

Abstract
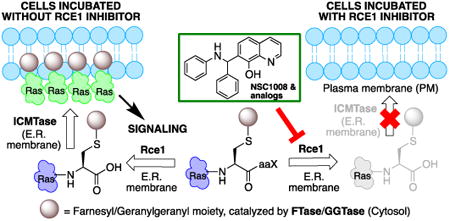
Ras converting enzyme 1 (Rce1) is an endoprotease that catalyzes processing of the C-terminus of Ras protein by removing -aaX from the CaaX motif. The activity of Rce1 is crucial for proper localization of Ras to the plasma membrane where it functions. Ras is responsible for transmitting signals related to cell proliferation, cell cycle progression, and apoptosis. The disregulation of these pathways due to constitutively active oncogenic Ras can ultimately lead to cancer. Ras, its effectors and regulators, and the enzymes that are involved in its maturation process are all targets for anticancer therapeutics. Key enzymes required for Ras maturation and localization are the farnesyltransferase (FTase), Rce1, and isoprenylcysteine carboxyl methyltransferase (ICMT). Among these proteins, the physiological role of Rce1 in regulating Ras and other CaaX proteins has not been as fully explored. Small-molecule inhibitors of Rce1 could be useful as chemical biology tools to understand further the downstream impact of Rce1 on Ras function and serve as potential leads for cancer therapeutics. Structure-activity relationship (SAR) analysis of a previously reported Rce1 inhibitor, NSC1011, has been performed to generate a new library of Rce1 inhibitors. The new inhibitors caused a reduction in Rce1 in vitro activity, exhibited low cell toxicity, and induced mislocalization of EGFP-Ras from the plasma membrane in human colon carcinoma cells giving rise to a phenotype similar to that observed with siRNA knockdowns of Rce1 expression. Several of the new inhibitors were more effective at mislocalizing K-Ras compared to a potent farnesyltransferase inhibitor (FTI), which is significant because of the preponderance of K-Ras mutations in cancer.
Graphical abstract
1. Introduction
Small molecule mediated inhibition of oncogenic Ras signaling is an emerging trend in the anticancer drug discovery field.1, 2 Overall, it is estimated that about 30% of human cancers involve activating Ras mutations.3 Ras proteins are membrane-associated small GTPases that mediate signal transduction events related to growth, differentiation, cytoskeletal organization, and membrane trafficking. Ras has a characteristic CaaX motif (where C is cysteine, a is an aliphatic amino acid, and X is one of several amino acids) at its C-terminus, which interacts sequentially with farnesyltransferase (FTase), Ras converting enzyme 1 endoprotease (Rce1), and isoprenylcysteine carboxyl methyltransferase (ICMT). All Ras isoforms localize to the plasma membrane where they are poised to mediate their signaling effects.4-6 There are multiple approaches to modulating Ras signaling. Chemotherapeutic targeting of mutant Ras proteins with guanine nucleotide mimics is perceived as impractical due to the picomolar binding affinity of Ras for GTP and GDP and the availability of GTP and GDP in micromolar concentrations within the cell.7, 8 Also, Ras proteins do not have accessible pockets on their surface. Despite these difficulties, a few recent studies report allosteric9-12 and covalent inhibitors13, 14 of mutant K-Ras. Additional approaches include inhibition of downstream effectors (e.g., kinases) and proteins essential for transformative growth in the presence of oncogenic Ras.15
An alternative strategy to inhibit oncogenic Ras signaling involves disruption of the Ras maturation process, which subsequently leads to the protein's mislocalization. Farnesyltransferase inhibitors (FTIs) progressed to late stage clinical trials (e.g., Tipifarnib, Lonafarnib, Salisarib), but the overall efficacy in patients with solid tumors was far less than expected, mainly due to alternative geranylgeranylation of K-Ras and N-Ras isoforms.16 Studies in cell culture showed that FTIs disrupt Ras localization. Ras is also mislocalized in the absence of Rce1 protease or ICMT activities.17, 18 The deletion of the gene encoding Rce1 markedly sensitizes tumor cells to FTI treatment,19 and the elimination of ICMT in fibroblasts blocks oncogenic K-Ras mediated transformation.20 Mice lacking the Rce1 gene die in early stages of embryonic development,17 whereas tissue-specific knockouts display context specific effects. Loss of Rce1 from heart tissue results in lethal cardiomyopathy, whereas a liver-specific knockout appears healthy and has normal hematopoietic function.21
Selective and potent inhibitors of Rce1 would be useful to further investigate the physiological role of Rce1 in regulating Ras and other CaaX proteins and to explore their potential as an anticancer chemotherapeutic strategy. Known Rce1 inhibitors22, 23 range from substrate mimics to small molecules.24-29 Selective inhibition of the Rce1 protease, however, continues to be a challenging problem. The ideal inhibitor must avoid simultaneous inhibition of the functionally related and evolutionarily distinct CaaX protease sterile mutant 24 (Ste24).30 Ste24 is essential for the maturation of lamin A, and defects in Ste24 activity, either by mutation or inhibition, lead to the development of laminopathies, such as progeria, muscular dystrophy, and lipodystrophy.31, 32
Rce1 is an integral membrane protein localized to the endoplasmic reticulum (ER).33, 34 The crystal structure of the human Rce1 (HsRce1) has not yet been elucidated, although topology data on Rce1 from Saccharomyces cerevisiae (ScRce1)35 and the crystal structure of a homolog from the archaea Methanococcus maripaludis36 are available. Originally thought to be a cysteine protease,37 then a metalloprotease,38 the crystal structure of the archea homolog suggests that it proteolyzes Ras through a novel mechanism involving an active site glutamate.36 The possibility of a cysteine protease mechanism has been eliminated,35 whereas a glutamate and two histidines are essential for proteolytic activity,39, 40 supporting the latter two mechanistic hypotheses. The sequence of Rce1 from M maripaludis (MmRce1) is only 15% identical to that of human Rce1 (HsRce1),36 so one should be cautious in assigning structure and function of the human enzyme based on MmRce1's structure.
NSC1011 (1, Figure 1) inhibits HsRce1 in an in vitro proteolysis assay (IC50 = 9 μM)29 and mislocalizes a GFP-Ras2p reporter in yeast cells.41 In this report, we present an investigation of the structure-activity relationships (SAR) of NSC1011 to improve potency and selectivity against HsRce1. Additionally, we show the ability of NSC1011 and some of its derivatives to mislocalize EGFP-Ras isoforms in a human colon carcinoma cell line (HCT-116).
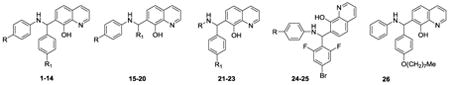
Figure 1. NSC1011 (1) and ring identification used to describe the SAR.

2. Results and Discussion
2.1 Chemistry
The preparation of the majority of the compounds was carried out using the classical Betti reaction or its modified versions (Scheme 1). The classical Betti reaction is a one pot multicomponent reaction between an amine, an aldehyde, and an electron rich bicyclic phenol.42-46 The lead molecule NSC1011 (1, Table 1) was synthesized by the reaction between p-aminobenzoic acid, benzaldehyde, and 8-hydroxyquinoline in ethanol at room temperature in the presence of a catalytic amount of pyridine for 10-15 days stirring to afford 1 in about 16% yield (protocol A). Heating the reaction to 120 °C in ethanol (protocol B) led to a decrease in reaction time to 12 hours and an improvement in yield to 55%. If carried out under microwave conditions at 140 °C, the reaction time could be reduced to approximately 15 min (protocol C) in higher yield (65%). Compounds 2-6, 9-10, 15-18, 21, 24-25, 37-38, and 40-42 were prepared by following any one of four protocols (A-D). Compounds 7 and 8 (NSC 1013 and 84093, respectively) were acquired from the National Cancer Institute's Developmental Therapeutics Program (DTP).
Scheme 1. General synthetic approach to NSC1011 analogsa.

aReagents and conditions: (i) See the experimental section for details of various reaction conditions.
Table 1. Enzymatic results for compounds synthesized, 1-26.
| ||||
|---|---|---|---|---|
|
| ||||
| No. | R | R1 | HsRcel Percent Activity Remaininga (10 μM) | HsRcel IC50 (μM)b |
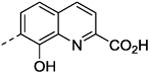
| lc | CO2H | H | 25 ± 0.05 | 6.9 ±1.06 |
| 2 | H | H | 39 ±1.3 | 8.9 ±1.08 |
| 3 | CO2H | CN | 76 ±3.1 | 16 ± 1.1 |
| 4 | CO2H | Me | 32 ±3.6 | 7.1 ± 1.0 |
| 5 | CO2H | Br | 23 ± 1.3 | 6.7±1.1 |
| 6 | CO2H | F | 32 ±0.01 | 8.2± 1.1 |
| 7d | H | NO2 | 85 ± 5.0 | 11 ±1.2 |
| 8d | Me | H | 54 ± 5.2 | 8.8± 1.1 |
| 9 | CN | H | 56 ± 0.2 | nd |
| 10 | NO2 | Br | 64 ± 1.2 | nd |
| 11 | CO2Et | H | 52 ± 3.2 | nd |
| 12 | CO2Et | CF3 | 89 ±3.8 | nd |
| 13 | CO2Et | F | 63 ±0.4 | nd |
| 14 | CO2Et | CI | 71 ±0.6 | nd |
| 15 | Co2H | 2-pyridine | 90 ±5.2 | 38 ± 1.1 |
| 16 | CO2H | 3-pyridine | 70 ±1.8 | 14 ±1.0 |
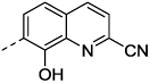
| 17 | CO2H | cyclohexyl | 34 ±4.8 | 9.8±1.1 |
| 18 | H | cyclohexyl | 59 ±0.03 | nd |
| 19 | CO2Et | cyclopentyl | 82 ±1.2 | nd |
| 20 | H | H | 82 ±0.49 | nd |
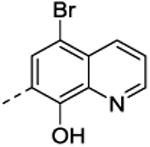
| 21 |
|
H | 83 ±5.7 | nd |
| 22 | Ph(CO) | H | 81 ±1.1 | nd |
| 23 | PhCH | H | 73 ±2.1 | nd |
| 24 | H | - | 76 ±1.7 | nd |
| 25 | CO2H | - | 64 ±3.7 | nd |
| 26 | - | - | 89 ±3.5 | nd |
The percent activity remaining values are the averages of replicates from one assay in the presence of inhibitor.
nd = Not determined.
Compound 1 (NSC1011) has previously been tested and data published,29 however it was resynthesized, retested, and included here for comparative purposes.
Compounds 7 and 8 were acquired from the NCI DTP and are included for SAR comparison.
Ester derivatives 11-14 and 19 were prepared by refluxing benzocaine, the corresponding aldehyde, and 8-hydroxyquinoline in a 10% aqueous solution of NaCl instead of ethanol.47 Benzamide derivative 22 was successfully obtained in 70% yield by refluxing a mixture of benzamide, benzaldehyde, and 8-hydroxyquinoline in dichloroethane in the presence of catalytic p-toluenesulfonic acid.48 Synthesis of imine analog 23 required heating a mixture of ammonium carbamate, benzaldehyde (2 equivalents), and 8-hydroxyquinoline in ethanol at 125 °C for 12-15 hours.49 Octyloxy analog 26 required the initial preparation of the aldehyde intermediate 26a50 by reacting p-hydroxybenzaldehyde with octyl bromide in the presence of potassium carbonate and catalytic potassium iodide (Scheme 2). Subsequently, a mixture of 26a, aniline, and 8-hydroxyquinoline was neatly heated at 130 °C for 24 hours to afford the Betti reaction product 26. Compound 39 was synthesized by first reacting 8-hydroxyquinoline-2-carboxylic acid and benzylic alcohol in the presence of triphenylphosphine and diisopropylazodicarboxylate in THF solvent at 0 to 10 °C in a modified Mitsunobu procedure51 to generate intermediate hydroxyquinoline 39a52 (Scheme 2), which was subsequently heated neatly with 8-hydroxyquinoline and benzaldehyde in the presence of catalytic pyridine at 120 °C to afford compound 39 in 15% yield. Quinoline derivative 43 was prepared in two steps (Scheme 3). The intermediate imine 43a was synthesized by heating a mixture of aniline and benzaldehyde neatly at 60 °C. Without further purification, 43a was added to the lithiated quinoline derivative, which was generated by reacting 7-bromoquinoline with n-butyllithium at -78 °C, to afford 43 in 7% yield.
Scheme 2. Synthesis of intermediates 26a and 39aa.

aReagents and conditions: (i) K2CO3, cat. KI, acetone, reflux; (ii) PPh3, DIAD, THF, 0 °C to rt, 1 hr.
Scheme 3. Synthetic approach to 43a.

aReagents and conditions: (i) neat, 60 °C; (ii) n-BuLi, THF, -78 °C; (iii) 43a, THF, -78 °C-rt, 3 h.
The 1-naphthol derivative 27 was synthesized in 95% yield by reacting 1-naphthol, aniline, and benzaldehyde in ethanol for 12-24 hours at room temperature in the presence of a catalytic amount of pyridine. The same conditions were used to yield the other 1-naphthol derivatives 28-31, 33, and 45-46, with the exception of 31, which was carried out in DMSO at room temperature by stirring for 36-48 hours. In contrast to the 1-naphthols, 2-naphthol derivatives 34 and 35 required heating the respective reaction mixtures at 100 °C. The diastereomeric 2-naphthol derivative 47 was synthesized by heating the reactants neatly at 85 °C for 12 hours. The product precipitated out as a crystalline solid upon discharge of ethanol and cooling to room temperature.53 Derivative 32 was prepared in modest yield by treating NSC1011 (1) with iodomethane in the presence of potassium hydroxide.
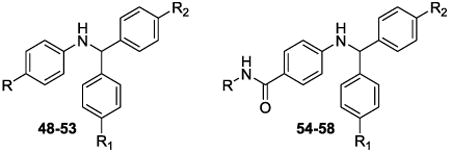
The diphenyl compounds, 48-58 were prepared by reductive amination of the appropriate benzophenone and benzocaine in the presence of titanium(IV) chloride and sodium cyanoborohydride (Scheme 4).54 Esters 48-50 were then hydrolyzed to acids 51-53 and the subsequent amidations with aniline, 2-aminonapthalene, and 3-aminobenzoic acid were carried out to give 54-58. Similarly, napthyl analog 44 was prepared via reductive amination using 2-benzoylnapthalene and aniline.
Scheme 4. Synthesis of analogs 48-58.

Reagents and conditions: (i) (a) i. TiCl4, DCM; 0 °C - rt; (b) NaCNBH3, MeOH; (ii), 5N NaOH (aq), dioxane/EtOH, rt; (iii) R-NH2, COMU, DIPEA, DMF, 0 °C - rt
All derivatives synthesized were tested as racemic mixtures in the biological assays.
2.2 Biological assays
2.2.1 CaaX proteolysis assay determines efficacy of Rce1 inhibition
An established in vitro fluorescence-based assay was used to measure the CaaX protease activity of the human Rce1 (HsRce1) following treatment with the inhibitors.29, 55, 56 The assay is based on the Rce1-dependent cleavage of a quenched fluorogenic K-Ras4b-derived peptide using membranes derived from yeast expressing HsRce1. Inhibitors were added at 10 μM and the percentage of remaining HsRce1 activity determined by comparison of initial rates (Tables 1-3). The lead compound NSC1011 (1) was resynthesized and included for comparison with the new derivatives. From the set of derivatives synthesized, certain structure-activity relationships were apparent.
Table 3. Enzymatic results for compounds 48-58.
| |||||
|---|---|---|---|---|---|
|
| |||||
| No. | R | R1 | R2 | HsRcel% Activity Remaining (10 μM)a | HsRcel IC50 (μM)b |
| 48 | CO2Et | H | H | 82 ±4.4 | nd |
| 49 | CO2Et | Br | H | 79 ± 3.4 | nd |
| 50 | CO2Et | Br | Br | 83 ± 7.6 | nd |
| 51 | CO2H | H | H | 90 ± 3.6 | nd |
| 52 | CO2H | Br | H | 86 ± 0.2 | nd |
| 53 | CO2H | Br | Br | 83 ± 2.6 | nd |
| 54 | Ph | H | H | 78 ± 1.0 | nd |
| 55 | Ph | Br | H | 78 ± 2.5 | nd |
| 56 | Ph | Br | Br | 80 ± 4.8 | nd |
| 57 |
|
H | H | 89 ± 3.5 | nd |
| 58 |
|
H | H | 72 ± 2.2 | nd |
The % activity remaining values are the averages of replicates from one assay in the presence of inhibitor.
nd = Not determined
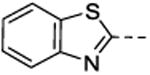
At 10 μM, NSC1011 (1) reduced the activity of HsRce1 to 25% of the uninhibited control (Table 1). NSC1008 (2), which lacks the 4-carboxylic acid functionality on ring A (Figure 1), was slightly less potent than NSC1011 (1), showing a slight increase in the remaining activity (39%). Other derivatives showed that a 4-carboxyate on the A-ring was slightly more inhibitory than an unsubstituted A-ring (e.g., 17 vs. 18, 24 vs. 25, 45 vs. 46). Replacement of the 4-carboxylate in the A-ring by either a methyl (8), a cyano (9), nitro (10), or alkyl ester (11-14, 19, 32) was not tolerated. Replacing the A-ring with a benzothioazolyl (21), benzoyl (22), or benzyl (23) group was detrimental to the inhibition of Rce1 proteolysis.
Some substituents in the para-position of the B-ring were tolerated well (Table 1); analogs containing 4-bromo (5), 4-fluoro (6), and 4-methyl (4) groups dropped the activity of Rce1 to 23-32% of the uninhibited control, whereas analogs containing 4-nitro (7), 4-cyano (3), or 4-octyloxy (26) groups did not inhibit HsRce1 activity significantly. Replacement of the B-ring phenyl group with 2-pyridyl (15), 3-pyridyl (16), or trihalogen-substituted rings designed to take advantage of halogen bonding (24, 25),57, 58 led to no significant reduction in Rce1 activity. Complete removal of the B-ring (20) had no significant effect on Rce1 proteolysis activity, although analogs with an aliphatic cyclic moiety such as a cyclohexyl group (17-19) showed similar activity to NSC1011 (1).
