

Abstract
The Pseudomonas aeruginosa type III secretion system (T3SS) is a primary virulence determinant and a potential target for antivirulence drugs. One candidate target is ExsA, a member of the AraC family of DNA-binding proteins required for expression of the T3SS. A previous study identified small molecules based on an N-hydroxybenzimidazole scaffold that inhibit the DNA-binding activity of several AraC proteins, including ExsA. In this study, we further characterized a panel of N-hydroxybenzimidazoles. The half-maximal inhibitory concentrations (IC50s) for the tested N-hydroxybenzimidazoles ranged from 8 to 45 μM in DNA-binding assays. Each of the N-hydroxybenzimidazoles protected mammalian cells from T3SS-dependent cytotoxicity, and protection correlated with reduced T3SS gene expression in a coculture infection model. Binding studies with the purified ExsA DNA-binding domain (i.e., lacking the amino-terminal self-association domain) confirmed that the activity of N-hydroxybenzimidazoles results from interactions with the DNA-binding domain. The interaction is specific, as an unrelated DNA-binding protein (Vfr) was unaffected by N-hydroxybenzimidazoles. ExsA homologs that control T3SS gene expression in Yersinia pestis, Aeromonas hydrophila, and Vibrio parahaemolyticus were also sensitive to N-hydroxybenzimidazoles. Although ExsA and Y. pestis LcrF share 79% sequence identity in the DNA-binding domain, differential sensitivities to several of the N-hydroxybenzimidazoles were observed. Site-directed mutagenesis based on in silico docking of inhibitors to the DNA-binding domain, and on amino acid differences between ExsA and LcrF, resulted in the identification of several substitutions that altered the sensitivity of ExsA to N-hydroxybenzimidazoles. Development of second-generation compounds targeted to the same binding pocket could lead to drugs with improved pharmacological properties.
INTRODUCTION
ESKAPE pathogens (Enterococcus faecalis, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) are a group of antibiotic-resistant, nosocomial pathogens identified as the most common causes of hospital-acquired infections (1–3). Among them, P. aeruginosa accounted for 7.5% of all hospital-acquired infections in 2009 to 2010 (4). P. aeruginosa infections are particularly challenging to treat due to a large number of intrinsic, acquired, and mutational mechanisms conferring antibiotic resistance (5). Clinical data from more than 200 U.S. hospitals showed that P. aeruginosa strains causing bloodstream infections and pneumonia had multidrug resistance (resistance to ≥3 drug classes) rates of 15% and 22%, respectively (6). Furthermore, multidrug resistance is associated with a >2-fold-increased risk for in-hospital mortality (7). Alternative therapeutic approaches are required as the range of effective antibiotics narrows. Antivirulence drugs are one promising approach. Rather than targeting an essential cellular process, antivirulence drugs target an essential pathogen-specific virulence function. In theory, antivirulence drugs could disrupt the expression, assembly, secretion, or activity of a virulence determinant.
Several antivirulence candidates target the P. aeruginosa type III secretion system (T3SS) (8–18). The T3SS is a primary virulence determinant of P. aeruginosa that functions by translocating effector proteins into host cells. The effector proteins possess antihost properties important for phagocytic avoidance and systemic spread of the organism (19). The T3SS regulon consists of ∼40 genes that encode the secretion and translocation machinery, regulatory factors, effectors, and effector-specific chaperones (20). The genes are organized within 10 transcriptional units, and each is under the direct transcriptional control of ExsA. Strains lacking exsA show a complete lack of T3SS gene expression and are significantly attenuated for T3SS-dependent cytotoxicity toward cultured mammalian cells and virulence in murine models of pneumonia (17, 21). ExsA-dependent expression of T3SS genes is induced under low-Ca2+ conditions or upon contact of P. aeruginosa with host cells (20). Both signals convert the assembled but inactive secretion machinery into a secretion-competent form through a poorly defined mechanism (22, 23). ExsA activity is intimately coupled to secretion by a partner-switching mechanism. The partner-switching mechanism involves three proteins in addition to ExsA: ExsC, ExsD, and ExsE. Both ExsC and ExsD have two potential binding partners. ExsD is an anti-activator that binds to the NTD of ExsA to form a 1:1 stoichiometric complex that inhibits both ExsA self-association and DNA-binding activity (22, 24, 25). ExsC forms a 2:2 stoichiometric complex with ExsD and functions as an anti-anti-activator (26). ExsC is also a T3SS chaperone and forms a 2:1 complex with ExsE (27–29). The ExsC-ExsE complex prevents ExsC from associating with ExsD (24). The current working model is that ExsA-dependent transcription is inactive under nonpermissive conditions (i.e., high Ca2+) because the binding equilibria favor formation of the inhibitory ExsD-ExsA and ExsC-ExsE complexes. The equilibria are altered under inducing conditions due to secretion and/or translocation of ExsE (27, 28, 30). The resulting decrease in the intracellular concentration of ExsE favors formation of the ExsD-ExsC complex (i.e., partner switching), thereby releasing ExsA to activate transcription.
N-Hydroxybenzimidazoles are a class of antivirulence compounds that inhibit the DNA-binding activities of some AraC proteins, including ExsA (31–33). AraC proteins represent a large family of transcriptional activators with important roles in virulence gene regulation, metabolism, and antibiotic resistance (34). The defining feature of AraC proteins is a conserved 99-amino-acid domain containing two helix-turn-helix DNA-binding motifs. The N-hydroxybenzimidazoles were first identified using an in silico screen for small molecules that interact with the DNA-binding domains of MarA and Rob, both AraC family proteins from E. coli (31). Following initial identification of lead compounds, in vitro experiments performed with the AraC family member SoxS confirmed the in silico prediction that N-hydroxybenzimidazoles interfere with DNA-binding activity, and several compounds with 50% inhibitory concentrations (IC50s) in the low micromolar range were identified. One of those compounds demonstrated activity against six AraC family proteins, including ExsA (31). N-Hydroxybenzimidazoles were specifically selected from the in silico analyses as a scaffold for further development based on their potential for chemical diversity (31). Subsequent studies led to the identification of several N-hydroxybenzimidazole analogs with increased activity against ExsA (32).
In the present study, we examined a panel of these inhibitors to better define their interaction with ExsA. The inhibitory activities of these compounds were confirmed by their abilities to decrease T3SS-mediated cytotoxicity toward Chinese hamster ovary (CHO) cells in vitro. Inhibition of T3SS activity occurs at the transcriptional level, and inhibition of T3SS gene expression as measured by a green fluorescent protein (GFP) reporter is decreased in the presence of each inhibitor. A putative N-hydroxybenzimidazole binding pocket is located in the ExsA DNA-binding domain. Amino acid substitutions in this pocket resulted in altered sensitivity to N-hydroxybenzimidazole inhibitors. The ability of these compounds to inhibit ExsA activity and DNA binding by several other T3SS activators (ExsA homologs in Yersinia pestis, Vibrio parahaemolyticus, and Aeromonas hydrophila) further demonstrates their ability to exert broad-spectrum activity as antivirulence therapeutics.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The bacterial strains used in this study are listed in Table S1 in the supplemental material. Escherichia coli strain DH5α was used for routine cloning and maintained on LB-Lennox (LB) agar plates with gentamicin (15 μg/ml) or ampicillin (100 μg/ml) as appropriate. E. coli strain Tuner (DE3) was used for protein purification and maintained on LB agar with ampicillin (100 μg/ml). P. aeruginosa strain PA103 and derivatives thereof were maintained on Vogel-Bonner minimal medium (VBM) with gentamicin (100 μg/ml) as necessary. The N-hydroxybenzimidazoles used in this study were previously reported (compounds 4816, 5330, 5631, and 5707) or obtained as a gift (compounds 5784 and 5816) from Paratek Pharmaceuticals (32, 33).
Plasmid construction and mutagenesis.
Expression vectors for purification of ExsAHis, LcrFHis, and VfrHis with amino-terminal 10× histidine tags were described previously (35–37). Expression vectors for AscAHis and ExsAVp-His carrying N-terminal 10× histidine tags were constructed by the Gibson Assembly method (New England BioLabs) using the PCR products indicated in Tables S1 and S2 in the supplemental material and pET16b digested with NdeI and BamHI as the destination vector. Expression vectors for the carboxy-terminal domains (ExsA-CTDHis and ExsAVp-CTDHis) were constructed in the same manner. Site-directed mutagenesis was also performed using Gibson Assembly. The pEB124 template was used to amplify two overlapping fragments containing the desired substitution, which were joined with the appropriate precut vector by incubation in a Gibson Assembly reaction. The ExsA and LcrF mutant expression vectors pAM196, pAM202-217, and pAM231 were constructed in the same manner (using primer pairs listed in Table S2) and joined with the pET16b vector.
