

Abstract
The OM43 clade within the family Methylophilaceae of Betaproteobacteria represents a group of methylotrophs that play important roles in the metabolism of C1 compounds in marine environments and other aquatic environments around the globe. Using dilution-to-extinction cultivation techniques, we successfully isolated a novel species of this clade (here designated MBRS-H7) from the ultraoligotrophic open ocean waters of the central Red Sea. Phylogenomic analyses indicate that MBRS-H7 is a novel species that forms a distinct cluster together with isolate KB13 from Hawaii (Hawaii-Red Sea [H-RS] cluster) that is separate from the cluster represented by strain HTCC2181 (from the Oregon coast). Phylogenetic analyses using the robust 16S-23S internal transcribed spacer revealed a potential ecotype separation of the marine OM43 clade members, which was further confirmed by metagenomic fragment recruitment analyses that showed trends of higher abundance in low-chlorophyll and/or high-temperature provinces for the H-RS cluster but a preference for colder, highly productive waters for the HTCC2181 cluster. This potential environmentally driven niche differentiation is also reflected in the metabolic gene inventories, which in the case of the H-RS cluster include those conferring resistance to high levels of UV irradiation, temperature, and salinity. Interestingly, we also found different energy conservation modules between these OM43 subclades, namely, the existence of the NADH:quinone oxidoreductase complex I (NUO) system in the H-RS cluster and the nonhomologous NADH:quinone oxidoreductase (NQR) system in the HTCC2181 cluster, which might have implications for their overall energetic yields.
INTRODUCTION
Methylotrophs are a taxonomically diverse group of microorganisms that use reduced one-carbon (C1) compounds as their sole carbon and energy sources (1, 2). Unlike methanotrophs, non-methane-utilizing methylotrophs (here simply denoted methylotrophs) cannot oxidize methane but instead degrade more oxidized C1 compounds, like methanol, methylamine, and formaldehyde, using different pathways for their oxidation, demethylation, and assimilation into biomass (1–3). Marine methylotrophs play an important role in the metabolism and assimilation of C1 compounds like methanol and methylated compounds containing amino, halide, and/or sulfur moieties in the oceans (4).
Many model methylotrophs have been cultured, which has facilitated studies on their genomic and functional characteristics and diversity (3). Among the marine methylotrophs, the OM43 clade belonging to the Betaproteobacteria occurs abundantly in productive aquatic environments from coastal waters to brackish and freshwater ecosystems (5, 6). The occurrence of close relatives of the marine OM43 clade in freshwater habitats, which form a closely related, but separate, lineage known as the LD28 clade (5, 7), also implies that OM43-like organisms have a broader biogeographical distribution. Surveys in the western Atlantic determined that OM43 clade members represented 5% of the bacterial clones retrieved (8). Furthermore, studies with specific fluorescence in situ hybridization (FISH) probes showed that about 4% of the 4′,6-diamidino-2-phenylindole (DAPI)-stained cells could be attributed to the OM43 clade in the North Sea, thus constituting an important fraction of the bacterioplankton in this area during spring but not in other seasons (9). In the northwestern Atlantic Ocean, 16S rRNA gene sequences belonging to OM43 bacteria were found during winter in a relative abundance of 3% (10). Other studies indicated that the abundance and distribution of this group seem to be associated with phytoplankton blooms (and consequently with primary productivity), where OM43 cell counts increased from below the limit of detection to 0.8 × 108 cells liter−1 on average, representing one of the dominant bacterial groups in surface bloom waters, which increased in population size almost 6-fold compared with that in nonbloom areas (11). Proteomic analyses showed the presence of OM43-like methanol dehydrogenase homologs (XoxF) in coastal environments such as the Gulf of Maine and the Chesapeake Bay (12) and the Oregon coast (13), where XoxF accounted for up to 2.3% of all identifiable peptides, suggesting that these bacteria indeed play an important role in the oxidation of methanol and other C1 compounds in coastal ecosystems (13).
The first OM43 isolate was obtained from the coastal waters off Oregon, using a dilution-to-extinction method with seawater-based low-nutrient medium (14). However, it has been noted that OM43 cells are difficult to maintain under laboratory conditions in seawater-based medium (5).
To date only two strains of the OM43 clade have been isolated and genome sequenced: strain HTCC2181, from the Oregon coast (5), and strain HIMB624 (also called KB13), isolated off the coast of Hawaii (15). These strains contain some of the smallest genomes among the free-living bacteria (∼1.3 Mbp), share more than two-thirds of their genomic repertoire, and belong to the family Methylophilaceae (15). How such a high level of genome (and probably functional) conservation is replicated in OM43 genotypes from contrasting oceanic provinces with diverse environmental conditions (i.e., from the tropics [15] to the Arctic [16]) remains to be elucidated. The Red Sea is a very unusual marine habitat with high UV irradiation and strong gradients in temperature and salinity, as well as generally low concentrations of nutrients (17), and thus represents an interesting study site to investigate the concomitant adaptations of marine microbes.
Here, we describe the isolation, basic physiology, and genomic traits of an OM43 isolate recovered from the ultraoligotrophic environment of the Red Sea (18) in comparison to those of the currently available reference OM43 genomes from divergent oceanic provinces and their closest freshwater relatives. Driven by the results of the genome comparisons, we also present an analysis of OM43 subgroup distribution and abundance retrieved from metagenomic data from disparate provinces, which gave valuable insights into the ecology of this clade. Functional differences in the genomes of the resulting two clusters are used to explain the ecotype differentiation.
MATERIALS AND METHODS
Water sample collection.
Seawater samples for medium preparation and inoculum were collected from the central Red Sea area (22°06.630N, 38°47.965E) at a depth of 20 m on 2 March, 2011, with a submersible pump. Collected samples were transported in 20-liter carboys that had been prewashed with distilled water and rinsed with seawater.
Culturing techniques.
Isolation was done by the dilution-to-extinction method, using a modified low-nutrient seawater-based medium (14, 19), consisting of a nonautoclaved, filtered (0.22 μm) seawater medium amended with the following nutrients: KH2PO4 (1 μM), (NH4)2SO4 (10 μM), 0.1× Gamborg's (1,000×) vitamins (20), and trace elements A (cupric sulfate, ferric citrate, sodium selenite, and zinc sulfate), B (ammonium molybdate, ammonium vanadate, manganese sulfate, nickel sulfate, sodium silicate, and stannous chloride), and C (aluminum chloride, barium acetate, cadmium chloride, chromic chloride, cobalt dichloride, germanium dioxide, potassium iodide, rubidium chloride, silver nitrate, sodium fluoride, and zirconyl chloride). The trace elements A, B, and C (catalog no. 99-182, 99-175, and 99-176, respectively, 1,000× concentration; Corning Cellgro) were used at a final concentration of 1× each. The medium was inoculated with seawater diluted to contain approximately 1 cell ml−1 and incubated at 28°C (simulating the in situ temperature) in five 96-well Teflon plates following the protocol of Stingl et al. (21).
Cell counts for the inoculum and screening of cultures were done by flow cytometry (Guava easyCyte flow cytometer; Millipore) as suggested for marine bacteria (22) using SYBR green (1× final concentration; Invitrogen, USA).
Screening, identification, and growth of isolates.
The cultures in each well were monitored and screened 4, 8, and 12 weeks postinoculation via cell counts and 16S rRNA gene-based PCR as described previously (23). A total of 48 wells out of five 96-well plates showed growth, from which we were able to identify 36 isolates based on 16S rRNA gene classification. The taxonomic identification was done using BLAST (24) and the RDP Classifier and SeqMatch tools (25). Twenty of these putative isolates (representing 56% of all wells identified) belonged to the order Methylophilales (based on the SeqMatch top scores, ranging from 0.952 to 0.992), and all shared 99.6 to 99.8% identity with the OM43 clade clone NB180505 (GenBank accession no. EU543497) based on NCBI BLASTN. The rest of the isolates recovered belonged to the orders Oceanospirillales (19%), Vibrionales (6%), Gammaproteobacteria and Rhodobacterales (5% each), and Pseudomonadales, Burkholderiales, and Rhizobiales (3% each). One OM43 culture was used as an inoculum for a second dilution-to-extinction experiment in order to obtain a pure culture, this time in autoclaved medium. From here, 16S rRNA gene amplicons from three putatively pure isolates of OM43 were purified with MinElute columns (Qiagen) and bidirectionally Sanger sequenced. These “subculture isolates,” named MBRS-F5, MBRS-G12, and MBRS-H7, were chosen after several rounds of transfers, purification steps, and success in growth. Subsequently, they were transferred into polycarbonate flasks for analysis of the growth rate and yield with (n = 3) and without (n = 3) the addition of methanol (100 μM) as previously described (5). Finally, cells were kept at −80°C in 10% glycerol and/or 5% dimethyl sulfoxide (DMSO) for long-term storage.
