

ABSTRACT
NADP+ is a vital cofactor involved in a wide variety of activities, such as redox potential and cell death. Here, we show that NADP+ negatively regulates an acetyltransferase from Myxococcus xanthus, Mxan_3215 (MxKat), at physiologic concentrations. MxKat possesses an NAD(P)-binding domain fused to the Gcn5-type N-acetyltransferase (GNAT) domain. We used isothermal titration calorimetry (ITC) and a coupled enzyme assay to show that NADP+ bound to MxKat and that the binding had strong effects on enzyme activity. The Gly11 residue of MxKat was confirmed to play an important role in NADP+ binding using site-directed mutagenesis and circular dichroism spectrometry. In addition, using mass spectrometry, site-directed mutagenesis, and a coupling enzymatic assay, we demonstrated that MxKat acetylates acetyl coenzyme A (acetyl-CoA) synthetase (Mxan_2570) at Lys622 in response to changes in NADP+ concentration. Collectively, our results uncovered a mechanism of protein acetyltransferase regulation by the coenzyme NADP+ at physiological concentrations, suggesting a novel signaling pathway for the regulation of cellular protein acetylation.
IMPORTANCE Microorganisms have developed various protein posttranslational modifications (PTMs), which enable cells to respond quickly to changes in the intracellular and extracellular milieus. This work provides the first biochemical characterization of a protein acetyltransferase (MxKat) that contains a fusion between a GNAT domain and NADP+-binding domain with Rossmann folds, and it demonstrates a novel signaling pathway for regulating cellular protein acetylation in M. xanthus. We found that NADP+ specifically binds to the Rossmann fold of MxKat and negatively regulates its acetyltransferase activity. This finding provides novel insight for connecting cellular metabolic status (NADP+ metabolism) with levels of protein acetylation, and it extends our understanding of the regulatory mechanisms underlying PTMs.
INTRODUCTION
The dynamic and reversible mechanism of protein acetylation is an important regulatory posttranslational modification (PTM), which controls numerous cellular processes in the three kingdoms of life (1–4). Recent studies have identified >4,500 acetylated proteins, ranging from transcription factors and ribosomal proteins to many metabolic enzymes that are related to glycolysis, gluconeogenesis, the tricarboxylic acid (TCA) cycle, and fatty acid, nitrogen, and carbon metabolism (3, 5–8). Following the discovery of acetylation of the Salmonella enterica acetyl coenzyme A (acetyl-CoA) synthetase in 2002 (9), this type of PTM has also emerged as an important metabolic regulatory mechanism in bacteria. In the last decade, lysine acetylation of proteins has also been reported in other organisms (6, 7, 10–16).
Protein lysine acetylation can occur via either enzymatic or nonenzymatic acetylation, such as chemical acetylation. Intracellular acetyl phosphate (AcP) plays a critical role in a chemical acetylation reaction; for example, AcP has been shown to chemically acetylate histones, serum albumin, and synthetic polylysine in vitro (17). In addition, recent research has shown that AcP can chemically acetylate lysine residues, and acetate metabolism can globally affect the level of protein acetylation in Escherichia coli. In addition, mutant cells that did not synthesize AcP or convert AcP to acetate, leading to its accumulation, showed significantly reduced or elevated acetylation levels in vivo, respectively (5). This finding suggests that the intracellular AcP concentration is correlated with protein acetylation levels (5, 7, 8).
The proportion of enzymatic acetylation reactions in the cell is relatively small compared to that of chemical acetylation reactions (7), and enzymatic acetylation in bacteria requires protein acetyltransferases and deacetylases. Protein acetyltransferases control the acetylation of specific proteins under various physiological conditions, whereas protein deacetylases play a role in removing the acetyl group from some acetylated proteins in response to changes in the cellular energy status via promptly sensing the intracellular NAD (NAD+)-to-NADH ratio (5, 7, 18). In addition to deacetylating the proteins acetylated by acetyltransferases, protein deacetylases also specifically deacetylate the lysines acetylated by AcP (18).
Gcn5-type N-acetyltransferase (GNAT) acetyltransferases catalyze the transfer of the acetyl group from the acetyl-CoA donor to a primary amine of small molecules and proteins that are involved in a wide variety of cellular processes. GNATs, named for the homology to the yeast GCN5 protein (yGCN5p), are characterized by signature sequence motifs and structural homology (19). GNATs are conserved in all domains of life and represent one of the largest protein superfamilies (20). GNATs are involved in the acetylation of antibiotics, hormones, tRNA, histones, metabolic enzymes, and transcription factors and are thus implicated in a wide variety of cellular processes (4, 20–23).
Transcriptional and posttranslational regulation mechanisms of GNAT protein acetyltransferases have been demonstrated. For instance, regulation of the expression of the acuA gene, which encodes an acetyltransferase in Bacillus subtilis, is under the control of CcpA, a global regulatory protein that is affected by the quality of the carbon source available to the cell (24). In E. coli, transcription of the acetyltransferase PatZ (also known as YfiQ or Pka) is controlled at the transcriptional level by intracellular cyclic AMP (cAMP) levels. The catabolite activator protein (CAP)-cAMP complex has been proposed to bind to two sites in the patZ promoter to induce the expression of genes that increase the overall acetylation of proteins (25, 26).
The activity of protein acetyltransferases is also allosterically controlled by small biological molecules. Recently, we found that the amino acid-binding (ACT) domain is fused to the GNAT acetyltransferase in Micromonospora aurantiaca (MaKat), which confers amino acid-induced allosteric regulation (11). Similarly, fusion of the cAMP domain to the GNAT domain can occur in response to various stress conditions (i.e., cAMP concentration) to modulate the activity of a GNAT protein acetyltransferase in mycobacteria (27–30). Here, we report a new acetyltransferase found in M. xanthus (MxKat), which is encoded by the gene Mxan_3215 and contains both GNAT- and NAD(P)-binding Rossmann-type fold domains, similar to the cAMP-GNAT and ACT-GNAT domain organizations reported previously (11).
Similar to amino acids and cAMP, the coenzyme NADP+ is a small molecule that plays a very important role in many biological processes. For instance, NADP+ is a required cofactor in the reductive biosynthesis of fatty acids, isoprenoids, and aromatic amino acids (31). Furthermore, the NAD(P) coenzyme may perform very important regulatory functions in various physiologic settings. For example, NADP+ can induce a surrounding signal, such as reactive oxygen species, to regulate function (32). As the reverse reaction product of acetylation, NAD+ is required for many bacterial NAD+-dependent deacetylases (33–35). Thus, we hypothesized that NAD(P)+ coenzymes are specific regulatory ligands that may modulate the acetylation activity of MxKat in M. xanthus.
