

SUMMARY
Mechanical allodynia, induced by normally innocuous low-threshold mechanical stimulation, represents a cardinal feature of neuropathic pain. Blockade or ablation of high-threshold small-diameter unmyelinated C-fibers has limited effects on mechanical allodynia1–4. While large myelinated A-fibers, in particular Aβ-fibers, have previously been implicated in mechanical allodynia5–7, an A-fiber-selective pharmacological blocker is still lacking. Here we report a new method for targeted silencing of A-fibers in neuropathic pain. We found that Toll-like receptor 5 (TLR5) is co-expressed with neurofilament-200 in large-diameter A-fiber neurons in the dorsal root ganglion (DRG). Activation of TLR5 with its ligand flagellin results in neuronal entry of the membrane impermeable lidocaine derivative QX-314, leading to TLR5-dependent blockade of sodium currents predominantly in A-fiber neurons of mouse DRGs. Intraplantar co-application of flagellin and QX-314 (flagellin/QX-314) dose-dependently suppressed mechanical allodynia following chemotherapy, nerve injury, and diabetic neuropathy, but this blockade is abrogated in Tlr5-deficient mice. In vivo electrophysiology demonstrated that flagellin/QX-314 co-application selectively suppressed Aβ-fiber conduction in naive and chemotherapy-treated mice. TLR5-mediated Aβ blockade but not capsaicin-mediated C-fiber blockade also reduced chemotherapy-induced ongoing pain without impairing motor function. Finally, flagellin/QX-314 co-application suppressed sodium currents in large-diameter human DRG neurons. Thus, our findings provide a new tool for targeted silencing of Aβ-fibers and neuropathic pain treatment.
Toll-like receptors (TLRs) are typically expressed in immune and glial cells to mediate innate and adaptive immunity8, 9. Recent studies have shown that TLRs such as TLR3, TLR4, and TLR7 are also expressed in primary sensory neurons especially C-fiber nociceptive neurons in the DRG to regulate sensory functions such as pain and itch10, 11. To determine if DRG expresses different TLRs, we first employed quantitative PCR to analyze 12 TLRs in mouse DRGs. As shown in Supplementary Fig. 1, TLR1-TLR9 and TLR13, but not TLR11 and TLR12, were expressed by mouse DRGs. Surprisingly we found a unique distribution pattern of TLR5 in DRG neurons. In situ hybridization revealed that Tlr5 mRNA is expressed in medium-large diameter mouse DRG neurons that co-express neurofilament-200 (NF200), a marker for myelinated A-fibers12 (Fig. 1a). Size frequency analysis revealed overlapping distribution patterns of Tlr5 mRNA-positive and NF200-immunoreactive (IR) neurons; both are present in medium to large neurons (Fig. 1b). Quantitative analysis demonstrated that 22% and 26% of DRG neurons express Tlr5 and NF200, respectively, and 20% of DRG neurons are positive for both Tlr5 and NF200. Further analysis showed that 91% of Tlr5+ neurons contain NF200 (Fig. 1c), and vice versa, 78% of NF200+ neurons express Tlr5. Tlr5 mRNA was also observed in a small portion of small-diameter neurons (Fig. 1a, 1c). Within the Tlr5+ population, 7.2% of neurons express calcitonin gene related peptide (CGRP), a marker for peptidergic C-fibers and some Aδ fibers13, and 7.7% of neurons express tyrosine hydroxylase (TH), a marker for low-threshold C-fibers14, but only 0.5% neurons bind isolectin B4 (IB4), a marker for non-peptidergic C-fibers (Fig. 1c, Supplementary Fig. 2). Immunohistochemistry confirmed the expression of TLR5 protein in NF200-IR DRG neurons in wild-type mice (Fig. 1d). TLR5-IR was lost in Tlr5 knockout (Tlr5−/−) mice, suggesting the selectivity of the TLR5 antibody (Fig. 1d).
Figure 1. TLR5 is co-localized with A-fiber marker NF200 in DRG neurons, skin nerve fibers, and spinal cord axonal terminals in mice.