Protein expression and purification.
E. coli Tuner (DE3) transformed with the histidine-tagged protein expression vectors was cultured overnight at 37°C on LB agar containing ampicillin (100 μg/ml) and then used to inoculate 100 ml of LB containing ampicillin (100 μg/ml) to an initial A600 of 0.1. The culture was grown shaking at 25°C until the A600 reached 0.5. Additional ampicillin was added to a final concentration of 300 μg/ml, and expression was induced by addition of 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After a 2-h incubation at 25°C, cells were harvested by centrifugation (10 min at 6,000 × g and 4°C) and resuspended in 10 ml of ExsA binding buffer (20 mM Tris-HCl [pH 7.9], 500 mM NaCl, 0.5% Tween 20, 20 mM imidazole, and 1 protease inhibitor cocktail tablet [complete mini, EDTA-free protease inhibitor cocktail; Roche Applied Science]). Cells were lysed by passage through a Microfluidizer (Microfluidics, Newton, MA). Lysates were cleared by two rounds of centrifugation (10 min at 20,000 × g and 4°C) and subjected to purification by Talon metal affinity resin (Clontech Laboratories). Briefly, 200 μl of packed Talon metal affinity resin was equilibrated with ExsA binding buffer. Cleared lysates were incubated with the resin for 20 min at 4°C with slow agitation. The unbound fraction was collected by removing the supernatant following centrifugation at 700 × g for 0.5 min. The resin was washed 3 times with 5 ml of ExsA binding buffer. His-tagged protein was eluted with 1 ml of ExsA elution buffer (20 mM Tris-HCl [pH 7.9], 500 mM NaCl, 0.5% Tween 20, and 500 mM imidazole) and collected in the supernatant fraction following centrifugation at 700 × g for 0.5 min to sediment the Talon metal affinity resin. After elution from the Talon metal affinity resin, fractions were analyzed on SDS-PAGE gels and stained with Coomassie brilliant blue. The eluate was dialyzed overnight in 4 liters of ExsA binding buffer (excluding imidazole) with 1 mM dithiothreitol (DTT) at 4°C with constant stirring. Protein concentrations were determined with the DC protein assay (Bio-Rad) using bovine serum albumin (BSA) protein standards.
EMSAs.
Probes for the exsC (200 bp), regA (275 bp), and algD (160 bp) promoter regions were amplified by PCR (primer pairs 22963127-49188917, 33075941-33075940, and 85333731-85333730, respectively) and end labeled with 10 μCi of [γ-32P]ATP as previously described (35). Electrophoretic mobility shift assay (EMSA) reaction mixtures containing 100 nM purified protein, ExsA DNA binding buffer (10 mM Tris [pH 7.5], 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 5% glycerol), 25 ng/μl of poly(2′-deoxyinosinic-2′-deoxycytidylic acid), and 100 μg/ml of bovine serum albumin were incubated with each inhibitor (concentrations indicated in the figure legends) in a total volume of 19 μl for 5 min at 25°C. Specific and nonspecific probes (0.05 nM) were added to a total volume of 20 μl and incubated for an additional 15 min at 25°C. Samples were analyzed by electrophoresis on 5% polyacrylamide glycine gels (10 mM Tris [pH 7.5], 380 mM glycine, 1 mM EDTA) at 4°C. Imaging was performed using an FLA-7000 phosphorimager (Fujifilm) and MultiGauge v3.0 software (Fujifilm). EMSA images contain a dashed line to indicate the removal of a lane; all samples were run on the same gel.
Flow cytometry.
P. aeruginosa strains carrying a green fluorescent protein (GFP) transcriptional reporter (PexoS-gfp) were assayed as follows. Cells were cultured overnight on VBM agar with gentamicin (100 μg/ml) at 37°C. The next day, bacteria were suspended in prewarmed Ham's F12 tissue culture medium at 1 × 106 CFU/ml and treated with the desired N-hydroxybenzimidazole (125 μM) or dimethyl sulfoxide (DMSO) (2.5%) for 15 min at 25°C. After pretreatment with the inhibitors, 1 ml of the bacterial suspension was added to each well of a 24-well plate containing 1 × 105 CHO cells (multiplicity of infection [MOI], 10:1). Plates were centrifuged at 500 × g for 5 min, and the infection was allowed to proceed for 4 h at 37°C. After incubation, the growth medium was aspirated, and nonadherent cells were removed by washing 3 times with 1 ml of phosphate-buffered saline (PBS). CHO cells and adherent P. aeruginosa were collected following a 5-min incubation with 1 ml of 0.1% Triton X-100 in PBS while nutating at 25°C, followed by a 5-min incubation in a sonicating water bath. GFP fluorescence was measured by counting 10,000 bacteria per sample on a Becton Dickinson LSR II at the University of Iowa Flow Cytometry Facility.
LDH release assays.
PA103 and PA103 ΔexsA strains were grown overnight on VBM agar. The following day, cells were suspended in Ham's F12 tissue culture medium at 1 × 106 CFU/ml and treated with 2.5% DMSO or N-hydroxybenzimidazoles as indicated in the figure legends for 15 min at 25°C. CHO cells seeded in a 96-well plate at 2 × 104 cells/well were washed once with 200 μl of Ham's F12 medium and cocultured with 200 μl of the bacterial suspension (MOI, 10:1). Plates were centrifuged at 500 × g for 5 min prior to incubation at 37°C. After a 90-min incubation, plates were centrifuged at 500 × g for 5 min, and lactate dehydrogenase (LDH) release was measured using the CytoTox 96 nonradioactive cytotoxicity assay (Promega). As a positive control, the kit-provided bovine heart LDH enzyme was diluted 1:5,000 in 10 ml of PBS with 1% BSA.
Transcriptional reporter assays.
Transcriptional reporter activity was assayed as follows. P. aeruginosa strains were grown overnight at 37°C on VBM agar with gentamicin (100 μg/ml for P. aeruginosa or 15 μg/ml for E. coli) as necessary. PA103 exsA::Ω miniPexsC-lacZ, PA103 exsA::Ω miniPexoT-lacZ, or GS162 miniPexoT-lacZ carrying an exsA expression vector (pEB124) was diluted to an A600 of 0.1 in Trypticase soy broth (TSB) with either 2.5% DMSO or a 125 μM concentration of the desired N-hydroxybenzimidazole and 0.1% arabinose to induce exsA expression. Cultures were grown with shaking at 30°C until the A600 reached 1.0, and β-galactosidase activity was assayed as previously described with the substrate 2-nitrophenyl-β-d-galactopyranoside (ONPG) (26). Miller units were calculated and are reported as the averages from at least three replicates, with error bars in figures representing the standard errors of the means (SEM). The statistical significance of experimental findings was determined by one-way analysis of variance (ANOVA) using Graphpad Prism version 5.0c for Mac OS X (GraphPad Software, La Jolla, CA).
RESULTS
N-Hydroxybenzimidazoles interact with the carboxy-terminal domain of ExsA to inhibit binding to the PexsC promoter.
Several N-hydroxybenzimidazoles that inhibit DNA binding by SoxS also have activity against other AraC family proteins, including ExsA (31). Several structurally related N-hydroxybenzimidazoles (Fig. 1) were tested for inhibition of DNA binding by ExsA using electrophoretic mobility shift assays (EMSAs) (31–33). Reactions were performed by incubating purified histidine-tagged ExsA with each N-hydroxybenzimidazole for 5 min prior to addition of the radiolabeled PexsC promoter probe, a previously characterized binding target of ExsA (35). After an additional 15 min, the samples were subjected to nondenaturing polyacrylamide gel electrophoresis and phosphorimaging. We previously reported that ExsA preferentially forms a complex referred to as shift product 2 when bound to the PexsC promoter probe. Shift product 2 represents the cooperative binding of two ExsA monomers to two distinct sites on the promoter probe (Fig. 2A, lane 2) (35). While ExsA treated with the vehicle alone (DMSO) (Fig. 2A, lane 3) had no effect on binding to the PexsC probe, each of the N-hydroxybenzimidazoles inhibited ExsA-PexsC complex formation to some degree (Fig. 2A, lanes 4 to 9). To quantitatively measure the inhibitory activities of the compounds, the half-maximal inhibitory concentration (IC50) for each N-hydroxybenzimidazole was determined by titration experiments. A representative example of ExsA binding to the PexsC probe in the presence of increasing concentrations of compound 5707 is shown in Fig. 2B and C. Compound 5631 was the most active inhibitor, with an IC50 of 8 μM, while the remaining compounds had IC50s ranging from 16 to 45 μM (Table 1). Inhibition by compounds 5631 and 5707 was reported previously (3 μM and 13 μM, respectively) and is comparable to the activity observed in our experiments (32).
