

Abstract
Generating sets of trial structures that sample the configurational space of crystal packing possibilities is an essential step in the process of ab initio crystal structure prediction (CSP). One effective methodology for performing such a search relies on low-discrepancy, quasi-random sampling, and our implementation of such a search for molecular crystals is described in this paper. Herein we restrict ourselves to rigid organic molecules and, by considering their geometric properties, build trial crystal packings as starting points for local lattice energy minimization. We also describe a method to match instances of the same structure, which we use to measure the convergence of our packing search toward completeness. The use of these tools is demonstrated for a set of molecules with diverse molecular characteristics and as representative of areas of application where CSP has been applied. An important finding is that the lowest energy crystal structures are typically located early and frequently during a quasi-random search of phase space. It is usually the complete sampling of higher energy structures that requires extended sampling. We show how the procedure can first be refined, through targetting the volume of the generated crystal structures, and then extended across a range of space groups to make a full CSP search and locate experimentally observed and lists of hypothetical polymorphs. As the described method has also been created to lie at the base of more involved approaches to CSP, which are being developed within the Global Lattice Energy Explorer (Glee) software, a few of these extensions are briefly discussed.
1. Introduction
The great majority of compounds synthesized by chemists exist at room temperature as solids, often in crystal form. Crystallization itself can be a challenging part of the synthetic process, further complicated by polymorphs (the existence of multiple crystal structures of a given compound), impurities, or the desire for crystals with particular structural properties.1 The physical properties of an organic molecular crystal must derive in part not only from those of its constituent molecules but also by the arrangement of molecules in a crystal and the intermolecular interactions that either drive, or result from, a particular crystal packing. Many of the molecules which the chemist is interested in synthesizing are chosen due to their solid-state properties, such as in the fields of organic semiconductors,2 pigments,3 and porous molecular materials.4−6 The selection and control of solid form is also vitally important in developing pharmaceutical molecules into tablets with satisfactory stability and bioavailability; the issues raised by polymorphism in pharmaceutical chemistry have been characterized extensively.7 A large proportion of organic molecules is known to be polymorphic,8 although the relationship between molecular characteristics and the existence of polymorphs is unclear.
For the above reasons, the importance of characterizing the crystal structure is key to rationalizing many properties. Many tools used to probe the molecular structure, such as X-ray diffraction, solid state NMR,9−11 or those of solid-state spectroscopy,12,13 are sensitive to the local and long-range structure of a molecule within the crystal. From a theoretical perspective, the prediction of crystal structures ab initio is a natural challenge to theoretical and computational chemists and has valuable applications in characterizing the landscape of possible crystal structures available to a given molecule. This challenge has drawn a community of researchers who seek to solve these structures from limited initial data and preferably from just the two-dimensional chemical diagram of the molecular structure.14−16 The progress that has been made by this community is clear from published studies on large and flexible molecules5,17−23 and can be tracked in a series of collaborative exercises in which active members of the field have been challenged to predict the structures of unpublished crystals.24,25 These ”blind tests” of crystal structure prediction (CSP) attempt to benchmark the successes and limitations of the different contemporary approaches, the progress that has been made, and that which is still required.
A fundamental concept in our approach to CSP is to represent the internal potential energy of a crystal structure as a function of the intra- and intermolecular coordinates, where the intramolecular structure, energy, and properties are calculated using quantum chemical methods, and intermolecular interactions are calculated using anisotropic atom–atom potentials.26 Although research in our group into methods for efficiently handling molecular flexibility, and for the calculation of free-energies,27 is active, this paper is concerned with characterizing the potential energy surface describing the crystal packing of rigid molecules. Thus, our configurational space is the union of the coordinates which describe the positions and orientations of the molecules in the crystal’s asymmetric unit and the degrees of freedom which determine the unit cell. These, along with the space group operations, define a crystal structure. Our potential energy surface (PES), which is a function of these variables, is a force field which comprises an exp-6 model of short-range and dispersion interactions combined with an atomic multipole electrostatic model derived from single molecule DFT calculations. We seek to characterize the resulting lattice energy surface by reliably locating and ranking all local minima within a certain energy of the global minimum. Each local minimum on the lattice energy surface could represent an observable polymorph of the molecule in question.
Efforts toward improving our ability to predict crystal structures largely focus on either the evaluation of more accurate lattice energies or the challenge of locating all possible structures through sampling of the PES. The problem of sampling phase space is the chief concern of this paper, and we describe methods that have been implemented in our Global Lattice Energy Explorer (Glee) software which is described herein. Each minimum on the PES will have an associated basin within which any structure will, when relaxed using local lattice energy minimization, be reached. The structure generator outlined here seeks to sample trial structures such that all local minima are located during the minimization step. We take an approach of ensuring as diverse a sampling as possible, because we are not only interested in the structure corresponding to the global lattice energy minimum but rather the entire ”landscape” of structures. For most applications of the prediction of molecular crystals, the landscape should be sampled as completely as possible within the energy range of expected polymorphism. Recent calculations on over 1000 crystal structures of known polymorphs show that, while the majority of polymorphs are separated by less than 2 kJ mol–1, occasional pairs of known polymorphs differ by 10 or more kJ mol–1.27 The number of distinct crystal packing alternatives within such an energy window above the global minimum usually amounts to many 10s and very frequently over 100 distinct crystal structures for small organic molecules.28 In the class of crystals which can support inclusion compounds, it may be that the structures which are higher in energy, and less perfectly packed from a purely energetic point of view, are the most interesting.29 A focus on too small a set of structures at, or around, the global lattice energy minimimum would be to risk losing the richness of the landscape and potential solid form diversity of a molecule, which must be considered in developing a molecule into a useful material.
Alternative approaches to crystal structure generation which have been applied to molecular crystals include simulated annealing30 and more sophisticated31 variants of Monte Carlo searches and genetic algorithms,32−34 as well as the early pioneering CSP studies using purely random or grid searches.35−37 In fact, these simplest methods have been remarkably successful, consistently performing well in the structure searching aspect of the blind tests of CSP.24,25 We follow previous groups who favor low-discrepancy, quasi-random sampling,38,39 as we require an algorithm that samples the phase-space completely and efficiently. Quasi-random sequences have attractive properties for locating local minima; in particular, at each step in the sequence the configurational space is as uniformly sampled as possible. Unlike a deterministic method such as a grid-based search, the convergence of the search can be continuously monitored and extended until one is suitably confident that all relevant local energy minima have been sampled. The problems of structure generation and lattice energy minimization can be programmed in this way to make full use of the computational resources available to us, to require very few pieces of input information, and to be repeatable.
The purpose here is to describe our implementation of a quasi-random search, whose use has already been demonstrated in studies of mechanochemical reactions40 and cocrystal formation,41 as well as to investigate and optimize the performance of our algorithm. While quasi-random CSP methods have been extensively applied to CSP, there have been few detailed studies of the performance of such a search. For the purposes of this study, we follow the convergence of finding a complete set of possible low energy crystal structures for three molecular systems. We investigate the coverage of packing space as a function of the number of trial structures that have been generated and lattice energy minimized and the influence of modifying the volume available to the molecule during the structure generation. We also describe the use of the separating axis theorem to relieve molecular clashes in generated structures, in place of rejection.
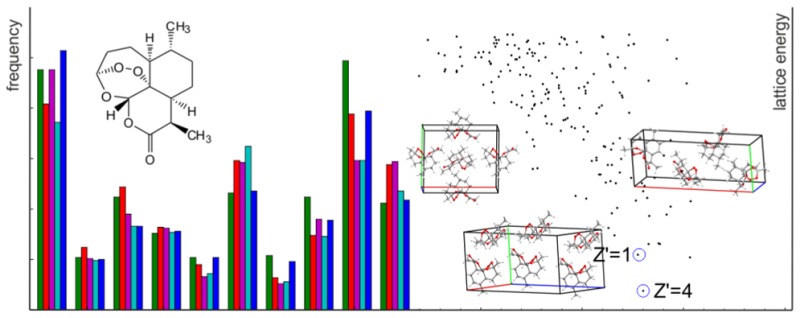
The three molecules studied, artemisinin, quinacridone, and an organic cage (Figure 1), were chosen for diversity in molecular characteristics, in terms of shape and intermolecular interactions, and from three areas where CSP has found applications. Artemisinin, whose discovery was honored by the 2015 Nobel prize in medicine, is a drug used in the treatment of malaria42 and, potentially, cancer.43 Quinacridone finds use in the pigment and semiconductor industries44 and has known polymorphism. The third molecule investigated, hereafter referred to as CC1, is one of a series of porous organic cages that we have studied previously using simulated annealing;21,45 these cages are of interest as solution processable porous materials, and CC1 has the interesting behavior of switching between porous and nonporous polymorphs.46 These three molecules not only exemplify the relevance of CSP in various application areas but also test and demonstrate the performance of the Glee code in cases with a range of known experimental polymorphs, molecular geometries, and intermolecular interactions.