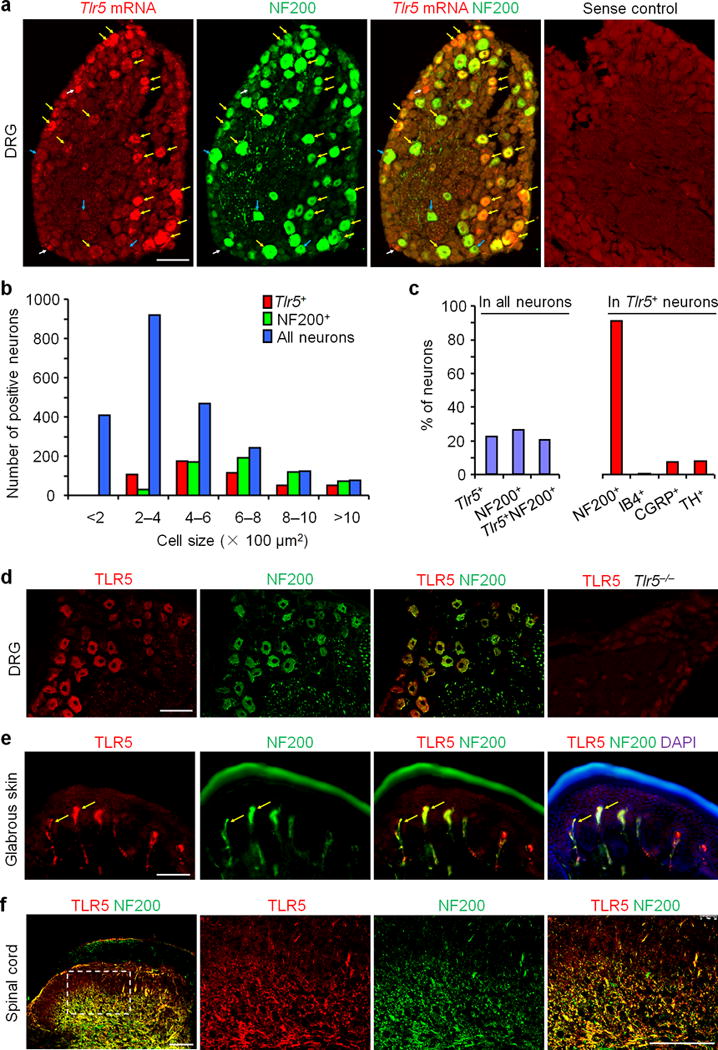
(a) Co-localization of Tlr5 mRNA and NF200-IR in DRG neurons. Yellow, white, and blue arrows show double-labeled neurons, Tlr5 single-labeled neurons, and NF200 single-labeled neurons, respectively. Control (sense probe) shows no signal. Scale, 100 μm. (b) Size distribution frequency of Tlr5 mRNA-positive (Tlr5+), NF200-IR (NF200+) neurons, and total neurons in DRG. A total of 2238 neurons from 3 mice were counted. (c) Percentage of total DRG neurons expressing Tlr5, NF200, and both Tlr5 and NF200, and percentage of Tlr5+ neurons expressing NF200, IB4, CGRP, and TH. A total of 1320 Tlr5+ neurons from 3 mice were counted. (d) Co-localization of TLR5-IR and NF200-IR in DRG neurons. Note the TLR5 staining is absent in Tlr5−/− mice. Scale, 100 μm. (e) Co-localization (shown by arrows) of TLR5 and NF200 in glabrous skin of hindpaw. Scale, 100 μm. (f) Co-localization of TLR5 and NF200 in deep laminae of spinal cord dorsal horn. Boxed region of the image on the left is enlarged in the images to the right. Scale bars, 100 μm.
Next we investigated whether TLR5 protein is also present in axons and axonal terminals in peripheral (skin) and central (spinal cord) sites, as a result of anterograde axonal transport of TLR5 from DRG neuronal cell bodies. We observed marked TLR5 expression in NF200-IR A-fibers in Meissner corpuscle-like structures in the dermis of hindpaw glabrous skin (Fig. 1e). We also found some TLR5+ and NF200+ double-labeled nerve fibers at the base of hair follicles, but no labeling was seen near Merkel cells (Supplementary Fig. 3a–c). A- and C-fibers project to distinct laminae of the spinal dorsal horn: unmyelinated C-fibers project to the superficial layers (laminae I-II), thin myelinated Aδ-fibers project to laminae I and V, whereas Aβ-fibers terminate in the deep laminae (III–V)15. TLR5-IR was only found in axonal terminals that co-express NF200 in the deep layers (Fig. 1f). No co-localization with CGRP was observed in central terminals (Supplementary Fig. 3d). There is less labeling of TLR5-IR fibers in the skin vs. spinal cord, in part due to lower sensitivity of the antibody in skin tissue. Given the unique peripheral projection to Meissner corpuscle-like structures in glabrous skin and central projections to the deep laminae of the dorsal horn, the TLR5-IR A-fibers should be primarily Aβ-fibers15.