Genome sequencing, assembly, and annotation.
For whole-genome sequencing, each isolate was grown in 5 liters of autoclaved low-nutrient medium using 9-liter polycarbonate carboys (Nalgene), continuously sparged with sterile air at 28°C for 1 month until they reached maximum cell density (∼107 cells ml−1). Each individual culture was filtered through a 0.1-μm-pore-size membrane filter (PALL Life Sciences Supor-100), and DNA was extracted from the filters using a phenol-chloroform protocol (26) as modified by Jimenez-Infante et al. (23).
Sequencing was done using the Illumina GAII platform at the KAUST Bioscience Core Lab. Raw reads were checked using FastQC (27) and quality trimmed using Trimmomatic (28) before being assembled using SPAdes version 3.1.1 (29). The assembled contigs were subsequently annotated using the INDIGO pipeline (30) as previously described (23). The same automated annotation pipeline was used for the draft assemblies of the reference OM43 genomes (HTCC2181 [5] and KB13/HIMB624 [15]) in order to harmonize the gene-calling and functional prediction steps prior to comparative genomics. Because the genomes of MBRS-F5 and MBRS-G12 are identical or nearly identical to those of MBRS-H7 (average nucleotide identities [ANIs] of 99.99% and 100%, respectively), the further analysis and description here focus on isolate MBRS-H7 only.
Phylogenetic analysis.
The evolutionary relationship of the OM43 clade was determined based on single marker genes such as rRNA genes, the internal transcribed spacer (ITS), and the genes encoding xanthorhodopsin (XR) and on a concatenated alignment of 764 conserved single-copy genes (CSCG) shared with other representative Methylophilaceae members with sequenced genomes (see Table S1 in the supplemental material), complementing previous phylogeny analyses (31).
Substitution models for nucleotide and protein-based trees were predetermined from the aligned sequences using jModelTest2 (32) and ProtTest3 (33), respectively.
The phylogenies of the full-length rRNA genes (16S and 23S) were inferred using the GAMMA+I+GTR nucleotide substitution model, while a maximum-likelihood tree of the XR proteins was obtained using the GAMMA+WAG amino acid substitution model, conducted with PhyML as implemented in GeneiousPro vR9 (Biomatters, Ltd.) (34) with 1,000 bootstraps. In the latter case, the evolutionary position of XR-encoding genes from the OM43 strains was placed within the context of other rhodopsin families classified by Vollmers et al. (35). For ITS phylogeny, whole ITS sequences were subjected to BLAST searches against GenBank (nr), genomic survey sequences (gss), and environmental samples (env_nt) in GeneiousPro R9. The best BLAST hits matching regions were retrieved and aligned with MUSCLE. Then, a maximum-likelihood tree was constructed in CLC Genomics Workbench v8.5.1 (CLC bio) with GTR+G+T as the nucleotide substitution model. Redundant sequences were not considered. The aligned region had a size of 431 to 753 bp, and the tree was constructed based on a 233- to 593-bp extracted region.
Prior to core gene phylogenetic inference using the concatenated CSCG, we first identified orthologous genes across 17 genomes of methylotrophic bacteria (see Table S1 in the supplemental material) using the automated phylogenomic pipeline Hal (36). The subsequent 764 core gene-based alignment was used for constructing a maximum-likelihood tree (100 bootstraps) with FastTree v2.1.5 as implemented in GeneiousPro v8.17 (34) with the settings Fastest and WAG and optimization of the Gamma20 likelihood.
Fragment recruitment analysis.
An approximation of the global abundance and distribution of the OM43 clade was determined by fragment recruitment analyses of the two reference OM43 genomes (HTCC2181 and KB13) and MBRS-H7 as the representative for the Red Sea OM43 genomes as queries against marine metagenomic data sets, including the Red Sea, Global Ocean Sampling (GOS), Hawaii Ocean Time Series (HOT), Bermuda Atlantic Time Series (BATS), and Eastern Tropical South Pacific (ETSP), among others (see Table S2 in the supplemental material). Prior to recruitment, the 5S, 16S, and 23S rRNA regions were masked in each of the reference genomes. Fragment recruitment was performed using BLASTN as described by Jimenez-Infante et al. (23) with the modification that the best matches were considered to have length and sequence identity values greater than 200 bp and 85%, respectively.
Comparative genomics.
Similarities and differences among the genome of MBRS-H7, the two previously sequenced genomes of marine OM43 strains HTCC2181 and KB13, and the closely related freshwater methylotrophs “Candidatus Methylopumilus planktonicus” MMS-2-53 and “Candidatus Methylopumilus turicensis” MMS-10A-171 were evaluated using the EDGAR comparative analysis tool (37). For metabolic characterization, the predicted proteins from the three available OM43 genomes were subjected to BLAST searches and visualized in the KEGG Automatic Annotation Server (KAAS) (38). Membrane transporters for each OM43 clade strain were determined using the TransportTP transporter prediction server (39) with an E value threshold of 10−5.
ANIs between all OM43 strains were determined using JSpecies (40) with default BLASTN parameters. Additionally, in silico DNA-DNA hybridizations (DDH) of all closely related reference genomes against H7 were obtained with the genome-to-genome calculation tool (GGDC 2.0 BLAST+ alignment; http://ggdc.dsmz.de/distcalc2.php) (41), and the results were analyzed based on the recommended parameters (formula 2).
Nucleotide sequence accession numbers.
The draft genome of MBRS-H7 has been deposited in GenBank as MBRSH7 under the accession number CP011002. The genome and corresponding annotations used in this study are available in INDIGO (http://www.cbrc.kaust.edu.sa/indigo/dataCategories.do) with database identifier MBRSH7. Other OM43 subcultures identical to MBRSH7 obtained in this study have genomes available in INDIGO as MBRSF5 and MBRSG12 and in GenBank under the accession numbers CP011001 and CP011003, respectively.
RESULTS
General characteristics of the Red Sea isolate.
This study describes an isolate belonging to the OM43 clade (here designated MBRS-H7), which was obtained using the dilution-to-extinction cultivation technique. MBRS-H7 was obtained from a surface water sample of the oligotrophic Red Sea and has cells that consist of curved rods ranging in size from 0.4 to 0.9 μm in length and from 0.15 to 0.3 μm in diameter (see Fig. S1 in the supplemental material). The 16S rRNA gene of MBRS-H7 has nucleotide identities of 99.9% and 96.5% to strains KB13 (from Hawaii) and HTCC2181 (from Oregon), respectively. MBRS-H7 showed a higher cell biomass in the presence of methanol (100 μM) of 1.3 × 107 cells ml−1 than with nonmethanol treatment (7.5 × 105 cells ml−1) (see Fig. S2 in the supplemental material), similar to previous findings for the strains HTCC2181 (5) and KB13 (15). Likewise, the growth rate was estimated to be greater with methanol at 1.5 ± 0.07 generation day−1 (± standard deviation [SD]) (n = 3), which is in a range similar to that for strain HTCC2181 (5, 42).
Evolutionary relationships of the OM43 clade with other methylotrophic bacteria.
Previous phylogenetic analyses based on the 16S rRNA genes positioned the OM43 members within the Methylophilaceae, which are characterized as obligate type I methylotrophs, with the closest cultured representatives among Methylotenera and Methylophilus species (5). In order to get a more refined phylogenetic placement of the marine OM43 clade members and these related Betaproteobacteria, we constructed a genome-based tree using a set of 764 conserved single-copy genes that we found to be universal in the sequenced genomes of 14 Methylophilaceae bacteria and the three OM43 genomes (Fig. 1; see also Table S1 in the supplemental material). Similar to the 16S and 23S rRNA gene-based phylogenies (see Fig. S3 in the supplemental material), the topology of this core gene tree supports the placement of OM43 clade members as a separate lineage diverging from the Methylophilaceae family and forming a group separate from that of their freshwater relatives. The marine OM43 clade also clustered in two subclades, encompassing isolates from the Hawaii and the Red Sea (or the Hawaii-Red Sea [H-RS] clade) and the HTCC2181 clade (Fig. 1; see also Fig. S3), which are closely related to “Ca. Methylopumilus planktonicus” MMS-253 (LD28 group). All current isolates within the OM43 clade originate from marine environments, have the smallest genomes among the sequenced methylotrophic bacteria, and harbor genes for the utilization of methanol and formaldehyde but not methylamine.