Figure 1.

Chemical diagrams of the three molecules studied here: a) artemisinin, b) quinacridone, and c) CC1.
2. Methods
For rigid molecules, the process of CSP involves the following general steps: (i) molecular geometry optimization; (ii) trial crystal structure generation; and (iii) local lattice energy minimization of trial structures. Clustering of structures is performed after their lattice energy minimization to remove duplicates and assess the completeness of the search.
All calculations presented here are performed with rigid molecular geometries after step (i), taken from isolated molecule geometry optimization using the B3LYP functional with a 6-311G** basis set within the Gaussian09 software.47
2.1. Crystal Structure Generation
2.1.1. Mapping Quasi-Random Numbers to Structural Parameters
Our sampling of the crystal packing configurational space is based on quasi-random, low-discrepancy sequences generated by the Sobol method,48 in a similar manner to Della Valle and co-workers38 and Pantelides and Adjiman.39,49 The present study is restricted to rigid molecules, for which the molecular geometry is kept fixed throughout the generation and optimization of crystal structures. In this approximation, each independent molecule in the asymmetric unit requires three parameters to determine its position and three for its orientation. The values of a further X (X = 1–6) parameters must be generated in order to specify the internal angles and lengths of the unit cell parallelepiped: X = 6 in the case of a triclinic cell, although fewer for lattices with restrictions on cell lengths and angles. Each parameter, pi, is associated with a quasi-random number, xi ∈ [0,1), although, as is discussed below, not all parameters are determined independently of each other.
Molecular Positions
The mapping from three random numbers to the three positions of a molecule’s centroid is trivial. Each number, xi, is taken as a position in fractional coordinates along a particular cell axis. To keep the method general, we include translation along all three lattice vectors in all space groups, regardless of whether the energy is invariant to particular translations in certain space groups. Molecular orientations relative to the global axis frame are sampled using the quaternion based Shoemake method,50 which has previously, for example, been used in the generation of molecular dimers.51 The positions and orientations of all molecules in the unit cell are then generated by applying space group symmetry operators to the asymmetric unit.
Unit Cell Sampling
Each unit cell angle, θj, that is not constrained by space group symmetry is sampled to give an even distribution in cos(θj) according to
| 1 |
with n = 2 and 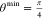 , and xi is the relevant element of
the Sobol vector. This choice samples
the range from
, and xi is the relevant element of
the Sobol vector. This choice samples
the range from 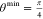 to
to 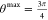 with a probability density that is highest
at its center of
with a probability density that is highest
at its center of 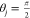 . The function
used to sample cell angles
was chosen to provide a balance between sampling a spread of angles
and avoiding problematic representations of structures. It is not
generally the case that a particular crystal structure has a unique
choice of lattice vectors. In triclinic and monoclinic systems, many
options for the unit cell have very acute or obtuse angles, which
are computationally awkward and inefficient to lattice energy minimize.
The chosen range for cell angles will not exclude any structures but
attempts to only generate versions of structures without flat unit
cells.
. The function
used to sample cell angles
was chosen to provide a balance between sampling a spread of angles
and avoiding problematic representations of structures. It is not
generally the case that a particular crystal structure has a unique
choice of lattice vectors. In triclinic and monoclinic systems, many
options for the unit cell have very acute or obtuse angles, which
are computationally awkward and inefficient to lattice energy minimize.
The chosen range for cell angles will not exclude any structures but
attempts to only generate versions of structures without flat unit
cells.
It is only at the stage of selecting bounds for the cell lengths that our algorithm includes specific information pertaining to the individual system. Our sampling is influenced by the ”box model” of Pidcock and Motherwell,52 which established relationships between molecular dimensions and unit cell lengths. We establish a target volume for the unit cell as the sum of the volumes of all molecules in the unit cell, multiplied by a constant, henceforth referred to as the target volume parameter (TVP). TVP takes a default value of 1.0 but is varied in a later section of this paper to investigate its influence on the performance of the method. The molecular volume is calculated as that of a box chosen to enclose all of its atoms. This box is defined by calculating the axes of inertia of each molecule and finding the maximum and minimum value of the projection of each of its atomic coordinates onto these axes, including standard van der Waals radii53 for each atom. In this section, the difference between the maximum and minimum value of the projections of atomic coordinates, with an appropriate consideration of each atom’s van der Waals radius, will be referred to as the molecule’s ”shadow” onto that axis. As a measure of the volume of the molecule, the product of these three shadows onto the molecule’s axes of inertia would be an overestimate when compared to a more usual measure of volume based on atomic volumes or the molecular van der Waals surface. However, when generating crystal structures we expect to start with a larger volume before allowing the cell to contract under intermolecular forces at a later stage of the process.
The bounds of the three cell lengths can be calculated by considering this target volume and also the projections of the atomic positions onto the lattice vectors. Given that the unit cell angles have been determined, we are able to fix the direction of each unit cell vector in a global axis frame and can calculate the shadow of each lattice vector onto each of our global axes. We must consider separately all molecules in the unit cell that differ by rotation and find the maximal and minimal values of molecular projection on each cell vector, j, which we denote sjmax and sj, respectively (Figure 2). To sample a physically realistic range of cell lengths, we choose the length of the first unit cell vector in the range from c · sjmin to c · Nmols · sj, where Nmols is the number of molecules in the unit cell, and c is a constant used to scale the entire range:
| 2 |
Figure 2.

Molecular projections onto lattice vectors, used to define the sampling range for unit cell lengths. The directions of the three lattice vectors, l1,2,3, are shown, and the molecular projections of two quinacridone molecules are shown onto lattice vector l2. Thin lines show the projection of the edges of the van der Waals radii of each atom onto the lattice vector. Bold red and blue lines show the molecular shadows onto l2. In this example, s2min = 9.13 Å and s2 = 9.57 Å.
The constant c is fixed at 0.75 in this study, reflecting the fact that molecular dimensions can extend past the length of unit cell dimensions.52 The second unit cell length is sampled in the same manner, using projections of the molecular dimensions onto the direction of the second vector and taking the next element of the Sobol vector to sample the relevant range. The third (final) cell length is chosen to give a normal distribution of cell volumes, whose mean is the target volume described above. Thus, the next element of the Sobol vector samples a normal distribution with a standard deviation of 0.15 sjmin, which we find yields a reasonable distribution of volumes. The only cases where this sampling of unit cell lengths is altered are (i) when Nmols = 1, where sj is increased by 50% to ensure a spread of unit cell lengths is sampled and (ii) in crystal systems that place restrictions on cell lengths, where fewer independent unit cell lengths must be determined. We also cycle through the permutations of possible orderings in which the cell lengths could be assigned, so as to avoid any possible systematic bias.
2.1.2. Screening of Unphysical Structures
Before the crystal structure’s parameters are optimized with respect to the lattice energy, unphysical structures, particularly those in which molecules overlap, should be rejected or adjusted. To do this quickly, the convex hull of the molecule is calculated,54 and the separating axis theorem55 is employed to calculate the overlap it has with its neighbors. The molecule’s convex hull is a polytope whose vertices are at atomic positions; these are defined such that the object is convex, and all atomic positions that are not vertices of the hull lie within its volume. A common analogy is to compare the convex hull of an object to its shape if it were wrapped in wrapping paper. As the number of vertices defining the convex hull grows more slowly than the total number of atoms in the molecule, it is an efficient object to deal with when molecules are large. Furthermore, in the case of rigid molecules, the convex hull needs only to be calculated once for each type of molecule, which is performed before generating structures. The convex hulls can be manipulated with the usual symmetry operators, and all neighboring molecular pairs are tested for overlap.
From the separating axis theorem we determine whether a pair of convex hulls overlap and the vector of minimum length which is required to separate the two objects. A set of vectors is taken, which are either normal to the faces of a hull or to an edge from each.56 Onto this set, the shadow of each convex hull is projected (as always, considering the finite size of the atoms by including the appropriate van der Waals radius), and the overlap of the two shadows is measured. The minimal length of overlap along any vector in this set yields the smallest vector required to separate the objects. If there is no overlap of the shadows on any axis in the set, the convex hulls do not overlap, as is the case in the example in Figure 3.
Figure 3.
Separating axis theorem test for molecular overlap. The separating axis theorem prescribes the vectors upon which to project the vertices of the convex hulls when testing polytopes for their overlap in space. An example for the cage molecule CC1 is shown with convex hulls overlaid on the molecular geometry. In the geometry shown there is a vector upon which the “shadows“ of their hulls, the blue and red vectors, do not overlap. If they did overlap, the set of overlapping blue and red vectors would determine the minimum displacement necessary to separate them in the direction of that vector.