QX-314 is a positively charged, membrane impermeable lidocaine derivative (MW: 263). Binshtok et al. have previously demonstrated that co-application of QX-314 and the C-fiber-specific activator capsaicin results in neuronal entry of QX-314 through TRPV1 channels, leading to a functional blockade of C-fibers and subsequent pain relief16. Activation of certain G-protein-coupled receptors (GPCR) also causes QX-314 influx through TRPV1 or TRPA1 and blockade of C-fibers via GPCR-TRPV1/A1 interactions17. Given the selective expression of TLR5 in A-fibers and the previously demonstrated coupling of TLRs with ion channels (e.g., TLR7/TRPA118), we hypothesized that activation of TLR5 might cause QX-314 entry and functional blockade of A-fibers when TLR5 is coupled with some ion channel. Flagellin, a principal component of bacterial flagella, is one of the best studied TLR5 ligands8, 19. To assess if flagellin can cause neuronal entry of QX-314, we recorded evoked transient sodium currents in dissociated primary cultures of mouse large-diameter A-fiber (>30 μm) and small-diameter C-fiber (<25 μm) DRG neurons via in vitro patch clamp electrophysiology. Co-application of flagellin (10 ng/ml ≈ 0.3 nM) with QX-314 (5 mM16) produced a substantial inhibition of sodium currents in A-fiber but not in C-fiber neurons (Fig. 2a). This effect was also time-dependent, with partial inhibition at 5 min and maximal inhibition at 10 min following the co-application, whereas flagellin or QX-314 alone produced no effects (Fig. 2b,c, Supplementary Fig. 4a,b). Importantly, this inhibition of sodium currents was compromised in neurons from Tlr5−/− mice, suggesting a specific effect of flagellin on TLR5 (Fig. 2a,b). In sharp contrast, co-application of capsaicin (1 μM) and QX-314 (5 mM) only blocked sodium currents in C-fiber but not A-fiber neurons (Supplementary Fig. 5a–c).
Figure 2. Co-application of flagellin and QX-314 blocks sodium currents in large-diameter A-fiber neurons of mouse and human DRGs.
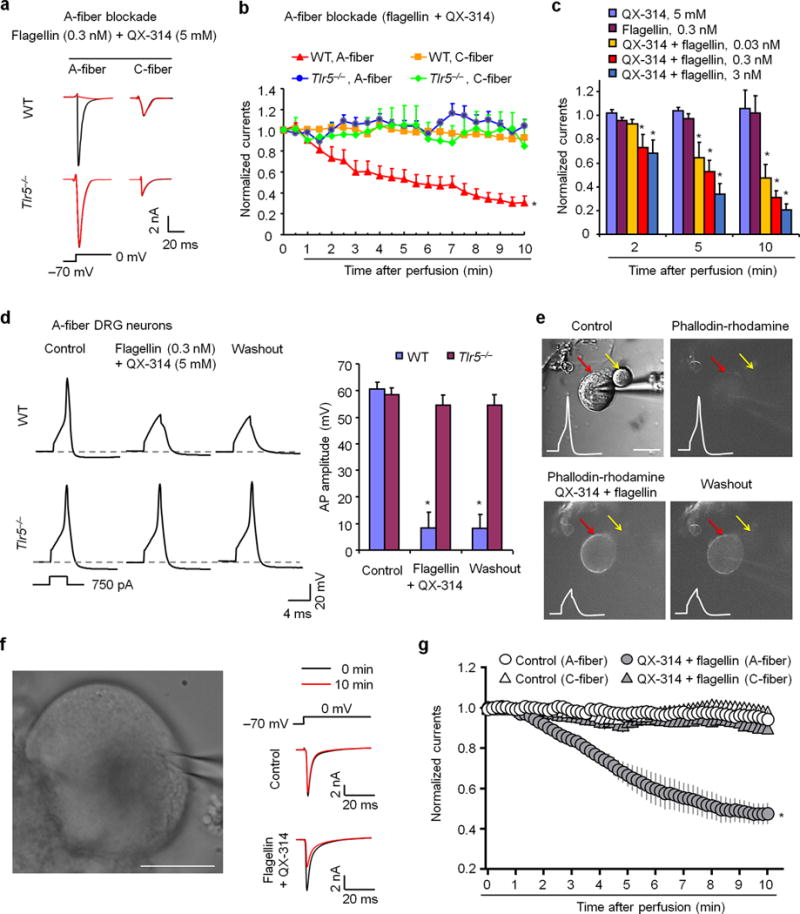
(a) Traces of transient sodium currents in A-fiber and C-fiber neurons of WT and Tlr5−/− mice. Transient sodium currents were evoked by a 200 ms voltage step from −70 mV to 0 mV (inset). (b) Time-course of sodium current amplitudes after perfusion with flagellin (0.3 nM) and QX-314 (5 mM) in mouse A-fiber neurons (30–55 μm, n = 20 from 7 WT mice, n = 5 from 5 Tlr5−/− mice) and C-fiber neurons (10–25 μm, n = 22 from 7 WT mice, n = 5 from 3 Tlr5−/− mice). *P<0.05, vs. WT C-fiber and