FIG 1.
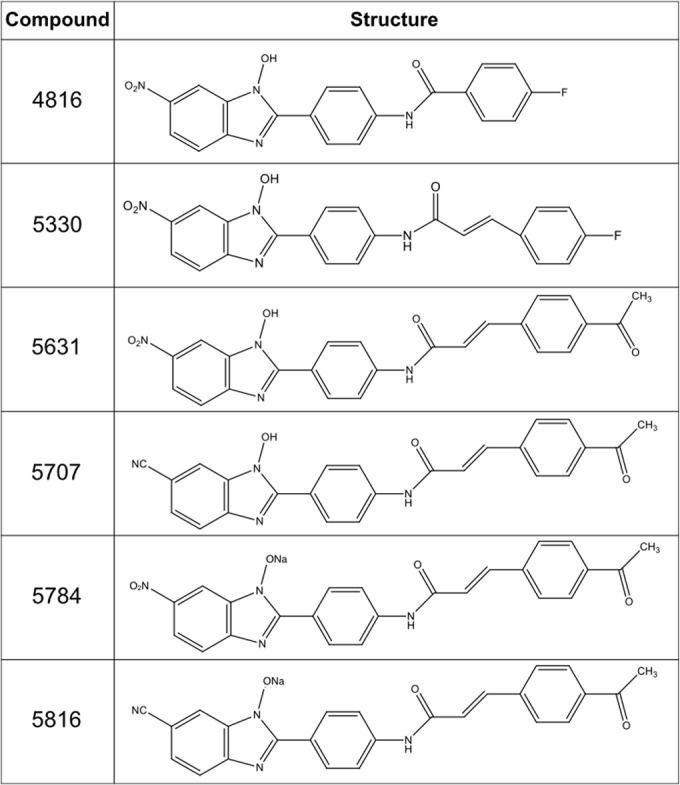
Structures of the N-hydroxybenzimidazoles used in this study. Note that compound 5784 is the sodium salt of compound 5631 and that compound 5816 is the sodium salt of compound 5707.
FIG 2.
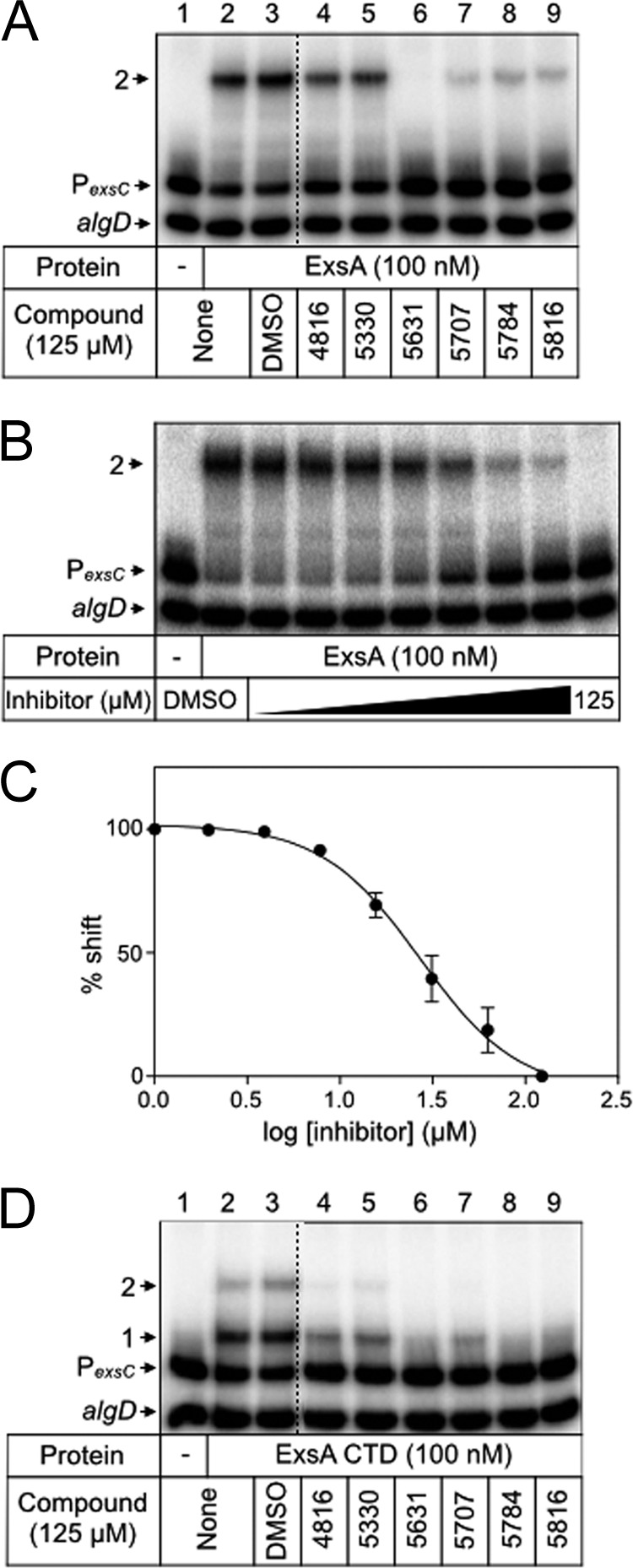
Inhibition of ExsA-DNA binding by N-hydroxybenzimidazoles. ExsA (100 nM) (A and B) or ExsACTD (100 nM) (D) was incubated with DMSO (2.5%) or the indicated N-hydroxybenzimidazole (125 μM) for 5 min prior to addition of an equimolar mixture of radiolabeled PexsC and nonspecific algD DNA probes (0.05 nM each). Binding reactions were allowed to proceed for 15 min at 25°C and then analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. The positions of shift products 1 and 2 are indicated. (C) Representative titration experiment used to determine the half-maximal inhibitory concentration (IC50) of each N-hydroxybenzimidazole required to inhibit ExsA-DNA binding. The percent shifted probe (y axis) was plotted as a function of N-hydroxybenzimidazole concentration (x axis).
TABLE 1.
IC50s of N-hydroxybenzimidazoles for ExsA and LcrF binding to the PexsC promoter probe
| N-Hydroxybenzimidazole | ExsA |
LcrF |
||
|---|---|---|---|---|
| Log IC50 (μM) | SE | Log IC50 (μM) | SE | |
| 4816 | 1.61 | 0.04 | 1.66 | 0.01 |
| 5330 | 1.65 | 0.03 | 1.63 | 0.03 |
| 5631 | 0.93 | 0.02 | 1.20 | 0.06 |
| 5707 | 1.40 | 0.03 | 2.63 | 0.08 |
| 5784 | 1.19 | 0.06 | 1.53 | 0.02 |
| 5816 | 1.20 | 0.03 | 2.61 | 0.11 |
N-Hydroxybenzimidazoles were originally identified through in silico docking simulations with MarA and Rob (31). Although most AraC family members have an amino-terminal domain involved in self-association and/or ligand binding, MarA is devoid of an amino-terminal domain and composed solely of a DNA-binding domain. It is reasonably assumed, therefore, that the inhibitory activity of the N-hydroxybenzimidazoles occurs through interactions with the DNA-binding domain, even for proteins with amino-terminal domains, including ExsA. To confirm this assumption, we tested the activities of the N-hydroxybenzimidazoles against the isolated DNA-binding domain from ExsA (carboxy-terminal residues 159 to 278), here referred to as ExsACTD (38). Whereas full-length ExsA preferentially forms shift product 2 upon binding to the PexsC promoter probe (Fig. 2A lane 2), ExsACTD more readily forms shift product 1 (i.e., binding of an ExsA monomer to a single site), owing to a lack of self-association between the ExsA monomers (Fig. 2D, lane 2) (39). Treatment of ExsACTD with each of the N-hydroxybenzimidazoles resulted in decreased formation of shift products 1 and 2 (Fig. 2D, lanes 4 to 9). These data demonstrate that the primary determinant for inhibition by N-hydroxybenzimidazoles is located in the DNA-binding domain of ExsA.