FIG 1.

Core genome phylogeny of Methylophilaceae bacteria based on maximum-likelihood phylogeny (n = 100 bootstraps) of 764 concatenated single-copy genes conserved in 17 representative genomes, including the OM43 isolates and closely related “Ca. Methylopumilus” strains. The main differences between the OM43 and closely related organisms are highlighted. The genome size is shown in parentheses. Accession numbers for each genome can be found in Table S1 in the supplemental material.
Like those for the other two strains in the OM43 clade, the genome of MBRS-H7 is predicted to encode xanthorhodopsin (XR), which is a variant of rhodopsin (light-driven proton-pumping proteins) with a potential for photoheterotropy (5). In relation to the overall Methylophilaceae bacteria described here, this genomic trait appeared to be exclusive to the OM43 clade and LD28 (“Ca. Methylopumilus planktonicus” MMS-2-53). Our BLAST analyses also identified proteorhodopsin genes for the freshwater isolates “Ca. Methylopumilus planktonicus” MMS-2-53 and “Ca. Methylopumilus turicensis” (Fig. 1; see also Fig. S4 in the supplemental material). Interestingly, the phylogenetic analysis of the XR proteins shows that the XR from the marine OM43 clade falls within the type II XR lineage (see Fig. S4), which is present in mesophilic and psychrophilic marine microbes and has the characteristic amino acid Leu105 for the absorption of green light (43). The type I XRs in contrast seem to be mostly found in mesophilic and thermophilic organisms (35).
Ecotype characterization and biogeography of the OM43 clade.
In order to elucidate whether OM43 members potentially exhibit a clade-specific biogeography distribution pattern that might be co-related to the physicochemical conditions of their specific environments, we conducted two analyses based first on the phylogeny of the 16S-23S internal transcribed spacer (ITS) region retrieved from OM43 genomes and environmental genomic sequence data sets and second on the genomic fragment recruitment from several other metagenome data sets from water columns of various oceans (see Table S2 in the supplemental material), including sequences from GOS (44, 45). In the first approach, using the ITS loci, which have been shown to give very good phylogenetic resolution of closely related species (46, 47), we found a more refined separation of the OM43 clade into two putative clusters with an average interclade sequence distance of 12.4%, namely, cluster A, which is further divided into two subgroups A1 (also containing strain HTCC2181) and the novel A2, and cluster B that contains the Red Sea and Hawaii strains (Fig. 2). BLAST searches of OM43-like ITS sequences in the public NCBI databases for environmental sequences indicated that very few homologues were present. The majority of the ITS sequences were affiliated with cluster A and included sequences from the surface and deep waters of the San Pedro Time Series (SPOT) and the northeast (NE) subarctic Pacific Ocean (Fig. 2). This pattern differs significantly from that of 16S rRNA genes, for which many OM43-like sequences have been published (data not shown), essentially reflecting the paucity of OM43 genomic fragments in public databases compared to those for PCR-based 16S rRNA gene analyses.
FIG 2.
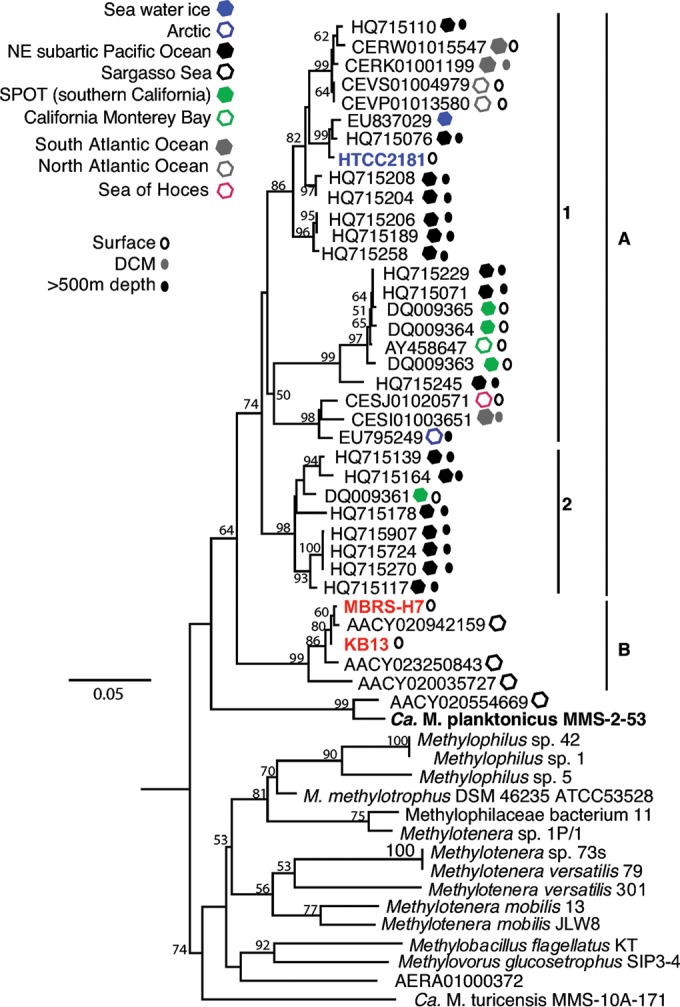
PhyML phylogenetic tree (n = 1,000 bootstraps) of the 16S-23S rRNA ITS region showing the relationships between the OM43 isolates, closely related isolates, and environmental samples. Closely related isolates are shown in bold. Red, H-RS cluster; blue, HTCC2181; black, LD28.
The microdiversity detected through ITS-based phylogeny was also reproduced in results based on fragment recruitment analyses of OM43 genomes against diverse surface water metagenomic data sets from GOS (Fig. 3; see also Fig. S5 in the supplemental material). Here, only very few OM43-like homologs could be recruited at ≥85% sequence identity against all three OM43 genomes (0.01 to 0.18% coverage per Mbp in 12 out of 88 samples), showing that the members of this clade are in low abundance in most pelagic habitats. Based on the few GOS samples in which OM43-like fragments were detected, we found the potential for an environmentally driven abundance and distribution of genotypes in subclades A and B (HTCC2181-like and the H-RS cluster); that is, HTCC2181 recruited better in samples with a high chlorophyll content but lower temperatures, whereas the recruitment results for genotypes of the H-RS cluster were greater in warmer areas (Fig. 3). Analyses based on samples covering multiple depths of the water columns from different oceanic provinces also proved that the OM43 clade members were rare (<0.01% per Mbp of metagenomic data) and showed a preference for the epipelagic zone, particularly for the H-RS “ecotype” (see Fig. S5 in the supplemental material). In all cases, the average identity of the recruited metagenomic reads was 85 to 90%, suggesting the presence of divergent genotypes in the samples.
FIG 3.
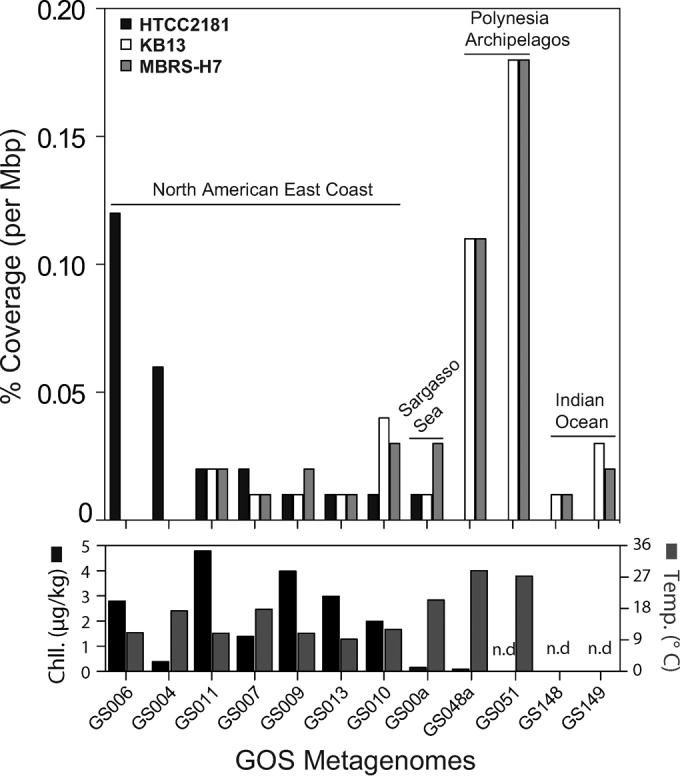
Fragment recruitment analysis comparing strains HTCC2181, KB13, and MBRS-H7 against metagenomes from the Global Ocean Sampling (GOS). The recruitment values were compared in parallel to chlorophyll (Chll.) concentrations and temperature. The GOS sampling sites and metadata can be found in Table S2 in the supplemental material. n.d, not determined.