To test the hypothesis, we screened four coenzymes (NAD+, NADH, NADP+, and NADPH) using Western blot analysis to evaluate their influence on the acetylation activity of MxKat. Furthermore, isothermal titration calorimetry (ITC) was used to analyze the interaction between each coenzyme and MxKat. We found that NADP+ specifically bound to MxKat and was strongly bound under physiologic concentrations. Further, we found that MxKat is an NADP+ concentration-dependent acetyltransferase. Through a site-directed mutant assay, we characterized the active site of NADP+ binding and acetylation of the acetyl-CoA synthetase MxAcs, a substrate of MxKat. These data will help elucidate the mechanism by which NADP+ regulates MxKat to affect the acetylation of specific proteins.
MATERIALS AND METHODS
Cloning, overexpression, and purification of proteins.
We amplified the Mxan_3215 and Mxan_2570 genes by PCR from the genomic DNA of M. xanthus DK 1622 using two primer pairs (5′-TAAGAATTCATGACGCCTCCTCTCGTCCTCC and TAAAAGCTTTCACAGGGTGAGGACCATCAACTCC-3′, and 5′-TAAGAATTCTTGTCCACGCTGGAGGAACG and TAAAAGCTTTCAGTCGTCGTTCTGCCTCAGC-3′). After digestion with the restriction enzymes EcoRI and HindIII, we cloned the genes encoding the Mxan_3215 and Mxan_2570 proteins into the pET-28a vector to generate PET28a-Mxan_3215 and PET28a-Mxan_2570, and we sequenced the clones for verification. E. coli strain BL21(DE3) was used for protein expression. We selected and cultured a single colony in a 3-ml overnight culture, which was used to inoculate 50 ml of Luria-Bertani medium with 1% kanamycin. The cells were cultivated at 37°C and then induced with 0.7 mM isopropyl-β-d-thiogalactoside at 20°C overnight. Next, we centrifuged the culture to obtain the cells, which were resuspended in phosphate-buffered saline (PBS) buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 [pH 7.4]) and incubated on ice for 15 min. The cells were sonicated in PBS buffer, and cell debris was removed by centrifugation at 8,000 rpm for 20 min. A nickel-nitrilotriacetic acid (Ni-NTA)–agarose column was used to purify the supernatant, and the bound protein was preequilibrated with the binding buffer. After the flowthrough was discarded, the column was washed with 10 ml of washing buffer (300 mM NaCl, 50 mM NaH2PO4, and 20 mM imidazole [pH 8.0]) to eliminate the hybrid proteins, and the bound proteins were eluted with a linear gradient of 20 to 250 mM imidazole in washing buffer. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to analyze the fractions, and fractions containing the desired protein were pooled and dialyzed against buffer P (37 mM NaCl, 10 mM Na2HPO4, 2.7 mM KCl, 1.8 mM KH2PO4, 5% glycerol [pH 7.9]). The His tag of Mxan_3215 was removed by digestion with thrombin (at 4°C overnight). The protein was concentrated using an Amicon Ultra-4 30,000-molecular-weight-cutoff centrifugal device (Millipore, Billerica, MA, USA). The bicinchoninic acid (BCA) assay was used to monitor the protein concentration, using buffer P as the control. The amount of protein after concentration was also analyzed by SDS-PAGE.
Analysis of the protein–small-molecule interaction using ITC.
ITC is a powerful approach in which the heat released or absorbed throughout a titration reaction is measured. Calorimetric measurements were performed at 25°C with an iTC200 system (MicroCal; GE Healthcare, USA). All solutions were thoroughly degassed before use by stirring them under vacuum conditions. MxKat protein and NADP+ were dissolved in 1 M PBS (pH 7.4) and then used for ITC to investigate the interaction between MxKat and NADP+. Calorimetric data were analyzed using Origin for ITC version 7.0383.
Phylogenetic analysis of the NAD(P)-binding domain in M. xanthus.
The boundaries of the NAD-binding domain in MxKat were identified by analysis in the Pfam database (http://pfam.xfam.org/). The sequence of the defined domain was used to identify orthologs in the Swiss-Prot database using BLASTp. Individual full-length sequences of the hits obtained were then analyzed using Pfam to identify their NAD-binding domains. Furthermore, the protein sequence analysis of MxKat using the Conserved Domain Database (CDD) (36) demonstrated that the NAD-binding domain of MxKat belonged to cd05266 (http://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=187576). All sequences were then aligned using Clustal W (EMBL). The alignments were edited using JalView, and phylogenetic and molecular evolutionary analyses were conducted using Molecular Evolutionary Genetics Analysis (MEGA) software version 6 (37).
Determination of the acetyltransferase activity of MxKat.
To successfully monitor the acetylation reaction activated by MxKat, a coupling enzyme activity reaction was established. One of the products of the acetylation reaction, CoA, is used by pyruvate dehydrogenase to convert NAD+ to NADH, which causes an increase in the absorbance at 340 nm (38, 39). The reaction mixture used to assay enzyme activity was optimized using 5 mM MgCl2, 2.4 mM pyruvate, 1 mM dithiothreitol (DTT), 0.2 mM thiamine PPi (TPP), 0.2 mM NAD, 50 μM acetyl-CoA, 13 μM acetyl-CoA synthetase (Acs), 0.03 U pyruvate dehydrogenase, 0.65 μM MxKat with or without coenzyme, 100 mM sodium acetate, 50 mM bis-Tris, and 50 mM Tris (pH 7.5) in a total volume of 300 μl. All components except for MxKat were mixed and incubated at 25°C for 10 min. The reaction was initiated by adding MxKat, and the reaction speed was determined by monitoring the production of NADH for 10 min. Each measurement was repeated three times.
In vitro protein acetylation assays.
To determine whether Mxan_2570 is a substrate of MxKat, 0.2 μM purified MxKat protein or bovine serum albumin (BSA) and 5 μM purified unacetylated MxAcs protein were added to a reaction mixture (200 μl total volume) containing 0.05 M HEPES buffer (pH 7.5), 200 μM Tris (2-carboxyethyl) phosphine hydrochloride, and 20 μM acetyl-CoA. The reaction mixtures were incubated at 37°C for 2 h (15). After the reaction, the MxAcs protein samples were divided into two portions: one was analyzed by SDS-PAGE and Western blotting, and the other was used for measurement of Acs activity. The acetylated MxAcs was isolated by SDS-PAGE and then analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
In vitro Acs assays.