The result of the separating axis theorem test can be used in one of two ways, each of which are investigated in this study. The simplest procedure is to reject any trial crystal structure that contains overlapping molecules. The proportion of rejected structures decreases as the target unit cell volume (as determined by the parameter TVP) is increased, which makes more efficient use of the Sobol sequence, at the expense of creating crystal structures that are farther from their final (post energy minimization) density and thus more expensive to lattice energy minimize. For this reason, we test the influence of our choice of TVP on the performance of the search. In this study, we test the structure generation procedure with rejection of trial structures using TVP = 1.0, 1.5, 2.0, and 2.5.
The second option that we have implemented is to adjust trial crystal structures to remove the overlap between molecules. We do this by expanding lattice vector lengths according to
| 3 |
The lattice vector indexed by i grows due to the relationship of the overlap vector, voverlap, and the vector between the centroids of the objects, vcentroid. When both vectors are given in fractional coordinates, the increase in cell length, Δlj, is given by the ratio of their components along that axis. For numerical stability, the cell is only expanded along axis j when vjcentroid > 0.05, and we add a parameter, η, to Δlj with the value 0.001 Å in this study. This lattice vector expansion procedure is iterated until the structure contains no overlapping molecules. In cases where molecules are positioned close to a space group symmetry element, the cell expansion required to relieve molecular overlap can lead to very large unit cells. We therefore place a limit on unit cell volume after lattice vector expansion, above which the trial structure is rejected. We define this limit (maximum volume parameter, MVP) in reference to the molecular volumes used to calculate the target volume parameter, TVP, as 2.5 times the sum of molecular volumes in the unit cell.
A typical use of the crystal structure generation procedure is to generate a set number of trial structures within a specified space group, to allow them to reach a minimum on the PES through lattice energy minimization, and then to monitor the results achieved to assess whether the sampling of possible structures is sufficiently complete. If more structures must be generated for a particular space group, then the search is continued, starting from the highest value of Sobol seed that has previously been used.
2.2. Lattice Energy Minimization
The crystal structure generator described above creates trial structures that could be lattice energy minimized by any method that can affordably be applied to the number of structures required to sample the PES. Currently, the Glee software is interfaced with the DMACRYS crystal structure modeling software,26 to make use of anisotropic atom–atom model potentials. All lattice energy minimizations reported here were performed using DMACRYS, which employs a quasi-Newton–Raphson, rigid-molecule optimization of molecular positions, orientations, and unit cell parameters with space group symmetry constrained. The intermolecular interaction energy between molecules M and N was modeled with an anisotropic model potential of the form
| 4 |
where i, k are atoms of type ι and κ belonging to molecules M and N, respectively, separated by the distance rik. The first two terms model the repulsive and attractive nonelectrostatic intermolecular interactions, whose parameters are taken from a revised version57,58 of the Williams99 force field.59 The final term, describing electrostatic interactions, is calculated from atom-centered multipoles up to rank 4 (hexadecapole) on all atoms, obtained from a distributed multipole analysis60 (DMA) of the B3LYP/6-311G** charge density. Charge–charge, charge–dipole, and dipole–dipole interactions were calculated using Ewald summation, while repulsion–dispersion interactions and all higher multipole–multipole interactions were truncated after a cutoff distance. The summation cutoff (for exp-6 interactions and higher-order multipole-multipole interactions) was set to 30 Å for CC1 and quinacridone and 15 Å for the more compact artemisinin molecule.
2.3. Clustering
Any method for structure prediction requires a procedure for comparing pairs of generated structures and determining whether they are, to within a set tolerance, identical. This step is essential to both remove duplicates from a data set and also to monitor the convergence of the completeness of the sampling. Clustering is only performed after lattice energy minimization of the trial structures. Various methods exist to perform this task in CSP, including the comparison of similarities of computed X-ray powder diffraction patterns61 and the Compack algorithm,62 which tests interatomic separations, and performs an overlay of molecules in order to quantify the similarity of the structures.
Structure comparison and clustering in this work has been processed with our in-house method, which is related to the Compack approach. A cluster of molecules is constructed surrounding each molecule in the asymmetric unit (we use clusters of 25 molecules in this work). We then construct a list of the displacements between atoms in the neighboring molecules and the centroid of the reference molecule. Two such lists can be compared, by positioning the origins of both clusters at the same position and testing whether, for every molecule in one cluster, a set of points occurs in the second which can be overlaid upon the first by the action of rotation only. An algorithm to calculate the optimal RMSD exists,63 and we use a tolerance for comparing pairs of structures, in Å, of 0.5 + 0.05 · r(c1), where r(c1) is the distance of the centroid of the molecule in the first list, to that of the reference molecule around which the cluster is built. If the centroid to origin distance of the molecule in the second structure, r(c2), is not within 20% of that of r(c1), or if the molecules contain different numbers of atoms (in the case of multicomponent crystals), the test fails automatically. If, under this criterion, the lists of clusters of atomic coordinates are determined to match for clusters around all molecules in the asymmetric unit, then the crystals are judged to have identical packings, corresponding to identical minima of the PES.
In order to build up the clusters in a robust and computationally efficient manner, we make use of the Niggli reduced cell64 representation of each crystal structure. The reduced cell vectors are calculated using an algorithm from the Computational Crystallography Toolbox.65 Furthermore, to improve the performance of the algorithm in comparing crystal structures whose molecules are large, the set of atomic positions used in the comparison is reduced to those atoms which comprise the convex hull of the molecule, after the hydrogen atoms have been removed.
A final point concerns molecular symmetry. The algorithm which calculates the optimal RMSD of the overlaid points is sensitive to the order of the coordinates, and hence up to S overlays may have to be performed, where S is the order of symmetry of the molecule. Without making assumptions about combinations of crystal and molecular symmetry operations, when building up lists of atomic positions, S such lists must be calculated: one for each set of coordinates that are equivalent under the internal symmetry of the molecule. We calculate the matrices for each of these operations, and by maintaining a consistent atomic labeling scheme and limiting ourselves, in this paper, to rigid molecules, this calculation is only required once. The CC1 cage is a particularly symmetric molecule, with S = 12, but even with this ”worst case” example the clustering is an inexpensive step in the entire CSP procedure.
3. Results and Discussion
We start with the results for a selection of molecule/space group combinations, choosing the space groups of the observed polymorphs of each molecule for detailed investigation of the convergence of the search for crystal structures. Since we must treat whole molecules in the crystal structure generation procedure, we consider the space groups of the observed structures after removing space group symmetry elements that correspond to intramolecular symmetry. Searches were performed with one molecule in the asymmetric unit (Z′ = 1) in P212121 for artemisinin; P1 and P21/c for CC1; and P21/c and P1̅ for quinacridone. Quinacridone has a known polymorph with two independent molecules in the asymmetric unit (Z′ = 2), so searches were also performed with Z′ = 2 in space group P1̅ .
We generated trial structures with each of the five variations on the structure generation procedure for each system (molecule/space group combination) with Z′ = 1. Four seaches employed rejection of trial structures with overlapping molecules, using different target cell volumes in the assignment of lattice parameters to the trial structures (TVP = 1.0; 1.5; 2.0; 2.5). A fifth search was performed for each system using the cell expansion method in place of rejection, with TVP = 1.0 and a maximum expansion of the unit cell volume (MVP) to a volume parameter of 2.5 (we refer to this method hereafter as SAT-expand). The SAT-expand method should be viewed as a variation on the simpler TVP = 1.0 search, but where structures with overlapping molecules are retained if this overlap can be relieved through expansion of the unit cell volume by up to 250%.
10000 trial structures were generated (after rejection) with each variation of the method for the searches with one molecule in the asymmetric unit. We expect this to be more structures than would generally be required per space group in a CSP study. This deliberate oversampling is performed to gather meaningful statistics. 50000 structures were generated for the quinacridone Z′ = 2 search with each method; a larger number is expected to be required to cover the higher dimensional space. A second, low symmetry (P1, Z′ = 4) polymorph of artemisinin is known. Therefore, to test if this structure could be located with our method, a 50000 structure search was performed for artemisinin in P1 with Z′ = 4 with the SAT-expand method only.
3.1. Convergence of the Number of Unique Crystal Structures
We first examine the efficiency with which each variation of the structure generation method uses the Sobol sequence. Since the low-discrepancy sampling is designed to uniformly sample phase space, we want a method that makes the best use of each point in the sequence; high rates of rejecting trial structures could undermine the uniformity of the search.
As expected, we find that fewer trial crystal structures are rejected when the target volume is increased (Table 1). The probability of molecular overlap is decreased as the volume per molecule is increased. Between 1 in 3 and 1 in 50 trial structures contain overlapping molecules when the target unit cell volume is chosen to just fit the molecules (TVP = 1.0) and there are large variations in the rejection rate between molecules. Quinacridone leads to the most rejected structures: this long, thin molecule clashes with neighbors in most orientations generated from a random sampling. Trial structures of the more isotropically shaped CC1 and artemisinin less frequently contain molecular clashes. We also find variations between space groups for a given molecule. P21/c generally leads to more rejected structures than simpler space groups with fewer symmetry elements (P21/c vs P1 for CC1, P21/c vs P1̅ for quinacridone), since more of the configurational space lies sufficiently close to a symmetry element such that symmetry generated molecules overlap with the original. The Z′ = 2 search is particularly problematic: the generation of 50000 accepted structures required almost 108 trial structures with TVP = 1.0, an acceptance rate of 0.05%.