Comparative genomics of the OM43 clade and freshwater relatives.
In order to elucidate potential metabolic differences that might explain the different levels of fitness of the two subclades, we analyzed all three available genomes of the OM43 clade. A summary of the genome characteristics of these isolates compared to those of their freshwater relatives is shown in Table 1. The draft genome of MBRS-H7 is similar in size to the KB13 genome (ca. 1.35 Mbp) and has the lowest G+C content compared to those of HTCC2181 and the freshwater MMS-2-53 (LD28) and MMS-10A-171. Relative to the existing reference marine OM43 genomes, we found that MBRS-H7 was more similar to KB13 (94.19% ANI) than to HTCC2181 (65.95% ANI). Given the high sequence similarity among the genomes of KB13 and MBRS-H7 that is just below the recommended value for species designation (95% ANI threshold) (48), we performed in silico DNA-DNA hybridization that was equivalent to 57%, which added further evidence to the hypothesis that they might represent different species. Obviously, physiological and biochemical tests need to be done in order to test the hypothesis.
TABLE 1.
General characteristics of isolates from the marine OM43 and related freshwater “Ca. Methylopumilus” strains
| Parameter | Result for strain: |
||||
|---|---|---|---|---|---|
| HTCC2181a | KB13a | MBRS-H7 | MMS-2-53b | MMS-10A-171b | |
| No. of contigs | 3 | 1 | 1 | 1 | 1 |
| Genome size (bp) | 1,304,428 | 1,334,326 | 1,352,785 | 1,356,428 | 1,754,988 |
| GC (%) | 37.9 | 35.4 | 35.5 | 36.97 | 44.52 |
| ORF | 1,320 | 1,382 | 1,377 | 1,389 | 1,757 |
| tRNAs | 36 | 36 | 36 | 36 | 39 |
| Isolation point | Oregon | Hawaii | Red Sea | Zurich | Zurich |
| Isolation source | Seawater | Seawater | Seawater | Lake | Lake |
| ANIc (%) | |||||
| With respect to HTCC2181 | 100 | 66.34 | 65.95 | 65.98 | 65.48 |
| With respect to KB13 | 66.26 | 100 | 94.19 | 65.83 | 66.14 |
| Growth rate (generations per day)d | 1.5 | NDe | 1.5 | 0.37 | ND |
Genomes were reannotated in the INDIGO pipeline. Open reading frame (ORF) and RNA predictions may vary from those in the original data.
MMS-2-53, “Ca. Methylopumilus planktonicus”; MMS-10A-171, “Ca. Methylopumilus turicensis” (7).
ANI, average nucleotide identity.
ND, not determined.
Core metabolic functions conserved among the OM43 clade and freshwater relatives.
Overall, the core genome of OM43, “Ca. Methylopumilus planktonicus,” and “Ca. Methylopumilus turicensis” is estimated to encompass a set of 643 protein-coding genes (Fig. 4; see also Table S3 in the supplemental material), which corresponds to 36 to 49% of their predicted genes.
FIG 4.

Comparative analysis of the predicted proteomes of the OM43 clade and their closest freshwater relatives. The Venn diagram shows the core, unique, and flexible genome components among strains HTCC2181, KB13, MBRS-H7 (Red Sea isolate), MMS-2-53, and MMS-10A-171.
As reported for the marine OM43 clade (5, 15) and freshwater relatives (7), MBRS-H7 is also predicted to encode an incomplete tricarboxylic acid (TCA) cycle, lacking putative genes coding for alpha-ketoglutarate dehydrogenase. In addition, it is predicted to encode a malate (quinone) dehydrogenase (EC 1.1.5.4) for oxaloacetate production, instead of the canonical malate dehydrogenase (EC 1.1.1.37). Glycolysis is predicted to occur via the Entner-Doudoroff pathway, as previously described in the reference genomes (5, 7, 15). HTCC2181, MBRS-H7, and MMS-10A-171 harbor genes for sulfate uptake and for the assimilatory reduction to sulfide through adenylyl sulfate (APS) into 3′-phosphoadenylyl sulfate (PAPS); strains KB13 and MMS-2-53 are predicted to lack the genes encoding APS kinase (cysC) (EC 2.7.1.25). The enzyme PAPS reductase (gene cysH) (EC 1.8.4.8) is present in all isolates, but this gene is nonorthologous in HTCC2181 to those in the KB13, MBRS-H7, and freshwater relatives (22 to 26% identity).
All members of the OM43 clade are predicted to possess a homologue of the methanol dehydrogenase (MDH) large subunit encoded by the xoxF gene. Its presence has been detected in all Methylophilaceae described so far (7, 31, 56), and its role in methanol oxidation has also been demonstrated in members of this family, such as Methylotenera mobilis JLW8, and even in more divergent model methylotrophs, such as Methylobacterium extorquens AM1 (49, 50). The predicted XoxF protein from the Red Sea strain MBRS-H7 is 86% identical (amino acid sequence level) to that in M. mobilis and is 84%, 88%, 90%, and 97% identical to those in MMS-10A-171, MMS-2-53, HTCC2181, and KB13, respectively. The key enzymes used for biomass incorporation via the ribulose monophosphate (RUMP) cycle are also part of their core genome, including 3-hexulose-6-phosphate synthase (EC 4.1.2.43) and 6-phospho-3-hexuloisomerase (EC 5.3.1.27). The transfer of the C1 moiety from formaldehyde can also occur via a tetrahydrofolate (H4F)-dependent pathway that is also present in the OM43 core genome.
Other conserved genes in the marine and freshwater strains include those putatively encoding the high-affinity phosphate-selective transporter (pstSCAB), a ferredoxin-dependent sulfite/nitrite reductase, the UvrABCD DNA repair system, and genes for the anaplerotic regeneration of oxaloacetate using pyruvate carboxylase (EC 6.4.1.1). All strains are also predicted to have the potential to synthesize all 20 amino acids.
Except for the RnfG subunit (which was not found in MMS-10A-171), the core genome also encodes a RnfABCDGE system (Fig. 5), used to translocate sodium generating an electrochemical gradient, which presumably acts coupled to different energy-generating systems found for each subcluster (described below).
FIG 5.

Proposed scheme for methanol oxidation coupled to the different energy generation complexes in HTCC2181 and Hawaii-Red Sea (H-RS) isolates. OM43 core elements in the electron transport chain (top panel) are shown in black and unique components (bottom panels) are shown in gray (H-RS cluster) and white (HTCC2181). *, MBRS-H7 is missing the nuoA subunit.
Flexible gene sets of the H-RS OM43 cluster.
As we deduced previously, the OM43 strains from Hawaii (KB13) and the Red Sea (MBRS-H7) form a phylogenomic lineage (H-RS cluster) separate from that of strain HTCC2181 (Fig. 1). Accordingly, we found 329 proteins that are exclusive to this cluster, which equate to about 24% of the predicted proteomes (see Table S4 in the supplemental material).
Functionally, this H-RS flexible genome is predicted to carry a complete operon for molybdate transport (modABC). It also harbors several genes encoding enzymes involved in combating osmotic stress, including those for ectoine biosynthesis (ectABC), and a transporter for proline uptake using a putative sodium-proline symporter (see Tables S4 and S5 in the supplemental material), as well as a pdxH gene for the synthesis of vitamin B6 (Table 2; see also Table S4).
TABLE 2.
Gene inventory differentiating marine OM43 and freshwater “Ca. Methylopumilus” relatives
| Component [gene(s)] | Results for straina: |
||||
|---|---|---|---|---|---|
| HTCC 2181 | KB13 | MBRS-H7 | MMS-2-53 | MMS-10A-171 | |
| Molybdate transport (modABC) | − | + | + | − | − |
| NADH:quinone oxidoreductase (nuoA to nuoN) | − | + | +b | +c | +c |
| Cytochrome bd (cydAB) oxidase complex | − | + | + | − | − |
| Ectoine biosynthesis (ectABC) | − | + | + | − | − |
| Pyridoxamine 5′-phosphate oxidase (pdxH) | − | + | + | − | − |
| Adenylyl-sulfate kinase | + | − | + | − | + |
| Phosphate starvation-inducible protein (psiE) | − | 1 | 2 | 1 | 1 |
| CzcA family heavy metal efflux protein | − | 1 | 2 | 1 | 1 |
| Co-Zn-Cd efflux system component | − | − | + | − | − |
| Copper-translocating P-type ATPase | − | − | + | − | − |
| Cold shock (and UV/salinity resistance) (cspD) | − | − | 1 | − | − |
| Cold shock protein (cspA) | 2 | 1 | 1 | 1 | 1 |
| Na+-pumping NADH-quinone oxidoreductase (nqrA to nqrF) | + | − | − | − | − |
| Uracil permease protein (pyrP) | + | − | − | − | − |
| Sodium-alanine symporter (agcS) | + | − | − | − | − |
| Sulfate permease YbaR (SulP family) transporter | 2 | 1 | − | − | − |
Presence or absence is indicated by a + or − symbol or with numeral values, when variable copies are present. Note that presence does not always imply orthology.