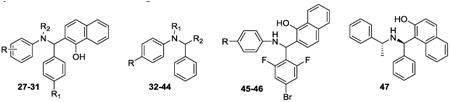
Replacing the hydroxyquinoline moiety with a 1-naphthol group (27-31, 33, 45-46) provided compounds with slightly better in vitro activities against HsRce1 than NSC1011 (1) (Table 2). In this series of analogs, A-ring substitutions (28-31, 45-46) induced similar levels of diminished Rce1 activity, and a bromine at the 4-position of the 1-naphthol ring system (33) retained inhibitory activity. In contrast, 2-naphthol derivatives 34, 35, and 47 were weaker inhibitors, and replacement of the quinoline with a phenyl ring (51) led to an inactive compound (Table 3).
Table 2. Enzymatic results for compounds 27-47.
| |||||
|---|---|---|---|---|---|
|
| |||||
| No. | R | R1 | R2 | HsRcel Percent Activity Remaining (10 μM)a | HsRcel IC50 (μM)b |
| 27 | H | H | H | 17 ±0.4 | 4.9±1.1 |
| 28 | p-t-butyl | H | H | 38 ±0.47 | 5.0±1.1 |
| 29 | p-CO2H | H | H | 19 ±2.4 | 4.2±1.1 |
| 30 | m-CO2H | H | H | 23 ±5.5 | 5.3 ±1.1 |
| 31 | p-CO2H | Br | H | 23 ±1.1 | 3.9 ±1.0 |
| 32 | CO2Me | H |
|
65 ±2.1 | nd |
| 33 | H | H |
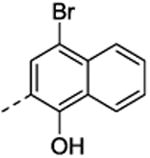
|
27 ±3.2 | 5.4 ±1.1 |
| 34 | H | H |
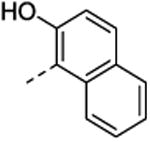
|
59 ±0.65 | nd |
| 35 | H | H |
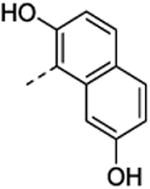
|
51 ±2.9 | nd |
| 36 | H | H |
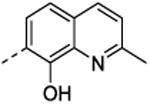
|
83 ±6.9 | nd |
| 37 | H | H |
|
83 ± 0.1 | nd |
| 38 | H | H |
|
77 ± 3.9 | nd |
| 39 | H | H |
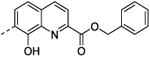
|
75 ± 2.9 | nd |
| 40 | H | H |
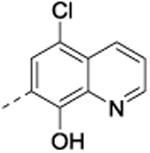
|
68 ± 0.7 | nd |
| 41 | H | H |
|
73 ± 3.5 | nd |
| 42 | H | H |
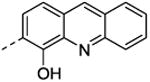
|
51 ± 2.0 | nd |
| 43 | H | H |
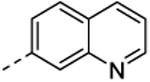
|
82 ± 0.3 | nd |
| 44 | H | H |
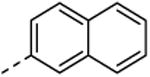
|
77 ± 2.8 | nd |
| 45 | H | - | - | 45 ±0.32 | 9.4 ±1.1 |
| 46 | CO2H | - | - | 21 ±2.4 | 3.8 ±1.1 |
| 47 | - | - | - | 63 ±0.08 | nd |
The % activity remaining values are the averages of replicates from one assay in the presence of inhibitor.
nd = Not determined
In general, adding substituents to the quinoline had a detrimental effect on the inhibitory properties relative to NSC1011 (1). Substitutions at the 2-position of the hydroxyquinoline moiety (36, 37, 38, 39) or a ring extension (42) did not reduce HsRce1 activity relative to the unsubstituted parent compound NSC1008 (2) (Table 2). Derivatives with halogen substitutions at the 5-position of the hydroxquinoline ring system were relatively inactive. The 8-hydroxy group is critical for inhibition of HsRce1; compounds lacking it (43, 44) or possessing an 8-methoxy group (32) have little inhibitory activity. This suggests that a hydrogen bond donor is necessary for inhibition of proteolysis.
Diphenyl analogs 48 and 51 were synthesized to investigate the importance of the bicyclic ring system (Rings C and D). Neither the ethyl ester 48 nor the acid derivative 51 showed any effect upon inhibition of the enzyme, and similarly, para-bromo substitution on either one or both of the A- and B-rings (49, 50, 52, 53) resulted in relatively inactive compounds (Table 3). Extension of these diphenyl derivatives via amidation at the para-carboxy moiety of ring A to yield phenyl amide counterparts 54-58 also did not provide compounds with significant inhibitory activity against HsRce1.
The IC50 values of a selection of analogs (1-8, 15-17, 27-31, 33, 45, and 46) were measured in the in vitro proteolysis assay (Figure 2).29, 55, 56 The set included thirteen derivatives that inhibited the activity of Rce1 to less than 40% of the control in the single-point assay and some of the less active analogs for comparison. The IC50 values are generally consistent with the single-point data, and indicate that the 1-naphthol derivatives 27, 29, and 31 (IC50 = 4.9, 4.2, and 3.9 μM, respectively) are marginally more potent compared to the corresponding 8-hydroxyquinoline analogs 1, 2, and 5 (IC50 = 6.9, 8.9, 6.7 μM, respectively) in the biochemical assay. The inhibitory activity of the 1-naphthol derivatives infers that HsRce1 may not be a zinc metalloprotease (8-hydroxyquinoline scaffold chelates zinc, whereas 1-naphthol scaffold does not), which agrees with structural data reported for MmRce1.36
Figure 2. HsRce1 IC50 values.

Inhibitors were evaluated using the fluorescence-based CaaX proteolysis assay measuring the percent activity remaining.
2.2.2 Analysis of inhibitor toxicity in human cells
To further investigate the role of Rce1 inhibitors in human cell lines and their effect on the localization of Ras isoforms, our secondary biological screening strategy was to remove any potentially cytotoxic compounds. Effective inhibitors, as determined by the IC50 values in the CaaX proteolysis assay, were tested in HCT-116 human colon carcinoma cells to initially assess the levels of toxicity in mammalian cell lines (Figure 3 and Supplemental Figure 1). The majority of compounds appeared relatively non-toxic after treatment of cells for 20 hours at 25 μM causing less than 10% cell death compared to DMSO-treated (0.4%) and untreated cells. Compounds 5 and 45 caused approximately 25% and 40% cell death, respectively.
Figure 3. Rce1 inhibitor toxicity in human cells.

Compounds were added to cells at 25 μM then incubated for 20 hours. UT = untreated, DMSO = DMSO (0.4%) treated cells. Cell viability was measured using the CellTiter-Blue assay (Promega). Each column is the average of three independent experiments. Error bars indicate SEM.
2.2.3 Cell-based assay of Ras mislocalization
Previous reports have shown that EGFP-Ras constructs are mislocalized from the plasma membrane (PM) in both Rce1-/- cells19 and in response to FTIs.59 To quantitatively investigate the impact of Rce1 inhibitors on the plasma membrane (PM) localization of Ras, HCT-116 cells were transiently transfected with EGFP conjugated to the N-terminus of H-, N-, or K-Ras (K-Ras4b), treated with compounds, and the cross-sectional fluorescence intensity of Ras analyzed similarly to the method previously described for yeast.41 Prior to testing the Rce1 inhibitors, the impact of reduced Rce1 expression on EGFP-Ras localization was validated in HCT-116 cells using an siRNA knock-down approach (Figure 4). Approximately 60-75% knock-down of Rce1 RNA levels was observed, and this led to mislocalization of all three Ras isoforms, albeit to different degrees, which nonetheless confirmed the expected phenotype.
Figure 4. Rce1 siRNA knock-down mislocalizes EGFP-Ras isoforms in human cells.
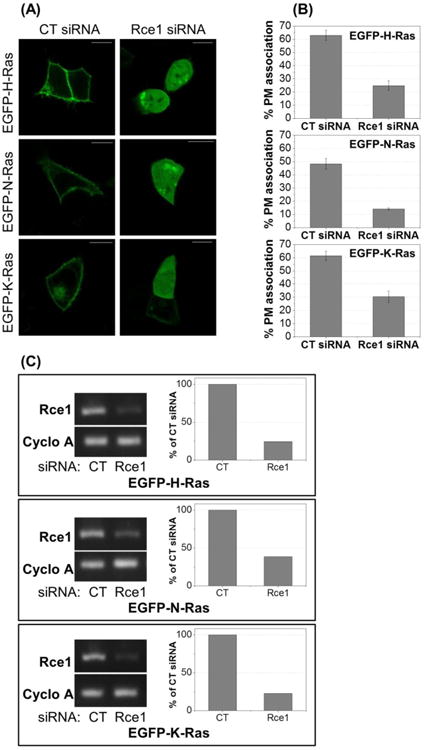
(A) HCT-116 cells were transfected with either control (CT) or Rce1 siRNA and with either EGFP-H-, N-, or K-Ras. Cells were imaged by confocal microscopy 24 hours post-transfection. Images show representative cells. Scale bars = 10 μm. (B) Quantification of EGFP-H-, N-, and K-Ras mislocalization (see Experimental Section for method). ∼30 cells per condition were analyzed. (C) HCT-116 cells were transfected with either CT or Rce1 siRNA and with either EGFP-H-, N-, or K-Ras. Cells were imaged by confocal microscopy 24 hours post-transfection, then lysed for mRNA analysis by semi-quantitative RT-PCR for Rce1 and cyclophilin A expression. Graphs report the densitometric analysis of the gels.
Live cell analysis in HCT-116 cells showed that EGFP constructs of H-, N-, and K-Ras were localized to the PM in untreated (data not shown) and DMSO-treated cells (Figure 5). Quantification of each EGFP-Ras isoform in DMSO-treated conditions, revealed that ∼50% of the total fluorescence was detected within 1 μm of the PM, as defined by the threshold boundaries in cross-sectional fluorescence profiles of individual cells (see experimental section for details). As proof of principle, cells were treated with 2 μM of the FTI L744,832,60 and a strong cytosolic localisation was observed for H-Ras and N-Ras (<10% of the total fluorescence at the PM). This localization was consistent with previous reports.61 Additionally, the relatively higher abundance of K-Ras at the PM (∼35%) was also consistent, as K-Ras can be alternatively prenylated by geranylgeranyl transferase (GGTase), which enables proper localization of K-Ras.59
Figure 5. Rce1 inhibitors mislocalize H-, N-, and K-Ras in human cells.
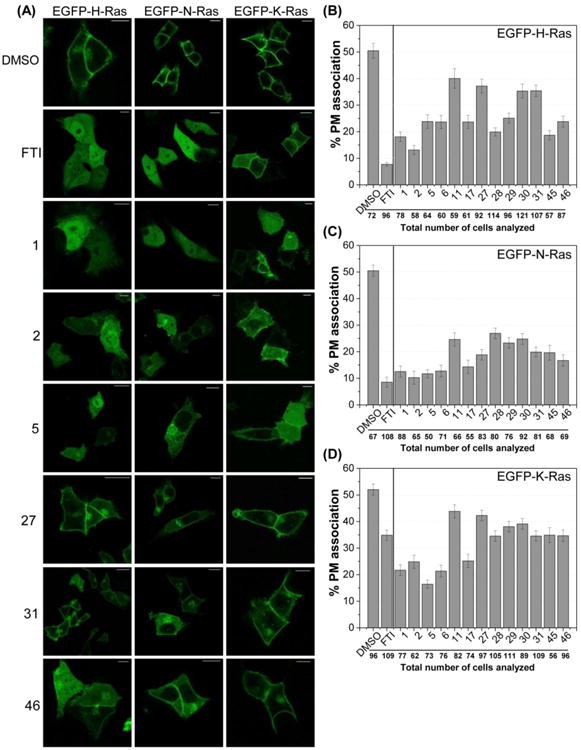
(A) HCT-116 cells were transiently transfected with EGFP fusion constructs of H-, N-, or K-Ras, and treated with either DMSO, FTI (2 μM), or Rce1 inhibitors (25 μM) for 20 hours. Images show representative cells. Scale bars = 10 μm. (B-D) Quantification of H-, N-, and K-Ras mislocalization (see supporting information for method). Percent PM association is reported with SEM bars.
Compounds 1, 2, 5, 6, 11, 17, 27-31, 45, and 46 were tested to quantify the level of EGFP-Ras PM association (Figure 5B-D). These compounds were selected because they reduced the activity of Rce1 to <40% of the uninhibited control in the CaaX proteolysis assay and showed low cell toxicity (<40% cell death after 20-hour treatment). Compound 11 was also included from the 8-hydroxyquinoline series even though it only reduced Rce1 activity to 52% in the proteolysis assay. Data showed that compound 2 was the most effective in mislocalizing H-Ras and N-Ras, with PM association as low as 14% and 10%, respectively. Rce1 inhibitors 1, 2, 5, 6, and 17 were more effective at mislocalizing K-Ras than the FTI, and the lowest PM association (16%) for K-Ras was observed with compound 5.
The mislocalization experiments demonstrate that the 8-hydroxyquinoline derivatives (1, 2, 5, 6, and 17) are marginally better at causing stronger cytoplasmic localization and reduced PM association of Ras as compared to the 1-napththol derivatives (27-31, 45, and 46). It is possible that 8-hydroxyquinoline derivatives have better cell permeability and availability on the endoplasmic reticulum membrane (ER) as compared to the 1-naphthol derivatives. Interestingly, the Rce1 inhibitors differentially mislocalized the Ras isoforms; greater mislocalization was observed for N-Ras as compared to H-Ras and K-Ras. This was also reflected in the results observed following Rce1 siRNA knockdown. It is not surprising that the compounds were least effective at mislocalizing K-Ras; others report that in Rce1-deficient mouse embryonic fibroblasts (MEFs), GFP-K-Ras4B still exhibits significant PM localization, although at lower levels compared to those observed in the wild-type MEFs.19, 59, 62 Similarly, it has been shown that deubiquitination of Rce1 by ubiquitin-specific protease 17 (USP17) reduces Rce1 activity in MEF cells, resulting in mislocalization of H-Ras and N-Ras, but not K-Ras.63 K-Ras mislocalization is less affected by Rce1 inhibition or genetic knockout, indicating that downstream processing of K-Ras to the PM differs from that of H- and N-Ras. The fact that several of the compounds were better at mislocalizing K-Ras than the FTI is noteworthy, because activating mutations in K-Ras are the most frequently observed in cancer relative to the other isoforms.3 The alternative prenylation by geranylgeranyl transferases (GGTases) when FTases are blocked is attributed to the inability of FTIs to significantly impact K-Ras function and the poor performance of FTIs in clinical trials.16, 64, 65
2.2.4 Inhibitors show selectivity for Rce1
A key requirement of an inhibitor of Rce1 is selectivity toward Rce1 over the functionally similar Ste24 protease. This is important due to the role that Ste24 plays in the maturation of lamin A, where inhibition of Ste24 leads to laminopathies.66 Compounds 1-8, 15-17, 27-31, 33, 45, and 46 were tested in a single-point (10 μM) CaaX proteolysis assay29, 55, 56 using membranes derived from yeast expressing human Ste24 enzyme67 (Figure 6A). We found that the majority of the compounds tested did not cause strong inhibition of Ste24. We also tested whether or not the Rce1 inhibitors inhibit farnesyltransferase (FTase). Compounds 2, 5, and 30, which have representative structures and activities against Rce1, were chosen and tested in a fluorescence-based FTase assay.68, 69 The data showed that FTase activity is not inhibited at concentrations as high as 50 μM (Figure 6B). These data suggest that the identified inhibitors of Rce1 do not inhibit the other known CaaX protease and the upstream enzyme in the Ras maturation pathway, strengthening the argument that the efficacy of the compounds in human cells results from the intracellular inhibition of Rce1 and not inhibition of FTase.
Figure 6. Efficacies of Rce1 inhibitors for Ste24 and FTase.

A) Compounds were evaluated at 10 μM using the fluorescence-based CaaX proteolysis assay measuring the percent activity remaining. Yeast cells expressed either HsRce1 or HsSte24. Error bars show SD. B) Indicated compounds were evaluated at 10, 25, and 50 μM using the fluorescence-based FTase assay. Error bars show SD.
3. Conclusion
We prepared derivatives of the Rce1 inhibitor NSC1011, some of which showed IC50 values in the low micromolar range in an in vitro fluorescent proteolysis assay. Compounds identified as inhibitors in the biochemical screen of Rce1 activity also caused the mislocalization of EGFP-labeled Ras in HCT-116 cells, which is a human colon carcinoma cell line. Compounds possessing a naphthol group instead of the hydroxyquinoline were more potent in the in vitro proteolysis assay, whereas the quinolinol derivatives were better able to mislocalize EGFP-tagged Ras in the cellular assay. The inhibitors were reasonably non-toxic in the short term (<20 hours), and they were more effective at disrupting the localization of N-Ras, by comparison to H-Ras and K-Ras. One might have expected to see similar levels of mislocalization among the isoforms, but using an siRNA to knock down Rce1 expression, we observed Ras mislocalization phenotypes among N-Ras, H-Ras, and K-Ras similar to the ones observed with the Rce1 inhibitors. Differential impact on the PM localization of the Ras isoforms is also observed in cells lacking the Rce1 gene, indicating that K-Ras is localized to the PM by a different path than H-Ras and N-Ras.19, 59, 62 Several Rce1 inhibitors were more effective at mislocalizing K-Ras compared to the FTI, which is an intriguing result given the preponderance of activating K-Ras mutations in certain forms of cancer.3 The results of this study further support the need for potent Rce1 inhibitors to better understand the biology and role of Rce1 in the processing of the Ras isoforms. In addition to observing the mislocalization of Ras isoforms, more study is required to determine the effect of the Rce1 inhibitors on Ras activity and signaling pathways downstream of Ras. Rce1 inhibition in the presence of oncogenic K-Ras in mouse haematopoietic cells worsens the myeloproliferative disease compared to oncogenic K-Ras alone.70 It will therefore be interesting to test the effect of Rce1 inhibition in other disease systems, which may be context dependent. Selective and potent Rce1 inhibitors would significantly aid these investigations.
4. Material and Methods
4.1 Chemistry
4.1.1 Materials
All reagents and solvents were purchased from commercial sources and used without further purification unless otherwise noted.