MxAcs was incubated with MxKat, acetyl-CoA (60 μM), and NADP+ (2 μM) at 37°C for 60 min. After the reaction, the acetylated MxAcs was isolated from the reaction mixture by gel filtration and ultrafiltration. The activity of acetylated MxAcs was measured in a coupled enzymatic assay in which CoA was used by malate dehydrogenase to convert NAD+ to NADH, resulting in an increase in absorbance at 340 nm. The standard reaction mixture contained 100 mM Tris-HCl (pH 7.7), 10 mM l-malate, 0.2 mM CoA, 8 mM ATP (pH 7.5), 1 mM NAD+, 10 mM MgCl2, 3 units of malate dehydrogenase, 0.4 units of citrate synthase, 100 mM potassium acetate, and various amounts of MxAcs (acetylated and nonacetylated).
Western blotting assays.
The sample concentrations were verified using a BCA protein assay kit (Tiangen). Protein samples were then separated by SDS-PAGE and transferred to a polyvinylidene fluoride membrane for 30 to 60 min at 100 V. The membrane was sealed at 24°C in sealing solution, which includes 1× TBST (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.1% Tween 20) and 5% nonfat dry milk (NFDM), for 120 min. Antiacetyllysine (anti-AcK) antibody was used at a dilution of 1:15,000 in TBST–0.5% NFDM. After incubation at 4°C overnight, the blot was washed three times with TBST. The membrane was incubated with 1 μg/ml horseradish peroxidase-conjugated anti-mouse IgG (in TBST with 3% BSA) at ambient temperature for 120 min. We used an enhanced chemiluminescence system (Pierce, USA) to detect the signal combined with a luminescent image analyzer (DNR Bio-Imaging Systems, Israel), according to the manufacturer's instructions.
Mass spectrometry for analysis of the acetylation site.
Protein bands were extracted from the gel and repeatedly washed with 50% ethanol to remove the stain. The gel pieces were dehydrated in acetonitrile and then dried in a centrifugal evaporator (Thermo Fisher Scientific, MA). The gel piece was then treated with 10 mM DTT in 50 mM ammonium bicarbonate and alkylated with 50 mM iodoacetamide in 50 mM ammonium bicarbonate. The pieces were washed with wash solution, dehydrated again, and then rehydrated in 50 mM ammonium bicarbonate with 10 ng/μl trypsin (Promega, Madison, WI); subsequently, digestion continued at 37°C for 12 h. Extraction buffer (75% acetonitrile and 0.1% trifluoroacetic acid) was added to the gel pieces to extract the peptides. The extracts were pooled and concentrated in a centrifugal evaporator.
The sample was dissolved using 4 μl of solvent A (0.1% [vol/vol] formic acid and 2% acetonitrile in water), and the solution was injected into a manually packed reversed-phase column and eluted with a 60-min gradient at a flow rate of 300 nl/min. The high-performance liquid chromatography eluate was directly electrosprayed into an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). The mass spectrometric analysis was carried out with an automatic switch between a full MS scan using Fourier transform mass spectroscopy in the Orbitrap device and an MS/MS scan using collision-induced dissociation in the dual linear ion trap. Full MS spectra with an m/z range of 350 to 1,700 were acquired with a resolution (full width at half maximum [FWHM]) of 240,000. The five most intense ions in each full MS spectrum were sequentially isolated for MS/MS fragmentation with a normalized collision energy of 35%. Automatic gain control was set at 3E4 for ion trap and at 1E6 for Orbitrap.
Thermo Proteome Discoverer (Thermo Fisher Scientific) was used to generate .mgf files that were subsequently checked against the Mxan_2570 sequence with the Mascot search engine (version 2.3.01; Matrix Science, United Kingdom). The search parameters were enzyme, trypsin; missed cleavage, 2; fixed modification, carbamidomethyl-C; variable modification, acetyl (protein N-term), oxidation-M, and acetylation-K; peptide mass tolerance, 10 ppm; fragment mass tolerance, 0.5 Da; and selected charge states, +2, +3, and + 4.
Site-directed mutagenesis and purification of Mxan_2570 and Mxan_3215.
The K622Q mutation was introduced into the recombinant plasmid PET28a-Mxan_2570 using the QuikChange mutagenesis kit (TransGen, Beijing, China). The G9A and G11A mutations in PET28a-Mxan_3215 were introduced in the same way. The mutations were confirmed by DNA sequencing. The expression and purification of the mutant proteins were performed according to the same procedures described above.
Circular dichroism spectrometry assay.
Purified wild-type (WT) Mxan_3215 protein and the G11A mutant protein were evaluated using circular dichroism spectrometry (Applied Photophysics, Leatherhead, United Kingdom) in the far-UV region (190 to 260 nm) at room temperature using a 10-mm cuvette. All samples were desalted, and the buffer was exchanged with PBS buffer (pH 7.4) using a Zeba desalt spin column (Thermo Scientific, Pierce, Rockford, IL). The sample solutions were diluted to a concentration of 3 μM. The circular dichroism spectrum scan of every sample was performed in triplicate.
RESULTS
MxKat showed an NAD(P)-GNAT domain organization and was confirmed to be a protein acetyltransferase.
Previous research demonstrated that proteins containing a GNAT domain can catalyze the acetylation of Acs to modulate its activity (10–12, 40). Furthermore, small molecules can bind to the multidomain acetyltransferase to regulate its activity in response to environmental signals. Here, we focused on the multidomain acetyltransferase Mxan_3215 (MxKat) derived from M. xanthus. Pfam version 27.0 (41) analysis showed that Mxan_3215 contains a GNAT domain and an NAD(P)-binding domain in the C and N termini at residues 273 to 437 and 5 to 185, respectively (Fig. 1A). The amino acid sequence of the GNAT domain in MxKat was aligned with that of MaKat (Micau_1670 in Micromonospora aurantiaca) (11), MsPat (MSMEG_5458 in Mycobacterium smegmatis) (27), Mt-PatA (Rv0998 in Mycobacterium tuberculosis) (27), 1Z4RA (human GCN5 acetyltransferase) (42), 1YGHB (yeast histone acetyltransferase) (43), and 1M1DA (tetrahymena GCN5 acetyltransferase) (44) to analyze the possible motif. The results showed a strong similarity between MxKat and several other types of acetyltransferases; they share the common core motif of the GNAT domain, R/Q-X-X-G-X-G/A (Fig. 1B). This motif was found to be important for acetyl-CoA recognition and binding (45). Using SWISS-MODEL (46) analysis, a three-dimensional model of the GNAT domain of MxKat was generated (see Fig. S1 in the supplemental material); the conserved pivotal residues, R366 and G369, were predicted to be in the junction of the α-helix and β-sheet, while the G371 residue was predicted to be at the end of the α-helix. In addition, phylogenetic analysis was conducted using MEGA with the GNAT domain sequences of various acetyltransferases, and the results showed that MxKat clustered with EcPatZ from E. coli (25), SePat from Salmonella enterica (47), RpPat from Rhodopseudomonas palustris (16), MaKat (11), Mt-PatA, and MsPat (48) (see Fig. S2 in the supplemental material). All of these acetyltransferases have been shown to acetylate Acs in different bacteria, which suggests that MxKat is an acetyltransferase in M. xanthus that may also acetylate Acs.