Missing nuoA.
nuoG is nonorthologous to OM43.
Interestingly, while the genome of HTCC2181 is predicted to encode an Na+-pumping NADH:quinone oxidoreductase (NQR), both KB13 and MBRS-H7 genomes putatively encode an operon of the analogous (but not homologous) H+-pumping NADH:quinone oxidoreductase complex I (NUO). KB13 harbors all 14 central nuo subunits (nuoA to nuoN) that are necessary to perform all bioenergetic functions, while the genome of MBRS-H7 is missing only the nuoA gene. This implies that the two OM43 subclades employ two different electron translocation pathways (from NADH to a ubiquinone) with the simple NQR system in the HTCC2181 clade and the more elaborate complex I system in the H-RS cluster. A similar NUO system was also identified in the freshwater strains MMS-2-53 and MMS-10A-171, with a nonorthologous subunit G.
In BLAST-based searches (data not shown), the NQR system from HTCC2181 had the best hits with members of Alphaproteobacteria from the Rhodobacteraceae family, suggesting that these systems may have been horizontally acquired. The opposite was found for the H-RS NUO system, which has the highest identities with members of the Methylophilaceae family within the Betaproteobacteria, as expected.
The electrons from both systems are transferred via the quinone pool to the low-affinity aa3-type heme copper cytochrome oxidase, which is also present in MMS-2-43 but not in MMS-10A-171. In the case of the H-RS clade, oxidative phosphorylation appears also to be complemented by the bd-type quinol oxidase (cydAB), which produces a proton motive force by using quinols to reduce oxygen to water and translocating protons to the periplasmic space (reference 51 and references therein) and was not present in the HTCC2181 or the “Ca. Methylopumilus” strains. A proposed scheme of the differences and similarities in the electron transport chain in the H-RS cluster and HTCC2181 is shown in Fig. 5.
Red Sea-specific OM43 genes.
The genome of the Red Sea strain MBRS-H7 possesses 115 genes that are unique, of which 41 are hypothetical genes, while the rest are predicted to confer resistance to different stress conditions (see Table S6 in the supplemental material), including those for copper and heavy metal transport, and high-temperature tolerance such as the cold shock protein (cspD), whose expression can be induced by increasing temperatures and also by UV exposure in psychrophilic organisms (52) or by osmotic stress in some pathogenic bacteria (53). Several proteins related to membrane and cell wall structure, including an array of glycosyl transferases and genes involved in lipopolysaccharide biosynthesis or transmembrane transport, are also unique to the Red Sea strains (see Table S6).
A small genomic island of 9,102 bp harboring genes encoding toxic metal resistance proteins was also detected using the Web tool IslandViewer (54). This “resistance island” contains heavy metal efflux proteins, a copper-translocating ATPase, a Co-Zn-Cd efflux protein, and the heavy metal transcriptional activator, MerR, in close proximity to a phage transcriptional regulator and a site-specific recombinase (see Table S6 in the supplemental material). Interestingly, this 9-kbp genomic island is highly identical (96% nucleotide identity with 78% coverage) to the plasmid in Shewanella sp. (GenBank accession no. CP000740), which implies that it may have been horizontally acquired and is integrated in the MBRS-H7 genome.
Additionally, two paralogous genes encoding a putative phosphate starvation-inducible gene E (psiE) were present in the Red Sea OM43 (but only one in KB13) (Table 2). Although its function remains undetermined, experimental validation in Escherichia coli showed that PhoB positively regulates psiE under phosphate starvation conditions (55). Other prominent differences in gene content among the marine OM43 isolates and their freshwater relatives are highlighted in Table 2, showing the presence/absence and variations in gene copy numbers of functionally important genes, thus reflecting possible adaptations to the specific environments where they were isolated.
DISCUSSION
In this study, we describe a novel methylotrophic OM43 isolate from the surface waters of the Red Sea and provide further genomic evidence for the differentiation of the OM43 clade into two subclades, one containing this isolate (and that from Hawaii) and the other one containing the Oregon coast isolate (HTCC2181).
Previous studies have shown that the methylotrophic bacteria of the family Methylophilaceae have great metabolic versatility among different groups (and even species) and show variations in their physiologic and genomic traits (56, 57). This family also encompasses the marine OM43 clades and their closest freshwater relatives LD28 (“Ca. Methylopumilus planktonicus” MMS-2-53) and PRD01a001B (“Ca. Methylopumilus turicensis” MMS-10A-171), whose core genomes (described here) retain the main characteristics of the Methylophilaceae (3), such as the presence of XoxF, a homolog of the methanol dehydrogenase acting on methanol oxidation, and genes for formaldehyde utilization.
Xanthorhodopsins, which were present in the OM43 core genome, were also found in LD28 but not in other Methylophilaceae or any other main methylotrophs, including marine and freshwater isolates encompassing diverse taxonomic groups (data not shown), potentially representing an exceptional trait of the marine OM43 and LD28 cells among methylotrophic bacteria. In particular, since XR constitutes only 1 to 2% of the total marine rhodopsins and seems to be much more abundant in freshwater ecosystems, hot springs, and hypersaline environments (35), this positions the OM43 and LD28 clades among the few that harbor this gene, presumably as a strategy for photoheterotrophic growth under oligotrophic conditions (58). This also corroborates the observation that OM43 members have originated from the freshwater lineages as proposed recently (7), and based on our phylogeny and genome comparisons, they originate most likely from the LD28 (“Ca. Methylopumilus planktonicus” MMS-2-53), sharing more predicted proteins in common (51 proteins) (Fig. 4), which are not present in the freshwater MMS-10A-171, including the XR ortholog. XR's function as a light-driven proton pump has been observed (with no concomitant growth advantage) by heterologous expression of Octadecabacter XR in Escherichia coli (35); nevertheless, its role in photoheterotrophy or survival in OM43 and other bacterioplankton groups remains unanswered.
ITS classification, including environmental sequences, showed a diversification within this clade, with HTCC2181-like members (subclade A1 and subclusters within; Fig. 2), a group with no culture representatives (subclade A2; Fig. 2), and the H-RS cluster (subclade B; Fig. 2). Such an ecotype diversification has been described for other members of the Methylophilaceae family (31) and may reflect a possible niche-specific adaptation of subgroups of OM43, as observed in other marine bacteria, such as the ubiquitous SAR11 clade (59–61) and Prochlorococcus marinus (62, 63), among others. For instance, some Prochlorococcus species with >97% identity at the 16S rRNA gene level may at the same time display significant genomic differences, forming ecotypes that differ in their habitat light regimes (high-light- versus low-light-adapted ecotypes), pigment contents, nitrogen utilization, and cyanophage specificity, among other characteristics defined by the components of the flexible genome (63).
At the genome level, several differences were found among these OM43 clade ecotype clusters, which might be responsible for the ecological success of the different subgroups in divergent marine provinces. Specifically, the H-RS cluster showed distinctive features for cofactor acquisition and/or biosynthesis (Table 2; see also Table S5 in the supplemental material). Molybdate is being transported by a high-affinity system (modABC) that may also be able to transport sulfate (64), giving an extra molybdenum and sulfur source for H-RS bacteria. Vitamin B6, a cofactor synthesized by pyridoxamine 5′-phosphate oxidase (gene pdxH), is involved in many biological reactions in the central metabolism, but its concentration in the marine environments can range from undetectable to picomoles per liter (reference 65 and references therein). It is also involved in tolerance to oxidative stress in some eukaryotes such as plants and yeast (66) and could play a protective role in the H-RS cluster.