4.1.2 Instrumentation
1H, 13C, and 19F NMR spectra were recorded on either a 600 MHz, 500 MHz, or 400 MHz spectrometer. Chemical shifts (δ) are expressed in ppm. High Resolution Mass spectroscopy (HRMS) was performed on Agilent Technologies 6540 UHD Accurate-Mass QTOF LC/MS system equipped with Agilent 1260 Infinity series HPLC. Purity of the final compounds was determined to be >95% (unless otherwise stated) using either PerkinElmer 2400 Series II elemental analyzer on CHN mode or reversed-phase HPLC. Elemental analysis values are reported as percentages. For HPLC purity, analysis was performed on either Agilent 1290 Infinity series uHPLC (Method-A) or Agilent 1260 Infinity series HPLC (Method-B) using a ZORBAX Extend-C18, Rapid Resolution HT (1.8 μm particle size, 50 × 2.1 mm dimensions) column with quantitation by area under the curve (AUC) at 254 nm (Agilent Diode Array Detector). We applied either of the following two methods for the analysis: Method-A (mobile phase A: water (pH 8.2 using aq. NH4OH) and mobile phase B: acetonitrile) starting from 5% B and ramping to 100% B over 10 min followed by 100% B hold for 11 min, then ramping to 5% B over 12 min and equilibrating prior to the next run; and Method-B (mobile phase A: water (with 0.1% formic acid) and mobile phase B: acetonitrile) starting from 5% B and ramping to 100% B over 10 min followed by 100% B hold for 11 min, then ramping to 5% B over 13 min and equilibrating prior to the next run. The injection volume was 1 μL and the flow rate was 0.3 mL/min with maximum pressure of about 600 Bar. Samples were dissolved in acetonitrile or they were initially dissolved in approximately 50 μL of DMSO and the resulting solution diluted with acetonitrile. X-ray diffraction data were collected at 100 K on a Bruker APEX II DUO diffractometer.
4.1.3 Synthetic procedures and characterization data
General procedures for the synthesis of compounds 1-6, 9-10, 15-18, 21, 24-25, 37-38, and 40-42: Protocol A, B, C, or D was followed.
Protocol A
4-(((8-Hydroxyquinolin-7-yl)(phenyl)methyl)amino)benzoic acid (1)
p-Aminobenzoic acid (0.27 g, 2.0 mmol) and benzaldehyde (0.21 g, 2.0 mmol) were suspended in ethanol (10 mL) followed by stirring at room temperature for about 5 min. To the mixture, 8-hydroxyquinoline (0.29 g, 2.0 mmol) was added followed by the addition of pyridine (0.2 mL). The reaction mixture was stirred at room temperature for 10-15 days. The reaction was monitored by TLC (3:2 acetonitrile/water) and LC-MS. A light yellow cream colored precipitate was isolated upon filtration, which was washed several times with ethanol to afford pure compound 1 as a cream colored amorphous solid (0.12 g, 0.32 mmol, 16%). 1H NMR (600 MHz, DMSO-d6, δ): 12.00 (br s, 1H), 10.11 (br s, 1H), 8.86 (dd, J = 1.4 Hz, 4.1 Hz, 1H), 8.28 (dd, J = 1.4 Hz, 8.3 Hz, 1H), 7.60 (d, J = 8.8 Hz, 2H), 7.54 (m, 2H), 7.39 (d, J = 8.2 Hz, 3H), 7.33 (t, J = 7.5 Hz, 2H), 7.25 (m, 2H), 6.66 (d, J = 8.7 Hz, 2H), 6.24 (d, J = 7.2 Hz, 1H); 13C NMR (150 MHz, DMSO-d6, δ): 167.4, 151.6, 149.9, 148.3, 142.0, 138.1, 136.0, 130.9, 128.4, 127.6, 127.4, 127.0, 126.1, 124.5, 121.8, 117.6, 117.6, 111.8, 53.9; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C23H18N2O3, 234.09189; found 234.09182. Anal. Calcd for C23H18N2O3: C, 74.58; H, 4.90; N, 7.56. Found: C, 74.43; H, 4.72; N, 7.47.
Protocol B
4-(((4-Bromophenyl)(8-hydroxyquinolin-7-yl)methyl)amino)benzoic acid (5)
p-Aminobenzoic acid (0.27 g, 2.0 mmol), and p-bromobenzaldehyde (0.37 g, 2.0 mmol) were suspended in ethanol (5 mL) followed by stirring at room temperature for about 5 min. To the mixture, 8-hydroxyquinoline (0.29 g, 2.0 mmol) was added and the reaction mixture was refluxed, and heated up to 120 °C for about 12 hours. The reaction was monitored by TLC (3:2 acetonitrile/water,) and LC-MS. The reaction mixture was later allowed to cool down and a cream colored precipitate was isolated upon filtration, which was washed several times with ethanol to afford pure compound 5 as a light cream colored amorphous solid (0.31 g, 35%). 1H NMR (600 MHz, DMSO-d6, δ): 12.02 (br s, 1H), 10.19 (br s, 1H), 8.86 (dd, J = 1.4 Hz, 4.1 Hz, 1H), 8.28 (dd, J = 1.4 Hz, 8.3 Hz, 1H), 7.62 (d, J = 8.8 Hz, 2H), 7.52 (m, 4H), 7.40 (d, J = 8.6 Hz, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.26 (d, J = 7.1 Hz, 1H), 6.66 (d, J = 8.7 Hz, 2H), 6.21 (d, J = 7.1 Hz, 1H); 13C NMR (150 MHz, DMSO-d6, δ): 167.3, 151.5, 150.0, 148.4, 141.5, 138.1, 136.1, 131.3, 130.9, 129.6, 127.7, 126.0, 124.0, 121.9, 120.1, 117.8, 117.7, 111.8, 53.5; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C23H17BrN2O3, 312.00240 and 314.00035, found 312.00230 and 314.00042. Anal. Calcd for C23H17BrN2O3: C, 61.48; H, 3.81; N, 6.23. Found: C, 61.32; H, 3.62; N, 6.19. HPLC (Method B) Rt, = 9.787 min (>95%).
Protocol C
7-((Benzo[d]thiazol-2-ylamino)(phenyl)methyl)quinolin-8-ol (21)
2-Aminobenzothiazole (0.30 g, 2.0 mmol), and benzaldehyde (0.21 g, 2.0 mmol) were taken in a 10-mL microwave reaction vessel equipped with protective septum cap, and charged with minimal volume of ethanol (not more than 2.5 mL). To the mixture, 8-hydroxyquinoline (0.29 g, 2.0 mmol) was added, and the reaction vessel was subjected to microwave heating at 140 °C in a Discovery-CEM closed reaction vessel microwave system, equipped with stirring, for about 15 min. After completion of the reaction, which was detected by LC-MS, the reaction mixture was allowed to cool down, and the solvents were removed under reduced pressure. The residual solid was taken into a 50-mL beaker and heated in the presence of hexane, and washed several times with ethanol to afford pure compound 21 as a colorless amorphous solid (0.69 g, 90%). 1H NMR (600 MHz, DMSO-d6, δ): 10.11 (br s, 1H), 8.95 (d, J = 8.3 Hz, 1H), 8.86 (d, J = 3.9 Hz, 1H), 8.30 (d, J = 8.2 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 7.56 (m, 2H), 7.43 (d, J = 8.6 Hz, 1H), 7.35 (m, 5H), 7.23 (t, J = 7.3 Hz, 1H,), 7.18 (t, J = 7.5 Hz, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H); 13C NMR (150 MHz, DMSO-d6, δ): 165.5, 152.2, 149.7, 148.4, 142.1, 138.1, 136.1, 130.5, 128.4, 127.7, 127.0, 126.1, 125.5, 124.4, 121.9, 121.1, 120.9, 118.3, 118.3, 117.5, 55.1; HRMS-ESI (m/z): [M + H]+ calcd for C23H17N3OS 384.11706, found 384.11726 and [M – C7H6NO2]+ calcd for C23H17N3OS, 234.09189, found 234.09189. Anal. Calcd for C23H17N3OS: C, 72.04; H, 4.47; N, 10.96. Found: C, 71.66; H, 4.34; N, 10.82; HPLC (Method B) Rt = 9.427 min (>95%).
Protocol D
3-(Phenyl(phenylamino)methyl)acridin-4-ol (42)
Aniline (0.37 g, 4.0 mmol), and benzaldehyde (0.42 g, 4.0 mmol) were dissolved in ethanol (10 mL) followed by stirring at room temperature for about 5 minutes. To the mixture, acridin-4-ol (0.78 g, 4.0 mmol) and catalytic pyridine (2 drops) was added and the reaction mixture was refluxed up to 100 °C for about 24 hours. The reaction was monitored by TLC (3:2 acetonitrile/water) and LC-MS. The reaction mixture was later allowed to cool down and a dark brownish yellow precipitate was isolated upon gravity filtration, which was washed several times with ethanol to afford pure compound as an amorphous solid (0.79 g, 42%). 1H NMR (500 MHz, CDCl3, δ): 8.70 (s, 1H), 8.19 (d, J = 8.7 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.77 (t, J = 7.5 Hz, 1H), 7.61 (d, J = 8.9 Hz, 1H), 7.54 (m, 3H), 7.49 (d, J = 8.9 Hz, 1H), 7.33 (t, J = 7.7 Hz, 2H), 7.25 (m, 2H), 7.11 (t, J = 7.5 Hz, 2H), 6.68 (m, 3H), 6.23 (s, 1H), 4.53 (s, 1H); 13C NMR (125 MHz, CDCl3, δ): 148.3, 147.7, 147.3, 142.7, 140.3, 136.1, 130.5, 129.3, 129.1, 128.9, 128.5, 127.6, 127.5, 127.4, 126.14, 126.10, 126.03, 122.9, 118.5, 117.9, 113.8, 56.8; HRMS-ESI (m/z): calculated for fragment ion [M – C6H6N]+ 284.10754 found 284.10760. Anal. Calcd for C26H20N2O: C, 82.95; H, 5.36; N, 7.44. Found: C, 82.47; H, 5.07; N, 7.28.
7-(Phenyl(phenylamino)methyl)quinolin-8-ol (2)
Protocol A, amorphous solid (0.13 g, 20%) 1H NMR (600 MHz, CDCl3, δ): 8.74 (m, 1H), 8.55 (br s, 1H), 8.10 (d, J = 8.2 Hz, 1H), 7.61 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 7.7 Hz, 2H), 7.39 (m, 1H), 7.31 (m, 3H), 7.25 (m, 1H), 7.10 (t, J = 7.9 Hz, 2H), 6.68 (t, J = 7.3 Hz, 1H), 6.63 (d, J = 7.9 Hz, 2H), 6.14 (d, J = 4.0 Hz, 1H), 4.47 (d, J = 2.9 Hz, 1H); 13C NMR (150 MHz, CDCl3, δ): 149.1, 148.2, 147.6, 142.7, 138.5, 136.2, 129.3, 128.9, 127.8, 127.6, 127.5, 126.8, 124.3, 121.9, 118.1, 117.9, 113.8, 57.0; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C22H18N2O, 234.09189, found 234.09185. Anal. Calcd for C22H18N2O: C, 80.96; H, 5.56; N, 8.58. Found: C, 81.02; H, 5.40; N, 8.60; HPLC (Method A) Rt = 4.086 min (>95%).
4-(((4-Cyanophenyl)(8-hydroxyquinolin-7-yl)methyl)amino)benzoic acid (3)
Protocol A, pale yellow amorphous solid (0.879 g, 44%). 1H NMR (500 MHz, DMSO-d6, δ): 12.08 (br s, 1H), 10.30 (br s, 1H), 8.88 (dd, J = 4.2 Hz, 1.5 Hz, 1H), 8.30 (dd, J = 8.3 Hz, 1.5 Hz, 1H), 7.82 (d, J = 8.3 Hz, 2H), 7.66-7.56 (m,5H), 7.49 (d, J = 8.6 Hz, 1H), 7.41 (d, J = 8.6 Hz, 1H), 7.34 (d, J = 7.3 Hz, 2H), 6.69 (d, J = 8.8 Hz, 2H), 6.34 (d, J = 7.2 Hz, 1H); 13C NMR (125 MHz, CDCl3, δ): 167.8, 151.8, 150.7, 149.0, 148.2, 138.6, 136.6, 133.0, 131.5, 128.8, 128.3, 126.5, 124.0, 122.5, 119.3, 118.6, 118.3, 112.4, 110.3, 54.4; HRMS-ESI (m/z): [M + H]+ calcd for C24H17N3O3 396.1343, found 396.1365. Anal. Calcd for C24H17N3O3: C, 72.90; H, 4.33; N, 10.63. Found: C, 72.63; H, 3.94; N, 10.42.
4-(((8-Hydroxyquinolin-7-yl)(p-tolyl)methyl)amino)benzoic acid (4)
Protocol A, yellow amorphous solid (0.416 g, 22%). 1H NMR (400 MHz, DMSO-d6, δ): 12.00 (br s, 1H), 10.10 (br s, 1H), 8.83 (dd, J = 4.1 Hz, 1.5 Hz, 1H), 8.26 (dd, J = 8.3 Hz, 1.3 Hz, 1H), 7.58 (d, J = 8.8 Hz, 2H), 7.53-7.48 (m, 2H), 7.37-7.51, (m, 1H), 7.26-7.20 (m,3H), 7.12-7.10 (m, 2H), 6.63 (d, J = 8.7 Hz, 2H), 6.17 (d, J = 7.0 Hz, 1H), 2.23 (s, 3H); 13C NMR (125 MHz, CDCl3, δ): 167.9, 152.2, 150.3, 148.8, 139.5, 138.6, 136.6, 136.5, 131.4, 129.4, 128.1, 127.8, 126.6, 125.2, 122.3, 118.02, 118.00 112.2, 54.1, 21.1. Anal. Calcd for C24H20N2O3: C, 74.98; H, 5.24; N, 7.29. Found: C, 74.99; H,4.85; N, 7.15.
4-(((4-Fluorophenyl)(8-hydroxyquinolin-7-yl)methyl)amino)benzoic acid (6)
Protocol A, off-white powder (0.409 g, 53%). 1H NMR (400 MHz, DMSO-d6, δ): 12.02 (br s, 1H), 10.17 (br s, 1H), 8.85 (d, J = 2.9 Hz, 1H), 8.27 (d, J = 8.2 Hz, 1H), 7.60 (d, J = 8.7 Hz, 2H), 7.55-7.49 (m, 2H), 7.41-7.38 (m, 3H), 7.26 (d, J = 7.2 Hz, 1H), 7.15 (t, J = 8.8 Hz, 2H), 6.64 (d, J = 8.6 Hz, 2H), 6.21 (d, J = 7.1 Hz, 1H); 13C NMR (150 MHz, CDCl3, δ): 167.8, 161.7 (d, 1J (19F, 13C) = 243.3 Hz, CFar), 152.0, 150.5, 148.9, 138.6 (d, 4J (19F, 13C) = 3.0 Hz, CFar), 136.6, 131.4, 129.8 (d, 3J (19F, 13C) = 8.2 Hz, CFar),, 128.2, 126.4, 124.8, 122.3, 118.2, 118.1, 115.6 (d, 2J (19F, 13C) = 21.3 Hz, CFar), 112.3, 53.8; 19F NMR (470 MHz, DMSO-d6, δ): -60.9; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C23H17FN2O3 252.08247, found 252.08319; Anal. Calcd for C23H17FN2O3: C, 71.13; H, 4.41; N, 7.21. Found: C, 70.88; H, 4.21; N, 7.25.
4-(((8-Hydroxyquinolin-7-yl)(phenyl)methyl)amino)benzonitrile (9)
Protocol C, amorphous solid (0.35 g, 50%). 1H NMR (600 MHz, CDCl3, δ): 8.77 (dd, J = 1.5 Hz, 4.2 Hz, 1H), 8.61 (br s, 1H), 8.12 (dd, J = 1.4 Hz, 8.3 Hz, 1H), 7.48 (d, J = 8.6 Hz, 1H), 7.43 (m, 3H), 7.34 (m, 5H), 7.28 (m, 1H), 6.60 (d, J = 8.8 Hz, 2H), 6.18 (d, J = 5.3 Hz, 1H), 5.05 (d, J = 5.2 Hz, 1H); 13C NMR (150 MHz, CDCl3, δ): 150.6, 149.3, 148.5, 141.3, 138.4, 136.3, 133.8, 129.1, 128.0, 127.5, 126.4, 122.7, 122.2, 120.5, 118.4, 114.6, 113.3, 99.6, 56.5; HRMS-ESI (m/z): [M – C7H5N2]+ calcd for C23H17N3O 234.09189, found 234.09168. Anal. Calcd for : C, 78.61; H, 4.88; N, 11.96. Found: C,78.03; H, 4.68; N, 12.09.
7-((4-Bromophenyl)((4-nitrophenyl)amino)methyl)quinolin-8-ol (10)
Protocol C, amorphous solid (0.40 g, 45%). 1H NMR (600 MHz, Acetone-d6, δ): 9.16 (br s, 1H), 8.85 (dd, J = 1.5 Hz, 4.2 Hz, 1H), 8.31 (dd, J = 1.5 Hz, 8.3 Hz, 1H), 8.01 (d, J = 9.3 Hz, 2H), 7.55 (m, 4H), 7.45 (m, 3H), 7.10 (d, J = 6.3 Hz, 1H), 6.83 (d, J = 9.2 Hz, 2H), 6.42 (d, J = 6.3 Hz, 1H); 13C NMR (150 MHz, acetone-d6, δ): 154.2, 150.7, 149.6, 141.7, 139.1, 138.9, 137.1, 132.6, 130.6, 129.0, 127.1, 126.7, 123.5, 123.2, 121.8, 119.1, 113.0, 55.7; HRMS-ESI (m/z): [M – C6H5N2O2]+ calcd for C22H16BrN3O3 312.00240 and 314.00035, found 312.00243 and 314.00054; HPLC (Method B) Rt = 11.240 min (>95%).