FIG 1.

MxKat is a protein acetyltransferase. (A) Illustration of the domain organization of Mxan_3215. The protein sequence of Mxan_3215 was analyzed in http://pfam.xfam.org/search/sequence, and the result indicated that Mxan_3215 has two domains, one for NAD(P) binding (positions 5 to 185) and one GNAT domain (positions 273 to 437). (B) Multiple sequence alignments of protein acetyltransferases in various species, including MaKat (M. aurantiaca), MsPat (M. smegmatis), Rv0998 (Mt-PatA, M. tuberculosis), 1YGHB (yeast histone acetyltransferase), 1Z4RA (human GCN5 acetyltransferase), 1M1DA (tetrahymena GCN5 acetyltransferase), and Mxan_3215 (MxKat, M. xanthus). The binding motif is boxed, and key amino acids of the Rossmann fold are marked with arrowheads. (C). Purified MxAcs was incubated with or without MxKat and acetyl (Ac)-CoA in vitro at 37°C for 2 h. After incubation, samples were collected and analyzed by SDS-PAGE, and the acetylation levels were determined by Western blotting using a specific anti-AcK antibody. (D) In vitro acetylation affected the activity of MxAcs. MxAcs enzyme activity was measured after incubation with or without acetyl-CoA in the presence of MxKat for the indicated times. The MxAcs activity is shown as a percentage of the maximum activity determined for MxAcs before acetylation. The data are expressed as the means ± standard deviations (SD) of the results from three identical assays.
To confirm whether MxKat, which contains a GNAT domain, actually functions as an acetyltransferase, we incubated purified MxKat with acetyl-CoA and recombinant MxAcs (Mxan_2570) in vitro. The Western blotting results demonstrated that MxKat has protein lysine acetyltransferase activity for acetylating Acs (Fig. 1C).
To investigate the effect of this acetylation on enzyme activity, MxAcs was incubated with MxKat in the presence or absence of acetyl-CoA for 2 h. In the presence of both acetyl-CoA and MxKat, MxAcs activity was reduced, indicating that lysine acetylation effectively decreased MxAcs activity (Fig. 1D).
Characterization of molecular binding with the regulatory NAD(P)-binding domain under physiological conditions.
The NAD(P)-binding domain of MxKat belongs to the cd05266 subfamily of the NAD(P)-binding family. To determine the specific coenzyme molecule that modulates MxKat activity, we evaluated four coenzymes, NAD, NADH, NADP+, and NADPH, using Western blot analysis. The acetylation was attenuated only when the component contained NADP+, which demonstrated that only NADP+ affected the acetylation function of MxKat on MxAcs (Fig. 2A). To further determine the binding affinity of NADP+ to MxKat, we performed ITC to measure the thermodynamic parameters of this binding reaction. The MxKat solution was loaded in the cell, and the NADP+ solution was titrated by injection into the cell. Figure 2B (upper panel) shows substantial heat released by the titration of NADP+ into MxKat; the power profile displayed an obvious heat peak, and a sigmoid-shaped curve of the enthalpy change during the titration process was observed (Fig. 2B, lower panel). This result confirmed that there was an exothermic reaction between NADP+ and MxKat. Thus, the ITC experiments confirmed the interaction of NADP+ with MxKat in terms of thermodynamics. The binding affinity can be quantified using the binding constant KB, which was 3.43 × 105 M−1 (KD [equilibrium dissociation constant], 2.92 μM) for NADP+ binding to MxKat. Previous research demonstrated that the absolute metabolite concentration range of NADP+ in E. coli is 0.14 to 31.1 μM (49). Therefore, the ITC data combined with Western blot results indicate that NADP+ can bind to MxKat at physiologic concentrations and may negatively regulate the function of MxKat to acetylate MxAcs.
FIG 2.
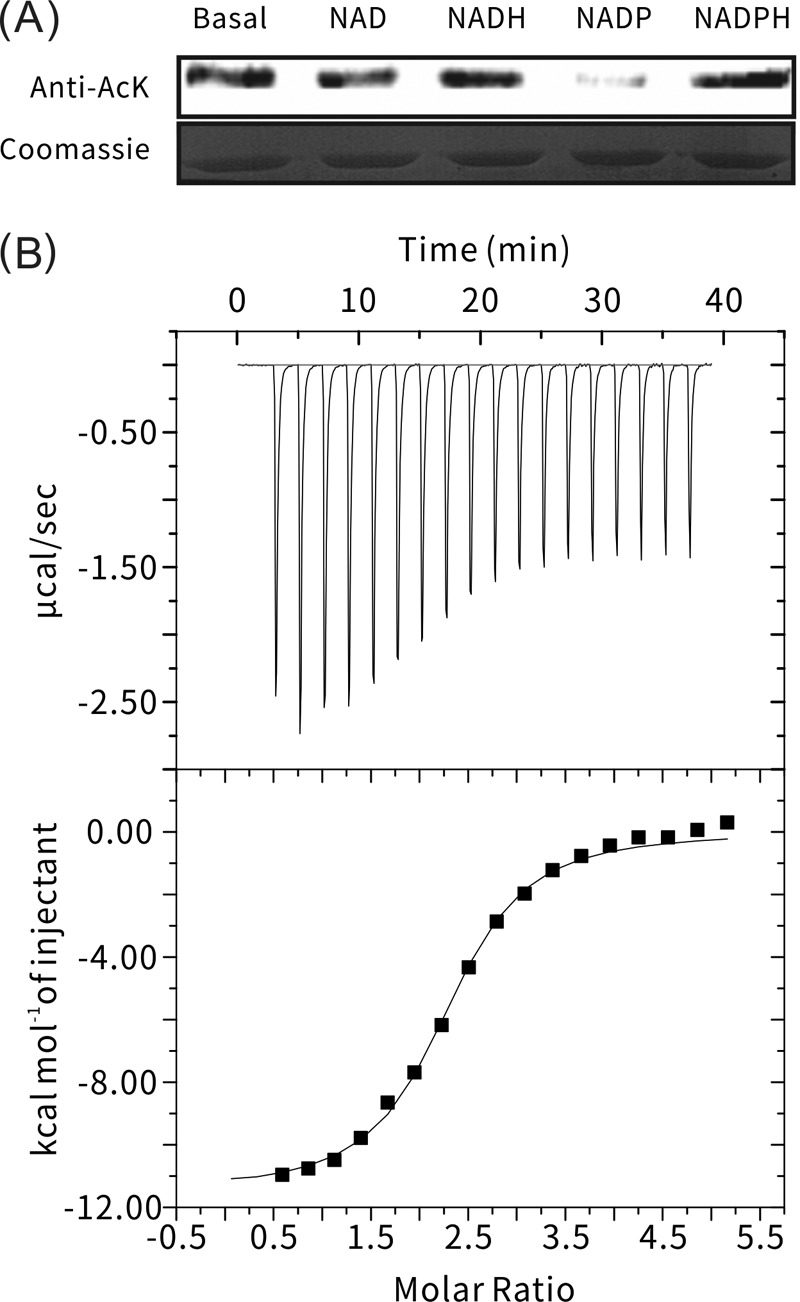
Screening of MxKat ligands. (A) Acetylation level was determined by Western blot analysis with acetyllysine antibodies after every coenzyme (NAD, NADH, NADP+, and NADPH) bound with MxKat (upper panel). At the same time, another protein gel was stained with Coomassie brilliant blue (lower panel). (B) NADP+ binding to MxKat was validated using ITC. Purified MxKat was dissolved in 1 M PBS (pH 7.4) to a final concentration of up to 38 μM. Meanwhile, NADP+ was dissolved in PBS to 1 mM and used to inject for titration. Deionized water was used as a blank control.