Other stress resistance-related genes uniquely found in the H-RS cluster are the ectABC genes for ectoine biosynthesis. These genes for the biosynthesis of this important osmoprotectant are mainly present in halophilic microorganisms from the Alphaproteobacteria, Gammaproteobacteria, and Actinobacteria and in a limited number of Beta-, Delta-, and Epsilonproteobacteria (67). Thus, organisms in the H-RS cluster represent some of the few betaproteobacterial organisms that are able to biosynthesize ectoine to cope with changing and/or extreme conditions in the marine environment. Apart from acting in osmoregulation, ectoine has other protective properties against temperature (high or freezing), UV radiation, and cytotoxins (reference 68 and references therein). These features may be of great importance for H-RS members in warm environments with high salinities such as the Red Sea (∼40 practical salinity units [psu]) and Kaneohe Bay, HI (∼35 psu) (69), in contrast to HTCC2181 isolated from the Oregon coast (<31 psu) (70), which has a significant input of freshwater from the Columbia River.
Nevertheless, even without biosynthetic pathways for ectoine (or other osmolytes), HTCC2181-like bacteria could theoretically still cope with the high-salinity conditions. This may be possible due to the presence of the agcS gene (sodium-alanine symporter AGCS family) (Fig. 5; Table 2) for the uptake of alanine (another common compatible solute). Furthermore, we presume that osmoregulation in HTCC2181 may be coupled to sodium translocation through the NQR system (nqrA to nqrF genes) (Fig. 5), which is exclusively found in marine and pathogenic bacteria, and was present in HTCC2181 only. Probably originating from the RnfABCDGE sodium-translocating system (71), the Na+-NQR complex generates energy by using an electrochemical sodium gradient, or sodium motive force (SMF), during oxidative phosphorylation, which helps the cells to maintain homeostasis in the saline marine environment (72).
On the contrary, the H-RS cluster possesses a type I respiratory complex also found in other bacteria and in mitochondria (nuoA to nuoN genes) (Fig. 5) that pumps protons against an electrochemical gradient, generating proton motive force (PMF) from which about 40% is used to synthesize ATP (reference 73 and references therein). While complex I is able to pump 3 to 4 H+/2 e−, the Na+-NQR complex pumps 2 Na+/2 e− (74), creating a lower electrochemical gradient. This indicates that members of the H-RS cluster have a (presumably) more efficient system for energy generation compared to that of HTCC2181. In addition, the presence of a cytochrome bd-I (cydAB) complex (Fig. 5) may provide additional energy generation for the H-RS cluster by the vectorial transport of protons through the membrane, generating a PMF (reference 51 and references therein). Given its high affinity to oxygen, it can allow colonization in oxygen-limited environments, and it also confers resistance to stressful conditions such as the presence of cyanide or nitric oxide (reference 75 and references therein). BLAST searches indicated that the cytochrome bd-I (cydAB) from the H-RS cluster has high amino acid identities with members of the Alpha- and Gammaproteobacteria, indicating a possible horizontal acquisition of this complex.
These differences between energy-generating systems in both OM43 subclusters might be important as niche-specific adaptations. For example, a high-energy yield is necessary in oligotrophic environments such as the Red Sea and Kaneohe Bay, where nutrients are scarce as opposed to coastal, highly productive areas where HTCC2181-like members seem to be more abundant.
With their very streamlined genomes, the HTCC2181-like bacteria are presumed to have undergone gene loss of the respiratory complex I (NUO) and have acquired a more simple system, the NQR. It is also presumed that NQR originates from the common ancestor of the Chlorobi and Bacteroidetes groups and has been horizontally transferred in multiple events among divergent taxa (71). Here, we report different energy-generation machineries in members of the same group (OM43). These differences may be important in ecotype formation and the occupation of different niches of each OM43 subcluster. Some advantages may be found in the H-RS cluster members possessing the NUO system, with putatively higher ATP yields that may be necessary for energy-demanding functions such as the ectoine biosynthesis (not found in HTCC2181), needed to cope better with more saline conditions in the marine environment.
Additional adaptive traits were found as part of the unique component of the Red Sea isolate MBRS-H7, for instance, the presence of genes associated with resistance to heat, UV radiation, and probably osmoprotection that help them to cope with an environment of high temperatures, UV exposure, and salinity (76). A “resistance” island with high similarity to a plasmid in Shewanella sp. may be the result of horizontal gene transfer as an adaptation to high concentrations of metals, such as Cu, Zn, and Cd, among others, that have been associated with the anthropogenic impact in the coastal areas of the Red Sea (77, 78) and may also affect the near offshore areas from which the strains were isolated.
These potential niche adaptations described through the comparative genomics of a few OM43 isolates were also reflected in the distribution and abundance of this bacterial clade along different marine provinces. Fragment recruitment analyses showed a clear dominance of members of the H-RS cluster in higher temperatures and less productive environments than HTCC2181, which showed an opposite trend. Nevertheless, the low recruitment in GOS and other metagenomic data sets from disparate provinces indicates that the OM43 clade and the H-RS cluster in particular may not be as abundant as previously thought. Additional isolates or single-cell genomes of marine OM43 populations from divergent marine habitats are needed to further test these hypotheses.
Supplementary Material
ACKNOWLEDGMENT
We thank the staff of the Bioscience Core Lab at KAUST for the library construction and sequencing.
Funding Statement
This study was sponsored by King Abdullah University and Technology (KAUST) through the CRG-2 program.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02852-15.
REFERENCES
- 1.Anthony C. 1982. The biochemistry of methylotrophs. Academic Press, London, United Kingdom. [Google Scholar]
- 2.Chistoserdova L, Lidstrom M. 2013. Aerobic methylotrophic prokaryotes, p 267–285. In Rosenberg E, DeLong E, Lory S, Stackebrandt E, Thompson F (ed), The prokaryotes. Springer, Berlin, Germany. [Google Scholar]
- 3.Chistoserdova L, Kalyuzhnaya MG, Lidstrom ME. 2009. The expanding world of methylotrophic metabolism. Annu Rev Microbiol 63:477–499. doi: 10.1146/annurev.micro.091208.073600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neufeld JD, Boden R, Moussard H, Schafer H, Murrell JC. 2008. Substrate-specific clades of active marine methylotrophs associated with a phytoplankton bloom in a temperate coastal environment. Appl Environ Microbiol 74:7321–7328. doi: 10.1128/AEM.01266-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giovannoni SJ, Hayakawa DH, Tripp HJ, Stingl U, Givan SA, Cho JC, Oh HM, Kitner JB, Vergin KL, Rappe MS. 2008. The small genome of an abundant coastal ocean methylotroph. Environ Microbiol 10:1771–1782. doi: 10.1111/j.1462-2920.2008.01598.x. [DOI] [PubMed] [Google Scholar]
- 6.Rappé MS, Vergin K, Giovannoni SJ. 2000. Phylogenetic comparisons of a coastal bacterioplankton community with its counterparts in open ocean and freshwater systems. FEMS Microbiol Ecol 33:219–232. doi: 10.1111/j.1574-6941.2000.tb00744.x. [DOI] [PubMed] [Google Scholar]
- 7.Salcher MM, Neuenschwander SM, Posch T, Pernthaler J. 2015. The ecology of pelagic freshwater methylotrophs assessed by a high-resolution monitoring and isolation campaign. ISME J 9:2442−2453. doi: 10.1038/ismej.2015.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rappe MS, Kemp PF, Giovannoni SJ. 1997. Phylogenetic diversity of marine coastal picoplankton 16S rRNA genes cloned from the continental shelf off Cape Hatteras, North Carolina. Limnol Oceanogr 42:811–826. doi: 10.4319/lo.1997.42.5.0811. [DOI] [Google Scholar]