N-Hydroxybenzimidazoles inhibit ExsA-dependent gene expression.
Each of the tested N-hydroxybenzimidazoles inhibits the DNA-binding activity of ExsA in vitro, leading to the hypothesis that ExsA-dependent gene expression would also be affected by these compounds. To rule out the possibility that N-hydroxybenzimidazoles affect growth of P. aeruginosa, and therefore gene expression, bacterial growth following N-hydroxybenzimidazole treatment was assayed. Wild-type P. aeruginosa grown overnight on VBM agar was suspended at an A600 of 0.001 in 5 ml of LB with each N-hydroxybenzimidazole (125 μM) or DMSO (2.5%) and grown to an A600 of 1.0 at 37°C with shaking. Compared to that of untreated P. aeruginosa, growth was not significantly affected by treatment with DMSO or any of the N-hydroxybenzimidazoles (see Fig. S1 in the supplemental material).
To determine whether N-hydroxybenzimidazoles affect ExsA-dependent gene expression, we tested the ability of each compound to reduce expression of the ExsA-dependent PexoS promoter fused to the gene encoding green fluorescent protein (PexoS-gfp reporter). For this assay, contact with CHO cells provided the inducing signal for T3SS gene expression. To minimize toxicity to CHO cells, we used a strain lacking the known T3SS effectors in strain PA103 (PA103 ΔexoU, exoT::Tc). P. aeruginosa was treated with each inhibitor for 15 min prior to incubation with CHO cells for 4 h at 37°C. Cocultures were washed, treated with a mild detergent, and water bath sonicated to detach adherent P. aeruginosa. GFP production by adherent P. aeruginosa was measured by flow cytometry. Whereas an exsA mutant demonstrated no activation of the GFP reporter following the 4-h coculture (see Fig. S2 in the supplemental material), approximately 80% of untreated PA103 ΔexoU exoT::Tc cells responded to host cell contact and activated transcription of the GFP reporter (Fig. 3; see also Fig. S2). While treatment with DMSO resulted in a small decrease in GFP expression (72.5% of the population expressed GFP) compared to that of untreated cells, treatment with each of the N-hydroxybenzimidazoles resulted in reporter activity reductions of approximately 50% compared to that in DMSO-treated cells (Fig. 3). Although statistically significant, inhibition by the N-hydroxybenzimidazoles was not complete, potentially indicating partial resistance by efflux, catabolism, or some other mechanism.
FIG 3.
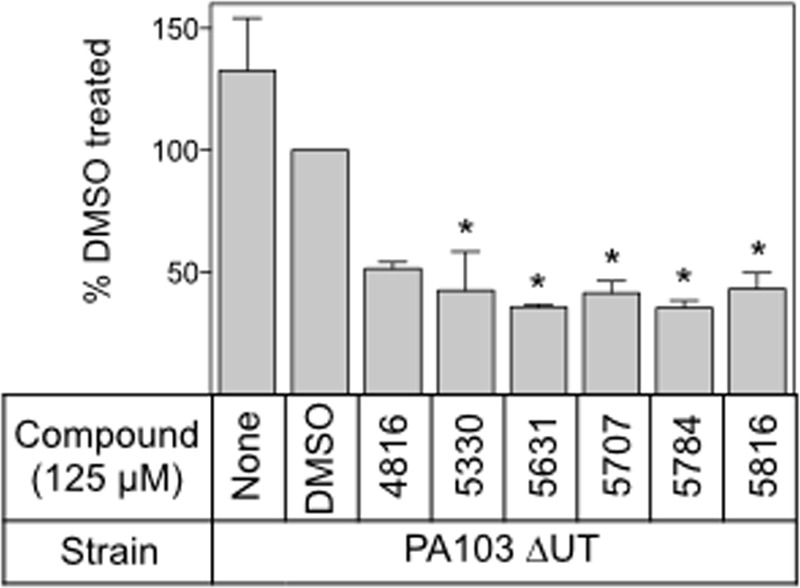
N-Hydroxybenzimidazoles inhibit ExsA-dependent gene expression. Shown are representative data from an exoU exoT mutant carrying a GFP transcriptional reporter (PA103 ΔexoU exoT::Tc PexoS-gfp) incubated with DMSO (2.5%) or the indicated N-hydroxybenzimidazole (125 μM) for 15 min prior to incubation with CHO cells for 4 h at 37°C. Reporter activity was measured by flow cytometry and is reported as the percentage of the total bacterial cell population that was GFP positive relative to that of DMSO-treated cells normalized to 100%. *, P < 0.01.
N-Hydroxybenzimidazoles inhibit ExsA-dependent cytotoxicity toward CHO cells.
To determine whether N-hydroxybenzimidazoles inhibit ExsA-dependent cytotoxicity, we examined their ability to protect CHO cells from lysis using lactate dehydrogenase (LDH) release assays. As a control, we first tested the panel of N-hydroxybenzimidazoles for direct inhibition of lactate dehydrogenase activity using the purified LDH enzyme. Neither DMSO nor the N-hydroxybenzimidazoles altered LDH activity (see Fig. S3 in the supplemental material).
To examine protection of CHO cells, wild-type strain PA103 was preincubated with the N-hydroxybenzimidazoles (125 μM) for 15 min prior to coculture with CHO cells at an MOI of 10 for 90 min. CHO cell lysis was quantified by measuring LDH release. Under these conditions, LDH release is dependent upon expression of the T3SS (compare LDH release for uninfected CHO cells and cells infected with the ΔexsA strain to that of cells infected with wild-type PA103 in Fig. 4). Compared to that in untreated cells or cells treated with the vehicle alone, treatment with each of the N-hydroxybenzimidazoles resulted in a significant reduction in LDH release by CHO cells infected with PA103 (Fig. 4).
FIG 4.
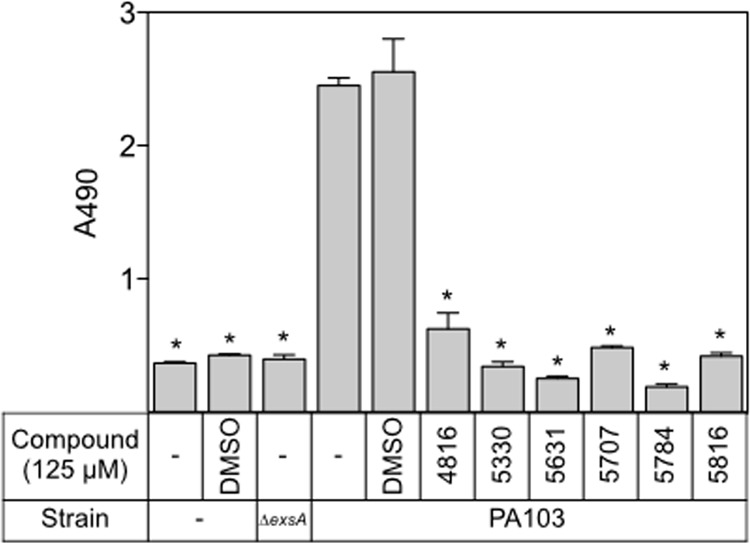
N-Hydroxybenzimidazoles inhibit T3SS-mediated cytotoxicity. PA103 was incubated with DMSO (2.5%) or the indicated N-hydroxybenzimidazole (125 μM) for 15 min at 25°C prior to incubation with CHO cells for 90 min at 37°C. CHO cell lysis was assayed by measuring LDH release. Statistical differences were determined by comparison to DMSO treatment. *, P < 0.01.
N-Hydroxybenzimidazoles do not inhibit ExsA-dependent transcription in broth-grown cultures.
We next examined the activity of ExsA-dependent transcriptional reporters (PexsC-lacZ and/or PexoT-lacZ) integrated at the CTX phage attachment site in cells grown in broth culture. Only compounds 5330 and 5784 significantly inhibited PexsC-lacZ reporter activity, and none of the N-hydroxybenzimidazoles inhibited PexoT-lacZ reporter activity (see Fig. S4A and B in the supplemental material). Because P. aeruginosa has a large repertoire of efflux pumps and catabolic pathways that might interfere with the activity of the N-hydroxybenzimidazoles, we switched to a heterologous system in E. coli strain GS162 with a PexoT-lacZ reporter integrated into the chromosome. Expression of the PexoT-lacZ reporter in E. coli requires introduction of an exsA expression plasmid. Compared to treatment with DMSO, however, the N-hydroxybenzimidazoles still failed to significantly inhibit reporter activity (see Fig. S4C).