MxKat may function as an NADP+-regulated protein acetyltransferase.
In silico analysis combined with the Western blotting assay described above demonstrated that MxKat is a protein lysine acetyltransferase that can acetylate MxAcs. As shown in Fig. 3A, the acetylation of MxAcs was observed during incubation with MxKat and acetyl-CoA, and acetylation significantly decreased in the presence of NADP+. The NADP+-binding domain fused to the acetyltransferase domain in MxKat. The observed decrease in the acetylation level of MxAcs in the presence of NADP+ indicates that the NADP+ domain may regulate the activity of the GNAT domain and that MxKat can function as an NADP+-regulated protein acetyltransferase. In support of this hypothesis, in the presence of NADP+, MxKat activity exhibited a ligand concentration-dependent decrease in protein acetylation (Fig. 3B).
FIG 3.

Regulation of MxKat activity for protein acetylation by NADP+. (A) Acetylation of MxAcs (Mxan_2570) by MxKat. MxAcs (10 μg) was incubated alone or in the presence of MxKat (0.2 μM), acetyl-CoA (60 μM), and NADP+ (2 μM) in a 100-μl volume at 37°C for 60 min, followed by SDS-PAGE analysis. The acetylation level was determined by Western blot analysis with acetyllysine antibodies (upper panel). At the same time, another protein electrophoresis gel was stained with Coomassie brilliant blue (lower panel). (B) Acetylation levels of MxAcs were measured using a Western blot assay. With fixed concentrations of MxAcs (1.5 μM), MxKat (0.2 μM), and acetyl-CoA (60 μM), various concentrations of NADP+ were added to the reaction system. The reaction lasted 60 min at 37°C and was followed by SDS-PAGE analysis. (C) Acetyltransferase activity of MxKat was measured using a coupled enzymatic assay. The initial rate of formation of NADH is shown in the presence or absence of NADP+. The control represents an assay in which PBS buffer was used instead of MxKat. (D) Enzymatic activity of MxAcs. In the presence of NADP+, the acetylation of MxAcs was attenuated; thus, the enzyme activity of MxAcs was higher in the presence than in the absence of NADP+. The control represents an assay in which PBS buffer was used in place of MxKat.
To further test whether NADP+ can regulate the acetylation activity of MxKat, we investigated the MxKat enzyme activity in the presence or absence of NADP+. The acetylation reaction of MxKat was monitored by a coupled enzymatic assay, in which the amount of CoA liberated following acetylation is measured according to the formation of reduced NADH from NAD+ by pyruvate dehydrogenase (38, 39). In the presence of NADP+, the enzyme activity of MxKat was significantly attenuated compared to that in the absence of NADP+ (Fig. 3C), which indicates that NADP+ negatively modulated MxKat. Furthermore, we were also interested in the effect of NADP+ binding on the enzyme activity of MxAcs upon MxKat acetylation. Therefore, we used a coupled enzymatic assay to continuously monitor the catalytic reaction of the acetylated MxAcs. As shown in Fig. 3D, in the presence of NADP+, low MxKat activity resulted in a low acetylation level of MxAcs, revealing a higher level of enzyme activity of MxAcs than that in the absence of NADP+. Taken together, these results demonstrate that MxKat acetylated MxAcs and that the acetylation was mediated by NADP+.
A previous study demonstrated that bacterial acetyltransferases recognize a specific lysine site for protein acetylation, and an acetylation motif (PXXXXGK) was proposed in AMP-forming Acs (9). In the present study, we confirmed that GNAT acetyltransferases can recognize this motif and acetylate its last lysine residue, such as Lys617 of MtAcs from M. tuberculosis, Lys610 of SlAcs from Streptomyces lividans, Lys606 of RpAcs from Rhodopseudomonas palustris, Lys609 of EcAcs from E. coli, and Lys609 of SeAcs from S. enterica (Fig. 4A).
FIG 4.

Lys622 residue of MxAcs is acetylated by MxKat. (A) Sequence alignment of acetyl-CoA synthetases (Acs); a conserved PKXXXXK motif (boxed region) in Acs was found in MxAcs (Mxan_2570). Key amino acids and the acetylated site are marked with arrowheads and an asterisk, respectively. (B) LC-MS/MS spectrum of a tryptic peptide with a mass/charge ratio (m/z) of 419.2 obtained from acetylated MxAcs. This spectrum matched that of the peptide (boxed sequence) in MxAcs (inset shows the sequence of the purified protein), with a mass shift in the b3 and y4 ions corresponding to acetylation at the lysine residue (arrowhead at position 622). (C) Western blot analysis of MxAcs upon acetylation of MxKat. Wild-type MxAcs or the MxAcsK622Q mutant was incubated with various components at 37°C for 60 min, followed by SDS-PAGE analysis. The reaction was performed in the presence and absence of NADP+.
To determine the specific site(s) used for acetylation in MxAcs, in vitro-acetylated MxAcs was separated by electrophoresis, and the band corresponding to MxAcs was analyzed by MS. LC-MS confirmed that a peptide with sequence SGKIMR (419.2 Da) contained the major acetylated lysine residue (Lys622) (Fig. 4B) in the protein, which corresponds to the last lysine residue of the PXXXXGK motif.