Table 1. Number of Trial Structures Required To Generate 10000 Accepted Crystal Structures (50000 for Z′ = 2 Quinacridone and Z′ = 4 Artemisinin) for Each Systema.
| system | TVP = 1.0 | TVP = 1.5 | TVP = 2.0 | TVP = 2.5 | SAT-expand |
|---|---|---|---|---|---|
| CC1 (P1) | 17863 (9581) | 17225 (8999) | 17215 (8274) | 17215 (7681) | 10090 (8514) |
| CC1 (P21/c) | 156918 (9723) | 38951 (9211) | 23395 (8843) | 18133 (8279) | 16022 (9059) |
| quinacridone (P1̅ ) | 251805 (9804) | 55400 (9827) | 30696 (9761) | 23262 (9651) | 25131 (9862) |
| quinacridone (P21/c) | 501181 (9767) | 78400 (9533) | 36348 (9315) | 24135 (9075) | 26617 (9353) |
| quinacridone (P1̅ , Z′ = 2) | 96693852 (32021) | 8325359 (46213) | 1057042 (46443) | 504452 (45714) | 480626 (43541) |
| artemisinin (P212121) | 39082 (9894) | 16866 (9584) | 13166 (9071) | 12018 (8490) | 11208 (9362) |
| artemisinin (P1, Z′ = 4) | 363185 (38510) |
Z′ = 1 unless otherwise stated. The number in parentheses is the number of accepted structures that lead to a successful lattice energy minimization.
Considering only Z′ = 1, the differences in rejection rate between space groups and between molecules nearly disappear at large target volumes, where the proportion of rejected structures is decreased. An increase of only 50% to the target volume (TVP = 1.5) has the largest impact on systems where rejection rates were very large (CC1 P21/c and both space groups for quinacridone). At TVP = 2.5, the acceptance rates are quite high: almost all trial structures of artemisinin are accepted, and acceptance rates are in the 40–50% range for CC1 and quinacridone. The acceptance rate for Z′ = 2 is also improved dramatically when a larger target volume is used, so that almost 1 in 10 structures is accepted for TVP = 2.5.
Increasing the volume of generated unit cells clearly makes more efficient use of the Sobol sequence. On the other hand, trial structures with smaller volumes are closer in cell parameters to the final densely packed, lattice energy minimized crystal structures. As a result, the proportion of accepted structures that results in successful lattice energy minimization is highest (96–99%) when the target volume is matched with the molecular volume (TVP = 1.0). Trial unit cells with large volumes prove more challenging for the lattice energy minimizer; up to 23% of accepted Z′ = 1 trial structures fail to find a local minimum using TVP = 2.5 (Table 1). Furthermore, it could be conjectured that making initial guesses that are close to the final, energy minimized structures are more likely to end up in narrow wells and thus more reliably locate all low energy structures than when TVP is set artificially high.
In searches where structures with overlapping molecules are rejected, there is a balance between efficient use of the Sobol sequence, which is best at high target volume, and ease of lattice energy minimization, which is best at smaller target volumes. The SAT-expand method compares favorably to the rejection-based methods on both criteria (final column, Table 1). The rate of accepting trial structures is as high as the TVP = 2.5 searches, since only those that require excessively large unit cell expansion to relieve molecular clashes are rejected. However, since structures in the SAT-expand approach are initially generated with TVP = 1.0 and many of these do not require significant expansion, many of the structures entering energy minimization are close to the densities of the final lattice energy minima. This results in higher success rates of lattice energy minimization than generating directly with large unit cells (e.g., TVP = 2.5).
As a first analysis of the convergence of the crystal structure searches, we monitored the number of unique, low energy lattice energy minima that had been located as the search progressed. For this analysis, we defined the low energy region as that within 15 kJ/mol of the global minimum. Figure 4 displays the results for artemisinin in P212121 (corresponding plots for the other systems can be found in the Supporting Information). The rate of finding new crystal structures is high at the beginning of the search but levels off to the point where no new crystal structures are being located. We observed that all of the methods converge to the same number of unique structures. The TVP = 1.0 method converges most slowly as a function of the number of Sobol vectors attempted but fastest as a function of the number of valid, lattice energy minimized structures. Given that lattice energy minimization is the most costly part of the process, this suggests a slight advantage of generating trial structures with small unit cell volumes. Again, the SAT-expand approach compares favorably with simple rejection, making efficient use of the Sobol sequence and converging quickly with respect to the number of lattice energy minimizations.
Figure 4.

Number of unique crystal structures, within 15 kJ/mol of the global minimum, for artemisinin in space group P212121, displayed (a) as a function of the current position in the Sobol sequence and (b) as a function of the total number of successfully energy minimized structures.
3.2. Energetic Assessment of Sampling Convergence
As well as monitoring how the total number of unique low energy crystal structures converges during a crystal structure search (Figure 4), it is useful to monitor the evolution of the energy of the lowest energy structure found during a search, as a function of the number of structures that have been energy minimized. Figure 5 displays the rate at which the energy of both the lowest individual structure, and the set of the lowest 10 structures, converges with respect to the number of successful energy minimizations for artemisinin (P212121) and CC1 (P21/c).
Figure 5.

Average lattice energy of the ten lowest energy structures is shown, as a function of the number of minimized structures generated in the experimentally observed space group for a) artemisinin in P212121 and b) CC1 in P21. The dashed lines indicate the energy of the single lowest energy structure, where the color relates to the same method in the legend. The data had converged after 1000 and 6500 minimizations for a) and b), respectively, so is not shown beyond this point for clarity.
Several points are immediately obvious. The lowest energy structure in the set is found rapidly, which we find to be true for all systems studied here (see the Supporting Information for results for the other systems). Once the lowest energy structure remains stable with respect to the number of energy minimized structures, we assume that this corresponds to the true global minimum on the lattice energy surface. We also monitor the mean energy of the 10 lowest energy structures that have been located, to see by which point in the search this larger set of low energy structures remains stable. We find that convergence of the set of the 10 lowest energy structures is about an order of magnitude slower than the rate of finding the global minimum and that the convergence is quicker for artemisinin than CC1. For artemisinin, the sampling in this space group appears to be complete for all variations of the search (different TVP and SAT-expand) well before 1000 successful lattice energy minimizations.
We observe that searches using a large TVP generally appear to converge more slowly than the smaller target volumes, with the SAT-expand method performing fairly well; this is in line with our expectations based on the convergence of the number of unique structures (Figure 4). The change in performance of the search upon changing TVP is particularly stark for CC1 in P21/c, where the entire set of 10 lowest energy structures converges slowly for TVP ≥ 1.5. On the basis of these results, the most satisfactory results are obtained when searching either with rejection-based sampling and a small target volume, recognizing that many trial structures will be rejected, or the SAT-expand method.
There is evidence66 and intuition behind the idea that the deeper wells on the PES may well also have a large watershed around them, and a quasi-random search seeks to take advantage of this. The rapid convergence of the set of lowest energy structures is a useful property when looking to make rapid searches in a wide range of space groups, as it should be possible to estimate the limit of the lowest energy structure in the set before completeness is achieved. Noting the number of structures needed to find the ten lowest energy structures gives us a ball park figure of the absolute minimum number of structures that we would wish to successfully lattice energy minimize in these space groups. In some cases, this can be as few as several hundred lattice energy minimizations, although the small computational expense of generating and minimizing structures means that we would generally afford ourselves several thousand structures in space groups whose lowest energy structure is within the lattice energy range of interest to us.
3.3. Rate of Sampling of Low Energy Structures
The number of times that each low energy predicted crystal structure is located can be studied in detail, to investigate how our attempt at uniform sampling of configurational space during trial structure generation translates into uniformity of sampling of local energy minima. Figure 6 displays the number of times that each of the 10 lowest energy crystal structures appear in each search for four of our systems. The sampling of individual low energy crystal structures is clearly uneven; each system has some structures that are more rarely located than others.
Figure 6.

Bar charts showing the frequency with which each low energy structure is located. For each of the lowest 10 unique structures, for the denoted systems, the energy above the minimum in the set is displayed on the horizontal axis, and the number of times that it was found in the search is read from the vertical axis. The five methods appear alongside each other, with the color of the bar signifying the method.
The case of CC1 in space group P1 (Figure 6) is very simple: the frequency of finding each minimum decreases as the lattice energy increases, and for all methods, well over half of the initial structures relax to the two lowest minima (4000–5000 hits to te global minimum, 1500–2000 hits to the second lowest energy structure). CC1 (P1) is also the system with the largest energy differences between structures, since there are few ways to achieve a low energy crystal packing when all molecules are related by translational symmetry only (space group P1). The five variations on the sampling method show very similar performance for CC1 (P1), with the searches that sample smaller volume trial structures leading to slightly more structures overall, as the rate of achieving successful lattice energy mimimization is slightly larger from these trial structures.