4-(((8-Hydroxyquinolin-7-yl)(pyridin-2-yl)methyl)amino)benzoic acid (15)
Protocol A, amorphous solid (0.562 g, 65%). 1H NMR (400 MHz, DMSO-d6, δ): 12.05 (br s, 1H), 10.28 (br s,1H), 8.87 (dd, J = 4.0, 1.3 Hz, 1H), 8.57 (d, J = 4.0 Hz, 1H), 8.26 (dd, J = 8.3, 1.3 Hz, 1H), 7.78 (td, J = 7.8, 1.7 Hz, 1H), 7.61 (d, J = 8.7 Hz, 2H), 7.56-7.49, (m, 3H), 7.39-7.27 (m,3H), 6.71 (d, J = 8.6 Hz, 2H), 6.33 (d, J = 7.0 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 168.1, 160.6, 151.8, 150.8, 149.6, 149.0, 138.8, 137.8, 136.7, 131.6, 128.4, 126.9, 124.7, 123.2, 122.8, 122.5, 121.8, 118.3, 112.5, 55.5; HRMS-ESI (m/z): [M + H]+ calcd for C22H17N3O3 372.1348, found 372.1349
4-(((8-Hydroxyquinolin-7-yl)(pyridin-3-yl)methyl)amino)benzoic acid (16)
Protocol A, pink amorphous solid solid (1.05 g, 57%). 1H NMR (400 MHz, DMSO-d6, δ): 12.06 (br s, 1H), 10.26 (br s, 1H), 8.83 (d, J = 3.1 Hz, 1H), 8.61 (d, J = 1.0 Hz, 1H), 8.44 (d, J = 4.0 Hz, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.74-7.70 (m, 1H), 7.63-7.60 (m, 2H), 7.56-7.50 (m, 2H), 7.40-7.32, (m, 3H) 6.66 (d, J = 8.6 Hz, 2H), 6.25 (d, J = 7.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 167.8, 151.9, 149.5, 148.9, 148.7, 138.6, 137.8, 136.6, 135.6, 131.5, 126.9, 128.7, 126.2, 124.1, 124.0, 122.4, 118.5, 118.3, 112.4, 52.7; HRMS-ESI (m/z): [M + H]+ calcd for C22H17N3O3 372.1343, found 372.1341. Anal. Calcd for C22H17N3O3: C, 71.15; H, 4.61; N, 11.31. Found: C, 70.82; H, 4.24; N, 11.15.
4-((Cyclohexyl(8-hydroxyquinolin-7-yl)methyl)amino)benzoic acid (17)
Protocol B, off-white amorphous solid (0.406 g, 54%). 1H NMR (500 MHz, DMSO-d6, δ): 11.89 (br s, 1H), 9.87 (br s, 1H), 8.86 (dd, J = 4.2, 1.6 Hz, 1H), 8.26 (dd, J = 8.3, 1.6 Hz, 1H), 7.54-7.51 (m, 3H), 7.46 (d, J = 8.5 Hz, 1H), 7.36 (d, J = 8.6 Hz, 1H), 6.99 (d, J = 8.2 Hz, 1H), 6.59 (d, J = 8.7 Hz, 2H), 4.77 (t, J = 8.4 Hz, 1H), 2.11 (d, J = 11.6 Hz, 1H), 1.80-1.61 (m, 4H), 1.29-1.05 (m, 6H); 13C NMR (150 MHz, DMSO-d6, δ): 167.8, 152.8, 150.6, 148.6, 138.3, 136.4, 131.4, 127.8, 126.3, 125.3, 122.0, 118.1, 117.2, 111.5, 55.1, 42.9, 30.4, 29.6, 26.12, 26.10; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C23H24N2O3 240.13884, found 240.13960. Anal. Calcd for C23H24N2O3: C, 73.38; H, 6.43. N, 7.44. Found: C, 73.28; H, 6.34; N, 7.42.
7-(Cyclohexyl(phenylamino)methyl)quinolin-8-ol (18)
Protocol B, off-white amorphous solid (0.257 g, 39%). 1H NMR (600 MHz, DMSO-d6, δ): 9.74 (br s, 1H), 8.84 (d, J = 2.7 Hz, 1H), 8.24 (d, J = 8.2 Hz, 1H), 7.51-7.49 (m, 2H), 7.32 (d, J = 8.5 Hz, 1H), 6.90 (t, J = 7.8 Hz, 2H), 6.56 (d, J = 7.9 Hz, 2H), 6.36 (t, J = 7.2 Hz, 1H), 6.13 (d, J = 8.4 Hz, 1H), 4.71 (t, J = 8.3 Hz, 1H), 2.12(d, J = 11.6 Hz, 1H), 1.78-1.72 (m, 2H), 1.64-1.57 (m, 2H), 1.32-1.00 (m, 6H); 13C NMR (150 MHz, DMSO-d6, δ): 150.1, 148.5, 148.0, 137.7, 135.9, 128.6, 127.1, 126.1, 125.7, 121.3, 117.3, 115.1, 112.1, 54.6, 42.7, 29.9, 29.2, 26.1, 25.8, 25.7; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C22H24N2O 240.13884, found 240.13909. Anal. Calcd for C22H24N2O: C, 79.48; H, 7.28; N, 8.43. Found: C, 79.09; H, 7.14; N, 8.36.
7-((4-Bromo-2,6-difluorophenyl)(phenylamino)methyl)quinolin-8-ol (24)
Protocol A, amorphous solid (0.26 g, 30%). 1H NMR (600 MHz, CDCl3, δ): 8.73 (m, 1H), 8.56 (bs, 1H), 8.10 (m, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.40 (m, 1H), 7.30 (d, J = 8.05 Hz, 1H), 7.15 (m, 2H), 7.05 (m, 2H), 6.72 (m, 3H), 6.54 (d, J = 8.5 Hz, 1H), 4.77 (d, J = 8.6 Hz, 1H); 13C NMR (150 MHz, CDCl3, δ): 160.9 (dd, 1J (19F, 13C) = 252.6 Hz, 3J (19F, 13C) = 9.3 Hz, CFar), 149.2, 148.1, 146.4, 138.0, 135.9, 129.3, 127.7, 126.1, 121.8, 121.3, 120.9 (t, 3J (19F, 13C) = 12.8 Hz, CBrar), 118.4, 117.5 (t, 2J (19F, 13C) = 17.5 Hz, CCar), 117.4, 115.8 (distorted dd, 2J (19F, 13C) = 23.9 Hz, CHar), 113.6, 47.5; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C22H15BrF2N2O 347.98356 and 349.98151, found 347.98341 and 349.98180. Anal. Calcd for C22H15BrF2N2O: C, 59.88; H, 3.43; N, 6.35. Found: C, 59.76; H, 3.16; N, 6.32.
4-(((4-Bromo-2,6-difluorophenyl)(8-hydroxyquinolin-7-yl)methyl)amino)benzoic acid (25)
Protocol A, amorphous solid (0.17 g, 18%). 1H NMR (600 MHz, DMSO-d6, δ): 12.08 (br s, 1H), 10.18 (br s, 1H), 8.83 (m, 1H), 8.29 (d, J = 8.2 Hz, 1H), 7.65 (d, J = 8.1 Hz, 3H), 7.53 (m, 1H), 7.46 (d, J = 8.0 Hz, 2H), 7.39 (d, J = 8.5 Hz, 1H), 7.34 (d, J = 6.5 Hz, 1H), 6.60 (d, J = 8.2 Hz, 2H), 6.35 (d, J = 6.4 Hz, 1H); 13C NMR (125 MHz, Acetone-d6, δ): 167.7, 162.2 (dd, 1J (19F, 13C) = 252.9 Hz, 3J (19F, 13C) = 9.0 Hz, CFar), 152.23, 152.17, 150.5, 149.4, 139.0, 137.0, 132.4, 129.0, 127.1, 123.1, 122.1 (merged t, CBrar), 119.9, 118.2, 116.8, 116.5 (merged t, CCar), 112.9 (distorted d, CHar), 47.5; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C23H15BrF2N2O3 347.98356 and 349.98151, found 347.98338 and 349.98151; Rt HPLC (Method-B) = 9.873 min (>95%).
8-Hydroxy-7-(phenyl(phenylamino)methyl)quinoline-2-carbonitrile (37)
Protocol D, amorphous solid (0.44 g, 25%). 1H NMR (500 MHz, DMSO-d6, δ): 10.56 (s, 1H), 8.54 (d, J = 8.4 Hz, 1H), 8.00 (d, J = 8.4 Hz, 1H), 7.81 (d, J = 8.5 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.42 (m, 2H), 7.33 (t, J = 7.8 Hz, 2H), 7.24 (t, J = 7.3 Hz, 1H), 7.01 (t, J = 7.9 Hz, 2H), 6.65 (d, J = 8.1 Hz, 2H), 6.52 (m, 2H), 6.18 (d, J = 7.1 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 150.1, 147.7, 142.3, 138.5, 138.0, 130.6, 129.6, 128.8, 128.41, 128.37, 128.0, 127.4, 126.9, 123.9, 117.8, 117.7, 116.3, 112.9, 54.0; HRMS-ESI (m/z): calculated for fragment ion [M – C6H6N]+ 259.08714 found 259.08709. Anal. Calcd for C23H17N3O: C, 78.61; H, 4.88; N, 11.96. Found: C, 77.59; H, 4.51; N, 11.63.
8-Hydroxy-7-(phenyl(phenylamino)methyl)quinoline-2-carboxylic acid (38)
Protocol D, amorphous solid (0.044g, 3%). 1H NMR (600 MHz, DMSO-d6, δ): 12.91 (br s, 1H), 10.45 (br s, 1H), 8.50 (d, J = 8.5 Hz, 1H), 8.11 (d, J = 8.5 Hz, 1H), 7.78 (d, J = 8.6 Hz, 1H), 7.52 (d, J = 8.6 Hz, 1H), 7.54 (m, 2H), 7.33 (t, J = 7.8 Hz, 2H), 7.23 (t, J = 7.4 Hz, 1H), 7.01 (m, 2H), 6.67 (d, J = 7.8 Hz, 2H), 6.54 (m, 1H), 6.50 (t, J = 7.3 Hz, 1H), 6.18 (d, J = 7.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 165.5, 150.7, 148.3, 144.8, 142.9, 138.8, 136.6, 129.5, 129.2, 128.8, 127.8, 127.4, 127.1, 120.2, 118.0, 116.8, 114.3, 113.4, 54.6; HRMS-ESI (m/z): calculated for fragment ion [M – C6H6N]+ 278.08172 found 278.08172.
5-Chloro-7-(phenyl(phenylamino)methyl)quinolin-8-ol (40)
Protocol D, amorphous solid (0.09g, 5%). 1H NMR (600 MHz, CDCl3, δ): 8.77 (d, J = 3.9 Hz, 1H), 8.52 (br s, 1H), 8.45 (d, J = 8.4 Hz, 1H), 7.74 (s, 1H), 7.48 (m, 3H), 7.33 (t, J = 7.7 Hz, 2H), 7.26 (m, 1H), 7.12 (t, J = 8.1 Hz, 2H), 6.71 (t, J = 7.3 Hz, 1H), 6.63 (d, J = 7.9 Hz, 2H), 6.10 (s, 1H), 4.42 (s, 1H); 13C NMR (150 MHz, CDCl3, δ): 148.5, 148.0, 147.2, 141.9, 138.7, 133.3, 129.2, 128.8, 127.6, 127.4, 126.1, 125.5, 124.8, 122.3, 120.9, 118.0, 113.6, 56.7; HRMS-ESI (m/z): calculated for fragment ion [M – C6H6N]+ 268.05292 found 268.05287.
5-Bromo-7-(phenyl(phenylamino)methyl)quinolin-8-ol (41)
Protocol D, amorphous solid (0.2 g, 10%). 1H NMR (500 MHz, CDCl3, δ): 8.73 (d, J = 3.9 Hz, 1H), 8.62 (br s, 1H), 8.40 (d, J = 8.5 Hz, 1H), 7.92 (s, 1H), 7.47 (m, 3H), 7.33 (t, J = 7.5 Hz, 2H), 7.25 (m, 1H), 7.12 (t, J = 7.9 Hz, 2H), 6.71 (t, J = 7.3 Hz, 1H), 6.63 (d, J = 8.0 Hz, 2H), 6.10 (s, 1H), 4.42 (s, 1H); 13C NMR (125 MHz, CDCl3, δ): 148.9, 148.7, 147.4, 142.1, 139.1, 136.0, 129.8, 129.4, 129.0, 127.8, 127.6, 127.0, 125.7, 122.8, 118.3, 113.8, 110.6, 56.9; HRMS-ESI (m/z): calculated for fragment ion [M – C6H6N]+ 312.00240 found 312.00245. Anal. Calcd for C22H17BrN2O: C, 65.20; H, 4.23; N, 6.91. Found: C, 65.01; H, 4.09; N, 6.74.
General procedure for the synthesis of compounds 11-14 and 19
Ethyl 4-(((8-hydroxyquinolin-7-yl)(phenyl)methyl)amino)benzoate (11)
Benzocaine (0.331 g, 2.0 mmol), benzaldehyde (0.212 g, 2.0 mmol) and 8-hydroxyquinoline (0.275 g, 2.0 mmol) were suspended in a 10% aqueous solution of NaCl (2 mL). This was heated to 100 °C overnight. Upon cooling, the resultant precipitate was filtered and washed with ethanol to yield 11 as a white amorphous solid (0.365 g, 46%). 1H NMR (600 MHz, DMSO-d6, δ): 10.13 (br s, 1H), 8.87 (d, J = 2.7 Hz, 1H), 8.29 (d, J = 8.2 Hz, 1H), 7.64 (d, J = 8.7 Hz, 2H), 7.60-7.52 (m, 2H), 7.40-7.32 (m, 6H), 7.34 (t, J = 7.7 Hz, 3H), 7.25 (t, J = 7.2 Hz, 1H), 6.69 (d, J = 8.5 Hz, 2H), 6.26 (d, J = 7.1 Hz, 1H), 4.17 (q, J = 7.0 Hz, 2H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6, δ): 165.7, 151.9, 149.9, 148.3, 141.9, 138.1, 136.0, 130.7, 128.4, 127.6, 127.4, 127.0, 126.1, 124.4, 121.8, 117.6, 116.8, 111.9, 59.5, 53.9, 14.3; HRMS-ESI (m/z): [M – C9H10NO2]+ calcd for C25H22N2O3 302.07927, found 302.08118. Anal. Calcd for C25H22N2O3: C, 75.36; H, 5.57; N, 7.03. Found: C, 75.62; H, 5.27; N, 7.03.
Ethyl 4-(((8-hydroxyquinolin-7-yl)(4-(trifluoromethyl)phenyl)methyl)amino)benzoate (12)
White solid (0.439 g, 47%). 1H NMR (600 MHz, DMSO-d6, δ): 10.27 (br s, 1H), 8.88 (d, J = 8.2 Hz, 1H), 8.30 (d, J = 8.2 Hz, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 8.6 Hz, 2H), 7.61 (d, J = 8.0 Hz, 2H), 7.57 (dd, J = 8.2, 4.1 Hz, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.42-7.41 (m, 2H), 6.71 (d, J = 8.5 Hz, 2H), 6.35 (d, J = 7.1 Hz, 1H), 4.18 (q, J = 7.1 Hz, 2H), 1.24 (t, J = 7.1 Hz, 3H); 13C NMR (150 MHz, DMSO-d6, δ): 165.7, 151.7, 150.1, 148.5, 146.7, 138.1, 136.1, 130.8, 128.1, 127.8, 126.0, 125.4, 123.6, 122.0, 117.8, 117.1, 112.0, 59.6, 53.7, 14.3; 19F NMR (564 MHz, DMSO-d6, δ): -60.9; HRMS-ESI (m/z): [M – C9H10NO2]+ calcd for C26H21F3N2O3 302.07927, found 302.08118. Anal. Calcd for C26H21F3N2O3: C, 66.95; H, 5.54; N, 6.01. Found: C, 66.69; H, 4.35; N, 5.91.
Ethyl 4-(((4-chlorophenyl)(8-hydroxyquinolin-7-yl)methyl)amino)benzoate (14)
Off-white powder (0.366 g, 42%). 1H NMR (600 MHz, DMSO-d6, δ): 10.20 (br s, 1H), 8.87 (d, 1H, J = 2.7 Hz), 8.30 (d, 1H, J = 8.2 Hz), 7.65 (d, 2H, J = 8.6 Hz), 7.56 (dd, 1H, J = 8.2, 4.1 Hz), 7.49 (d, 1H, J = 8.5 Hz), 7.40 (s, 5H), 7.35 (d, 1H, J = 7.1 Hz), 6.70 (d, 2H, J = 8.5 Hz), 6.25 (d, 1H, J = 7.1 Hz), 4.18 (q, 2H, J = 7.0 Hz), 1.23 (t, 3H, J = 7.1 Hz); 13C NMR (150 MHz, DMSO-d6, δ): 166.2, 152.2, 150.5, 148.9, 141.4, 138.6, 136.6, 132.1, 131.3, 129.7, 128.9, 128.2, 126.5, 124.4, 122.4, 118.2, 117.5, 112.4, 60.1, 53.9, 14.8; HRMS-ESI (m/z): [M – C9H10NO2]+ calcd for C25H21ClN2O3 268.05292, found 268.05451. Anal. Calcd for C25H21ClN2O3: C, 69.36; H, 4.89; N, 6.47. Found: C, 68.99; H, 4.67; N, 6.36.
Ethyl 4-((cyclopentyl(8-hydroxyquinolin-7-yl)methyl)amino)benzoate (19)
White powder (0.296 g, 38%). 1H NMR (600 MHz, CDCl3, δ): 9.89 (br s, 1H), 8.86 (br s, 1H), 8.25 (d, 1H, J = 8.1 Hz), 7.55-7.51 (m, 4H), 7.36 (d, 1H, J = 8.4 Hz), 7.12 (d, 1H, J = 7.2 Hz), 6.61 (d, 2H, J = 7.7 Hz), 4.79 (t, 1H, J = 8.3 Hz), 4.14-4.12 (m, 2H), 2.41-2.37 (m, 1H), 2.01-1.94 (m, 1H), 1.70-1.44 (m, 5H), 1.32-1.24 (m, 2H), 1.20 (m, 3H); 13C NMR (150 MHz, DMSO-d6, δ): 165.7, 152.2, 149.8, 148.2, 137.8, 135.9, 130.7, 127.3, 125.7, 121.5, 117.8, 115.9, 112.4, 59.4, 53.9, 45.5, 30.5, 28.8, 25.0, 24.7, 14.3; HRMS-ESI (m/z): [M – C9H10NO2]+ calcd for C24H26N2O3 226.12319, found 226.12274. Anal. Calcd for C24H26N2O3: C, 73.82; H, 6.71; N, 7.17. Found: C, 73.66; H, 6.54; N, 7.15.