Tlr5−/− mice, two-Way ANOVA. (c) Dose-dependent inhibition of sodium currents by A-fiber blockade in mouse A-fiber neurons (n = 5, 20, and 8 for 0.03, 0.3, and 3 nM, respectively). Note flagellin (n = 6 neurons) or QX-314 (n = 5 neurons) alone has no effects. *P<0.05, vs. QX-314 alone, Two-Way ANOVA. (d) Left panel, traces of action potentials in WT and Tlr5−/− DRG neurons before, during, and after flagellin (0.3 nM) + QX-314 (5 mM) application. Inset indicates the current injection protocol used for action potential generation. Dashed-lines indicate resting membrane potential. Right panel, action potential amplitudes of WT neurons (n = 17) and Tlr5−/− neurons (n = 18). *P<0.05, vs. control. (e) Flagellin/QX-314 treatment causes entry of phallodin-rhodamine (10 μM) into large (red arrow) but not small (yellow arrow) neuron. Scale, 30 μm. Upper left, DIC image of a recorded mouse DRG neuron. Subsequent panels show fluorescence images (red filter, 540 nm) of the same neuron upon flagellin/QX-314-mediated entry of phallodin-rhodamine. Insets show action potential blockade by flagellin/QX-314 application. (f, g) Flagellin/QX-314 blocks sodium currents (50 ms step, from −70 mV to 0 mV) in large-diameter A-fiber neurons of human DRGs. (f) Left, DIC image of a recorded human DRG neuron. Scale, 30 μm. Right, traces of transient sodium currents from vehicle control and QX-314 (12 mM)/flagellin (30 ng/ml = 0.9 nM) treated neurons. (g) Time course of flagellin/QX-314-induced inhibition of sodium currents in human DRG neurons with large diameters (55–80 μm, A-fiber) but not small-diameters (< 50 μm, C-fiber). *P<0.05, Two-Way ANOVA. For control (no treatment) groups, n = 6 C-fiber, 7 A-fiber neurons. For QX-314 + flagellin groups, n = 8 C-fiber, 8 A-fiber neurons. Human DRG cultures were prepared from 4 donors. All data were expressed as mean ± s.e.m.
Sodium channels are essential for the induction of action potentials in sensory neurons. We next investigated if this method of A-fiber blockade might also suppress action potentials in DRG neurons. Co-application of flagellin/QX-314 blocked action potentials in wild-type but not Tlr5-deficient A-fiber neurons, confirming a TLR5-mediated mechanism (Fig. 2d). The scaffold protein MyD88 (myeloid differentiation primary response 88) mediates canonical signaling of TLR5 to activate the NFκB pathway and is present in DRG neurons20. However, the A-fiber blockade by flagellin/QX-314 was not compromised in Myd88−/− A-fiber DRG neurons (Supplementary Fig. 5d,e).