To further validate the function of Lys622 in the acetylation of MxAcs, we established substitution mutations at the Lys622 position to generate the K622Q variant of MxAcs. Glutamine lacks a positive charge and therefore serves as a structural mimic for acetyllysine (12). MxAcsK622Q and MxAcsWT were incubated with the MxKat enzyme and acetyl-CoA. Western blotting was conducted to detect the acetylation level of MxAcsWT and MxAcsK622Q with an anti-acetyllysine antibody. As shown in Fig. 4C, the acetylation of MxAcsWT only was observed. No acetylation of MxAcsK622Q was observed, which indicated that MxKat modified the conserved lysine residue, Lys622, of the major active site in MxAcs.
Identification of residues in the NADP+-binding domain associated with NADP+ binding.
The NAD(P)-binding domain is widely present in the short-chain dehydrogenase/reductase (SDR) family, whose members contain a single domain with a structurally conserved Rossmann fold, an NAD(P)(H)-binding region, and a structurally diverse C-terminal region. The Rossmann fold is a common structural motif found in proteins that bind to nucleotides, especially the cofactor NAD(P). The structure with two repeats is composed of six parallel β-strands linked to two pairs of α-helices in the topological order β-α-β-α-β. The Rossmann fold is one of the three most highly represented folds in the Protein Data Bank (PDB), and it often can be identified by the short consensus amino acid sequence motif GX1–2GXXG (50). The NADP+-binding domain of MxKat was analyzed using the CDD (36) (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). As shown in Fig. 5A, MxKat has only one motif similar to the GX1–2GXXG motif and two Gly residues distributed at the 9 and 11 positions. In the three-dimensional protein model, we found that the G9 residue is likely located at the end of the β-sheet, whereas the G11 residue may be located between the α-helix and β-sheet (see Fig. S3 in the supplemental material). To determine the roles of these residues in NADP+ binding, site-directed mutagenesis was performed to convert the Gly to Ala at residues 9 and 11. The acetylation activities of the wild-type and mutant proteins in the presence of NADP+ were also determined using a coupled acetyltransferase enzyme assay and Western blot analysis.
FIG 5.

NADP+-binding sites of MxKat. (A) Binding motif of the NAD(P) domain of MxKat. (B and C) Initial rates of NADH formation and Western blot analysis are shown for the MxKat mutants (the G9A and G11A mutants, respectively). The basal level represents the enzyme activity of wild-type MxKAT. (Inset) MxAcs was acetylated by MxKat G9A and MxKat G11A in the presence or absence of NADP+. The acetylation level was determined by Western blotting with acetyllysine antibodies (upper panel). At the same time, another protein gel was stained with Coomassie brilliant blue (lower panel). (D) Circular dichroism assays showed that the G11A mutant does not perturb the NADP+-binding domain structure of MxKat.
When Gly9 was mutated to Ala, the enzyme activity of MxKatG9A was still negatively regulated by NADP+, as observed for wild-type MxKat (Fig. 5B). However, when Gly11 was mutated to Ala, no change in the activity of acetyltransferase was observed in the presence or absence of NADP+, which indicated that MxKatG11A was no longer modulated by NADP+ (Fig. 5C). The CD spectra (Fig. 5D) of MxKat and the G11A mutant were nearly identical in the far-UV region (200 to 260 nm), in which the signals arose from the MxKat chain structure. These data showed that the G11A mutation likely did not perturb the structure of the NAD+-binding domain of MxKat. Taken together, these results indicated that Gly11 of MxKat is involved in the interaction with NADP+ and is part of the active site for the NADP+-mediated regulation of MxKat.
DISCUSSION
Many deacetylases require coenzyme NAD+ to effectively remove the acetyl group from an acetylated protein. Our study further uncovered a new biochemical mechanism whereby the coenzyme NADP+ negatively regulates an acetyltransferase to reduce the acetylation level of specific intracellular proteins. These findings help deepen our understanding of the biochemical mechanism of how acetylation controls the activity/function of a metabolic enzyme by inducing a change in the NADP+ coenzyme.
NADP+ synthesis proceeds through two major mechanisms: NADP+ can be generated de novo from NAD+ through the action of NAD+ kinases (NADKs), or NADP+ might be formed from NADPH via the action of multiple NADPH-dependent enzymes, such as glutathione reductase (51). Although NADP+ is known to play several important regulatory roles, its regulatory mechanism with respect to acetylation has received little attention thus far. The major known biological function of NADP+ is as a precursor for NADPH formation (52). Furthermore, the mechanism and subcellular localization of NADP+ and NADPH generation appear to critically influence the physiological state of a cell and, therefore, cell survival. Furthermore, NADKs have been shown to regulate the formation of NADP+ from NAD+, which can be inhibited by NADPH (53). In this study, we found that NADPH did not affect the ability of MxKat to acetylate MxAcs (Fig. 2A), and the NADP+ modulation of MxKat was concentration dependent (Fig. 3B). Therefore, the homeostasis of NADP+ plays a crucial role in regulating MxKat, similar to the balance of NAD+ and NADH in mediating deacetylases.
Another study showed that the activities of protein acetyltransferases and deacetylases are carefully regulated in response to a change in the intracellular signals that control the acetylation of specific proteins (such as the level of acetyl-CoA or NAD+), which in turn affects the metabolic network (11). Acetyl-CoA and NAD+ are key indicators of cellular energy status, and protein lysine acetylation serves as a link that connects cellular energy levels to protein acetylation/deacetylation activity. In mycobacteria, cAMP directly activates MsKat and MtKat by binding to the cyclic nucleotide-binding (CNB) domain of two protein acetyltransferases, indicating that the levels of protein acetylation can also be modulated in response to changes in intracellular cAMP levels (54). This work demonstrated that the NADP+ domain is linked to GNAT acetyltransferase, which confirms NADP+-induced regulation of this enzyme. Therefore, protein acetyltransferases with an NADP+-GNAT domain organization might be a novel mechanism in response to the homeostasis of intracellular coenzymes.
The coenzyme NADP+ can be transformed to NADPH to maintain the intracellular redox potential by glucose-6-phosphate dehydrogenase (G6PD), 6-glyconate phosphate dehydrogenase (6GPD), NADP+-dependent isocitrate dehydrogenases (IDPs), and NADP+-dependent malic enzymes (MEPs), which are involved in glycolysis, gluconeogenesis, glycogen metabolism, and the TCA cycle. Moreover, these sugar metabolism-related enzymes have been shown to be regulated by lysine acetylation, which results in a reduction in enzyme activity (5–7). Low acetylation levels of the associated enzymes result in a high intracellular NADP+-to-NADPH ratio. Since NADP+ was found to modulate the protein acetyltransferase MxKat, it is possible that a high NADP+-to-NADPH ratio would influence the NADP+-mediated acetylation of some proteins, including enzymes involved in the conversion between NADP+ and NADPH. Future work will focus on the identification of all substrates of the MxKat enzyme and aim to reveal the complete landscape of NADP+-mediated acetylation.