The tendency for the lowest energy structures within a space group to be frequently located is repeated for all other systems, with the global minimum always one of the most sampled structures; this finding explains the rapid convergence of the global minimum energy shown in Figure 5. However, the lattice energy surfaces of most systems are more detailed than that of CC1 (P1), with more low-lying energy minima, and a less clear relationship between the energy of a local minimum and the frequency of finding it in a search. For example, the structure search for quinacridone in space group P1̅ leads to many energetically similar crystal structures, with similar layered packings of the planar molecule. The first two structures are found frequently, but we observe that it is much harder to locate all of the others (Figure 6). The differing frequencies of obtaining each minimum are difficult to explain, as only 6.68 kJ/mol separate the set, and they are structurally very similar; as will be noted later, we also have concerns with regard to our force-field for this system.
It is those crystal structures that are located infrequently that are most concerning, as they could easily be missed if sampling is stopped too early, and most systems that we studied have such structures on their landscapes. The fourth lowest energy structure in CC1 (P21/c), with a relative lattice energy of 4.15 kJ/mol is one such example (Figure 6b), as are structures 4 (1.81 kJ/mol) and 8–10 for quinacridone in P1̅ (Figure 6b) and, to a lesser extent, structures 2 (0.58 kJ/mol), 5 (2.27 kJ/mol), and 7 (2.72 kJ/mol) for artemisinin (Figure 6d). In most cases, the rate at which these challenging structures are found decreases as TVP is increased, meaning that the rejection-based methods with large unit cell volumes have a high risk of missing some low energy structures. An advantage of the SAT-expand method is that a range of initial volumes are covered during the search, and we find this method performs well on the challenging, infrequently sampled crystal structures.
Another way of examing the sampling of low energy structures is to keep track of where each occurrence of each low energy crystal structure was generated in the original Sobol sequence (Figure 7). This representation reassures us that the Sobol sequence is evenly exploring the configurational space, as the points leading to each low energy structure are evenly distributed along the series. This representation of how well the low energy structures are sampled is useful for monitoring a calculation as it proceeds, since it provides an immediate picture of the state of completeness. Again, we clearly see that increasing TVP hinders the sampling of some low energy structures (Figure 7b) and that a more even sampling is achieved with the SAT-expand method (Figure 7c).
Figure 7.

Hits to the 10 lowest ranked crystal structures of quinacridone P1̅ based on the combined complete search of the five methods. Each point represents a lattice energy minimization from a trial structure, showing the step in the Sobol sequence where the trial structure was generated and the lattice energy minimum to which it optimizes.
3.4. Multiple Independent Molecules
Our calculations on Z′= 2 crystal structures of quinacridone demonstrate the increased difficulty of predicting crystal structures with multiple independent molecules in the asymmetric unit. The inclusion of a second independent molecule can greatly increase the number of Sobol seeds needed to generate the desired number of crystal structures. This can be seen in Table 1, where far higher values in the Sobol sequence must be used for quinacridone P1̅ Z′ = 2 across all TVP values compared to the same space group with Z′ = 1. The larger Sobol sequences are needed as a large proportion of structures is rejected due to overlap of molecules. This is a particular problem with TVP = 1.0; large numbers of rejected structures lead to much more time spent on the structure generation as a whole.
The other challenging aspect of Z′ = 2 is the higher dimensionality of the energy surface and, hence, the smaller relative volume of configurational space that is expected to lattice energy minimize to any particular crystal structure. Although the number of unique Z′ = 2 crystal structures in the low energy region is small, some of our searches do not find the full set of structures until well over 10000 lattice energy minimizations have been completed (Figure 8); the TVP = 2.0 search has not located one of the low energy structures, even as 50000 lattice energy minimizations are approached. The SAT-expand method performs well in finding all low energy structures in a relatively low number of lattice energy minimizations but still suffers from very infrequent sampling of some structures (Figure 8b).
Figure 8.

Convergence and sampling of the quinacridone P1̅ Z′ = 2 search for crystal structures. a) The number of unique structures within 15 kJ/mol of the global minimum as a function of the total number of successful lattice energy minimizations, using each variation of the structure generation method. b) Hits of the 10 lowest energy crystal structures througout the Sobol sequence, using the SAT-expand method.
A second polymorph is known for artemisinin, with four independent molecules in the asymmetric unit. Ensuring complete searches of high Z′ structures is known to be difficult67 due to the very high dimensionality of search space, and we know of few previous studies which have successfully located Z′ = 368 and Z′ = 469 polymorphs in CSP studies. As a test of our methods, we generated 50000 structures in the relevant space group, P1 with Z′ = 4, and found the known crystal structure to be the lowest energy structure of all. However, there were only 3 matches to the experimentally observed structure from 38510 valid lattice energy minimizations. As with Z′ = 2 quinacridone, the search required a large number of steps in the Sobol sequence (Table 1), due to a high proportion of unphysical structures. The results demonstrate that high Z′ CSP is possible, albeit challenging.
3.5. Full Searches
The SAT-expand method, with a maximum volume parameter of 2.5, showed some of the best characteristics in the above tests and was used in an extended search across a range of space groups. Currently, 95 space groups are available to be searched in the Glee program, but we restrict ourselves here to a subset of the most commonly observed symmetries for organic molecular crystals. For chiral, enantiomerically pure artemisinin, 5000 structures were generated in each of 8 space groups (P1, P21, C2, P21212, P212121, C2221, P41212, and R3). These space groups were searched for CC1 and quinacridone, in addition to P1̅, Cc, P21/c, C2/c, Pna21, Pbcn, Pbca, and Pnma, all with 5000 accepted structures. All calculations were performed with one molecule in the asymmetric unit, except for the case of a search for the Z′ = 4 polymorph of artemisinin which has been included as a special example. Lattice energy minimization and clustering were performed using the same procedures as have been employed throughout.
Results are summarized in Figure 9, where each structure is represented by its calculated lattice energy and density. A central assumption of crystal structure prediction by global lattice energy minimization is that the most likely structure to be observed experimentally is that with the lowest free energy of formation. Although free energy contributions associated with the dynamics of molecules about the equilibrium positions can be significant,27,70 in this study we have focused on the lattice energy, which is the largest contribution to the free energy difference between crystal structures. The existence of polymorphs indicates that the process of crystallization is more subtle than a simple drive toward the single lowest lattice energy structure, but our methodology is predicated upon the assumption that all solvent-free, stable crystal structures can be located in a set of low-lying, lattice energy ordered structures determined from a quasi-random search.
Figure 9.

Lattice energy vs density plots for artemisinin, quinacridone, and CC1. Each point corresponds to a distinct crystal structure (a unique minimum on the PES). For the case of quinacridone, two sets of data have been calculated, and the basis set used in generating the electrostatic model is included in parentheses in the subcaption. The α polymorph was located at too high a lattice energy to appear on the graph in the case of the 6-311G** basis set. For artemisinin the Z′ = 4 structure is added. Predicted structures that geometrically match the experimental structures (see Table 2) are circled and labeled.
For artemisinin, we find that the second lowest energy structure from the full search of Z′ = 1 structures corresponds to the known crystal form (Figure 9a), to within 1.6% in lattice dimensions (Table 2). The structure is only 0.15 kJ/mol above the global minimum within the constraint of Z′ = 1. As described above, the second known artemisinin polymorph, in P1 with Z′ = 4, was located in our search, as the global minimum in P1 with Z′ = 4 and lower in energy than any other crystal structure that was generated in our full search (Figure 9a). These results suggest that this low symmetry crystal structure results from lowering the lattice energy, rather than being kinetically trapped as an “incomplete“ crystallization.71
Table 2. Matches from the Full CSP to Experimentally Determined Structures of the Observed Polymorphsa.