N-Hydroxybenzimidazoles inhibit DNA binding by ExsA homologs.
We previously found that ExsA homologs from Yersinia pestis (LcrF), Aeromonas hydrophila (AscA), Photorhabdus luminescens (LscA), and Vibrio parahaemolyticus (ExsA, here referred to as ExsAVp) complement a P. aeruginosa exsA mutant for T3SS gene expression when expressed in trans (35, 36). The DNA-binding domains of AscA, LscA, LcrF, and ExsAVp show 90%, 87%, 79%, and 67% identity to the corresponding region in ExsA (residues 159 to 278), respectively (Fig. 5A). Although these homologs share a high degree of sequence identity with ExsA, there are some differences in the sequence of recognition helix 1 (an α-helix known to mediate contacts between ExsA and promoter DNA) and several other regions throughout this domain (40). We hypothesized that each homolog might show differential responses to the panel of N-hydroxybenzimidazoles and that those differences could provide insight into the N-hydroxybenzimidazole interaction site. Each homolog was expressed and purified as a histidine-tagged fusion protein (LscA was insoluble and omitted from the analyses). The proteins were treated with the panel of N-hydroxybenzimidazoles for 5 min prior to incubation with the PexsC promoter probe. Like for ExsA, binding of LcrF and ExsAVp to the PexsC promoter probe resulted in the predominant formation of shift product 2 (Fig. 5B, C, and E, lanes 2). LcrF was much less sensitive to compounds 5707 and 5816 (5816 is a sodium salt of 5707) than was ExsA (Fig. 5C, lanes 7 and 9). Incubation of AscA with the PexsC promoter probe resulted in formation not of a distinct band but rather a broad, diffuse band likely representing an unstable complex (Fig. 5D, lane 2). Nevertheless, AscA binding is specific to PexsC, as shifting of the nonspecific control probe was not observed. ExsAVp was strongly inhibited by all the N-hydroxybenzimidazoles tested (Fig. 5E, lanes 4 to 9). The ability of the N-hydroxybenzimidazoles to affect DNA binding by each ExsA homolog suggested that the interaction occurs through a conserved element, while differences in sensitivity toward these compounds indicates that the binding determinants display some variability between homologs. To further confirm that the inhibitory effect of the N-hydroxybenzimidazoles was specific to the DNA-binding domain, ExsAVp-CTD was purified and examined for DNA binding in the presence of each inhibitor. As expected, the N-hydroxybenzimidazoles effectively inhibited DNA binding by ExsAVp-CTD (Fig. 6A).
FIG 5.

N-Hydroxybenzimidazoles inhibit the DNA-binding activity of other AraC family proteins. (A) Amino acid sequence alignment of the ExsA DNA-binding domain and homologs in Vibrio parahaemolyticus (ExsAVp), Yersinia pestis (LcrF), Aeromonas hydrophila (AscA), and Photorhabdus luminescens (LscA). Bold lines above the sequences indicate the recognition helices, RH1 and RH2. Boxes outline amino acids that were mutagenized in ExsA. ExsA (B), LcrF (C), AscA (D), and ExsAVp (E) (100 nM) were incubated with DMSO (2.5%) or the indicated N-hydroxybenzimidazole (125 μM) for 5 min prior to addition of radiolabeled specific (PexsC) and nonspecific (algD) probes (0.05 nM each). Binding reactions were allowed to proceed for 15 min at 25°C and then analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. The positions of unshifted probes and shifted protein-DNA complexes are indicated. Quantitation of the percent shift normalized to DMSO-treated protein (100%) is indicated, and statistical differences were determined by comparison to DMSO treatment. *, P < 0.01.
FIG 6.
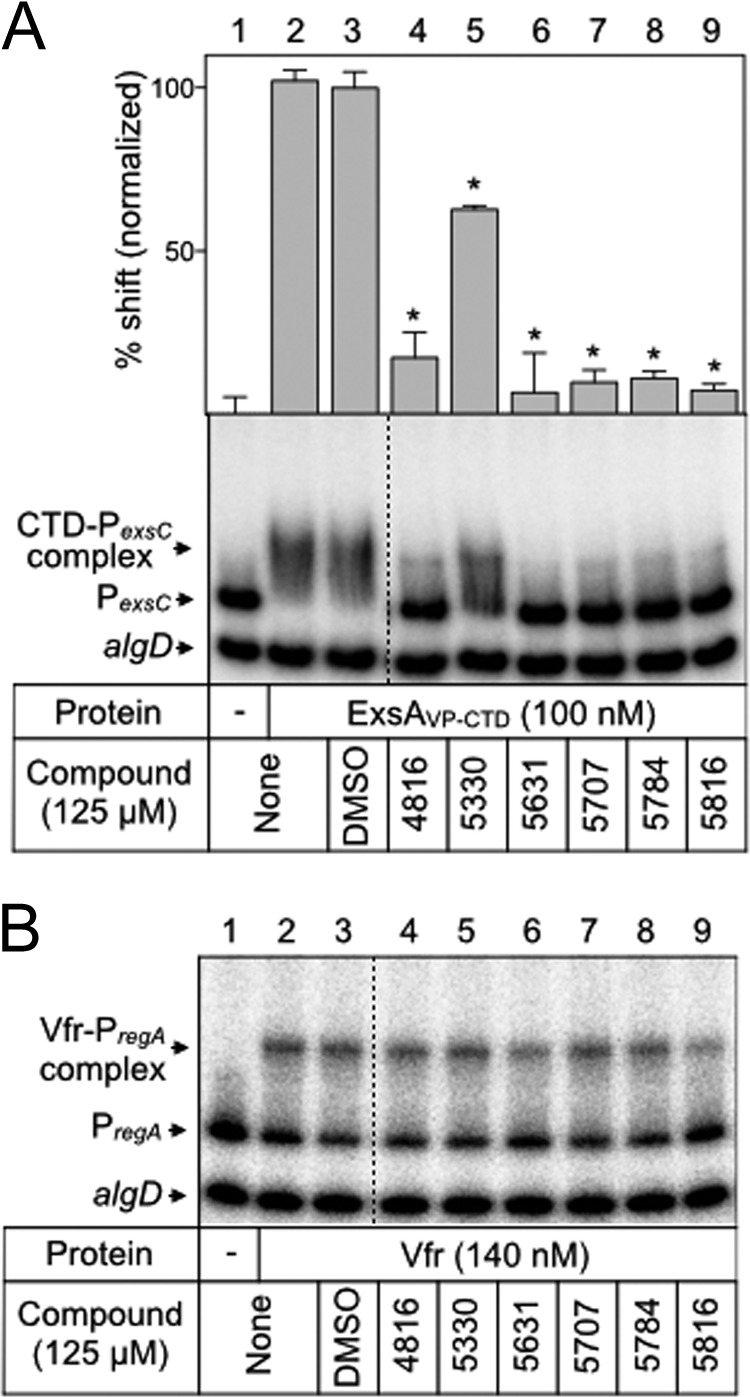
N-Hydroxybenzimidazoles specifically inhibit the DNA-binding activity of AraC family regulators. ExsAVp-CTD (A) and Vfr (B) were incubated with DMSO (2.5%) or the indicated N-hydroxybenzimidazole (125 μM) for 5 min prior to addition of radiolabeled specific (PexsC or PregA) and nonspecific (algD) probes (0.05 nM each). Binding reactions were allowed to proceed for 15 min at 25°C and then analyzed by native polyacrylamide gel electrophoresis and phosphorimaging. The positions of unshifted probes and shifted protein-DNA complexes are indicated. Quantitation of the percent shift normalized to DMSO-treated ExsAVp-CTD (100%) is indicated. Statistical differences were determined by comparison to DMSO treatment. *, P < 0.01.
The above-described findings are consistent with previous reports that N-hydroxybenzimidazoles have broad-spectrum activity against AraC proteins (31). As a further test of specificity, we examined inhibition of Vfr DNA binding by the N-hydroxybenzimidazoles. Vfr is a member of the CRP family of transcription regulators and distinct from the AraC family. Purified Vfr was treated with each N-hydroxybenzimidazole prior to incubation with a PregA promoter probe, a known Vfr target (41). Inhibition of Vfr DNA binding was not observed, supporting the conclusion that N-hydroxybenzimidazoles inhibit DNA binding by interacting specifically with the DNA-binding domain of ExsA and its homologs (Fig. 6B).
Comparison of ExsA and LcrF residues involved in N-hydroxybenzimidazole binding.