Apart from QX-314, we also examined flagellin-mediated neuronal entry of a larger fluorescence molecule phallodin-rhodamine (Mw: 1231 Da). Co-incubation of flagellin with phallodin-rhodamine (10 μM) resulted in entry to large DRG neurons, and phallodin-rhodamine fluorescence was correlated with QX-314-elicited inhibition of action potentials (Fig. 2e). Co-application of flagellin with phallodin-rhodamine in whole DRG preparations also lead to phallodin-rhodamine influx into NF200+ A-fiber neurons, and this influx was absent in DRG preparations from Tlr5−/− mice (Supplementary Fig. 6a–e).
To determine the translational potential of our observations in mouse, we also examined the effects of flagellin/QX-314 in human DRG neurons, dissociated from postmortem L3-L5 DRGs of 4 non-diseased donors (67-year-old male, 52-year-old female, 50-year-old female, and 29-year-old male). Notably, co-application of flagellin (30 ng/ml = 0.9 nM) and QX-314 (12 mM) time-dependently suppressed sodium currents in large-diameter A-fiber (55–80 μm) human DRG neurons, without affecting the currents in small-diameter C-fiber neurons (30–50 μm, Fig. 2f,g; Supplementary Fig. 4c,d). Notably, flagellin/QX-314 produced consistent inhibition of sodium currents in large-diameter DRG neurons from different age and sex groups (Fig. 2g). Size distribution analysis indicated that in human DRGs TLR5 is primarily expressed by large-diameter neurons that co-express NF-200 (Supplementary Fig. 7a,b).
To investigate A-fiber blockade in the peripheral axons, we conducted in vivo electrophysiology in anesthetized mice. Compound action potentials evoked by electrical stimulation of the hind paw were recorded from mouse sciatic nerves. Co-application of flagellin (0.3 μg ≈ 10 pmol) and QX-314 (2% ≈ 60 mM) by intraplantar injection produced a significant inhibition of Aβ-conduction, without affecting the conduction of Aα- and Aδ-fibers (Fig. 3a,b). As a positive control, 2% lidocaine blocked the conduction of all types of the fibers (Fig. 3a,b). Peripheral neuropathy and neuropathic pain induced by chemotherapy agents is a key limiting factor of chemotherapy as an anti-cancer treatment21. We therefore tested the blocking effects of flagellin/QX-314 in a mouse model of chemotherapy-induced neuropathic pain. Notably, one week after treatment with the chemotherapy agent paclitaxel (PAX, 6 mg/kg, i,p.), flagellin/QX-314 treatment inhibited the conduction of Aβ-fibers but not Aα- and Aδ-fibers (Fig. 3c,d). However, PAX did not change the pattern or percentages of TLR5+ and NF200+ neurons (Supplementary Fig. 8a). Consistently, in DRG neurons obtained from PAX-treated mice flagellin/QX-314 application only blocked sodium currents in large but not small diameter neurons (Supplementary Fig. 8b,c).
Figure 3. Co-application of flagellin and QX-314 selectively inhibits Aβ-fiber conduction in sciatic nerves of naive and chemotherapy-treated mice.