| |
cell
lengths |
cell
angles |
||||||
|---|---|---|---|---|---|---|---|---|
| crystal structure | a | b | c | α | β | γ | RMSD30 | |
| artemisinin (P212121) | expt | 24.066 | 9.439 | 6.354 | 90.00 | 90.00 | 90.00 | |
| pred | 24.456 | 9.399 | 6.386 | 90.00 | 90.00 | 90.00 | 0.131 | |
| artemisinin (P1) ,Z′ = 2 | expt | 9.881 | 9.891 | 15.343 | 93.28 | 90.92 | 102.99 | |
| pred | 9.892 | 10.020 | 15.164 | 90.81 | 93.64 | 102.32 | 0.247 | |
| CC1 (R3, β′) | expt | 21.015 | 21.015 | 10.491 | 90.00 | 90.00 | 120.00 | |
| pred | 21.623 | 21.602 | 10.851 | 90.02 | 90.02 | 119.98 | 0.603 | |
| CC1 (P21/c, α′) | expt | 12.810 | 10.910 | 36.810 | 90.00 | 97.49 | 90.00 | |
| pred | 13.425 | 11.156 | 37.761 | 90.00 | 94.45 | 90.00 | 0.812 | |
| quinacridone (P21/c, γ) | expt | 13.697 | 3.881 | 13.402 | 90.00 | 100.44 | 90.00 | |
| pred (6-31G**) | 12.847 | 4.251 | 13.370 | 90.00 | 97.08 | 90.00 | 0.288 | |
| pred (6-311G**) | 13.397 | 4.115 | 13.002 | 90.00 | 98.21 | 90.00 | 0.439 | |
| quinacridone (P21/c, β) | expt | 5.692 | 3.975 | 30.020 | 90.00 | 96.76 | 90.00 | |
| pred (6-31G**) | 5.746 | 4.110 | 29.565 | 90.00 | 93.90 | 90.00 | 0.369 | |
| pred (6-311G**) | 8.972 | 5.296 | 34.750 | 90.00 | 123.27 | 90.00 | 0.492 | |
| quinacridone (P1̅, αI) | expt | 3.802 | 6.612 | 14.485 | 100.68 | 94.40 | 102.11 | |
| pred (6-31G**) | 4.331 | 6.203 | 13.632 | 97.49 | 97.07 | 98.00 | 0.451 | |
| pred (6-311G**) | 4.620 | 6.372 | 12.530 | 95.25 | 97.82 | 103.62 | 1.245 | |
| quinacridone (P1̅, αII) | expt | 14.934 | 3.622 | 12.935 | 91.39 | 107.13 | 92.84 | |
| pred (6-31G**) | 13.684 | 4.369 | 13.239 | 90.00 | 115.39 | 90.00 | 0.219 | |
| pred (6-311G**) | 13.397 | 4.115 | 13.002 | 90.00 | 98.21 | 90.00 | 1.019 | |
RMSD30 is the deviation in atomic positions of a cluster of 30 molecules taken from predicted and experimental structures, not including hydrogen atoms. CC1 (R3) was generated in the P1 space group, which reduces to R3 on account of intramolecular symmetry, hence the cell angles differ at the second decimal place. The experimental structures of CC1 also contained residual solvent, which was removed for purposes of comparison. All structures were converted to their reduced unit cell for comparison. Å and degrees are used throughout.
Our results for quinacridone were surprisingly sensitive to the basis set used in generating the electrostatic model for intermolecular interactions. Among the quinacridone structures generated in the full search using B3LYP/6-311G** electrostatics, the γ polymorph (encircled Figure 9c) is the lowest energy experimentally known structure located, being the fifth lowest structure in energy 3.4 kJ/mol above the global minimum. The β (encircled Figure 9c) and αI polymorphs sit at 9.9 and 16.9 kJ/mol above the global minimum, respectively. These energy rankings are surprisingly high, and there is no reason to believe that these polymorphs are truly high energy crystal forms. Furthermore, the predicted structures are geometrically in fairly poor agreement with the structures determined from X-ray diffraction (Table 2).
To investigate the sensitivity of these results to the electrostatic model used in the force field model, all predicted crystal structures were reoptimized using atomic multipoles derived from a smaller basis set (6-31G**). The ranking of observed structures within the predictions changes significantly; β is now the global minimum (encircled Figure 9d) with γ and αI 3.4 and 5.68 kJ/mol above the minimum. While no full search was performed for Z′ = 2, the proposed structure of αII has been located in each of the preliminary CSP searches that have been used above to analyze the performance of TVP values and the SAT-expand method, with both 6-311G** and 6-31G** basis sets. However, as reported by Paulus et al.,72 we observe that the Z′ = 2 αII structure relaxes to the Z′ = 1 γ structure during lattice energy minimization. These two polymorphs seem to correspond to the same minimum on the lattice energy surface.
Throughout this study we have maintained the same methodology for generating a force field for all systems, but while the success has been fairly strong for the cases of artemisinin and CC1, it was not so for quinacridone. Previous work by Leusen72 located the known polymorphs of quinacridone among the lowest structures generated and minimized with a simple, isotropic atom force field, but Kraft has demonstrated the importance of anisotropic force fields in modeling the PES of polyaromatic hydrocarbons.73 We include an ab initio anisotropic electrostatic model, but it is highly sensitive to the underlying DFT calculations. The results for quinacridone demonstrate important differences in how the atomic multipoles model intermolecular electrostatics, which may be due to the strong basis set dependence of the original distributed multipole analysis algorithm that we employed here.74 When we change the basis set used for the electrostatic model, we use a empirically fitted exp-6 parameter set for all other intermolecular interactions, although not one that is fitted to polyaromatic hydrocarbons specifically. Even so, we would not expect such large changes in lattice energy and polymorph ordering as have been observed in this case. We also note that, even at the level of Hückel theory, the electronic structure of a π system will change as multiple rings are fused together, and, of course, this molecule is semiconducting in the solid state, indicating its unusual character. From the literature on enhanced π van der Waals interactions in aromatics, Grimme has suggested that stronger dispersion effects arise in systems beyond three fused rings,75 which would include quinacridone, but few systems from which our potential has been fitted. This study has, at the least, highlighted the need for more work to produce a transferable, accurate force field for these systems, while also tested the search methodology in a difficult case.
Finally, the results for CC1 agree with our earlier study of this molecule,21 which used a Monte Carlo simulated annealing approach to generating trial crystal structures. The two polymorphs are found in the low energy region of the landscape and are good geometrical matches to the structures determined by X-ray diffraction. The crystal structures of this organic cage are obtained by desolvation of solvate structures in which guest solvent molecules fill the voids within and between cage molecules. The structure-directing effect of the included solvent has been shown to be so strong that polymorph transformation can be achieved through exposure to solvent vapor.46 In this situation of strong solvent directing effects, it is unsurprising that the observed structures do not correspond to the lowest energy possibilities on the solvent-free energy landscape. Indeed, re-evaluation of the energies of the predicted structures of CC1 using dispersion-corrected solid state DFT shows little energetic reranking,21 providing further confidence in the force field based relative energies.
4. Conclusions
This paper outlines a method of generating trial structures of molecular crystals, which is an essential part of an ab initio crystal structure prediction methodology. A core idea of our methodology is to consider the shape of the molecules but to use as few other restrictions as possible in our quasi-random search. We have demonstrated that this is an effective method of determining the full set of low energy crystal packing possibilities of a molecule, which includes the experimentally observed polymorphs in the cases that we have studied here. We find that the global lattice energy minimum is typically located early in a search and sampled frequently throughout a quasi-random search. This is an important finding, as it suggests that short quasi-random searches can be applied to rapidly evaluate a molecule’s crystal packing preferences, which can be extended to complete, converged searches if desired. However, we also find that some low energy crystal structures are more infrequently sampled, making the frequency of locating such difficult structures rate-limiting when a complete crystal structure search is required.
We examined the influence of increasing the target unit cell volume in generating trial crystal structures, but large target volumes led to less reliable sampling of structures. Our use of the separating axis theorem allows us to quickly rule out unphysical trial structures, and by expanding the cell to relieve clashes, we can keep a larger proportion of trial structures. This is important, as the Sobol sequence is designed to cover the manifold of random numbers in an efficient way and helps us to rapidly consider a wide range of potential structures in the search. This SAT-expand approach has the best characterists, overall, of the variations on our method that we have investigated.
This tool not only provides us with a method to conduct a crystal structure prediction study for rigid molecules but also provides a platform upon which further functionality can be built. We have a robust method of exploring the PES that we show to be effective for a set of different molecules and structures with multiple molecules in the asymmetric unit (Z′ = 2 and 4). The search methodology also finds polymorphs whose crystallization is determined by solvent templating rather than the principle of close packing (CC1) or even in a case for which our energy model is not optimized for a particular case (quinacridone). The principle of a pseudorandom number search, when coupled to our code base, provides us with a lot of flexibility. We are currently extending this methodology to include further functionality, such as molecular flexibility, and are also incorporating these tools into high-througput screening of molecules for discovering molecular crystals with targeted properties.
Acknowledgments
We thank the European Research Council for funding under grant ERC-StG-2012-ANGLE-307358, as well as the EPSRC via grant EP/J01110X/1. We acknowledge the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work. All data supporting this study are openly available from the University of Southampton repository at http://dx.doi.org/10.5258/SOTON/385297.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.5b01112.