General procedure for the synthesis of compounds 27-31, 33, and 45-46
2-(Phenyl(phenylamino)methyl)naphthalen-1-ol (27)
Aniline (0.37 g, 4.0 mmol), and benzaldehyde (0.42 g, 4.0 mmol) were dissolved in ethanol (10 mL) followed by stirring at room temperature for about 5 min. To the mixture, 1-naphthol (0.57 g, 4.0 mmol) was added followed by the addition of catalytic pyridine (0.2 mL) and the reaction was stirred at room temperature for 24 hours. The reaction was monitored by TLC (3:2 acetonitrile/water) and LC-MS. A colorless precipitate was isolated upon filtration, which was washed several times with ethanol to afford pure compound 27 as a colorless amorphous solid (1.2 g, 95%). 1H NMR (600 MHz, CDCl3, δ): 10.82 (s, 1H), 8.27 (m, 1H), 7.75 (m, 1H), 7.46 (m, 2H), 7.41 (m, 2H), 7.31 (m, 4H), 7.17 (m, 2H), 7.08 (d, J = 8.4 Hz, 1H), 6.90 (t, J = 7.4 Hz, 1H), 6.83 (d, J = 7.7 Hz, 2H), 5.59 (s, 1H), 4.23 (s, 1H); 13C NMR (150 MHz, CDCl3, δ): 152.7, 146.8, 141.8, 134.1, 129.6, 129.5, 128.6, 127.9, 127.5, 126.6, 126.3, 125.8, 125.4, 122.5, 121.8, 119.8, 117.7, 116.7, 66.5; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C23H19NO 233.09664, found 233.09661. Anal. Calcd for C23H19NO: C, 84.89; H, 5.89; N, 4.30. Found: C, 84.73; H, 5.91; N, 4.45.
2-(((4-(tert-Butyl)phenyl)amino)(phenyl)methyl)naphthalen-1-ol (28)
Crystals (1.29 g, 85%), m.p. 157-158 °C (hexane). 1H NMR (600 MHz, CDCl3, δ): 11.11 (s, 1H), 8.27 (m, 1H), 7.74 (m, 1H), 7.46 (m, 2H), 7.41 (m, 2H), 7.34 (m, 3H), 7.30 (m, 1H), 7.20 (d, J = 7.5 Hz, 2H), 7.09 (d, J = 8.3 Hz, 1H,), 6.80 (d, J = 7.6 Hz, 2H), 5.56 (s, 1H), 4.17 (s, 1H), 1.24 (s, 9H); 13C NMR (150 MHz, CDCl3, δ): 152.6, 144.5, 144.1, 141.8, 133.8, 129.2, 128.3, 127.7, 127.3, 126.3, 126.2, 126.1, 125.6, 125.1, 122.3, 119.5, 117.5, 116.2, 66.8, 34.1, 31.4; HRMS-ESI (m/z): [M – C10H14N]+ calcd for C27H27NO 233.09664, found 233.09670. Anal. Calcd for C27H27NO: C, 85.00; H, 7.13; N, 3.67. Found: C, 84.32; H, 7.05; N, 3.64.
4-(((1-Hydroxynaphthalen-2-yl)(phenyl)methyl)amino)benzoic acid (29)
Amorphous solid (0.35 g, 24%). 1H NMR (600 MHz, DMSO-d6, δ): 12.00 (s, 1H), 9.64 (s, 1H), 8.25 (d, J = 7.95 Hz, 1H), 7.79 (m, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.44 (m, 6H), 7.32 (m, 2H), 7.23 (m, 2H), 6.70 (d, J = 8.8 Hz, 2H), 6.34 (d, J = 7.1 Hz, 1H); 13C NMR (150 MHz, DMSO-d6, δ): 167.4, 151.8, 149.4, 142.5, 133.4, 130.9, 128.3, 127.6, 127.4, 126.9, 125.9, 125.4, 125.2, 125.0, 123.7, 122.1, 119.8, 117.5, 111.9, 54.0; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C24H19NO3 233.09664, found 233.09618. Anal. Calcd for C24H19NO3: C, 78.03; H, 5.18; N, 3.79. Found: C, 78.30; H, 5.24; N, 3.79.
3-(((1-Hydroxynaphthalen-2-yl)(phenyl)methyl)amino)benzoic acid (30)
Amorphous solid (0.32 g, 22%). 1H NMR (600 MHz, DMSO-d6, δ): 12.59 (bs, 1H), 9.68 (s, 1H), 8.24 (d, J = 8.0 Hz, 1H); 7.78 (d, J = 7.7 Hz, 1H); 7.39 (m, 9H), 7.21 (m, 1H), 7.12 (m, 2H), 6.91 (m, 1H), 6.74 (d, J = 7.1 Hz, 1H); 6.28 (d, J = 7.0 Hz, 1H); 13C NMR (150 MHz, DMSO-d6, δ): 167.8, 149.4, 148.1, 142.9, 133.4, 131.3, 128.8, 128.3, 127.6, 127.4, 126.8, 125.8, 125.5, 125.2, 125.0, 123.9, 122.1, 119.7, 117.2, 117.2, 113.9, 54.5; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C24H19NO3 233.09664, found 234.09696. Anal. Calcd for C24H19NO3: C, 78.03; H, 5.18; N, 3.79. Found: C, 77.88; H, 5.35; N, 3.70.
4-(((4-Bromophenyl)(1-hydroxynaphthalen-2-yl)methyl)amino)benzoic acid (31)
The reaction was performed in DMSO solvent and followed the same procedure as discussed for compound 26. Amorphous solid (0.98 g, 55%). 1H NMR (600 MHz, DMSO-d6, δ): 12.10 (bs, 1H), 9.71 (bs, 1H), 8.26 (d, J = 8.0 Hz, 1H), 7.79 (d, J = 7.7 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.43 (m, 8H), 7.22 (d, J = 6.9 Hz, 1H), 6.70 (d, J = 8.1 Hz, 2H), 6.32 (d, J = 6.9 Hz, 1H); 13C NMR (150 MHz, DMSO-d6, δ): 167.5, 151.7, 149.5, 142.0, 133.6, 131.3, 131.0, 129.7, 127.7, 126.1, 125.3, 125.2, 123.3, 122.2, 120.0, 120.0, 117.8, 112.7, 112.0, 53.6; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C24H18BrNO3 311.00715 and 313.00511, found 311.00690 and 313.00503. Anal. Calcd for C24H18BrNO3: C, 64.30; H, 4.05; N, 3.12. Found: C, 64.48; H, 4.23; N, 3.25.
4-Bromo-2-(phenyl(phenylamino)methyl)naphthalen-1-ol (33)
Amorphous solid (1.45 g, 90%). 1H NMR (600 MHz, CDCl3, δ): 11.07 (s, 1H), 8.30 (d, J = 8.4 Hz, 1H), 8.11 (d, J = 8.4 Hz, 1H), 7.59 (m, 1H), 7.51 (m, 1H), 7.36 (m, 6H), 7.20 (m, 2H), 6.94 (t, J = 7.4 Hz, 1H), 6.84 (d, J = 7.8 Hz, 2H), 5.51 (s, 1H), 4.22 (s, 1H); 13C NMR (150 MHz, CDCl3, δ): 152.5, 146.3, 141.1, 131.9, 129.5, 129.4, 129.4, 128.7, 127.7, 127.7, 126.9, 126.6, 125.9, 122.8, 121.9, 118.4, 116.5, 112.3, 66.2; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C23H18BrNO 311.00715 and 313.00511, found 311.00811 and 313.00622. Anal. Calcd for C23H18BrNO: C, 68.33; H, 4.49; N, 3.46. Found: C, 68.18; H, 4.23; N, 3.38.
2-((4-Bromo-2,6-difluorophenyl)(phenylamino)methyl)naphthalen-1-ol (45)
Amorphous solid (1.49 g, 85%). 1H NMR (600 MHz, CDCl3, δ): 10.02 (s, 1H), 8.33 (m, 1H), 7.74 (m, 1H), 7.48 (m, 2H), 7.24 (m, 3H), 7.12 (d, J = 8.0 Hz, 2H), 6.93 (m, 3H), 6.74 (d, J = 8.5 Hz, 1H), 6.42 (d, J = 10.5 Hz, 1H), 4.57 (d, J = 10.7 Hz, 1H); 13C NMR (150 MHz, CDCl3, δ): 161.4 (dd, 1J (19F, 13C) = 252.2 Hz, 3J (19F, 13C) = 9.2 Hz, CFar), 152.9, 145.0, 134.5, 129.8, 127.4, 126.9, 125.6, 125.6, 124.3, 122.6, 122.4 (t, 3J (19F, 13C) = 12.8 Hz, CBrar), 122.2, 119.4, 117.1, 116.3 (distorted dd, 2J (19F, 13C)= 26.2 Hz, CHar), 115.5, 115.1 (t, 2J (19F, 13C) = 17.0 Hz, CCar), 53.2; HRMS-ESI (m/z): [M –C6H6N]+ calcd for C23H16BrF2NO 346.98831 and 348.98626, found 346.98740 and 348.98551. Anal. Calcd for C23H16BrF2NO: C, 62.74; H, 3.66; N, 3.18. Found: C, 62.54; H, 3.33; N, 3.17.
4-(((4-Bromo-2,6-difluorophenyl)(1-hydroxynaphthalen-2-yl)methyl)amino)benzoic acid (46)
p-Aminobenzoic acid (0.68 g, 5.0 mmol), and 4-bromo-2,6-difluorobenzaldehyde (1.10 g, 5.0 mmol) were suspended in ethanol (about 15 mL) followed by stirring at room temperature for about 5 min. To the mixture, 1-naphthol (0.72 g, 5.0 mmol) was added and the resulting mixture was stirred at room temperature for about 24 hours. A cream colored precipitate was isolated upon filtration, which was washed several times with ethanol and warm hexane to afford pure 37 as a light cream colored amorphous solid (0.67 g, 28%). 1H NMR (600 MHz, DMSO-d6, δ): 12.07 (bs, 1H), 9.67 (bs, 1H), 8.20 (m, 1H), 7.81 (m, 1H), 7.66 (d, J = 8.9 Hz, 2H), 7.56 (d, J = 8.6 Hz, 1H), 7.46 (m, 4H), 7.41 (d, J = 8.6 Hz, 1H), 7.29 (d, J = 6.8 Hz, 1H), 6.64 (d, J = 8.8 Hz, 2H), 6.42 (d, J = 6.8 Hz, 1H); 13C NMR (150 MHz, DMSO-d6, δ): 167.4, 160.6 (dd, 1J (19F, 13C) = 252.4 Hz, 3J (19F, 13C) = 9.3 Hz, CFar), 151.2, 149.5, 133.7, 131.1, 127.6, 125.9, 125.1, 125.0, 124.9, 121.7, 121.3, 120.6 (t, 3J (19F, 13C) = 13.0 Hz, CBrar), 119.0, 118.0, 117.5 (t, 2J (19F, 13C) = 17.4 Hz, CCar), 115.7 (distorted d, 2J (19F, 13C) = 29.3, CHar), 111.5, 46.3; HRMS-ESI (m/z): [M – C7H6NO2]+ calcd for C24H16BrF2NO3 346.98831 and 348.98626, found 346.98806 and 348.98620. Anal. Calcd for C24H16BrF2NO3: C, 59.52; H, 3.33; N, 2.89. Found: C, 59.33; H, 3.10; N, 2.86.
General procedure for the synthesis of compounds 34-35
1-(Phenyl(phenylamino)methyl)naphthalen-2-ol (34)
Aniline (0.18 g, 2.0 mmol), and benzaldehyde (0.21 g, 2.0 mmol) were dissolved in ethanol (5 mL) followed by stirring at room temperature for about 5 min. To the mixture, 2-naphthol (0.29 g, 2.0 mmol) and 2-3 drops of pyridine were added and the reaction mixture was refluxed at 100 °C for about 12 hours. The reaction was monitored by TLC (3:2 acetonitrile/water) and LC-MS. The reaction mixture was later allowed to cool down and a colorless precipitate was isolated upon filtration, which was washed with ethanol to afford pure compound 34 as a colorless amorphous solid (0.51 g, 80%). 1H NMR (600 MHz, CDCl3, δ): 11.49 (s, 1H), 7.78 (m, 2H), 7.74 (d, J = 8.9 Hz, 1H), 7.48 (m, 2H), 7.36 (m, 3H), 7.29 (m, 2H), 7.15 (m, 3H), 6.92 (t, J = 7.4 Hz, 1H), 6.77 (d, J = 7.7 Hz, 2H), 6.17 (s, 1H), 4.14 (br s, 1H); 13C NMR (150 MHz, CDCl3, δ): 156.4, 146.9, 141.2, 131.7, 130.2, 129.6, 129.6, 129.3, 129.2, 128.8, 128.2, 127.0, 123.0, 122.1, 121.6, 120.2, 116.5, 113.9, 63.0; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C23H19NO 233.09664, found 233.09656; HPLC (Method B) Rt = 10.972 min (>95%).
1-(Phenyl(phenylamino)methyl)naphthalene-2,7-diol (35)
Amorphous solid (0.50 g, 74%). 1H NMR (600 MHz, CDCl3, δ): 11.51 (s, 1H), 7.65 (m, 2H), 7.44 (d, J = 7.0 Hz, 2H), 7.31 (m, 3H), 7.15 (t, J = 7.6 Hz, 2H), 7.06 (s, 1H), 6.98 (m, 1H), 6.92 (t, J = 7.2 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 6.76 (d, J = 7.8 Hz, 2H), 6.00 (s, 1H), 5.00 (s, 1H), 4.12 (s, 1H); 13C NMR (150 MHz, CDCl3, δ): 157.1, 154.4, 146.9, 140.9, 133.2, 131.1, 130.0, 129.7, 129.6, 128.8, 128.2, 124.7, 122.1, 117.8, 116.5, 114.4, 112.6, 104.7, 63.0; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C23H19NO2 249.09155, found 249.09185. Anal. Calcd for C23H19NO2: C, 80.92; H, 5.61; N, 4.10. Found: C, 80.45; H, 5.42; N, 3.93.
General procedure for the synthesis of compounds 48-50
Ethyl 4-(benzhydrylamino)benzoate (48)
Benzophenone (0.500 g, 2.74 mmol) was dissolved in dry CH2Cl2 (10 mL) and TiCl4 (1M solution in CH2Cl2, 1.1 mL, 1.1 mmol) was added to it. The mixture was cooled to 0 °C followed by the addition of benzocaine (0.996 g, 6.03 mmol) in dry CH2Cl2 (10 mL) and stirred for about 3 hours at room temperature under N2. The reaction was then quenched with NaBH3CN (1M solution in THF, 3.29 mL, 3.29 mmol) followed by the addition of 10 mL of methanol and stirring for an additional 3 hours at room temperature. The mixture was made basic (pH ∼ 10) using 10% w/v aqueous NaOH and then filtered. The filtrate was extracted with CH2Cl2, and the organic extract was washed with brine and dried (MgSO4). The crude product was purified by flash chromatography (0% to 10% EtOAc/hexane) to give the product as a white amorphous solid (0.626 g, 73%). 1H NMR (600 MHz, DMSO-d6, δ): 7.65 (d, J = 8.9 Hz, 2H), 7.39 (m, 4H), 7.34 (t, J = 7.7 Hz, 4H), 7.29 (d, J = 7.2 Hz, 1H), 7.25 (t, J = 7.3 Hz, 2H), 6.72 (d, J = 8.8 Hz, 2H), 5.79 (d, J = 7.2 Hz, 1H), 4.19 (q, J = 7.0 Hz, 2H), 1.25 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6, δ): 165.8, 151.8, 142.6, 130.6, 128.5, 127.3, 127.0, 116.7, 112.1, 60.2, 59.5, 14.3; HRMS-ESI (m/z): [M – C9H10NO2]+ calcd for C22H21NO2 167.08608, found 167.08041; [M + Na]+ calcd for C22H21NO2 354.14670, found 234.14412. Anal. Calcd for C22H21NO2: C, 79.73; H, 6.39; N, 4.23. Found: C, 79.38; H, 6.23; N, 4.17.
Ethyl 4-(((4-bromophenyl)(phenyl)methyl)amino)benzoate (49)
White amorphous solid (0.310 g, 28%). 1H NMR (500 MHz, DMSO-d6, δ): 7.64 (d, J = 8.9 Hz, 2H), 7.54 (d, J = 8.4, 2H), 7.38-7.37 (m, 6H), 7.30-7.26 (m, 2H), 6.69 (d, J = 8.8 Hz, 2H), 5.80 (d, J = 7.2 Hz, 1H), 4.19 (q, J = 7.8 Hz, 2H), 1.25 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, DMSO-d6, δ): 166.2, 152.1, 142.6, 142.5, 131.9, 131.1, 130.0, 129.0, 127.9, 127.7, 120.6, 117.4, 112.7, 60.0, 59.9, 14.8; HRMS-ESI (m/z): [M + H]+ calcd for C22H20BrNO2 410.07557, found 410.07401; HPLC (Method B) Rt = 7.374 min (>95%).
Ethyl 4-((bis(4-bromophenyl)methyl)amino)benzoate (50)
White amorphous solid (0.250 g, 37%). 1H NMR (500 MHz, DMSO-d6, δ): 7.65 (d, J = 8.8 Hz, 2H), 7.55 (d, J = 8.4 Hz, 4H), 7.33-7.28 (m, 5H), 6.70 (d, J = 8.7 Hz, 2H), 5.82 (d, J = 7.3 Hz, 1H), 4.19 (q, J = 7.1 Hz, 2H), 1.25 (t, J = 7.1 Hz, 3H); 13C NMR (125 MHz, DMSO-d6, δ): 166.2, 152.0, 142.0, 132.0, 131.2, 130.1, 120.9, 117.7, 112.7, 60.1, 59.2, 14.8; HRMS-ESI (m/z): [M]+ calcd for C22H19Br2NO2 486.97825, found 486.98092; HPLC (Method B) Rt = 7.929 min (>95%).
General procedure for the synthesis of compounds 51-53
4-(Benzhydrylamino)benzoic acid (51). Ester, 48
(1.54 g, 4.65 mmol) was dissolved in dioxane (5 mL) followed by addition of 5 M aqueous NaOH (5 mL) and ethanol (5 mL). The mixture was stirred at 80 °C for 8 h. The crude mixture was concentrated yielding a white precipitate in the aqueous layer. Water was added until the precipitate completely dissolved. The mixture was then made acidic (pH 6) with 3N HCl (aq) from which the desired product precipitated. This was filtered, washed with hexane, and dried to give the product as a cream-colored amorphous solid (1.24 g, 88%). 1H NMR (500 MHz, DMSO-d6, δ): 12.0 (bs, 1H), 7.61 (d, J = 8.9 Hz, 2H), 7.39-7.37 (m, 4H), 7.35-7.32 (m, 4H), 7.25-7.19 (m, 3H), 6.68 (d, J = 8.8 Hz, 2H), 5.76 (d, J = 7.1 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 167.9, 152.1, 143.2, 131.3, 129.0, 127.8, 127.5, 118.1, 112.5, 60.7; HRMS-ESI (m/z): [M + Na]+ calcd for C20H17NO2 326.11570; found 326.11580; HPLC (Method B) Rt = 5.431 min (>90%).