To our knowledge, this study is the first to demonstrate the allosteric regulation of a protein acetyltransferase by NADP+, which indicates that the intracellular acetylation levels of proteins might be modulated in response to changes in the NADP+-to-NADPH ratio. This finding might provide the basis for uncovering the molecular mechanism underlying the homeostasis of protein acetylation/deacetylation in response to changes in intracellular signals.
Supplementary Material
ACKNOWLEDGMENT
We thank Yue-zhong Li of Shandong University for kindly providing the M. xanthus DK1622 strain.
Funding Statement
This work was also supported by grants from the National Key Technologies R&D Programs (2014AA021502), Fundamental Research Funds for the Central Universities, and the Shanghai Natural Science Foundation (14ZR1409600).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00661-15.
REFERENCES
- 1.Hu LI, Lima BP, Wolfe AJ. 2010. Bacterial protein acetylation: the dawning of a new age. Mol Microbiol 77:15–21. doi: 10.1111/j.1365-2958.2010.07204.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim GW, Yang XJ. 2011. Comprehensive lysine acetylomes emerging from bacteria to humans. Trends Biochem Sci 36:211–220. doi: 10.1016/j.tibs.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Hentchel KL, Escalante-Semerena JC. 2015. Acylation of biomolecules in prokaryotes: a widespread strategy for the control of biological function and metabolic stress. Microbiol Mol Biol Rev 79:321–346. doi: 10.1128/MMBR.00020-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thao S, Escalante-Semerena JC. 2011. Control of protein function by reversible Nε-lysine acetylation in bacteria. Curr Opin Microbiol 14:200–204. doi: 10.1016/j.mib.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinert BT, Iesmantavicius V, Wagner SA, Schölz C, Gummesson B, Beli P, Nyström T, Choudhary C. 2013. Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol Cell 51:265–272. doi: 10.1016/j.molcel.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Zhang K, Tian S, Fan E. 2013. Protein lysine acetylation analysis: current MS-based proteomic technologies. Analyst 138:1628–1636. doi: 10.1039/c3an36837h. [DOI] [PubMed] [Google Scholar]
- 7.Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, Lima BP, Scholle M, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ. 2014. Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One 9:e94816. doi: 10.1371/journal.pone.0094816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schilling B, Christensen D, Davis R, Sahu AK, Hu LI, Walker-Peddakotla A, Sorensen DJ, Zemaitaitis B, Gibson BW, Wolfe AJ. 2015. Protein acetylation dynamics in response to carbon overflow in Escherichia coli. Mol Microbiol 98:847–863. doi: 10.1111/mmi.13161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC. 2002. Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298:2390–2392. doi: 10.1126/science.1077650. [DOI] [PubMed] [Google Scholar]
- 10.You D, Yao LL, Huang D, Escalante-Semerena JC, Ye B-C. 2014. Acetyl coenzyme A synthetase is acetylated on multiple lysine residues by a protein acetyltransferase with a single Gcn5-type N-acetyltransferase (GNAT) domain in Saccharopolyspora erythraea. J Bacteriol 196:3169–3178. doi: 10.1128/JB.01961-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J-Y, You D, Leng P-Q, Ye B-C. 2014. Allosteric regulation of a protein acetyltransferase in Micromonospora aurantiaca by the amino acids cysteine and arginine. J Biol Chem 289:27034–27045. doi: 10.1074/jbc.M114.579078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tucker AC, Escalante-Semerena JC. 2013. Acetoacetyl-CoA synthetase activity is controlled by a protein acetyltransferase with unique domain organization in Streptomyces lividans. Mol Microbiol 87:152–167. doi: 10.1111/mmi.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu X, Vellaichamy A, Wang D, Zamdborg L, Kelleher NL, Huber SC, Zhao Y. 2013. Differential lysine acetylation profiles of Erwinia amylovora strains revealed by proteomics. J Proteomics 79:60–71. doi: 10.1016/j.jprot.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okanishi H, Kim K, Masui R, Kuramitsu S. 2013. Acetylome with structural mapping reveals the significance of lysine acetylation in Thermus thermophilus. J Proteome Res 12:3952–3968. doi: 10.1021/pr400245k. [DOI] [PubMed] [Google Scholar]
- 15.Lee DW, Kim D, Lee YJ, Kim JA, Choi JY, Kang S, Pan JG. 2013. Proteomic analysis of acetylation in thermophilic Geobacillus kaustophilus. Proteomics 13:2278–2282. doi: 10.1002/pmic.201200072. [DOI] [PubMed] [Google Scholar]
- 16.Crosby HA, Pelletier DA, Hurst GB, Escalante-Semerena JC. 2012. System-wide studies of N-lysine acetylation in Rhodopseudomonas palustris reveal substrate specificity of protein acetyltransferases. J Biol Chem 287:15590–15601. doi: 10.1074/jbc.M112.352104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramponi G, Manao G, Camici G. 1975. Nonenzymatic acetylation of histones with acetyl phosphate and acetyl adenylate. Biochemistry 14:2681–2685. doi: 10.1021/bi00683a018. [DOI] [PubMed] [Google Scholar]
- 18.AbouElfetouh A, Kuhn ML, Hu LI, Scholle MD, Sorensen DJ, Sahu AK, Becher D, Antelmann H, Mrksich M, Anderson WF, Gibson BW, Schilling B, Wolfe AJ. 2015. The E. coli sirtuin CobB shows no preference for enzymatic and nonenzymatic lysine acetylation substrate sites. Microbiologyopen 4:66–83. doi: 10.1002/mbo3.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw KJ, Rather PN, Hare RS, Miller GH. 1993. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol Rev 57:138–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vetting MW, S de Carvalho LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. 2005. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys 433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 21.Ikeuchi Y, Kitahara K, Suzuki T. 2008. The RNA acetyltransferase driven by ATP hydrolysis synthesizes N4-acetylcytidine of tRNA anticodon. EMBO J 27:2194–2203. doi: 10.1038/emboj.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spange S, Wagner T, Heinzel T, Kramer OH. 2009. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 41:185–198. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 23.Thao S, Chen CS, Zhu H, Escalante-Semerena JC. 2010. Nε-Lysine acetylation of a bacterial transcription factor inhibits its DNA-binding activity. PLoS One 5:e15123. doi: 10.1371/journal.pone.0015123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grundy FJ, Turinsky AJ, Henkin TM. 1994. Catabolite regulation of Bacillus subtilis acetate and acetoin utilization genes by CcpA. J Bacteriol 176:4527–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Castaño-Cerezo S, Bernal V, Blanco-Catala J, Iborra JL, Canovas M. 2011. cAMP-CRP co-ordinates the expression of the protein acetylation pathway with central metabolism in Escherichia coli. Mol Microbiol 82:1110–1128. doi: 10.1111/j.1365-2958.2011.07873.x. [DOI] [PubMed] [Google Scholar]