Intermolecular potential parameters and convergence graphs not included in the main text (PDF)
Crystallographic structure files (CIF format) with all low energy predicted crystal structures of artemisinin from the Z′ = 1 full search, including calculated lattice energies (CIF)
Crystallographic structure files (CIF format) with all low energy predicted crystal structures of CC1 from the Z′ = 1 full search, including calculated lattice energies (CIF)
Crystallographic structure files (CIF format) with all low energy predicted crystal structures of quinacridone from the Z′ = 1 full search using 6-31G** electrostatics, including calculated lattice energies (CIF)
Crystallographic structure files (CIF format) with all low energy predicted crystal structures of quinacridone from the Z′ = 1 full search using 6-311G** electrostatics, including calculated lattice energies (CIF)
Crystallographic structure files (CIF format) with all low energy predicted crystal structures of artemisinin from the Z′ = 4 search, including calculated lattice energies (CIF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tong H.; Ma W.; Wang L.; Wan P.; Hu J.; Cao L. Biomaterials 2004, 25, 3923–3929. 10.1016/j.biomaterials.2003.10.038. [DOI] [PubMed] [Google Scholar]
- Mas-Torrent M.; Rovira C. Chem. Rev. 2011, 111, 4833–4856. 10.1021/cr100142w. [DOI] [PubMed] [Google Scholar]
- Hunger K. Rev. Prog. Color. Relat. Top. 1999, 29, 71–84. 10.1111/j.1478-4408.1999.tb00129.x. [DOI] [Google Scholar]
- Tozawa T.; Jones J. T. A.; Swamy S. I.; Jiang S.; Adams D. J.; Shakespeare S.; Clowes R.; Bradshaw D.; Hasell T.; Chong S. Y.; Tang C.; Thompson S.; Parker J.; Trewin A.; Bacsa J.; Slawin A. M. Z.; Steiner A.; Cooper A. I. Nat. Mater. 2009, 8, 973–978. 10.1038/nmat2545. [DOI] [PubMed] [Google Scholar]
- Jones J. T. A.; Hasell T.; Wu X.; Bacsa J.; Jelfs K. E.; Schmidtmann M.; Chong S. Y.; Adams D. J.; Trewin A.; Schiffman F.; Cora F.; Slater B.; Steiner A.; Day G. M.; Cooper A. I. Nature 2011, 474, 367–371. 10.1038/nature10125. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Presly O.; White F.; Oppel I. M.; Mastalerz M. Angew. Chem., Int. Ed. 2014, 53, 1516–1520. 10.1002/anie.201308924. [DOI] [PubMed] [Google Scholar]
- Vippagunta S. R.; Brittain H. G.; Grant D. J. Adv. Drug Delivery Rev. 2001, 48, 3–26. 10.1016/S0169-409X(01)00097-7. [DOI] [PubMed] [Google Scholar]
- Cruz-Cabeza A. J.; Reutzel-Edens S. M.; Bernstein J. Chem. Soc. Rev. 2015, 44, 8619–8635. 10.1039/C5CS00227C. [DOI] [PubMed] [Google Scholar]
- Harris R. K. J. Pharm. Pharmacol. 2007, 59, 225–239. 10.1211/jpp.59.2.0009. [DOI] [PubMed] [Google Scholar]
- Hamaed H.; Pawlowski J. M.; Cooper B. F.; Fu R.; Eichhorn S. H.; Schurko R. W. J. Am. Chem. Soc. 2008, 130, 11056–11065. 10.1021/ja802486q. [DOI] [PubMed] [Google Scholar]
- Baias M.; Widdifield C. M.; Dumez J.-N.; Thompson H. P. G.; Cooper T. G.; Salager E.; Bassil S.; Stein R. S.; Lesage A.; Day G. M.; Emsley L. Phys. Chem. Chem. Phys. 2013, 15, 8069–8080. 10.1039/c3cp41095a. [DOI] [PubMed] [Google Scholar]
- Day G. M.; Zeitler J. A.; Jones W.; Rades T.; Taday P. F. J. Phys. Chem. B 2006, 110, 447–456. 10.1021/jp055439y. [DOI] [PubMed] [Google Scholar]
- Parrott E. P.; Zeitler J. A. Appl. Spectrosc. 2015, 69, 1–25. 10.1366/14-07707. [DOI] [PubMed] [Google Scholar]
- Day G. M. Crystallogr. Rev. 2011, 17, 3–52. 10.1080/0889311X.2010.517526. [DOI] [Google Scholar]
- Pantelides C. C.; Adjiman C. S.; Kazantsev A. V.. Prediction and Calculation of Crystal Structures; Spring International Publishing: Switzerland, 2014; pp 25–58. [Google Scholar]
- Price S. L. Chem. Soc. Rev. 2014, 43, 2098–2111. 10.1039/C3CS60279F. [DOI] [PubMed] [Google Scholar]
- Kazantsev A. V.; Karamertzanis P. G.; Adjiman C. S.; Pantelides C. C.; Price S. L.; Galek P. T.; Day G. M.; Cruz-Cabeza A. Int. J. Pharm. 2011, 418, 168–178. 10.1016/j.ijpharm.2011.03.058. [DOI] [PubMed] [Google Scholar]
- Gorbitz C. H.; Dalhus B.; Day G. M. Phys. Chem. Chem. Phys. 2010, 12, 8466–8477. 10.1039/c004055j. [DOI] [PubMed] [Google Scholar]
- Price L. S.; McMahon J. A.; Lingireddy S. R.; Lau S.-F.; Diseroad B. A.; Price S. L.; Reutzel-Edens S. M. J. Mol. Struct. 2014, 1078, 26–42. 10.1016/j.molstruc.2014.01.014. [DOI] [Google Scholar]
- Kendrick J.; Stephenson G. A.; Neumann M. A.; Leusen F. J. J. Cryst. Growth Des. 2013, 13, 581–589. 10.1021/cg301222m. [DOI] [Google Scholar]
- Pyzer-Knapp E. O.; Thompson H. P. G.; Schiffmann F.; Jelfs K. E.; Chong S. Y.; Little M. A.; Cooper A. I.; Day G. M. Chem. Sci. 2014, 5, 2235–2245. 10.1039/c4sc00095a. [DOI] [Google Scholar]
- Vasileiadis M.; Pantelides C. C.; Adjiman C. S. Chem. Eng. Sci. 2015, 121, 60–76. 10.1016/j.ces.2014.08.058. [DOI] [Google Scholar]
- Neumann M. A.; van de Streek J.; Fabbiani F. P. A.; Hidber P.; Grassmann O. Nat. Commun. 2015, 6, 7793. 10.1038/ncomms8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day G. M.; Cooper T. G.; Cruz-Cabeza A. J.; Hejczyk K. E.; Ammon H. L.; Boerrigter S. X. M.; Tan J. S.; Della Valle R. G.; Venuti E.; Jose J.; Gadre S. R.; Desiraju G. R.; Thakur T. S.; van Eijck B. P.; Facelli J. C.; Bazterra V. E.; Ferraro M. B.; Hofmann D. W. M.; Neumann M. A.; Leusen F. J. J.; Kendrick J.; Price S. L.; Misquitta A. J.; Karamertzanis P. G.; Welch G. W. A.; Scheraga H. A.; Arnautova Y. A.; Schmidt M. U.; van de Streek J.; Wolf A. K.; Schweizer B. Acta Crystallogr., Sect. B: Struct. Sci. 2009, 65, 107–125. 10.1107/S0108768109004066. [DOI] [PubMed] [Google Scholar]
- Bardwell D. A.; Adjiman C. S.; Arnautova Y. A.; Bartashevich E.; Boerrigter S. X. M.; Braun D. E.; Cruz-Cabeza A. J.; Day G. M.; Valle R. G. D.; Desiraju G. R.; van Eijck B. P.; Facelli J. C.; Ferraro M. B.; Grillo D.; Habgood M.; Hofmann D. W. M.; Hofmann F.; Jose K. V. J.; Karamertzanis P. G.; Kazantsev A. V.; Kendrick J.; Kuleshova L. N.; Leusen F. J. J.; Maleev A. V.; Misquitta A. J.; Mohamed S.; Needs R. J.; Neumann M. A.; Nikylov D.; Orendt A. M.; Pal R.; Pantelides C. C.; Pickard C. J.; Price L. S.; Price S. L.; Scheraga H. A.; van de Streek J.; Thakur T. S.; Tiwari S.; Venuti E.; Zhitkov I. K. Acta Crystallogr., Sect. B: Struct. Sci. 2011, 67, 535–551. 10.1107/S0108768111042868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price S. L.; Leslie M.; Welch G. W. A.; Habgood M.; Price L. S.; Karamertzanis P. G.; Day G. M. Phys. Chem. Chem. Phys. 2010, 12, 8478–8490. 10.1039/c004164e. [DOI] [PubMed] [Google Scholar]
- Nyman J.; Day G. M. CrystEngComm 2015, 17, 5154–5165. 10.1039/C5CE00045A. [DOI] [Google Scholar]