- 9.Sekar R, Fuchs BM, Amann R, Pernthaler J. 2004. Flow sorting of marine bacterioplankton after fluorescence in situ hybridization. Appl Environ Microbiol 70:6210–6219. doi: 10.1128/AEM.70.10.6210-6219.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Georges AA, El-Swais H, Craig SE, Li WK, Walsh DA. 2014. Metaproteomic analysis of a winter to spring succession in coastal northwest Atlantic Ocean microbial plankton. ISME J 8:1301–1313. doi: 10.1038/ismej.2013.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris RM, Longnecker K, Giovannoni SJ. 2006. Pirellula and OM43 are among the dominant lineages identified in an Oregon coast diatom bloom. Environ Microbiol 8:1361–1370. doi: 10.1111/j.1462-2920.2006.01029.x. [DOI] [PubMed] [Google Scholar]
- 12.Hanson BT, Hewson I, Madsen EL. 2014. Metaproteomic survey of six aquatic habitats: discovering the identities of microbial populations active in biogeochemical cycling. Microb Ecol 67:520–539. doi: 10.1007/s00248-013-0346-5. [DOI] [PubMed] [Google Scholar]
- 13.Sowell SM, Abraham PE, Shah M, Verberkmoes NC, Smith DP, Barofsky DF, Giovannoni SJ. 2011. Environmental proteomics of microbial plankton in a highly productive coastal upwelling system. ISME J 5:856–865. doi: 10.1038/ismej.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Connon SA, Giovannoni SJ. 2002. High-throughput methods for culturing microorganisms in very-low-nutrient media yield diverse new marine isolates. Appl Environ Microbiol 68:3878–3885. doi: 10.1128/AEM.68.8.3878-3885.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huggett MJ, Hayakawa DH, Rappe MS. 2012. Genome sequence of strain HIMB624, a cultured representative from the OM43 clade of marine Betaproteobacteria. Stand Genomic Sci 6:11–20. doi: 10.4056/sigs.2305090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins RE, Rocap G, Deming JW. 2010. Persistence of bacterial and archaeal communities in sea ice through an Arctic winter. Environ Microbiol 12:1828–1841. doi: 10.1111/j.1462-2920.2010.02179.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ngugi DK, Antunes A, Brune A, Stingl U. 2012. Biogeography of pelagic bacterioplankton across an antagonistic temperature-salinity gradient in the Red Sea. Mol Ecol 21:388–405. doi: 10.1111/j.1365-294X.2011.05378.x. [DOI] [PubMed] [Google Scholar]
- 18.Berninger UG, Wickham SA. 2005. Response of the microbial food web to manipulation of nutrients and grazers in the oligotrophic Gulf of Aqaba and northern Red Sea. Mar Biol 147:1017–1032. doi: 10.1007/s00227-005-1565-1. [DOI] [Google Scholar]
- 19.Button DK, Schut F, Quang P, Martin R, Robertson BR. 1993. Viability and isolation of marine bacteria by dilution culture: theory, procedures, and initial results. Appl Environ Microbiol 59:881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rappé MS, Connon SA, Vergin KL, Giovannoni SJ. 2002. Cultivation of the ubiquitous SAR11 marine bacterioplankton clade. Nature 418:630–633. doi: 10.1038/nature00917. [DOI] [PubMed] [Google Scholar]
- 21.Stingl U, Tripp HJ, Giovannoni SJ. 2007. Improvements of high-throughput culturing yielded novel SAR11 strains and other abundant marine bacteria from the Oregon coast and the Bermuda Atlantic Time Series study site. ISME J 1:361–371. doi: 10.1038/ismej.2007.49. [DOI] [PubMed] [Google Scholar]
- 22.Tripp HJ. 2008. Counting marine microbes with Guava Easy-Cyte 96 well plate reading flow cytometer. Protoc Exch doi: 10.1038/nprot.2008.29. [DOI] [Google Scholar]
- 23.Jimenez-Infante F, Ngugi DK, Alam I, Rashid M, Baalawi W, Kamau AA, Bajic VB, Stingl U. 2014. Genomic differentiation among two strains of the PS1 clade isolated from geographically separated marine habitats. FEMS Microbiol Ecol 89:181–197. doi: 10.1111/1574-6941.12348. [DOI] [PubMed] [Google Scholar]
- 24.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 25.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141−D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lueders T, Manefield M, Friedrich MW. 2004. Enhanced sensitivity of DNA- and rRNA-based stable isotope probing by fractionation and quantitative analysis of isopycnic centrifugation gradients. Environ Microbiol 6:73–78. doi: 10.1046/j.1462-2920.2003.00536.x. [DOI] [PubMed] [Google Scholar]
- 27.Andrews S. 2010. A quality control tool for high throughput sequence data. http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/.
- 28.Lohse M, Bolger AM, Nagel A, Fernie AR, Lunn JE, Stitt M, Usadel B. 2012. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res 40:W622−W627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. 2012. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alam I, Antunes A, Kamau AA, Ba Alawi W, Kalkatawi M, Stingl U, Bajic VB. 2013. INDIGO—integrated data warehouse of microbial genomes with examples from the Red Sea extremophiles. PLoS One 8:e82210. doi: 10.1371/journal.pone.0082210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beck DA, McTaggart TL, Setboonsarng U, Vorobev A, Kalyuzhnaya MG, Ivanova N, Goodwin L, Woyke T, Lidstrom ME, Chistoserdova L. 2014. The expanded diversity of methylophilaceae from Lake Washington through cultivation and genomic sequencing of novel ecotypes. PLoS One 9:e102458. doi: 10.1371/journal.pone.0102458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. doi: 10.1093/bioinformatics/btr088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vollmers J, Voget S, Dietrich S, Gollnow K, Smits M, Meyer K, Brinkhoff T, Simon M, Daniel R. 2013. Poles apart: Arctic and Antarctic Octadecabacter strains share high genome plasticity and a new type of xanthorhodopsin. PLoS One 8:e63422. doi: 10.1371/journal.pone.0063422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robbertse B, Yoder RJ, Boyd A, Reeves J, Spatafora JW. 2011. Hal: an automated pipeline for phylogenetic analyses of genomic data. PLoS Curr 3:RRN1213. doi: 10.1371/currents.RRN1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blom J, Albaum SP, Doppmeier D, Puhler A, Vorholter FJ, Zakrzewski M, Goesmann A. 2009. EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics 10:154. doi: 10.1186/1471-2105-10-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. 2007. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35:W182−W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H, Benedito VA, Udvardi MK, Zhao PX. 2009. TransportTP: a two-phase classification approach for membrane transporter prediction and characterization. BMC Bioinformatics 10:418. doi: 10.1186/1471-2105-10-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Richter M, Rossello-Mora R. 2009. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106:19126–19131. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meier-Kolthoff JP, Auch AF, Klenk HP, Goker M. 2013. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halsey KH, Carter AE, Giovannoni SJ. 2012. Synergistic metabolism of a broad range of C1 compounds in the marine methylotrophic bacterium HTCC2181. Environ Microbiol 14:630–640. doi: 10.1111/j.1462-2920.2011.02605.x. [DOI] [PubMed] [Google Scholar]
- 43.Man D, Wang W, Sabehi G, Aravind L, Post AF, Massana R, Spudich EN, Spudich JL, Beja O. 2003. Diversification and spectral tuning in marine proteorhodopsins. EMBO J 22:1725–1731. doi: 10.1093/emboj/cdg183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, Wu DY, Eisen JA, Hoffman JM, Remington K, Beeson K, Tran B, Smith H, Baden-Tillson H, Stewart C, Thorpe J, Freeman J, Andrews-Pfannkoch C, Venter JE, Li K, Kravitz S, Heidelberg JF, Utterback T, Rogers YH, Falcon LI, Souza V, Bonilla-Rosso G, Eguiarte LE, Karl DM, Sathyendranath S, Platt T, Bermingham E, Gallardo V, Tamayo-Castillo G, Ferrari MR, Strausberg RL, Nealson K, Friedman R, Frazier M, Venter JC. 2007. The Sorcerer II Global Ocean Sampling expedition: northwest Atlantic through eastern tropical Pacific. PLoS Biol 5:e77. doi: 10.1371/journal.pbio.0050077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yooseph S, Sutton G, Rusch DB, Halpern AL, Williamson SJ, Remington K, Eisen JA, Heidelberg KB, Manning G, Li W, Jaroszewski L, Cieplak P, Miller CS, Li H, Mashiyama ST, Joachimiak MP, van Belle C, Chandonia JM, Soergel DA, Zhai Y, Natarajan K, Lee S, Raphael BJ, Bafna V, Friedman R, Brenner SE, Godzik A, Eisenberg D, Dixon JE, Taylor SS, Strausberg RL, Frazier M, Venter JC. 2007. The Sorcerer II Global Ocean Sampling expedition: expanding the universe of protein families. PLoS Biol 5:e16. doi: 10.1371/journal.pbio.0050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown MV, Fuhrman JA. 2005. Marine bacterial microdiversity as revealed by internal transcribed spacer analysis. Aquat Microb Ecol 41:15–23. doi: 10.3354/ame041015. [DOI] [Google Scholar]