One striking observation from the ExsA homolog studies was the difference between ExsA and LcrF sensitivities to compounds 5707 and 5816, despite the high degree of similarity between these proteins. To confirm this observation, we determined the IC50s for each N-hydroxybenzimidazole compound against LcrF as described earlier for ExsA. Compared to ExsA, compounds 5707 and 5816 were at least 5- and 7-fold less effective inhibitors of LcrF DNA-binding activity, with IC50s of >125 μM (Table 1). The remaining N-hydroxybenzimidazoles had similar IC50s against ExsA and LcrF.
The DNA-binding domain of AraC family proteins consists of two helix-turn-helix motifs, and each motif contains a recognition helix that makes base-specific contacts with the DNA target site. The ExsA recognition helices encompass residues L198 to Y209 and Q248 to F259 (Fig. 5A) (40). Within these regions, ExsA and LcrF differ by only one amino acid (S207 in ExsA). If the N-hydroxybenzimidazoles inhibit DNA binding by interacting with or near the recognition helices, then a substitution at S207 in ExsA might render ExsA less sensitive to compounds 5707 and 5816. To test this hypothesis, the DNA-binding activity of purified ExsAS207T in the presence of each N-hydroxybenzimidazole was examined by EMSA as previously described (Fig. 7B). Compared to that of wild-type ExsA, compounds 5707 and 5816 had reduced activity against ExsAS207T (Fig. 7A and B; lanes 7 and 9). As compound 5816 seemed to be the most affected by this substitution, the IC50 for this compound was determined by a titration experiment as previously described for wild-type ExsA (see Fig. S5 in the supplemental material). The calculated IC50 of 208 μM confirmed that there was a reduction in the inhibitory activity of compound 5816 for the ExsAS207T mutant (compared to an IC50 of 14 μM for 5816 against wild-type ExsA as observed in a side-by-side titration experiment). The corresponding mutation (T203S) was made in LcrF to determine whether this substitution would have the opposite effect on N-hydroxybenzimidazole activity. However, only slight differences were observed in LcrFT203S-PexsC complex formation in the presence of N-hydroxybenzimidazoles compared to wild-type LcrF (Fig. 7C and D, lanes 4 to 9). The finding that the S207T substitution in ExsA, but not the corresponding T203S substitution in LcrF, significantly affects N-hydroxybenzimidazole activity suggests that other regions of the DNA-binding domain contribute to the N-hydroxybenzimidazole interaction.
FIG 7.

Amino acid substitution S207T in recognition helix 1 alters ExsA-DNA binding in the presence of several N-hydroxybenzimidazoles. ExsA (A), ExsAS207T (B), LcrF (C), and LcrFT203S (D) (100 nM) was incubated with DMSO (2.5%) or each N-hydroxybenzimidazole (125 μM) for 5 min before incubation with specific (PexsC) and nonspecific (algD) radiolabeled probes (0.05 nM each) for 15 min at 25°C. DNA-binding reactions were visualized by native polyacrylamide gel electrophoresis and phosphorimaging. Protein-DNA complexes and unshifted specific and nonspecific probes are indicated. Quantitation of the percent shift normalized to DMSO-treated protein (100% shift) is indicated. Statistical differences were determined by comparison to DMSO treatment. *, P < 0.01.
As an alternative approach, we used in silico predictions of ligand-binding sites and identified a putative binding pocket located between the recognition helices of ExsA (Fig. 8). To examine the possibility that residues in the predicted ligand-binding site represent an N-hydroxybenzimidazole-binding site, residues in the binding site were replaced with glutamic acid with the expectation that a large residue might impair inhibitor binding. Several substitution mutants were not soluble (M177E, W185E, R221E, and F245E). The remaining mutants were assayed via EMSA for the ability to bind PexsC in the presence of each N-hydroxybenzimidazole. The Y181E, S247E, and Y250E substitution mutants no longer bound promoter DNA, and the effects of these substitutions on inhibitor binding could not be determined (data not shown). Glutamic acid substitutions at residues H180, N183, and I217 reduced the ability of several compounds to inhibit ExsA-PexsC formation (Table 2; see also Fig. S6 in the supplemental material). To further examine the effect of amino acid substitutions in the putative inhibitor-binding site, a second set of residues predicted to surround the perimeter of the binding pocket was selected for alanine mutagenesis. Five substitutions (E184A, K186A, R214A, M241A, and S246A) resulted in altered sensitivities to several inhibitors (Table 2; see also Fig. S6). The P213A substitution mutant remained relatively similar to the wild type. Several attempts to combine single alanine substitutions were made, but the resulting mutant proteins either were insoluble or no longer bound DNA (data not shown). Together, these substitution mutants suggest that residues in the putative binding pocket play a role in N-hydroxybenzimidazole inhibition of ExsA-DNA binding, but whether or not they are involved in direct contact with these compounds is yet to be determined.
FIG 8.

Structural model of a putative inhibitor-binding pocket in the ExsA DNA-binding domain predicted by the FTSite server. The amino acid residues in the predicted binding site that were mutagenized are indicated, and the recognition helices that make base-specific contacts with DNA are shown in blue.
TABLE 2.
Percent of the PexsC promoter probe shifted by wild-type ExsA and the indicated mutants in the presence of the N-hydroxybenzimidazoles
| ExsA | % of probe shifted in the presence of the indicated N-hydroxybenzimidazolea |
|||||
|---|---|---|---|---|---|---|
| 4816 | 5330 | 5631 | 5707 | 5784 | 5816 | |
| Wild type | 34 ± 4 | 60 ± 4 | 8 ± 5 | 3 ± 2 | 7 ± 4 | 7 ± 5 |
| H180E mutant | 45 ± 4 | 50 ± 5 | 11 ± 1 | 4 ± 1 | 9 ± 4 | 32 ± 5* |
| N183E mutant | 40 ± 2 | 59 ± 3 | 15 ± 2 | 51 ± 6* | 45 ± 8* | 41 ± 4* |
| I217E mutant | 26 ± 9 | 71 ± 10 | 2 ± 1 | 35 ± 4* | 69 ± 1* | 2 ± 1 |
| E184A mutant | 3 ± 1* | 59 ± 6 | 7 ± 1 | 33 ± 4* | 28 ± 4* | 5 ± 4 |
| K186A mutant | 55 ± 5 | 76 ± 2 | 15 ± 3 | 30 ± 7* | 16 ± 3 | 42 ± 3* |
| P213A mutant | 13 ± 6 | 59 ± 6 | 3 ± 1 | 2 ± 1 | 2 ± 1 | 3 ± 1 |
| R214A mutant | 2 ± 1* | 39 ± 1 | 2 ± 1 | 3 ± 1 | 38 ± 1* | 2 ± 1 |
| M241A mutant | 63 ± 6* | 51 ± 10 | 2 ± 1 | 6 ± 4 | 71 ± 4* | 63 ± 11* |
| S246A mutant | 28 ± 4 | 72 ± 9 | 2 ± 1 | 13 ± 5* | 25 ± 7 | 29 ± 5* |
The N-hydroxybenzimidazoles were used at a concentration of 125 μM. Each horizontal data set represents the percent of the PexsC promoter probe shifted following treatment with each N-hydroxybenzimidazole relative to an untreated control normalized to 100%. *, P < 0.01.
DISCUSSION
Previous studies identified T3SS inhibitors that target the activity of effector proteins, translocation of effectors, assembly of the secretion apparatus, and gene expression (8–18, 31, 32). The N-hydroxybenzimidazoles inhibit the DNA-binding activity of AraC family proteins, including ExsA (32, 33, 42). The IC50s for the N-hydroxybenzimidazole inhibitors used in this study ranged from 8 to 45 μM for ExsA. Inhibition of DNA-binding activity in vitro correlated with reduced ExsA-dependent gene expression in vivo and a significant decrease in T3SS-dependent cytotoxicity in coculture experiments with Chinese hamster ovary cells. The level of cytotoxicity following N-hydroxybenzimidazole treatment was nearly the same as seen with an exsA mutant, strongly suggesting that the protective effect resulted from inhibition of ExsA. Nevertheless, the potential for N-hydroxybenzimidazoles to exert broad-spectrum activity suggests that other virulence functions controlled by additional AraC family proteins (aside from ExsA) might also contribute to the protective effect. The genome sequence of P. aeruginosa strain PAO1 encodes at least 52 AraC family proteins. Questions regarding specificity and off-target effects will be the subject of future studies.