- Day G. M.; Chisholm J.; Shan N.; Motherwell W. D. S.; Jones W. Cryst. Growth Des. 2004, 4, 1327–1340. 10.1021/cg0498148. [DOI] [Google Scholar]
- Cruz-Cabeza A. J.; Day G. M.; Jones W. Chem. - Eur. J. 2009, 15, 13033–13040. 10.1002/chem.200901703. [DOI] [PubMed] [Google Scholar]
- Karfunkel H.; Gdanitz R. J. Comput. Chem. 1992, 13, 1171–1183. 10.1002/jcc.540131002. [DOI] [Google Scholar]
- Pillardy J.; Arnautova Y. A.; Czaplewski C.; Gibson K. D.; Scheraga H. A. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 12351–12356. 10.1073/pnas.231479298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Orendt A. M.; Ferraro M. B.; Facelli J. C. J. Comput. Chem. 2009, 30, 1973–1985. 10.1002/jcc.21189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q.; Oganov A. R.; Glass C. W.; Stokes H. T. Acta Crystallogr., Sect. B: Struct. Sci. 2012, 68, 215–226. 10.1107/S0108768112017466. [DOI] [PubMed] [Google Scholar]
- Lund A. M.; Pagola G. I.; Orendt A. M.; Ferraro M. B.; Facelli J. C. Chem. Phys. Lett. 2015, 626, 20–24. 10.1016/j.cplett.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzyabchenko A. V. J. Struct. Chem. 1984, 25, 416–420. 10.1007/BF00749334. [DOI] [Google Scholar]
- Williams D. E. Acta Crystallogr., Sect. A: Found. Crystallogr. 1996, 52, 326–328. 10.1107/S0108767395016679. [DOI] [Google Scholar]
- van Eijck B. P.; Kroon J. J. Comput. Chem. 1999, 20, 799–812. . [DOI] [PubMed] [Google Scholar]
- Della Valle R. G.; Venuti E.; Brillante A.; Girlando A. J. Chem. Phys. 2003, 118, 807. 10.1063/1.1527896. [DOI] [Google Scholar]
- Karamertzanis P. G.; Pantelides C. C. J. Comput. Chem. 2005, 26, 304–324. 10.1002/jcc.20165. [DOI] [PubMed] [Google Scholar]
- Bygrave P. J.; Case D. H.; Day G. M. Faraday Discuss. 2014, 170, 41–57. 10.1039/C3FD00162H. [DOI] [PubMed] [Google Scholar]
- Hoxha K.; Case D. H.; Day G. M.; Prior T. J. CrystEngComm 2015, 17, 7130–7141. 10.1039/C5CE01286D. [DOI] [Google Scholar]
- White N. J. J. Clin. Invest. 2004, 113, 1084–1092. 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J.; Wang D.; Zhang R.; Wang H. Clin. Cancer Res. 2008, 14, 5519–5530. 10.1158/1078-0432.CCR-08-0197. [DOI] [PubMed] [Google Scholar]
- Głowacki E. D.; Irimia-Vladu M.; Kaltenbrunner M.; Gsiorowski J.; White M. S.; Monkowius U.; Romanazzi G.; Suranna G. P.; Mastrorilli P.; Sekitani T.; Bauer S.; Someya T.; Torsi L.; Sariciftci N. S. Adv. Mater. 2013, 25, 1563–1569. 10.1002/adma.201204039. [DOI] [PubMed] [Google Scholar]
- Hasell T.; Culshaw J. L.; Chong S. Y.; Schmidtmann M.; Little M. A.; Jelfs K. E.; Pyzer-Knapp E. O.; Shepherd H.; Adams D. J.; Day G. M.; Cooper A. I. J. Am. Chem. Soc. 2014, 136, 1438–1448. 10.1021/ja409594s. [DOI] [PubMed] [Google Scholar]
- Jones J. T. A.; Holden D.; Mitra T.; Hasell T.; Adams D. J.; Jelfs K. E.; Trewin A.; Willock D. J.; Day G. M.; Bacsa J.; Steiner A.; Cooper A. I. Angew. Chem., Int. Ed. 2011, 50, 749–753. 10.1002/anie.201006030. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian09 Revision D.01; 2009; Gaussian Inc.: Wallingford, CT, 2009. [Google Scholar]
- Sobol I.USSR Computational Mathematics and Mathematical Physics 1976, 16, 236–242. [Google Scholar]
- Vasileiadis M.; Kazantsev A. V.; Karamertzanis P. G.; Adjiman C. S.; Pantelides C. C. Acta Crystallogr., Sect. B: Struct. Sci. 2012, 68, 677–685. 10.1107/S0108768112045636. [DOI] [PubMed] [Google Scholar]
- Shoemake K. In Graphics Gems III; Kirk D., Ed.; Academic Press Professional, Inc.: San Diego, CA, USA, 1992; Chapter Uniform Random Rotations, pp 124–132. [Google Scholar]
- Misquitta A. J.; Welch G. W.; Stone A. J.; Price S. L. Chem. Phys. Lett. 2008, 456, 105–109. 10.1016/j.cplett.2008.02.113. [DOI] [Google Scholar]
- Pidcock E.; Motherwell W. D. S. Cryst. Growth Des. 2004, 4, 611–620. 10.1021/cg034216z. [DOI] [Google Scholar]
- Bondi A. J. Phys. Chem. 1964, 68, 441–451. 10.1021/j100785a001. [DOI] [Google Scholar]
- O’Rourke J.Computational Geometry in C, 2nd ed.; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Gottschalk S.Separating axis theorem; Technical Report TR96-024; Department of Computer Science, UNC Chapel Hill: 1996.
- Torquato S.; Jiao Y. Phys. Rev. E 2009, 80, 876–879. [Google Scholar]
- Pyzer-Knapp E. O. Ph.D. Thesis, University of Cambridge, 2014. [Google Scholar]
- Thompson H. P. G. Ph.D. Thesis, University of Cambridge, 2014. [Google Scholar]
- Williams D. E. J. Comput. Chem. 2001, 22, 1154–1166. 10.1002/jcc.1074. [DOI] [Google Scholar]
- Stone A.; Alderton M. Mol. Phys. 2002, 100, 221–233. 10.1080/00268970110089432. [DOI] [Google Scholar]
- Karfunkel H.; Rohde B.; Leusen F.; Gdanitz R.; Rihs G. J. Comput. Chem. 1993, 14, 1125–1135. 10.1002/jcc.540141002. [DOI] [Google Scholar]
- Motherwell S.; Chrisholm J. J. Appl. Crystallogr. 2005, 38, 228–231. 10.1107/S0021889804027074. [DOI] [Google Scholar]
- Coutsias E. A.; Seok C.; Dill K. A. J. Comput. Chem. 2004, 25, 1849–1857. 10.1002/jcc.20110. [DOI] [PubMed] [Google Scholar]
- Grosse-Kunstleve R. W.; Sauter N. K.; Adams P. D. Acta Crystallogr., Sect. A: Found. Crystallogr. 2004, 60, 1–6. 10.1107/S010876730302186X. [DOI] [PubMed] [Google Scholar]
- Grosse-Kunstleve R. W.; Sauter N. K.; Moriarty N. W.; Adams P. D. J. Appl. Crystallogr. 2002, 35, 126–136. 10.1107/S0021889801017824. [DOI] [Google Scholar]
- Massen C. P.; Doye J. P. K. Phys. Rev. E 2007, 75, 037101. 10.1103/PhysRevE.75.037101. [DOI] [PubMed] [Google Scholar]
- Day G. M.; Motherwell W. D. S.; Jones W. Phys. Chem. Chem. Phys. 2007, 9, 1693–1704. 10.1039/b612190j. [DOI] [PubMed] [Google Scholar]
- Oswald I. D. H.; Allan D. R.; Day G. M.; Motherwell W. D. S.; Parsons S. Cryst. Growth Des. 2005, 5, 1055–1071. 10.1021/cg049647b. [DOI] [Google Scholar]
- van de Streek J.; Neumann M. A. CrystEngComm 2011, 13, 7135–7142. 10.1039/c1ce05881a. [DOI] [Google Scholar]
- Reilly A. M.; Tkatchenko A. J. Chem. Phys. 2013, 139, 024705. 10.1063/1.4812819. [DOI] [PubMed] [Google Scholar]
- Desiraju G. R. CrystEngComm 2007, 9, 91–92. 10.1039/B614933B. [DOI] [Google Scholar]
- Paulus E. F.; Leusen F. J. J.; Schmidt M. U. CrystEngComm 2007, 9, 131–143. 10.1039/B613059C. [DOI] [Google Scholar]
- Totton T. S.; Misquitta A. J.; Kraft M. J. Chem. Theory Comput. 2010, 6, 683–695. 10.1021/ct9004883. [DOI] [PubMed] [Google Scholar]
- Stone A. J. J. Chem. Theory Comput. 2005, 1, 1128–1132. 10.1021/ct050190+. [DOI] [PubMed] [Google Scholar]
- Grimme S. Angew. Chem., Int. Ed. 2008, 47, 3430–3434. 10.1002/anie.200705157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.