- 26.Hentchel KL, Thao S, Intile PJ, Escalante-Semerena JC. 2015. Deciphering the regulatory circuitry that controls reversible lysine acetylation in Salmonella enterica. mBio 6(4):e00891-15. doi: 10.1128/mBio.00891-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nambi S, Basu N, Visweswariah SS. 2010. cAMP-regulated protein lysine acetylases in mycobacteria. J Biol Chem 285:24313–24323. doi: 10.1074/jbc.M110.118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HJ, Lang PT, Fortune SM, Sassetti CM, Alber T. 2012. Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat Struct Mol Biol 19:811–818. doi: 10.1038/nsmb.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nambi S, Badireddy S, Visweswariah SS, Anand GS. 2012. Cyclic AMP-induced conformational changes in mycobacterial protein acetyltransferases. J Biol Chem 287:18115–18129. doi: 10.1074/jbc.M111.328112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Podobnik M, Siddiqui N, Rebolj K, Nambi S, Merzel F, Visweswariah SS. 2014. Allostery and conformational dynamics in cAMP-binding acyltransferases. J Biol Chem 289:16588–16600. doi: 10.1074/jbc.M114.560086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen LL, Liu LX, Ling ZQ, Hu FJ, Sun YP, Zhang JY, Yang C, Yang Y, Xiong Y, Guan KL, Ye D. 2014. Regulation of G6PD acetylation by KAT9/SIRT2 modulates NADPH homeostasis and cell survival during oxidative stress. EMBO J 33:1304–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim SY, Lee SM, Tak JK, Choi KS, Kwon TK, Park JW. 2007. Regulation of singlet oxygen-induced apoptosis by cytosolic NADP+-dependent isocitrate dehydrogenase. Mol Cell Biochem 302:27–34. doi: 10.1007/s11010-007-9421-x. [DOI] [PubMed] [Google Scholar]
- 33.Mikulik K, Felsberg J, Kudrnáčová E, Bezoušková S, Setinová D, Stodůlková E, Zídková J, Zídek V. 2012. CobB1 deacetylase activity in Streptomyces coelicolor. Biochem Cell Biol 90:179–187. doi: 10.1139/o11-086. [DOI] [PubMed] [Google Scholar]
- 34.Denu JM. 2005. The Sir2 family of protein deacetylases. Curr Opin Chem Biol 9:431–440. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Zhao X, Allison D, Condon B, Zhang FY, Gheyi T, Zhang AP, Ashok S, Russell M, MacEwan I, Qian YW, Jamison JA, Luz JG. 2013. The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem 56:963–969. doi: 10.1021/jm301431y. [DOI] [PubMed] [Google Scholar]
- 36.Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41:D348–D352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berndsen CE, Denu JM. 2005. Assays for mechanistic investigations of protein/histone acetyltransferases. Methods 36:321–331. doi: 10.1016/j.ymeth.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Kim Y, Tanner KG, Denu JM. 2000. A continuous, nonradioactive assay for histone acetyltransferases. Anal Biochem 280:308–314. doi: 10.1006/abio.2000.4546. [DOI] [PubMed] [Google Scholar]
- 40.Zhang J, Sprung R, Pei J, Tan X, Kim S, Zhu H, Liu CF, Grishin NV, Zhao Y. 2009. Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol Cell Proteomics 8:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, Sonnhammer EL, Tate J, Punta M. 2014. Pfam: the protein families database. Nucleic Acids Res 42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuetz A, Bernstein G, Dong A, Antoshenko T, Wu H, Loppnau P, Bochkarev A, Plotnikov AN. 2007. Crystal structure of a binary complex between human GCN5 histone acetyltransferase domain and acetyl coenzyme A. Proteins 68:403–407. [DOI] [PubMed] [Google Scholar]
- 43.Trievel RC, Rojas JR, Sterner DE, Venkataramani RN, Wang L, Zhou J, Allis CD, Berger SL, Marmorstein R. 1999. Crystal structure and mechanism of histone acetylation of the yeast GCN5 transcriptional coactivator. Proc Natl Acad Sci U S A 96:8931–8936. doi: 10.1073/pnas.96.16.8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poux AN, Cebrat M, Kim CM, Cole PA, Marmorstein R. 2002. Structure of the GCN5 histone acetyltransferase bound to a bisubstrate inhibitor. Proc Natl Acad Sci U S A 99:14065–14070. doi: 10.1073/pnas.222373899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neuwald AF, Landsman D. 1997. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem Sci 22:154–155. doi: 10.1016/S0968-0004(97)01034-7. [DOI] [PubMed] [Google Scholar]
- 46.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Cassarino TG, Bertoni M, Bordoli L, Schwede T. 2014. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42:W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starai VJ, Escalante-Semerena JC. 2004. Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica. J Mol Biol 340:1005–1012. doi: 10.1016/j.jmb.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 48.Xu H, Hegde SS, Blanchard JS. 2011. Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry 50:5883–5892. doi: 10.1021/bi200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. 2009. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol 5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kleiger G, Eisenberg D. 2002. GXXXG and GXXXA motifs stabilize FAD and NAD(P)-binding Rossmann folds through C(alpha)-H… O hydrogen bonds and van der Waals interactions. J Mol Biol 323:69–76. [DOI] [PubMed] [Google Scholar]
- 51.Pollak N, Dölle C, Ziegler M. 2007. The power to reduce: pyridine nucleotides—small molecules with a multitude of functions. Biochem J 402:205–218. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ying W. 2008. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10:179–206. doi: 10.1089/ars.2007.1672. [DOI] [PubMed] [Google Scholar]
- 53.Zerez CR, Moul DE, Gomez EG, Lopez VM, Andreoli AJ. 1987. Negative modulation of Escherichia coli NAD kinase by NADPH and NADH. J Bacteriol 169:184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nambi S, Gupta K, Bhattacharyya M, Ramakrishnan P, Ravikumar V, Siddiqui N, Thomas AT, Visweswariah SS. 2013. Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J Biol Chem 288:14114–14124. doi: 10.1074/jbc.M113.463992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.