There are several potential mechanisms that might account for inhibition of DNA binding by the N-hydroxybenzimidazoles. Inhibition may result from steric hindrance wherein the N-hydroxybenzimidazoles directly interfere with the interaction between ExsA and promoter DNA. Possible N-hydroxybenzimidazole interaction sites for a steric mechanism include the recognition helices that make base-specific contacts with the promoter region. The recognition helices are very similar in ExsA and its homologs (LcrF, AscA, and ExsAVP) (Fig. 5A), and targeting this conserved region could explain why the N-hydroxybenzimidazoles inhibit each of the AraC family proteins tested in this study. For ExsA and LcrF, in particular, there is only one amino acid difference (S207 in ExsA is a threonine in LcrF) in the recognition helices. A S207T substitution in recognition helix 1 did alter the sensitivity pattern of ExsA to more closely resemble that of LcrF. Several pieces of data, however, argue against the recognition helices being the primary interaction site. First, a previous study found one N-hydroxybenzimidazole with high activity against ExsA, Salmonella enterica serovar Typhimurium Rma, Proteus mirabilis PqrA, and E. coli MarA, SoxS, and Rob. Those six proteins share only 3 conserved amino acids out of the 24 combined residues that define recognition helices 1 and 2 (see Fig. S7 in the supplemental material). It seems unlikely that this low level of identity would support specific inhibition by the N-hydroxybenzimidazoles. Second, many of the mutations in the putative binding pocket also alter susceptibility to the N-hydroxybenzimidazoles (Table 2) but are located on the protein face opposite the DNA-binding domain. While these combined observations do not rule out a potential role for the recognition helices, they do suggest that other interaction determinants exist.
The alternative possibility is an allosteric mechanism whereby N-hydroxybenzimidazole binding induces a structural alteration in the DNA-binding domain that inhibits ExsA-DNA complex formation. One potential allosteric binding site is located between the recognition helices. N-hydroxybenzimidazole binding to the predicted pocket could negatively affect the spacing or orientation of the helices required for DNA binding. Single amino acid substitutions in the predicted inhibitor-binding pocket resulted in altered sensitivity to some of the N-hydroxybenzimidazoles. Some amino acid substitutions increased the sensitivity of ExsA to the inhibitors (P213A and R214A) while other mutations increased ExsA resistance (H180E, N183E, I217E, E184A, K186A, R214A, M241A, and S246A) (Table 2). No single substitution altered ExsA sensitivity to the entire panel of N-hydroxybenzimidazoles or resulted in complete resistance to any of the N-hydroxybenzimidazoles. Attempts to combine multiple substitutions were unsuccessful, as the resultant proteins lacked DNA-binding activity. Although substitutions in the predicted binding pocket altered the activity of many of the inhibitors in DNA-binding assays, it is important to note that those assays cannot distinguish between changes in ExsA-inhibitor interactions or changes in the ExsA structure that indirectly affect inhibitor binding or activity; therefore, the possibility that the inhibitors bind ExsA elsewhere cannot be ruled out.
The residues that constitute the predicted binding pocket are highly conserved in ExsA, LcrF, AscA, and ExsAVP, and that conservation is consistent with the finding that each is sensitive to inhibition by N-hydroxybenzimidazoles (Fig. 5). Harder to reconcile is that those same residues, and in fact the entire DNA-binding domain, are poorly conserved in ExsA, Rma, PqrA, MarA, SoxS, and Rob, and yet each is also sensitive to N-hydroxybenzimidazoles. How, then, do the N-hydroxybenzimidazoles exhibit such broad-spectrum activity? We propose that the inhibitor-binding pocket is structurally conserved in AraC proteins and that the pocket itself, rather than the precise amino acids, is the critical determinant. If the interaction between AraC proteins and N-hydroxybenzimidazoles is dependent on the shape of the pocket, rather than specific interactions with amino acids, then single amino acid changes might not completely prevent ExsA–N-hydroxybenzimidazole interactions. Consistent with that thought, we did find that some amino acid changes altered activity to some of the N-hydroxybenzimidazoles. Our mutagenesis experiments, therefore, do not necessarily point to specific amino acids as being involved in the N-hydroxybenzimidazoles interaction but rather reinforce the conclusion that these amino acids are involved in formation of a binding site pocket. Structural studies to better define the binding pocket could lead to the identification and/or development of second-generation compounds that retain this broad-spectrum activity.
Supplementary Material
Funding Statement
A.E.M. was supported by NIH training grant T32 AI07511.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02242-15.
REFERENCES
- 1.Rice LB. 2008. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: no ESKAPE. J Infect Dis 197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 2.Pendleton JN, Gorman SP, Gilmore BF. 2013. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther 11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- 3.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK, National Healthcare Safety Network Team, Participating National Healthcare Safety Network Facilities . 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol 29:996–1011. doi: 10.1086/591861. [DOI] [PubMed] [Google Scholar]
- 4.Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S. 2013. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol 34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- 5.Rice LB. 2006. Challenges in identifying new antimicrobial agents effective for treating infections with Acinetobacter baumannii and Pseudomonas aeruginosa. Clin Infect Dis 43(Suppl 2):S100–S105. doi: 10.1086/504487. [DOI] [PubMed] [Google Scholar]
- 6.Zilberberg MD, Shorr AF. 2013. Prevalence of multidrug-resistant Pseudomonas aeruginosa and carbapenem-resistant Enterobacteriaceae among specimens from hospitalized patients with pneumonia and bloodstream infections in the United States from 2000 to 2009. J Hosp Med 8:559–563. doi: 10.1002/jhm.2080. [DOI] [PubMed] [Google Scholar]
- 7.Nathwani D, Raman G, Sulham K, Gavaghan M, Menon V. 2014. Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: a systematic review and meta-analysis. Antimicrob Resist Infect Control 3:32. doi: 10.1186/2047-2994-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee VT, Pukatzki S, Sato H, Kikawada E, Kazimirova AA, Huang J, Li X, Arm JP, Frank DW, Lory S. 2007. Pseudolipasin A is a specific inhibitor for phospholipase A2 activity of Pseudomonas aeruginosa cytotoxin ExoU. Infect Immun 75:1089–1098. doi: 10.1128/IAI.01184-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnoldo A, Curak J, Kittanakom S, Chevelev I, Lee VT, Sahebol-Amri M, Koscik B, Ljuma L, Roy PJ, Bedalov A, Giaever G, Nislow C, Merrill AR, Lory S, Stagljar I. 2008. Identification of small molecule inhibitors of Pseudomonas aeruginosa exoenzyme S using a yeast phenotypic screen. PLoS Genet 4:e1000005. doi: 10.1371/journal.pgen.1000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aiello D, Williams JD, Majgier-Baranowska H, Patel I, Peet NP, Huang J, Lory S, Bowlin TL, Moir DT. 2010. Discovery and characterization of inhibitors of Pseudomonas aeruginosa type III secretion. Antimicrob Agents Chemother 54:1988–1999. doi: 10.1128/AAC.01598-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bowlin NO, Williams JD, Knoten CA, Torhan MC, Tashjian TF, Li B, Aiello D, Mecsas J, Hauser AR, Peet NP, Bowlin TL, Moir DT. 2014. Mutations in the Pseudomonas aeruginosa needle protein gene pscF confer resistance to phenoxyacetamide inhibitors of the type III secretion system. Antimicrob Agents Chemother 58:2211–2220. doi: 10.1128/AAC.02795-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams JD, Torhan MC, Neelagiri VR, Brown C, Bowlin NO, Di M, McCarthy CT, Aiello D, Peet NP, Bowlin TL, Moir DT. 2015. Synthesis and structure-activity relationships of novel phenoxyacetamide inhibitors of the Pseudomonas aeruginosa type III secretion system (T3SS). Bioorg Med Chem 23:1027–1043. doi: 10.1016/j.bmc.