4-(((4-Bromophenyl)(phenyl)methyl)amino) benzoic acid (52)
Cream-colored amorphous solid (0.175 g, 73%). 1H NMR (500 MHz, DMSO-d6, δ): 12.0 (bs, 1H), 7.62 (d, J = 8.7 Hz, 2H), 7.54 (d, J = 8.4 Hz, 2H), 7.38-7.31 (m, 6H), 7.27-7.21 (m, 2H), 6.68 (d, J = 8.7 Hz, 2H), 5.79 (d, J = 7.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 167.9, 151.9, 142.7, 142.6, 131.8, 131.3, 130.0, 129.0, 127.9, 127.7, 120.6, 118.3, 112.6, 60.0; HRMS-ESI (m/z): [M + Na]+ calcd for C20H16BrNO2 404.02621; found 404.02610; HPLC (Method B) Rt = 6.514 min (>95%).
4-((bis(4-Bromophenyl)methyl)amino)benzoic acid (53)
Cream-colored amorphous solid (0.110 g, 61%). 1H NMR (500 MHz, DMSO-d6, δ): 12.1 (bs, 1H), 7.63 (d, J = 8.8 Hz, 2H), 7.56-7.54 (m, 4H), 7.34-7.32 (m, 4H), 7.22 (d, J = 7.3 Hz, 1H), 6.67 (d, J = 8.8 Hz, 2H), 5.81 (d, J = 73 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 167.8, 151.7, 142.1, 131.9, 131.4, 130.1, 120.8, 118.5, 112.6, 59.3; HRMS-ESI (m/z): [M]+ calcd for C20H15Br2NO2 458.94695; found 458.94454; HPLC (Method B) Rt = 6.113 min (>90%).
General procedure for the synthesis of compounds 54-58
4-(Benzhydrylamino)-N-phenylbenzamide (54)
Acid 51 (0.100 g, 0.33 mmol) was dissolved in DMF (5 mL). The solution was cooled to 0 °C and COMU (0.142 g, 0.33 mmol) was added, followed by DIPEA (0.11 mL, 0.66 mmol). Aniline (0.150 mL, 1.65 mmol) was then added and the mixture stirred for 3 hours, during which time the temperature was allowed to rise to room temperature. Upon reaction completion, CH2Cl2 (20 mL) was added to the mixture, which was washed successively with saturated sodium bicarbonate and brine (twice). The organic portion was then dried over MgSO4 and concentrated under reduced pressure. The crude mixture was purified by flash chromatography (0% to 50% EtOAc/hexane) to give the product as a cream-colored amorphous solid (0.065 g, 52%). 1H NMR (500 MHz, DMSO-d6, δ): 9.75 (s, 1H), 7.73-7.69 (m, 4H), 7.36-7.33 (m, 4H), 7.31-7.28 (m, 3H), 7.26-7.23 (m, 2H), 7.10 (d, J = 7.2 Hz, 1H), 7.05-7.02 (m, 1H), 6.74 (d, J = 8.8 Hz, 2H), 5.79 (d, J = 7.1 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 165.8, 151.2, 143.4, 140.2, 129.4, 129.0, 128.9, 127.8, 127.5, 123.4, 122.3, 120.5, 112.6, 60.8; HRMS-ESI (m/z): [M + H]+ calcd for C26H22N2O 379.18104; found 379.18043; HPLC (Method B) Rt = 6.389 min (>95%).
4-(((4-Bromophenyl)(phenyl)methyl)amino)-N-phenylbenzamide (55)
Light brown amorphous solid (0.070 g, 53%). 1H NMR (500 MHz, DMSO-d6, δ): 9.76 (s, 1H), 7.73-7.69 (m, 4H), 7.55-7.53 (m, 2H), 7.41-7.34 (m, 6H), 7.31-7.25 (m, 3H), 7.11 (d, J = 7.3 Hz, 1H), 7.05-7.02 (m, 1H), 6.73 (d, J = 8.8 Hz, 2H), 5.81 (d, J = 7.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 165.8, 164.2, 151.0, 142.8, 140.2, 131.8, 130.1, 129.5, 129.0, 128.9, 127.9, 127.7, 123.4, 122.6, 120.6, 120.5, 112.6, 60.1; HRMS-ESI (m/z): [M + H]+ calcd for C26H21BrN2O 457.09155, 459.08950; found 457.09321, 459.09173; HPLC (Method B) Rt = 6.919 min (>95%).
4-((bis(4-Bromophenyl)methyl)amino)-N-phenylbenzamide (56)
Brown amorphous solid (0.030 g, 43%). 1H NMR (500 MHz, DMSO-d6, δ): 7.67-7.65 (m, 3H), 7.59-7.57 (m, 2H), 7.48-7.46 (m, 4H), 7.33 (t, J = 8.4 Hz, 2H), 7.19-7.17 (m, 4H), 7.12-7.09 (m, 1H), 6.55-6.53 (m, 2H), 6.50 (d, J = 4.1 Hz, 1H), 4.57 (d, J = 4.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 165.3, 149.5, 140.4, 138.3, 132.2, 129.1, 129.0, 128.7, 124.1, 124.0, 121.9, 120.0, 113.0, 61.4; HRMS-ESI (m/z): [M + H]+ calcd for C26H20Br2N2O 537.00002; found 537.00067; HPLC (Method B) Rt = 7.392 min (>90%).
4-(Benzhydrylamino)-N-(naphthalen-2-yl)benzamide (57)
Cream-colored amorphous solid (0.200 g, 25%). 1H NMR (500 MHz, DMSO-d6, δ): 9.97 (s, 1H), 8.40 (s, 1H), 7.85-7.75 (m, 6H), 7.48-7.34 (m, 10H), 7.25 (t, J = 7.3 Hz, 2H), 7.13 (d, J = 7.2 Hz, 1H), 6.76 (d, J = 8.7 Hz, 1H), 5.80 (d, J = 7.1 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 166.0, 151.3, 143.3, 137.8, 133.9, 130.1, 129.5, 129.0, 128.4, 127.9, 127.8, 127.7, 127.5, 126.7, 124.9, 122.2, 121.4, 116.3, 112.6, 60.8; HRMS-ESI (m/z): [M + H]+ calcd for C30H24N2O 429.19669; found 429.19666, [2M + H]+ calcd for C60H48N4O2 857.38555; found 857.38571; HPLC (Method B) Rt = 7.029 min (>95%).
3-(4-(Benzhydrylamino)benzamido)benzoic acid (58)
Cream-colored amorphous solid (0.050 g, 18%). 1H NMR (500 MHz, DMSO-d6, δ): 12.96, 1.3 Hz, 1H), 7.44-7.40 (m, 5H), 7.36-7.33 (m, 4H), 7.26-7.23 (m, 2H), 7.13 (d, J = 7.15 Hz, 1H), 6.74 (d, J = 8.8 Hz, 2H), 5.80 (d, J = 7.2 Hz, 1H); 13C NMR (125 MHz, DMSO-d6, δ): 167.8, 165.9, 151.3, 143.3, 140.4, 131.6, 129.5, 129.1, 129.0, 127.8, 127.5, 124.5, 124.2, 121. 9, 121.3, 112.6, 60.8; HRMS-ESI (m/z): [M + H]+ calcd for C27H22N2O3 423.17087; found 423.17208, [2M + H]+ calcd for C54H44N4O6 845.33391; found 845.33656; HPLC (Method B) Rt = 7.980 min (>90%).
Procedures for the synthesis of compounds 20, 22, 23, 26, 32, 36, 39, 43-44, and 47
7-((Phenylamino)methyl)quinolin-8-ol (20)
Aniline (0.37 g, 4.0 mmol) and paraformaldehyde (0.12 g, 4.0 mmol) were dissolved in ethanol (10 mL) followed by stirring at room temperature for about 5 min. To the mixture, 8-hydroxyquinoline (0.58 g, 4.0 mmol) was added and the reaction mixture was refluxed at 120 °C (Note: the reaction temperature was achieved through gradual increase during the course of 3 hours) for about 12 hours. The reaction was monitored by TLC (3:2 acetonitrile/water) and LC-MS. The reaction mixture was allowed to cool and a colorless precipitate was isolated upon filtration, which was washed several times with ethanol to afford pure compound 20 as a colorless amorphous solid (0.52 g, 52%). 1H NMR (600 MHz, acetone-d6, δ): 8.96 (br s, 1H), 8.81 (d, J = 3.8 Hz, 1H), 8.26 (d, J = 8.2 Hz, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.50 (m, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.06 (m, 2H), 6.72 (d, J = 7.9 Hz, 2H), 6.56 (t, J = 7.2 Hz, 1H), 5.52 (br s, 1H), 4.59 (distorted doublet, 2H); 13C NMR (150 MHz, acetone-d6, δ): 150.5, 149.8, 149.1, 139.0, 137.0, 129.8, 128.6, 128.6, 122.7, 122.5, 118.3, 117.4, 113.5, 42.4; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C16H14N2O 158.06059, found 158.06038; HPLC (Method-B) R = 6.980 min (>95%).
N-((8-Hydroxyquinolin-7-yl)(phenyl)methyl)benzamide (22)
Benzamide (0.67 g, 5.5 mmol), benzaldehyde (0.53 g, 5.0 mmol), and 8-hydroxyquinoline (0.72 g, 5.0 mmol) were dissolved in dichloroethane (12 mL). To the mixture, p-toluenesulphonic acid (95 mg, 0.5 mmol) was added as catalyst and the reaction mixture was refluxed at 125 °C for 12 hours. After completion of the reaction, which was detected by LC-MS, the reaction mixture was allowed to cool. A colorless precipitate was isolated upon filtration, which was taken in ethanol and heated for 15 min. The resulting solid was filtered and washed with ethanol to afford pure compound 22 as a colorless amorphous solid (1.22 g, 70%). 1H NMR (600 MHz, CDCl3, δ): 8.73 (m, 1H), 8.14 (m, 2H), 7.88 (d, J = 7.3 Hz, 2H), 7.55 (d, J = 8.5 Hz, 1H), 7.49 (m, 1H), 7.40 (m, 7H), 7.30 (m, 2H), 7.23 (m, 1H), 6.76 (d, J = 8.8 Hz, 1H); 13C NMR (150 MHz, CDCl3, δ): 166.6, 149.4, 148.4, 141.6, 138.6, 136.3, 134.7, 131.7, 128.9, 128.8, 128.7, 127.9, 127.5, 127.3, 127.4, 122.6, 122.1, 118.4, 55.7; HRMS-ESI (m/z): [M + H]+ calcd for C23H18N2O2 355.14465, found 355.14484 and [M – C7H6NO]+ calcd for C23H18N2O2 234.09189, found 234.09171. Anal. Calcd for C23H18N2O2: C, 77.95; H, 5.12; N, 7.90. Found: C, 77.48; H, 4.97; N, 7.98; HPLC (Method B) Rt = 8.960 min (>95%).
(E)-7-((Benzylideneamino)(phenyl)methyl)quinolin-8-ol (23)
Ammonium carbamate (1.17 g, 15.0 mmol), 8-hydroxyquinoline (0.72 g, 5.0 mmol), and benzaldehyde (1.06 g, 10.0 mmol) were suspended in ethanol (20 mL). The resulting mixture was refluxed at 125 °C for 12 hours. After completion of the reaction, which was detected by LC-MS, the solvent was removed through rotary evaporation, and the resulting residue was extracted with a dichloromethane/water mixture. The organic extracts were concentrated in a rotary evaporator and the residual solid was taken in a 50-mL beaker, suspended in hexane, and heated to remove any unreacted starting materials. The resulting free flowing yellow powder was compound 23 (0.090 g, 30%). 1H NMR (600 MHz, CDCl3, δ): 9.15 (bs, 1H), 8.76 (m, 1H), 8.56 (s, 1H), 8.07 (m, 1H), 7.85 (m, 2H), 7.65 (d, J = 8.6 Hz, 1H), 7.51 (d, J = 7.5 Hz, 2H), 7.38 (m, 4H), 7.30 (m, 3H), 7.22 (m, 1H), 6.28 (s, 1H); 13C NMR (150 MHz, CDCl3, δ): 161.9, 149.4, 148.3, 143.6, 138.9, 136.3, 136.1, 131.2, 128.8, 128.7, 128.6, 127.9, 127.8, 127.6, 127.2, 125.3, 121.7, 118.0, 71.8; HRMS-ESI (m/z): [M – C7H6N]+ calcd for C23H18N2O 234.09189, found 234.09163. Anal. Calcd for C23H18N2O: C, 81.63; H, 5.36; N, 8.28. Found: C, 81.10; H, 5.20; N, 8.23.
4-(Octyloxy)benzaldehyde (26a).50
4-Hydroxybenzaldehyde (0.61 g, 5 mmol) was dissolved in acetone. Potassium carbonate (0.759 g, 5.5 mmol) was added and the resulting mixture was stirred for 5 minutes at room temperature. To the mixture, 1-bromooctane (0.965 g, 5 mmol) and potassium iodide (0.83 g, 5 mmol) were added, followed by refluxing for 6 hours. TLC showed the completion of the reaction. Solvent was removed in vacuo and the resulting residue was extracted with ethyl acetate (2 × 10 mL), treated with brine, and dried over sodium sulfate. Flash chromatography using 5% ethyl acetate in hexane afforded a gummy colorless compound (0.94 g, 80%). 1H NMR (600 MHz, CDCl3, δ): 9.88 (s, 1H), 7.83 (d, J = 8.7 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H), 4.04 (t, J = 6.5 Hz, 2H), 1.81 (quin., J1 = 6.6 Hz, J2 = 7.2 Hz, 2H), 1.46 (quin., J1 = 6.8 Hz, J2 = 7.6 Hz, 2H), 1.32 (m, 8H), 0.89 (t, J = 6.8 Hz, 3H).
7-((4-(Octyloxy)phenyl)(phenylamino)methyl)quinolin-8-ol (26)
A mixture of aniline (0.186 g, 2.0 mmol), aldehyde 26a (0.47 g, 2.0 mmol), and 8-hydroxyquinoline (0.29 g, 2.0 mmol) was heated neatly up to 130 °C for about 24 hours. The reaction was monitored by TLC (4:1 acetonitrile/water) and LC-MS. Reverse phase chromatography (C18 column) using 4:1 acetonitrile/water afforded compound 26 as a colorless gummy solid (0.045 g, 5%). 1H NMR (500 MHz, CDCl3, δ): 8.74 (d, J = 3.4 Hz, 1H), 8.10 (d, J = 8.3 Hz, 1H), 7.62 (d, J = 8.6 Hz, 1H), 7.38 (m, 3H), 7.30 (d, J = 8.6 Hz, 1H), 7.10 (t, J = 7.9 Hz, 2H), 6.83 (d, J = 8.6 Hz, 2H), 6.67 (t, J = 7.3 Hz, 1H), 6.61 (d, J = 8.0 Hz, 2H), 6.08 (s, 1H), 4.42 (br s, 1H), 3.90 (t, J = 6.5 Hz, 2H), 1.74 (p, J1 = 6.6 Hz, J2 = 7.1 Hz, 2H), 1.41 (p, J1 = 6.8 Hz, J2 = 7.8 Hz, 2H), 1.28 (m, 8H), 0.87 (t, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3, δ): 158.4, 148.9, 148.0, 147.5, 138.2, 136.0, 134.4, 129.1, 128.5, 127.6, 126.6, 124.4, 121.6, 117.8, 117.6, 114.6, 113.5, 68.0, 56.3, 31.8, 29.36, 29.29, 29.25, 26.1, 22.7, 14.1; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C30H34N2O2 362.21200 found 362.21204. Anal. Calcd for C30H34N2O2: C, 79.26; H, 7.54; N, 6.16. Found: C, 78.51; H, 7.48; N, 5.34.
Methyl 4-(((8-methoxyquinolin-7-yl)(phenyl)methyl)amino)benzoate (32)
Compound 1 (0.100 g, 0.270 mmol) and KOH (0.068 g, 1.22 mmol) were suspended in DMF (5 mL). To this was added MeI (0.038 g, 0.270 mmol) and the mixture allowed to stir at room temperature overnight. The solvent was removed in vacuo and the crude residue purified by flash chromatography (EtOAc/hexane 0 → 10%) to yield 31 as white gummy solid (0.035 g, 33%). 1H NMR (600 MHz, CDCl3, 5): 8.93 (d, J = 2.5 Hz, 1H), 8.13 (d, J = 8.32 Hz, 1H), 7.80 (d, J = 8.6 Hz, 2H), 7.56 (q, J = 8.5 Hz, 2H), 7.42-7.38 (m, 3H), 7.33 (t, J = 7.6 Hz, 2H), 6.58 (d, J = 8.7 Hz, 2H), 6.28 (d, J = 5.4 Hz, 1H), 4.87 (d, J = 5.1 Hz, 1H), 3.98 (s, 3H), 3.81 (s, 3H); 13C NMR (150 MHz, DMSO-d6, δ): 167.2, 153.5, 150.8, 149.7, 142.8, 141.9, 136.2, 134.0, 131.5, 129.4, 128.8, 127.7, 127.6, 125.7, 123.7, 121.3, 119.0, 112.3, 62.5, 56.7, 51.5; HRMS-ESI (m/z): [M + H]+ calcd for C25H22N2O3 399.17087, found 399.17381. HPLC (Method B) Rt = 8.76 min (>95%).
