. J Cell Sci. 2013;126:4396–4405. doi: 10.1242/jcs.127365. [DOI] [PubMed] [Google Scholar]
- Shimogawa MM, Wargacki MM, Muller EG, Davis TN. Laterally attached kientochores recruit the checkpoint protein Bub1, but satisfy the spindle checkpoint. Cell Cycle. 2010;9:10. doi: 10.4161/cc.9.17.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shou WY, Seol JH, Shevchenko A, Baskerville C, Moazed D, Chen ZWS, Jang J, Shevchenko A, Charbonneau H, Deshaies RJ. Exit from mitosis is triggered by Tem1-dependent release of the protein phosphatase Cdc14 from nucleolar RENT complex. Cell. 1999;97:233–244. doi: 10.1016/s0092-8674(00)80733-3. [DOI] [PubMed] [Google Scholar]
- Spencer F, Gerring SL, Connelly C, Hieter P. Mitotic chromosome transmission fidelity mutants in Saccharomyces cerevisiae. Genetics. 1990;124:237–249. doi: 10.1093/genetics/124.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegmeier F, Visintin R, Amon A. Separase, polo kinase, the kinetochore protein Slk19, and Spo12 function in a network that controls Cdc14 localization during early anaphase. Cell. 2002;108:207–220. doi: 10.1016/s0092-8674(02)00618-9. [DOI] [PubMed] [Google Scholar]
- Tanaka TU, Rachidi N, Janke C, Pereira G, Galova M, Schiebel E, Stark MJ, Nasmyth K. Evidence that the Ipl1-Sli15 (Aurora kinase-INCENP) complex promotes chromosome bi-orientation by altering kinetochore-spindle pole connections. Cell. 2002;108:317–329. doi: 10.1016/s0092-8674(02)00633-5. [DOI] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- van Heusden GP. 14-3-3 Proteins: insights from genome-wide studies in yeast. Genomics. 2009;94:287–293. doi: 10.1016/j.ygeno.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Vanoosthuyse V, Hardwick KG. A novel protein phosphatase 1-dependent spindle checkpoint silencing mechanism. Curr Biol. 2009;19:1176–1181. doi: 10.1016/j.cub.2009.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernieri C, Chiroli E, Francia V, Gross F, Ciliberto A. Adaptation to the spindle checkpoint is regulated by the interplay between Cdc28/Clbs and PP2A(Cdc55) J Cell Biol. 2013;202:765–778. doi: 10.1083/jcb.201303033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visintin R, Hwang ES, Amon A. Cfi1 prevents premature exit from mitosis by anchoring Cdc14 phosphatase in the nucleolus. Nature. 1999;398:818–823. doi: 10.1038/19775. [DOI] [PubMed] [Google Scholar]
- Wang H, Liu D, Wang Y, Qin J, Elledge SJ. Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev. 2001;15:1361–1372. doi: 10.1101/gad.893201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Burke DJ. Checkpoint genes required to delay cell division in response to nocodazole respond to impaired kinetochore function in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1995;15:6838–6844. doi: 10.1128/mcb.15.12.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Burke DJ. Cdc55p, the B-type regulatory subunit of protein phosphatase 2A, has multiple functions in mitosis and is required for the kinetochore/spindle checkpoint in Saccharomyces cerevisiae. Mol Cell Biol. 1997;17:620–626. doi: 10.1128/mcb.17.2.620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ng TY. Phosphatase 2A Negatively Regulates Mitotic Exit in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17:80–89. doi: 10.1091/mbc.E04-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury EL, Morgan DO. Cdk and APC activities limit the spindle-stabilizing function of Fin1 to anaphase. Nat Cell Biol. 2007a;9:106–112. doi: 10.1038/ncb1523. [DOI] [PubMed] [Google Scholar]
- Woodbury EL, Morgan DO. The role of self-association in Fin1 function on the mitotic spindle. J Biol Chem. 2007b;282:32138–32143. doi: 10.1074/jbc.M705344200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.