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zetterström CE, Hasselgren J, Salin O, Davis RA, Quinn RJ, Sundin C, Elofsson M. 2013. The resveratrol tetramer (−)-hopeaphenol inhibits type III secretion in the gram-negative pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa. PLoS One 8:e81969. doi: 10.1371/journal.pone.0081969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahmood F, Hakimiyan A, Jayaraman V, Wood S, Sivaramakrishnan G, Rehman T, Reuhs BL, Chubinskaya S, Shafikhani SH. 2013. A novel human antimicrobial factor targets Pseudomonas aeruginosa through its type III secretion system. J Med Microbiol 62:531–539. doi: 10.1099/jmm.0.051227-0. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Dong Y, Zhou T, Liu X, Deng Y, Wang C, Lee J, Zhang LH. 2013. Pseudomonas aeruginosa cytotoxicity is attenuated at high cell density and associated with the accumulation of phenylacetic acid. PLoS One 8:e60187. doi: 10.1371/journal.pone.0060187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rzhepishevska O, Hakobyan S, Ekstrand-Hammarstrom B, Nygren Y, Karlsson T, Bucht A, Elofsson M, Boily JF, Ramstedt M. 2014. The gallium(III)-salicylidene acylhydrazide complex shows synergistic anti-biofilm effect and inhibits toxin production by Pseudomonas aeruginosa. J Inorg Biochem 138:1–8. doi: 10.1016/j.jinorgbio.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Sawa T, Yahr TL, Ohara M, Kurahashi K, Gropper MA, Wiener-Kronish JP, Frank DW. 1999. Active and passive immunization with the Pseudomonas V antigen protects against type III intoxication and lung injury. Nat Med 5:392–398. doi: 10.1038/7391. [DOI] [PubMed] [Google Scholar]
- 18.Warrener P, Varkey R, Bonnell JC, DiGiandomenico A, Camara M, Cook K, Peng L, Zha J, Chowdury P, Sellman B, Stover CK. 2014. A novel anti-PcrV antibody providing enhanced protection against Pseudomonas aeruginosa in multiple animal infection models. Antimicrob Agents Chemother 58:4384–4391. doi: 10.1128/AAC.02643-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hauser AR. 2009. The type III secretion system of Pseudomonas aeruginosa: infection by injection. Nat Rev Microbiol 7:654–665. doi: 10.1038/nrmicro2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaz MR, King JM, Yahr TL. 2011. Intrinsic and extrinsic regulation of type III secretion gene expression in Pseudomonas aeruginosa. Front Microbiol 2:89. doi: 10.3389/fmicb.2011.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith RS, Wolfgang MC, Lory S. 2004. An adenylate cyclase-controlled signaling network regulates Pseudomonas aeruginosa virulence in a mouse model of acute pneumonia. Infect Immun 72:1677–1684. doi: 10.1128/IAI.72.3.1677-1684.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCaw ML, Lykken GL, Singh PK, Yahr TL. 2002. ExsD is a negative regulator of the Pseudomonas aeruginosa type III secretion regulon. Mol Microbiol 46:1123–1133. doi: 10.1046/j.1365-2958.2002.03228.x. [DOI] [PubMed] [Google Scholar]
- 23.Vallis AJ, Yahr TL, Barbieri JT, Frank DW. 1999. Regulation of ExoS production and secretion by Pseudomonas aeruginosa in response to tissue culture conditions. Infect Immun 67:914–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brutinel ED, Vakulskas CA, Yahr TL. 2010. ExsD inhibits expression of the Pseudomonas aeruginosa type III secretion system by disrupting ExsA self-association and DNA binding activity. J Bacteriol 192:1479–1486. doi: 10.1128/JB.01457-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thibault J, Faudry E, Ebel C, Attree I, Elsen S. 2009. Anti-activator ExsD forms a 1:1 complex with ExsA to inhibit transcription of type III secretion operons. J Biol Chem 284:15762–15770. doi: 10.1074/jbc.M109.003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dasgupta N, Lykken GL, Wolfgang MC, Yahr TL. 2004. A novel anti-anti-activator mechanism regulates expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol 53:297–308. doi: 10.1111/j.1365-2958.2004.04128.x. [DOI] [PubMed] [Google Scholar]
- 27.Urbanowski ML, Lykken GL, Yahr TL. 2005. A secreted regulatory protein couples transcription to the secretory activity of the Pseudomonas aeruginosa type III secretion system. Proc Natl Acad Sci U S A 102:9930–9935. doi: 10.1073/pnas.0504405102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogelaar NJ, Jing X, Robinson HH, Schubot FD. 2010. Analysis of the crystal structure of the ExsC. ExsE complex reveals distinctive binding interactions of the Pseudomonas aeruginosa type III secretion chaperone ExsC with ExsE and ExsD. Biochemistry (Mosc) 49:5870–5879. doi: 10.1021/bi100432e. [DOI] [PubMed] [Google Scholar]
- 30.Urbanowski ML, Brutinel ED, Yahr TL. 2007. Translocation of ExsE into Chinese hamster ovary cells is required for transcriptional induction of the Pseudomonas aeruginosa type III secretion system. Infect Immun 75:4432–4439. doi: 10.1128/IAI.00664-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowser TE, Bartlett VJ, Grier MC, Verma AK, Warchol T, Levy SB, Alekshun MN. 2007. Novel anti-infection agents: small-molecule inhibitors of bacterial transcription factors. Bioorg Med Chem Lett 17:5652–5655. doi: 10.1016/j.bmcl.2007.07.072. [DOI] [PubMed] [Google Scholar]
- 32.Grier MC, Garrity-Ryan LK, Bartlett VJ, Klausner KA, Donovan PJ, Dudley C, Alekshun MN, Tanaka SK, Draper MP, Levy SB, Kim OK. 2010. N-Hydroxybenzimidazole inhibitors of ExsA MAR transcription factor in Pseudomonas aeruginosa: in vitro anti-virulence activity and metabolic stability. Bioorg Med Chem Lett 20:3380–3383. doi: 10.1016/j.bmcl.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 33.Kim OK, Garrity-Ryan LK, Bartlett VJ, Grier MC, Verma AK, Medjanis G, Donatelli JE, Macone AB, Tanaka SK, Levy SB, Alekshun MN. 2009. N-Hydroxybenzimidazole inhibitors of the transcription factor LcrF in Yersinia: novel antivirulence agents. J Med Chem 52:5626–5634. doi: 10.1021/jm9006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallegos MT, Schleif R, Bairoch A, Hofmann K, Ramos JL. 1997. Arac/XylS family of transcriptional regulators. Microbiol Mol Biol Rev 61:393–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brutinel ED, Vakulskas CA, Brady KM, Yahr TL. 2008. Characterization of ExsA and of ExsA-dependent promoters required for expression of the Pseudomonas aeruginosa type III secretion system. Mol Microbiol 68:657–671. doi: 10.1111/j.1365-2958.2008.06179.x. [DOI] [PubMed] [Google Scholar]
- 36.King JM, Schesser Bartra S, Plano G, Yahr TL. 2013. ExsA and LcrF recognize similar consensus binding sites, but differences in their oligomeric state influence interactions with promoter DNA. J Bacteriol 195:5639–5650. doi: 10.1128/JB.00990-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuchs EL, Brutinel ED, Klem ER, Fehr AR, Yahr TL, Wolfgang MC. 2010. In vitro and in vivo characterization of the Pseudomonas aeruginosa cyclic AMP (cAMP) phosphodiesterase CpdA, required for cAMP homeostasis and virulence factor regulation. J Bacteriol 192:2779–2790. doi: 10.1128/JB.00168-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brutinel ED, Vakulskas CA, Yahr TL. 2009. Functional domains of ExsA, the transcriptional activator of the Pseudomonas aeruginosa type III secretion system. J Bacteriol 191:3811–3821. doi: 10.1128/JB.00002-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marsden AE, Schubot FD, Yahr TL. 2014. Self-association is required for occupation of adjacent binding sites in Pseudomonas aeruginosa type III secretion system promoters. J Bacteriol 196:3546–3555. doi: 10.1128/JB.01969-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.King JM, Brutinel ED, Marsden AE, Schubot FD, Yahr TL. 2012. Orientation of Pseudomonas aeruginosa ExsA monomers bound to promoter DNA and base-specific contacts with the PexoT promoter. J Bacteriol 194:2573–2585. doi: 10.1128/JB.00107-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fuchs EL, Brutinel ED, Jones AK, Fulcher NB, Urbanowski ML, Yahr TL, Wolfgang MC. 2010. The Pseudomonas aeruginosa Vfr regulator controls global virulence factor expression through cyclic AMP-dependent and -independent mechanisms. J Bacteriol 192:3553–3564. doi: 10.1128/JB.00363-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garrity-Ryan LK, Kim OK, Balada-Llasat JM, Bartlett VJ, Verma AK, Fisher ML, Castillo C, Songsungthong W, Tanaka SK, Levy SB, Mecsas J, Alekshun MN. 2010. Small molecule inhibitors of LcrF, a Yersinia pseudotuberculosis transcription factor, attenuate virulence and limit infection in a murine pneumonia model. Infect Immun 78:4683–4690. doi: 10.1128/IAI.01305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.