2-Methyl-7-(phenyl(phenylamino)methyl)quinolin-8-ol (36)
Aniline (0.465 g, 5.0 mmol) and benzaldehyde (0.53 g, 5.0 mmol) were dissolved in ethanol (10 mL) followed by stirring at room temperature for about 5 minutes. To the mixture, 2-methyl-8-quinolinol (0.795 g, 5.0 mmol) and catalytic pyridine (2 drops) was added and the reaction mixture was refluxed up to 100 °C for about 24 hours. The reaction was monitored by TLC (3:2 acetonitrile/water) and LC-MS. Ethanol was removed by rotary evaporation to afford a crude solid, which was purified by flash chromatography using 2% ethyl acetate in hexane. The column fractions containing the compound were kept overnight in the hood to obtain needle shaped crystals (0.92 g, 54%). 1H NMR (500 MHz, CDCl3, δ): 7.97 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 8.5 Hz, 1H), 7.48 (m, 2H), 7.31 (m, 2H), 7.24 (m, 3H), 7.10 (m, 2H), 6.67 (m, 1H), 6.63 (m, 2H), 6.12 (d, J = 3.4 Hz, 1H), 4.49 (s, 1H), 2.69 (s, 3H); 13C NMR (125 MHz, CDCl3, δ): 157.3, 148.6, 147.7, 142.8, 137.8, 136.2, 129.3, 128.8, 127.6, 127.4, 125.8, 125.7, 124.2, 122.7, 117.86, 117.78, 113.8, 57.0, 25.1; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C23H20N2O 248.10754, found 248.10762. Anal. Calcd for C23H20N2O: C, 81.15; H, 5.92; N, 8.23. Found: C, 80.88; H, 5.89; N, 8.17. HPLC (Method-B) Rt = 2.770 (>95%).
Benzyl 8-hydroxyquinoline-2-carboxylate (39a).52
8-Hydroxyquinoline-2-carboxylic acid (0.945 g, 5 mmol), triphenyl phosphine (1.965 g, 7.5 mmol), and benzyl alcohol (0.54 g, 5 mmol) were dissolved in THF (50 mL) and the resulting mixture was stirred at 0 °C in an ice bath. Diisopropyl azodicarboxylate (1.515 g, 7.5 mmol) was added dropwise over a period of 5 min and stirring was continued for 30 minutes. Solvents were removed in vacuo and the resultant residue solid was triturated with diethyl ether (repeated 3 times) to remove most of the triphenyl phosphine oxide byproduct as a solid. The filtrate was treated with brine and dried over sodium sulfate. Reverse phase chromatography (C18 column) using 40% acetonitrile in water afforded a light yellow amorphous marerial (0.56 g, 40%). 1H NMR (500 MHz, CDCl3, δ): 8.69 (br s, 1H), 8.25 (m, 1H), 8.16 (m, 1H), 7.55 (m, 1H), 7.50 (m, 2H), 7.41 (m, 2H), 7.36 (m, 2H), 7.22 (d, J = 8.5 Hz, 1H), 5.49 (s, 2H); 13C NMR (125 MHz, CDCl3, δ): 165.0, 153.6, 145.4, 138.0, 137.4, 135.6, 130.5, 129.9, 128.9, 128.76, 128.71, 121.8, 117.8, 111.4, 67.9; HRMS-ESI (m/z): [M + H]+ calcd for C17H13NO3 280.09682, found 280.09732. HPLC (Method-B) Rt = 5.466 (>95%).
Benzyl 8-hydroxy-7-(phenyl(phenylamino)methyl)quinoline-2-carboxylate (39)
A mixture of 39a (0.42 g, 1.5 mmol), aniline (0.14 g, 1.5 mmol), and benzaldehyde (0.16 g, 1.5 mmol) was heated neatly at 120 °C for 12 hours. Reverse phase column chromatography (C18 column) using 40% acetonitrile in water afforded a light yellow solid compound. The solid compound was again washed with ethanol to afford a light yellow amorphous compound (0.1 g, 15%). 1H NMR (600 MHz, CDCl3, δ): 8.88 (br s, 1H), 8.20 (d, J = 8.5 Hz, 1H); 8.14 (d, J = 8.5 Hz, 1H), 7.76 (d, J = 8.5 Hz, 1H), 7.48 (m, 4H), 7.40 (t, J = 7.6 Hz, 2H), 7.35 (m, 2H), 7.31 (t, J = 7.7 Hz, 2H), 7.24 (t, J = 3.6 Hz, 1H), 7.10 (t, J = 8.1 Hz, 2H), 6.68 (t, J = 7.3 Hz, 1H), 6.62 (d, J = 7.9 Hz, 2H), 6.15 (s, 1H), 5.47 (s, 2H), 4.48 (br s, 1H); 13C NMR (150 MHz, CDCl3, δ): 165.0, 150.2, 147.5, 145.6, 142.4, 137.9, 137.3, 135.6, 129.36, 129.32, 129.0, 128.91, 128.89, 128.75, 128.68, 127.6, 125.6, 121.6, 118.0, 117.8, 113.7, 67.9, 56.9; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C30H24N2O3 368.12867 found 368.12867.
N-(Phenyl(quinolin-7-yl)methyl)aniline (43)
Aniline (0.37 g, 4.0 mmol) and benzaldehyde (0.42 g, 4.0 mmol) were heated together at 60 °C for 4 hours to generate imine 43a. In a separate vessel, n-butyl lithium (2.5 mL of 1.6 M in hexane, 0.256 g, 4 mmol) was added slowly via a dry syringe at -78 °C to a solution of 7-bromoquinoline (0.416 g, 2 mmol) in THF. The mixture was stirred at -78 °C for 45 minutes. A solution of 43a (0.362 g, 2 mmol) in dry THF was then added slowly via syringe and the reaction mixture was allowed to warm up to room temperature. The reaction was continued for 3 hours followed by the addition of an aqueous saturated solution of ammonium chloride. Solvents were removed in vacuo and extracted with ethyl acetate. Flash chromatography using 30% ethyl acetate in hexane afforded 43 as a light cream-colored amorphous solid (0.045 g, 7%). 1H NMR (500 MHz, CDCl3, δ): 8.88 (dd, J1 = 4.2 Hz, J2 = 1.7 Hz, 1H), 8.12 (m, 2H), 7.77 (d, J = 8.4 Hz, 1H), 7.58 (dd, J1 = 8.4 Hz, J2 = 1.7 Hz, 1H), 7.40 (m, 2H), 7.34 (m, 3H), 7.26 (m, 1H), 7.10 (m, 2H), 6.69 (t, J = 7.3 Hz, 1H), 6.59 (d, J = 8.0 Hz, 2H), 5.70 (d, J = 3.8 Hz, 1H), 4.37 (s, 1H); 13C NMR (125 MHz, CDCl3, δ): 150.7, 148.5, 147.1, 144.5, 142.4, 135.8, 129.2, 128.9, 128.4, 127.69, 127.65, 127.60, 127.5, 126.3, 121.1, 117.9, 113.6, 63.0; HRMS-ESI (m/z): [M + H]+ calcd for C22H18N2 311.15482 found 311.15479. HPLC (Method-A) Rt = 8.786 (>95%).
N-(naphthalen-2-yl(phenyl)methyl)aniline (44)
2-Naphthylphenyl ketone (0.232 g, 1 mmol) was dissolved in dry dichloromethane and TiCl4 (1.1 mL of 1M solution in CH2Cl2, 0.208 g, 1.1 mmol) was added to it. The mixture was cooled to 0 °C followed by the addition of aniline (0.195 g, 2.1 mmol) and stirred for about 3 hours at room temperature under N2. The reaction mixture was then quenched with NaBH3CN (1.2 mL of 1M solution in THF, 0.0754 g, 1.2 mmol) followed by the addition of 2 mL of methanol and stirred for an additional 1 hour at the same temperature. The mixture was made basic (pH ∼ 10) using 5M aqueous solution of NaOH and filtered through celite. The filtrate was extracted with ethyl acetate (2 × 5 mL), organic layers washed with brine and dried over sodium sulfate. Flash chromatography using hexane afforded a colorless amorphous solid (0.26 g, 85%). 1H NMR (500 MHz, CDCl3, δ): 7.84 (s, 1H), 7.79 (m, 3H), 7.45 (m, 3H), 7.39 (m, 2H), 7.32 (m, 2H), 7.26 (m, 1H), 7.11 (m, 2H), 6.69 (m, 1H), 6.58 (m, 2H), 5.65 (d, J = 3.6 Hz, 1H), 4.30 (d, J = 3.3 Hz, 1H); 13C NMR (125 MHz, CDCl3, δ): 147.6, 143.0, 140.4, 133.7, 133.0, 129.4, 129.0, 128.8, 128.2, 127.9, 127.7, 126.4, 126.14, 126.10, 125.9, 117.9, 113.7, 63.5; HRMS-ESI (m/z): [M – C6H6N]+ calcd for C23H19N 217.10173, found 217.10178. Anal. Calcd for C22H17BrN2O: C, 89.28; H, 6.19; N, 4.53. Found: C, 88.83; H, 5.97; N, 4.47. HPLC (Method-B) Rt = 7.261 (>95%).
1-((R-Phenyl(((R-1-phenylethyl)amino)methyl)naphthalen-2-ol (47)
R-Phenethylamine (0.66 g, 5.5 mmol) and benzaldehyde (0.58 g, 5.5 mmol) were stirred at room temperature for about 5 min. To the mixture, 2-naphthol (0.72 g, 5.0 mmol) was added and the reaction mixture was heated neatly at 85 °C for about 12 hours under nitrogen atmosphere. The reaction was monitored by TLC (5% methanol in dichloromethane) and LC-MS. After 12 hours heating, the reaction was dispersed at room temperature with ethanol (5 mL) and kept undisturbed for 12 hours. A colorless crystalline solid settled down, which was filtered and washed several times with ethanol and hexane to afford compound 47 (0.79 g, 45%) as a colorless crystalline solid, m.p. 154-155 °C (ethanol), literature m.p. 155-156 °C (EtOAc-hexane).53 1H NMR (600 MHz, CDCl3, δ): 13.70 (s, 1H), 7.71 (m, 2H), 7.36 (m, 4H), 7.19 (m, 10H), 5.45 (s, 1H), 3.88 (m, 1H), 2.27 (d, J = 9.6 Hz, 1H), 1.48 (d, J = 6.8 Hz, 3H); 13C NMR (150 MHz, CDCl3, δ): 157.28, 143.09, 141.49, 132.59, 129.71, 129.05, 128.94, 128.74, 128.68, 127.95, 127.88, 127.67, 126.68, 126.36, 122.37, 121.09, 120.05, 113.06, 60.27, 56.63, 22.96; HRMS-ESI (m/z): [M + H]+ calcd for C25H23NO 354.18579, found 354.18593 and [M – C8H10N]+ calcd for C25H23NO 233.09664, found 233.09681. Anal. Calcd for C25H23NO: C, 84.95; H, 6.56; N, 3.96. Found: C, 84.61; H, 6.54; N, 3.98.
4.1.4 X-ray diffractometry
Data were recorded on Bruker APEX-II DUO diffractometer using Cu Kα radiation (wavelength = 1.54178 Å) and a graphite monochromator.
X-ray crystal structure of 28
CCDC deposition number 1437906. The structure was refined using the program SHELXL-2014-1. Crystal data: C27H27NO, M = 381.49, triclinic, space group P -1, a = 9.1165(2) Å, b = 11.0940(3) Å, c = 11.2998(3) Å; α = 76.621(1)°, β = 72.212(1)°, γ = 72.021(1)°; V = 1023.14(5) Å3; Z = 2; T = 100.01 K; μ = 0.571 mm-1; Crystal size = 0.10 × 0.05 × 0.05 mm3. Of 16,805 reflections measured to theta range 4.157 to 66.621°, 3,507 were independent (Rint = 0.0000). Final R1 = 0.0350 (I > 2σ (I)), wR2 = 0.0924.
X-ray crystal structure of 36
CCDC deposition number 1437907. The structure was refined using the program SHELXL 2014-1. Crystal data: C23H20N2O, M = 340.41, monoclinic, space group P 1 21/n 1, a = 14.0529(7) Å, b = 5.5734(3) Å, c = 22.5326(11) Å; α = 90°, β = 99.465(2)°, γ = 90°; V = 1740.78(15) Å3; Z = 4; T = 100 K; μ = 0.627 mm-1; Crystal size = 0.18 × 0.18 × 0.07 mm3. Of 52,480 reflections measured to theta range 3.469 to 66.655°, 3,080 were independent (Rint = 0.0000). Final R1 = 0.0345 (I > 2σ (I)), wR2 = 0.0908.
4.2 Biological assays
4.2.1 Yeast strains
The yeast strain used in this study was SM3614 (MATa trp1 leu2 ura3 his4 can1 ste24∷LEU2 rce1∷TRP1).71 Plasmid-bearing versions of the strain were generated according to published methods.72 Strains were routinely grown at 30 °C using synthetic complete dropout (SC–Ura) medium.73 The over-expression plasmid encoding human Rce1p (pWS335) has been reported.39 The over-expression plasmid encoding human ZmpSte24 (pWS1275; 2μ URA3 PPGK-HA-HsSTE24) was constructed by conventional cloning methods starting with previously described plasmid pWS183.39
4.2.2 In vitro fluorescence-based CaaX proteolysis assay
An established fluorescence-based assay was used to monitor CaaX protease activity in the presence of inhibitors.29, 55, 56 The assay involved the mixing of a quenched fluorogenic substrate with yeast membranes enriched for the appropriate CaaX protease. The Rce1 fluorogenic substrate ABZ-KSKTKC (farnesyl)QLIM and the Ste24 substrate ABZ-KSKTKC(farnesyl)VIQL were purchased from AnaSpec (San Jose, CA) and are based on the K-Ras4b C-terminal sequence. ABZ is 2-aminobenzoic acid, and QL is (S)-2-amino-6-((2,4-dinitrophenyl)amino)hexanoic acid. The substrate was diluted from a 1-mM stock prepared in 4% DMSO. Membranes used as the source of protease activity were isolated from yeast according to our reported methods.25, 39, 67 The membranes and substrate were diluted with assay buffer (200 mM Tris, pH 7.5, 200 mM NaCl, 1 mg/mL BSA) immediately before use. Compounds were prepared as 50× stocks in DMSO. Assay assembly involved the dispensing of 50-μL aliquots of diluted membranes into the wells of a black, clear-bottom, 96-well microplate. This was followed by the addition of each compound to duplicate wells and a 15-min pretreatment incubation at 30 °C. After pretreatment, the assay was initiated by adding 50 μL of diluted substrate, resulting in final membrane and substrate concentrations of 0.25 mg/mL and 20 μM substrate, respectively. The sample fluorescence was measured every 30-60 s over a 60-min time course at 30 °C using a BioTek Synergy HT microplate fluorometer equipped with a 320/420-nm excitation/emission filter set. The collected data were exported to Microsoft Excel, graphed as a function of fluorescence vs time, and initial velocities determined. These values were used to calculate the percentage activities relative to a DMSO-treated sample, which was included as a control in each reaction set. To correct for compound-induced effects on fluorescence readout, samples were completely proteolyzed with trypsin (10 μg/mL) at the end of each experiment to determine the maximum possible fluorescence in the presence and absence (i.e., DMSO control) of compounds. These values were used to normalize raw reaction rates. IC50 experiments were conducted using a range of compound concentrations (0.25-250 μM final); the final DMSO concentration was the same in all reactions. IC50 values calculated with Prism 6.0 (GraphPad Software, Inc.) from values obtained for initial velocities.
4.2.3 Fluorescence-based FTase assay
To determine whether the compounds are inhibitors of the protein farnesyltransferase (FTase), a previously reported fluorescence-based FTase assay68 was adapted for use in a microtiter plate format.69 Briefly, 100 μL of a concentrated enzyme assay solution (5 μM Dansyl-GCVLS, 12.5 mM DTT, 125 mM Tris·HCl, pH 7.5, 25 mM MgCl2, 25 μM ZnCl2, 0.5% octyl-β-D-glucopyranoside, 25 μM FPP) was added to wells of a black 96-well plate (Nunc #237105). To each well, water (117.5 μL) was also added. Varying volumes of the different inhibitors dissolved in DMSO were added to the wells followed by addition of DMSO to a final volume of 12.5 μL DMSO in each well. Three different inhibitor concentrations (10, 25, 50 μM) were tested in the assay. The plate was first screened for initial fluorescence using a Beckman Coulter DTX 880 Multimode Detector using the A340/10 filter for excitation at 340 nm and F535/25 filter to monitor for emission. Afterward, 20 μL of enzyme solution (62.5 nM rat FTase, 50 mM Tris·HCl, pH 7.5, 50 μM ZnCl2, 5 mM MgCl2, 20 mM KCl, 1 mM DTT, 1 mg/mL BSA) was added to each well to initiate the enzymatic reaction; final assay volume was 250 μL. Fluorescence was monitored in the same way as for the initial fluorescence measurement but taking a fluorescence reading every 40 s for a total of 1 hour. Fluorescence data was exported to Microsoft Excel and the rate was determined by converting the rate obtained in the first 10 min of the reaction from fluorescence intensity units (FIU/s) to nM/s using the equation νi=RP/ΔF, where νi is the rate of the reaction in nM/s, R is the rate of the reaction in FIU/s, P is the amount of product in nM and ΔF is the difference in fluorescence intensity between the start of the assay and the maximal fluorescence intensity obtained.
4.2.4 Materials for cell based assays
Tissue culture medium was supplied by Sigma, and fetal bovine serum (FBS) were obtained from PAA laboratories. All other chemicals were supplied by Sigma unless stated otherwise.
4.2.5 Plasmids
All plasmids were propagated using E.coli DH5α and purified using the GenElute HP plasmid maxiprep kit (Sigma). EGFP-H-Ras, EGFP-N-Ras, and EGFP-K-Ras4b where obtained from Mark Philips (NYU).
4.2.6 Cell culture
HCT-116 cells (received from Rana Al Assah, NYUAD) were grown in Dulbecco's Minimal Essential Medium (D6429) plus 10% FBS, 1% non-essential amino acids (NEAA), and maintained at 37 °C, 5% CO2. For confocal fluorescence microscopy, HCT-116 cells were plated on 24-well glass-bottom plates (Greiner Bio-One) at 4 × 104 cells per well in 0.5 mL medium. The cells were transfected with the appropriate plasmid 24 hours post-seeding using TrueFect (United Biosystems) in accordance with the manufacturer's recommendations. The optimised ratio of DNA:TrueFect used for transfection of HCT-116 cells in a 24-well plate was 0.5 μg DNA per 1 μL TrueFect. For CellTiter-Blue assays, HCT-116 cells were plated on 96-well plates (Corning) at 7 × 103 cells per well in 100 μL medium. For Rce1 siRNA knock-down confocal microscopy and RT-PCR experiments, HCT-116 cells were plated on 35-mm glass-bottom dishes (Greiner Bio-One) at 2 × 105 cells per dish in medium (2 mL). The cells were transfected with 100 pmol of either Silencer® Cy™3-labeled Negative Control No. 1 siRNA (Life Technologies) or AccuTarget™ Premade siRNA Rce1 (Bioneer) and 1 μg of either EGFP-H-Ras, EGFP-N-Ras, or EGFP-K-Ras4b using Lipofectamine® RNAiMAX transfection reagent (3 μL) and Opti-MEM® (Life technologies) in accordance with the manufacturer's recommendations 24 h post-plating.