- 47.Ngugi DK, Stingl U. 2012. Combined analyses of the ITS loci and the corresponding 16S rRNA genes reveal high micro- and macrodiversity of SAR11 populations in the Red Sea. PLoS One 7:e50274. doi: 10.1371/journal.pone.0050274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 49.Nakagawa T, Mitsui R, Tani A, Sasa K, Tashiro S, Iwama T, Hayakawa T, Kawai K. 2012. A catalytic role of XoxF1 as La3+-dependent methanol dehydrogenase in Methylobacterium extorquens strain AM1. PLoS One 7:e50480. doi: 10.1371/journal.pone.0050480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skovran E, Palmer AD, Rountree AM, Good NM, Lidstrom ME. 2011. XoxF is required for expression of methanol dehydrogenase in Methylobacterium extorquens AM1. J Bacteriol 193:6032–6038. doi: 10.1128/JB.05367-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. 2011. The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta 1807:1398–1413. doi: 10.1016/j.bbabio.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mojib N, Andersen DT, Bej AK. 2011. Structure and function of a cold shock domain fold protein, CspD, in Janthinobacterium sp. Ant5-2 from East Antarctica. FEMS Microbiol Lett 319:106–114. doi: 10.1111/j.1574-6968.2011.02269.x. [DOI] [PubMed] [Google Scholar]
- 53.Schmid B, Klumpp J, Raimann E, Loessner MJ, Stephan R, Tasara T. 2009. Role of cold shock proteins in growth of Listeria monocytogenes under cold and osmotic stress conditions. Appl Environ Microbiol 75:1621–1627. doi: 10.1128/AEM.02154-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhillon BK, Chiu TA, Laird MR, Langille MG, Brinkman FS. 2013. IslandViewer update: improved genomic island discovery and visualization. Nucleic Acids Res 41:W129−W132. doi: 10.1093/nar/gkt394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim SK, Kimura S, Shinagawa H, Nakata A, Lee KS, Wanner BL, Makino K. 2000. Dual transcriptional regulation of the Escherichia coli phosphate-starvation-inducible psiE gene of the phosphate regulon by PhoB and the cyclic AMP (cAMP)-cAMP receptor protein complex. J Bacteriol 182:5596–5599. doi: 10.1128/JB.182.19.5596-5599.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chistoserdova L. 2011. Modularity of methylotrophy, revisited. Environ Microbiol 13:2603–2622. doi: 10.1111/j.1462-2920.2011.02464.x. [DOI] [PubMed] [Google Scholar]
- 57.Vorobev A, Beck DA, Kalyuzhnaya MG, Lidstrom ME, Chistoserdova L. 2013. Comparative transcriptomics in three Methylophilaceae species uncover different strategies for environmental adaptation. PeerJ 1:e115. doi: 10.7717/peerj.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.DeLong EF, Beja O. 2010. The light-driven proton pump proteorhodopsin enhances bacterial survival during tough times. PLoS Biol 8:e1000359. doi: 10.1371/journal.pbio.1000359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carlson CA, Morris R, Parsons R, Treusch AH, Giovannoni SJ, Vergin K. 2009. Seasonal dynamics of SAR11 populations in the euphotic and mesopelagic zones of the northwestern Sargasso Sea. ISME J 3:283–295. doi: 10.1038/ismej.2008.117. [DOI] [PubMed] [Google Scholar]
- 60.Field KG, Gordon D, Wright T, Rappe M, Urback E, Vergin K, Giovannoni SJ. 1997. Diversity and depth-specific distribution of SAR11 cluster rRNA genes from marine planktonic bacteria. Appl Environ Microbiol 63:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vergin KL, Beszteri B, Monier A, Cameron Thrash J, Temperton B, Treusch AH, Kilpert F, Worden AZ, Giovannoni SJ. 2013. High-resolution SAR11 ecotype dynamics at the Bermuda Atlantic time-series study site by phylogenetic placement of pyrosequences. ISME J 7:1322–1332. doi: 10.1038/ismej.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EMS, Chisholm SW. 2006. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science 311:1737–1740. doi: 10.1126/science.1118052. [DOI] [PubMed] [Google Scholar]
- 63.Rocap G, Larimer FW, Lamerdin J, Malfatti S, Chain P, Ahlgren NA, Arellano A, Coleman M, Hauser L, Hess WR, Johnson ZI, Land M, Lindell D, Post AF, Regala W, Shah M, Shaw SL, Steglich C, Sullivan MB, Ting CS, Tolonen A, Webb EA, Zinser ER, Chisholm SW. 2003. Genome divergence in two Prochlorococcus ecotypes reflects oceanic niche differentiation. Nature 424:1042–1047. doi: 10.1038/nature01947. [DOI] [PubMed] [Google Scholar]
- 64.Aguilar-Barajas E, Diaz-Perez C, Ramirez-Diaz MI, Riveros-Rosas H, Cervantes C. 2011. Bacterial transport of sulfate, molybdate, and related oxyanions. Biometals 24:687–707. doi: 10.1007/s10534-011-9421-x. [DOI] [PubMed] [Google Scholar]
- 65.Sañudo-Wilhelmy SA, Cutter LS, Durazo R, Smail EA, Gomez-Consarnau L, Webb EA, Prokopenko MG, Berelson WM, Karl DM. 2012. Multiple B-vitamin depletion in large areas of the coastal ocean. Proc Natl Acad Sci U S A 109:14041–14045. doi: 10.1073/pnas.1208755109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mooney S, Leuendorf JE, Hendrickson C, Hellmann H. 2009. Vitamin B6: a long known compound of surprising complexity. Molecules 14:329–351. doi: 10.3390/molecules14010329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pastor JM, Salvador M, Argandona M, Bernal V, Reina-Bueno M, Csonka LN, Iborra JL, Vargas C, Nieto JJ, Canovas M. 2010. Ectoines in cell stress protection: uses and biotechnological production. Biotechnol Adv 28:782–801. doi: 10.1016/j.biotechadv.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 68.Schwibbert K, Marin-Sanguino A, Bagyan I, Heidrich G, Lentzen G, Seitz H, Rampp M, Schuster SC, Klenk HP, Pfeiffer F, Oesterhelt D, Kunte HJ. 2011. A blueprint of ectoine metabolism from the genome of the industrial producer Halomonas elongata DSM 2581T. Environ Microbiol 13:1973–1994. doi: 10.1111/j.1462-2920.2010.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yeo SK, Huggett MJ, Eiler A, Rappe MS. 2013. Coastal bacterioplankton community dynamics in response to a natural disturbance. PLoS One 8:e56207. doi: 10.1371/journal.pone.0056207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fortunato CS, Crump BC. 2011. Bacterioplankton community variation across river to ocean environmental gradients. Microb Ecol 62:374–382. doi: 10.1007/s00248-011-9805-z. [DOI] [PubMed] [Google Scholar]
- 71.Reyes-Prieto A, Barquera B, Juarez O. 2014. Origin and evolution of the sodium-pumping NADH: ubiquinone oxidoreductase. PLoS One 9:e96696. doi: 10.1371/journal.pone.0096696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Juárez O, Barquera B. 2012. Insights into the mechanism of electron transfer and sodium translocation of the Na+-pumping NADH:quinone oxidoreductase. Biochim Biophys Acta 1817:1823–1832. doi: 10.1016/j.bbabio.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sazanov LA. 2014. The mechanism of coupling between electron transfer and proton translocation in respiratory complex I. J Bioenerg Biomembr 46:247–253. doi: 10.1007/s10863-014-9554-z. [DOI] [PubMed] [Google Scholar]
- 74.Bogachev AV, Murtazina RA, Skulachev VP. 1997. The Na+/e− stoichiometry of the Na+-motive NADH:quinone oxidoreductase in Vibrio alginolyticus. FEBS Lett 409:475–477. doi: 10.1016/S0014-5793(97)00536-X. [DOI] [PubMed] [Google Scholar]
- 75.Giuffrè A, Borisov VB, Mastronicola D, Sarti P, Forte E. 2012. Cytochrome bd oxidase and nitric oxide: from reaction mechanisms to bacterial physiology. FEBS Lett 586:622–629. doi: 10.1016/j.febslet.2011.07.035. [DOI] [PubMed] [Google Scholar]
- 76.Thompson LR, Field C, Romanuk T, Kamanda Ngugi D, Siam R, El Dorry H, Stingl U. 2013. Patterns of ecological specialization among microbial populations in the Red Sea and diverse oligotrophic marine environments. Ecol Evol 3:1780–1797. doi: 10.1002/ece3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abu-Zied RH, Basaham AS, El Sayed MA. 2013. Effect of municipal wastewaters on bottom sediment geochemistry and benthic foraminifera of two Red Sea coastal inlets, Jeddah, Saudi Arabia. Environ Earth Sci 68:451–469. doi: 10.1007/s12665-012-1751-7. [DOI] [Google Scholar]
- 78.Basaham AS, Rifaat AE, El-Mamoney MH, El Sayed MA. 2009. Re-evaluation of the impact of sewage disposal on coastal sediments of the southern Corniche, Jeddah, Saudi Arabia. JKAU Mar Sci 20:109–126. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.