4.2.7 Treatment of cells with compounds
Cells were transfected with plasmid as described above then treated with the stated concentration of FTI L-744,832 (Calbiochem), Rce1 inhibitors, or DMSO as the vehicle control. Cells were then incubated at 37 °C, 5% CO2 for an additional 20 hours before imaging or determining cell viability.
4.2.8 Cell viability assay
The CellTiter-Blue assay (Promega) was used to assess cell viability of HCT-116 cells following treatment with Rce1 inhibitors. Cells were plated and treated with compounds as described above. CellTiter-Blue was added to the cells 20 hours post-treatment per manufacturer's instructions. After 4 hours of incubation, fluorescence was measured using 560/590 nm filters on a BioTek Synergy microplate reader. Three independent experiments were performed, averaged, and plotted with SEM bars. The data is represented relative to untreated cells.
4.2.9 Confocal microscopy
Live cell imaging was carried out on HCT-116 cells in 24-well glass-bottom plates (for testing compounds) or 35-mm glass-bottom dishes (for Rce1 siRNA experiments) in a humidified CO2 incubator (at 37 °C, 5% CO2) 24 h post-transfection, using an Olympus IX81 with a 30× oil immersion objective (NA = 1.05). Excitation of EGFP was performed using an Argon ion laser at 488 nm. Emitted light was reflected through a 500-600 nm filter from a dichroic mirror. Data capture and extraction was carried out with FluoView10-ASW version 4.0 software (Olympus). At least 40 cells were imaged per condition. The relative distribution of EGFP-Ras isoforms was determined with Fiji image analysis platform74 and analyzed by a method adapted from Manandhar et al.41 A ROI line was drawn across the cell to measure the fluorescence intensity. The fluorescence intensity values were normalised (0 to 1) and used to calculate the percentage of the total fluorescence associated with the PM (% PM association). The PM width was set to 1 μm, (which was the average of several EGFP-Ras PM localized cells) and mislocalized fluorescence was defined as any fluorescence 1 μm or more from the PM.
4.2.10 Rce1 siRNA knock-down and RT-PCR
Following imaging by confocal microscopy, cells were lysed and the RNA extracted using the High Pure RNA isolation kit (Roche) according to manufacturer's instructions. The RNA concentration and purity was quantified using a NanoDrop 2000c UV-Vis spectrophotometer (Thermo Scientific). The absorbance was measured at 260 and 280 nm. All RNA was of sufficient purity with a A260/A280 ratio between 1.9-2.1 used in the reverse transcriptase reaction. Total RNA from each sample (2 μg) was reverse transcribed using the SuperScript® VILO™ cDNA synthesis kit (Life Technologies). For PCR amplification the cDNA samples were diluted 10-fold and 5 μL of each cDNA sample was used per reaction combined with a mastermix consisting of 1× NH4 reaction buffer (Bioline), 80 μM dNTPs (Bioline), 2 mM MgCl2, 20 pmol of each primer (5′-3′) Rce1 forward GTCCTGGTGGTGTCCAGTCT, Rce1 reverse GGAACACCAGCTCCTCTGTC, cyclophilin A forward CACCGTGTTCTTCGACATTG, cyclophilin A reverse GCCATCCAACCACTCAGTCT and 1 U Biotaq DNA polymerase (Bioline), made up to a final volume of 25 μL with dH2O. The cDNA was amplified using MultiGene™ OptiMax thermal cycler (Labnet International) with cycling parameters as follows: initial denaturation at 94 °C for 2 min, 30 cycles of 94 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min, and hold at 72 °C for 5 min. Products were resolved by 1.2% agarose gel electrophoresis containing SYBR® Safe DNA gel stain (Life Technologies) against HyperLadder™ 50 bp (Bioline).
Supplementary Material
Figure S1 showing toxicity data, Figures S2 and S3 showing ORTEP diagram for compounds 28 and 36, proteolysis assay IC50 determinations, and NMR spectra.
Acknowledgments
We thank the National Institutes of Health (GM67092) for partial support of this work. We also thank Matteo Lusi and Liang Li for performing X-ray crystallographic structure determinations, Rana Al Assah for providing HCT-116 cells, and Mark Philips for providing the EGFP-Ras constructs. The research was carried out using Core Technology Platform resources at New York University Abu Dhabi and mass spectrometry resources at the Proteomics and Mass Spectrometry Core Facility at the University of Georgia.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spiegel J, Cromm PM, Zimmermann G, Grossmann TN, Waldmann H. Nat Chem Biol. 2014;10:613. doi: 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- 2.van Hattum H, Waldmann H. Chem Biol. 2014;21:1185. doi: 10.1016/j.chembiol.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Prior IA, Lewis PD, Mattos C. Cancer Res. 2012;72:2457. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hancock JF, Peterson H, Marshall CJ. Cell. 1990;63:133. doi: 10.1016/0092-8674(90)90294-o. [DOI] [PubMed] [Google Scholar]
- 5.Linder ME, Deschenes RJ. Nat Rev Mol Cell Biol. 2007;8:74. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 6.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Nat Rev Mol Cell Biol. 2012;13:39. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gysin S, Salt M, Young A, McCormick F. Genes Cancer. 2011;2:359. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baines AT, Xu D, Der CJ. Future Med Chem. 2011;3:1787. doi: 10.4155/fmc.11.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waldmann H, Karaguni IM, Carpintero M, Gourzoulidou E, Hermann C, Brockmann C, Oschkinat H, Mueller O. Angew Chem, Int Ed. 2004;43:454. doi: 10.1002/anie.200353089. [DOI] [PubMed] [Google Scholar]
- 10.Palmioli A, Sacco E, Abraham S, Thomas CJ, Di Domizio A, De Gioia L, Gaponenko V, Vanoni M, Peri F. Bioorg Med Chem Lett. 2009;19:4217. doi: 10.1016/j.bmcl.2009.05.107. [DOI] [PubMed] [Google Scholar]
- 11.Rosnizeck IC, Graf T, Spoerner M, Traenkle J, Filchtinski D, Herrmann C, Gremer L, Vetter IR, Wittinghofer A, Koenig B, Kalbitzer HR. Angew Chem, Int Ed. 2010;49:3830. doi: 10.1002/anie.200907002. [DOI] [PubMed] [Google Scholar]
- 12.Maurer T, Garrenton LS, Oh A, Pitts K, Anderson DJ, Skelton NJ, Fauber BP, Pan B, Malek S, Stokoe D, Ludlam MJC, Bowman KK, Wu J, Giannetti AM, Starovasnik MA, Mellman I, Jackson PK, Rudolph J, Wang W, Fang G. Proc Natl Acad Sci USA. 2012;109:5299. doi: 10.1073/pnas.1116510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim SM, Westover KD, Ficarro SB, Harrison RA, Choi HG, Pacold ME, Carrasco M, Hunter J, Kim ND, Xie T, Sim T, Jaenne PA, Meyerson M, Marto JA, Engen JR, Gray NS. Angew Chem, Int Ed. 2014;53:199. doi: 10.1002/anie.201307387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD. Proc Natl Acad Sci USA. 2014;111:8895. doi: 10.1073/pnas.1404639111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Downward J. Clin Cancer Res. 2015;21:1802. doi: 10.1158/1078-0432.CCR-14-2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, Bishop WR, Pai JK. J Biol Chem. 1997;272:14459. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 17.Kim E, Ambroziak P, Otto JC, Taylor B, Ashby M, Shannon K, Casey PJ, Young SG. J Biol Chem. 1999;274:8383. doi: 10.1074/jbc.274.13.8383. [DOI] [PubMed] [Google Scholar]
- 18.Bergo MO, Leung GK, Ambroziak P, Otto JC, Casey PJ, Young SG. J Biol Chem. 2000;275:17605. doi: 10.1074/jbc.C000079200. [DOI] [PubMed] [Google Scholar]
- 19.Bergo MO, Ambroziak P, Gregory C, George A, Otto JC, Kim E, Nagase H, Casey PJ, Balmain A, Young SG. Mol Cell Biol. 2002;22:171. doi: 10.1128/MCB.22.1.171-181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bergo MO, Gavino BJ, Hong C, Beigneux AP, McMahon M, Casey PJ, Young SG. J Clin Invest. 2004;113:539. doi: 10.1172/JCI18829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bergo MO, Lieu HD, Gavino BJ, Ambroziak P, Otto JC, Casey PJ, Walker QM, Young SG. J Biol Chem. 2004;279:4729. doi: 10.1074/jbc.M310081200. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt WK, Dore TM. In: The Enzymes. Tamanoi F, Hrycyna CA, Bergo M, editors. 30B. Elsevier/Academic Press; New York: 2011. p. 231. [Google Scholar]
- 23.Dore TM, Schmidt WK. In: Handbook of Proteolytic Enzymes. Rawlings ND, Salvesen GS, editors. Vol. 2. Elsevier; Oxford, UK: 2013. p. 1720. [Google Scholar]
- 24.Ma YT, Gilbert BA, Rando RR. Biochemistry. 1993;32:2386. doi: 10.1021/bi00060a033. [DOI] [PubMed] [Google Scholar]
- 25.Dechert AM, MacNamara JP, Breevoort SR, Hildebrandt ER, Hembree NW, Rea AC, McLain DE, Porter SB, Schmidt WK, Dore TM. Bioorg Med Chem. 2010;18:6230. doi: 10.1016/j.bmc.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Y, Ma Yt, Rando RR. Biochemistry. 1996;35:3227. doi: 10.1021/bi952529s. [DOI] [PubMed] [Google Scholar]
- 27.Chen Y. Cancer Lett. 1998;131:191. doi: 10.1016/s0304-3835(98)00146-3. [DOI] [PubMed] [Google Scholar]
- 28.Schlitzer M, Winter-Vann A, Casey PJ. Bioorg Med Chem Lett. 2001;11:425. doi: 10.1016/s0960-894x(00)00685-5. [DOI] [PubMed] [Google Scholar]
- 29.Manandhar SP, Hildebrandt ER, Schmidt WK. J Biomol Screen. 2007;12:983. doi: 10.1177/1087057107307226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pryor EE, Jr, Horanyi PS, Clark KM, Fedoriw N, Connelly SM, Koszelak-Rosenblum M, Zhu G, Malkowski MG, Wiener MC, Dumont ME. Science. 2013;339:1600. doi: 10.1126/science.1232048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Quigley A, Dong YY, Pike ACW, Dong L, Shrestha L, Berridge G, Stansfeld PJ, Sansom MSP, Edwards AM, Bountra C, von Delft F, Bullock AN, Burgess-Brown NA, Carpenter EP. Science. 2013;339:1604. doi: 10.1126/science.1231513. [DOI] [PubMed] [Google Scholar]
- 32.Liu B, Zhou Z. Histol Histopathol. 2008;23:747. doi: 10.14670/HH-23.747. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt WK, Tam A, Fujimura-Kamada K, Michaelis S. Proc Natl Acad Sci USA. 1998;95:11175. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bracha-Drori K, Shichrur K, Lubetzky TC, Yalovsky S. Plant Physiol. 2008;148:119. doi: 10.1104/pp.108.120477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hildebrandt ER, Davis DM, Deaton J, Krishnankutty RK, Lilla E, Schmidt WK. Biochemistry. 2013;52:6601. doi: 10.1021/bi400647c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manolaridis I, Kulkarni K, Dodd RB, Ogasawara S, Zhang Z, Bineva G, O'Reilly N, Hanrahan SJ, Thompson AJ, Cronin N, Iwata S, Barford D. Nature. 2013;504:301. doi: 10.1038/nature12754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dolence JM, Steward LE, Dolence EK, Wong DH, Poulter CD. Biochemistry. 2000;39:4096. doi: 10.1021/bi9923611. [DOI] [PubMed] [Google Scholar]
- 38.Pei J, Grishin NV. Trends Biochem Sci. 2001;26:275. doi: 10.1016/s0968-0004(01)01813-8. [DOI] [PubMed] [Google Scholar]
- 39.Plummer LJ, Hildebrandt ER, Porter SB, Rogers VA, McCracken J, Schmidt WK. J Biol Chem. 2006;281:4596. doi: 10.1074/jbc.M506284200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mokry DZ, Manandhar SP, Chicola KA, Santangelo GM, Schmidt WK. Eukaryotic Cell. 2009;8:1891. doi: 10.1128/EC.00169-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manandhar SP, Hildebrandt ER, Jacobsen WH, Santangelo GM, Schmidt WK. Yeast. 2010;27:327. doi: 10.1002/yea.1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips JP, Barrall EM. J Org Chem. 1956;21:692. [Google Scholar]
- 43.Phillips JP. Chem Rev. 1956;56:271. [Google Scholar]
- 44.Betti M. Gazz Chim Ital. 1900;30:310. [Google Scholar]
- 45.Betti M. Gazz Chim Ital. 1901;31:377. [Google Scholar]
- 46.Pirrone F. Gazz Chim Ital. 1940;70:520. [Google Scholar]
- 47.Venugopala KN, Krishnappa M, Nayak SK, Subrahmanya BK, Vaderapura JP, Chalannavar RK, Gleiser RM, Odhav B. Eur J Med Chem. 2013;65:295. doi: 10.1016/j.ejmech.2013.04.061. [DOI] [PubMed] [Google Scholar]
- 48.Khodaei MM, Khosropour AR, Moghanian H. Synlett. 2006:916. [Google Scholar]
- 49.Szatmari I, Fulop F. Synthesis. 2009:775. [Google Scholar]
- 50.Tai CH, Wu HC, Li WR. Org Lett. 2004;6:2905. doi: 10.1021/ol049120s. [DOI] [PubMed] [Google Scholar]
- 51.Amstutz R, Bold G, Cotesta S, Jahnke W, Marzinzik A, Mueller-Hartwieg C, Ofner S, Stauffer F, Zimmermann J. WO 2009/106586 A1. 2009:103. [Google Scholar]
- 52.Miyake Y, Hosoda A, Mori H, Takagaki M, Nomura E, Taniguchi H. Kenkyu Hokoku - Wakayama-ken Kogyo Gijutsu Senta. 2007:19. [Google Scholar]
- 53.Cimarelli C, Mazzanti A, Palmieri G, Volpini E. J Org Chem. 2001;66:4759. doi: 10.1021/jo0101205. [DOI] [PubMed] [Google Scholar]
- 54.Rahman O, Kihlberg T, Långström B. Org Biomol Chem. 2004;2:1612. doi: 10.1039/b403481c. [DOI] [PubMed] [Google Scholar]
- 55.Porter SB, Hildebrandt ER, Breevoort SR, Mokry DZ, Dore TM, Schmidt WK. Biochim Biophys Acta. 2007;1773:853. doi: 10.1016/j.bbamcr.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hollander I, Frommer E, Mallon R. Anal Biochem. 2000;286:129. doi: 10.1006/abio.2000.4795. [DOI] [PubMed] [Google Scholar]
- 57.Riley KE, Murray JS, Fanfrlik J, Rezac J, Sola RJ, Concha MC, Ramos FM, Politzer P. J Mol Model. 2011;17:3309. doi: 10.1007/s00894-011-1015-6. [DOI] [PubMed] [Google Scholar]
- 58.Wilcken R, Zimmermann MO, Lange A, Joerger AC, Boeckler FM. J Med Chem. 2013;56:1363. doi: 10.1021/jm3012068. [DOI] [PubMed] [Google Scholar]
- 59.Roberts PJ, Mitin N, Keller PJ, Chenette EJ, Madigan JP, Currin RO, Cox AD, Wilson O, Kirschmeier P, Der CJ. J Biol Chem. 2008;283:25150. doi: 10.1074/jbc.M800882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sepp-Lorenzino L, Ma Z, Rands E, Kohl NE, Gibbs JB, Oliff A, Rosen N. Cancer Res. 1995;55:5302. [PubMed] [Google Scholar]
- 61.Abate-Pella D, Zeliadt NA, Ochocki JD, Warmka JK, Dore TM, Blank DA, Wattenberg EV, Distefano MD. ChemBioChem. 2012;13:1009. doi: 10.1002/cbic.201200063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Michaelson D, Ali W, Chiu VK, Bergo M, Silletti J, Wright L, Young SG, Philips M. Mol Biol Cell. 2005;16:1606. doi: 10.1091/mbc.E04-11-0960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burrows JF, Kelvin AA, McFarlane C, Burden RE, McGrattan MJ, De la Vega M, Govender U, Quinn DJ, Dib K, Gadina M, Scott CJ, Johnston JA. J Biol Chem. 2009;284:9587. doi: 10.1074/jbc.M807216200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sebti SM, Hamilton AD. Oncogene. 2000;19:6584. doi: 10.1038/sj.onc.1204146. [DOI] [PubMed] [Google Scholar]
- 65.Berndt N, Hamilton AD, Sebti SM. Nat Rev Cancer. 2011;11:775. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Davies BSJ, Fong LG, Yang SH, Coffinier C, Young SG. Annu Rev Genomics Hum Genet. 2009;10:153. doi: 10.1146/annurev-genom-082908-150150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt WK, Tam A, Michaelis S. J Biol Chem. 2000;275:6227. doi: 10.1074/jbc.275.9.6227. [DOI] [PubMed] [Google Scholar]
- 68.Pompliano DL, Gomez RP, Anthony NJ. J Am Chem Soc. 1992;114:7945. [Google Scholar]
- 69.Dozier JK, Khatwani SL, Wollack JW, Wang YC, Schmidt-Dannert C, Distefano MD. Bioconjugate Chem. 2014;25:1203. doi: 10.1021/bc500240p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wahlstrom AM, Cutts BA, Karlsson C, Andersson KME, Liu M, Sjogren AKM, Swolin B, Young SG, Bergo MO. Blood. 2007;109:763. doi: 10.1182/blood-2006-05-024752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tam A, Nouvet FJ, Fujimura-Kamada K, Slunt H, Sisodia SS, Michaelis S. J Cell Biol. 1998;142:635. doi: 10.1083/jcb.142.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Elble R. BioTechniques. 1992;13:18. [PubMed] [Google Scholar]
- 73.Michaelis S, Herskowitz I. Mol Cell Biol. 1988;8:1309. doi: 10.1128/mcb.8.3.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Nat Methods. 2012;9:676. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 showing toxicity data, Figures S2 and S3 showing ORTEP diagram for compounds 28 and 36, proteolysis assay IC50 determinations, and NMR spectra.