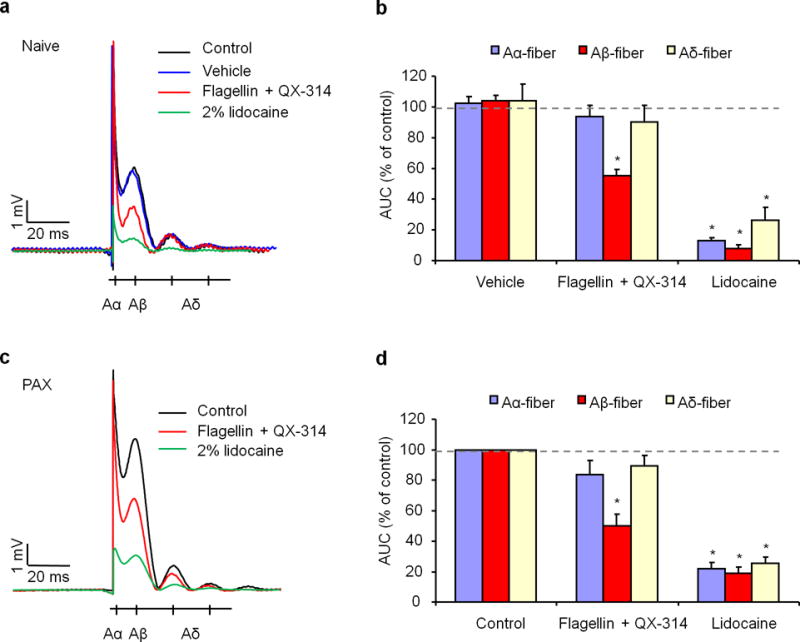
(a) In vivo sciatic nerve recordings in naïve mice show traces of compound potentials evoked by hindpaw stimulation and the effects of 0.3 μg (0.3 μM in 30 μl) flagellin/60 mM QX-314 or 2% lidocaine applied via intraplantar injection of the hindpaw. Schematic beneath the traces indicates the relative timing of Aα, Aβ, and Aδ potentials. Control animals received no treatment, and PBS was used as vehicle. (b) Relative amplitude of Aα, Aβ, and Aδ potentials, expressed as area under curve (AUC), being normalized to control. *P<0.05, vs. vehicle control, n = 5 (blockade) and 6 (vehicle) mice/group. (c) In vivo sciatic nerve recordings of paclitaxel (PAX)-treated mice at 1 week show traces of compound potentials and the effects of 0.3 μg flagellin/60 mM QX-314 or 2% lidocaine. (d) Relative amplitude of Aα, Aβ, and Aδ potentials. *P<0.05, vs. control, n = 8 mice/group. Note a specific inhibition on Aβ-conduction by flagellin/QX-314. All data were expressed as mean ± s.e.m.
Since Nav1.8-expressing afferents (C/Aδ-fibers) are dispensable for the development of chemotherapy-induced mechanical allodynia2, we hypothesized that Aβ-fiber blockade would inhibit chemotherapy-induced mechanical allodynia. As previously reported20, systemic injection of PAX elicited a rapid (<1 d) and persistent (>3 weeks) mechanical allodynia, demonstrated by a reduction in hind paw withdrawal thresholds in response to stimulation with von Frey hairs. Strikingly, intraplantar co-application of QX-314 (0.2%, ≈ 6 mM) with flagellin (0.1, 0.3, and 0.9 μg, i.e. 3–28 pmol) produced a dose-dependent reversal of mechanical allodynia 1 w after the PAX injection; this reversal peaked at 0.5 h, maintained at 3 h, but recovered at 24 h (Fig. 4a). Intraplantar injection of flagellin alone did not reduce PAX-induced mechanical allodynia in WT mice (data not shown). For comparison, 2% lidocaine only produced a very transient (<30 min) reversal of PAX-induced mechanical allodynia (Supplementary Fig. 9a) after recovery from initial motor impairment (<10 min). We further investigated PAX-induced neuropathic pain in WT and Tlr5−/− mice. Compared with WT mice, both baseline pain and PAX-elicited mechanical allodynia were unaltered in Tlr5−/− mice (Fig. 4b). However, QX-314/flagellin application failed to inhibit mechanical allodynia in Tlr5−/− mice (Fig. 4b), although this A-fiber blockade reversed allodynia in Myd88−/− mice (Supplementary Fig. 9b). Hence, flagellin/QX-314 blocks mechanical allodynia via TLR5 but not MyD88. By contrast, C-fiber blockade by intraplantar co-application of QX-314 (0.2%) with capsaicin (10 μg ≈ 33 nmol22, which is 1000 times higher than the flagellin dose for A-fiber blockade) had no effects on PAX-induced mechanical allodynia (Fig. 4a, Supplementary Fig. 9c). Local flagellin/QX-314 treatment is well tolerated by animals. Repeated applications of flagellin/QX-314, once a day for 5 days, produced persistent inhibition of PAX-elicited allodynia without showing signs of antinociceptive tolerance (Fig. 4c). Neither did the repeated applications affect overall wellbeing of animals and cause local inflammation in hindpaw (Supplementary Fig. 10).
Figure 4. A-fiber blockade by co-application of flagellin/QX-314 inhibits mechanical allodynia and ongoing pain in different neuropathic pain conditions.
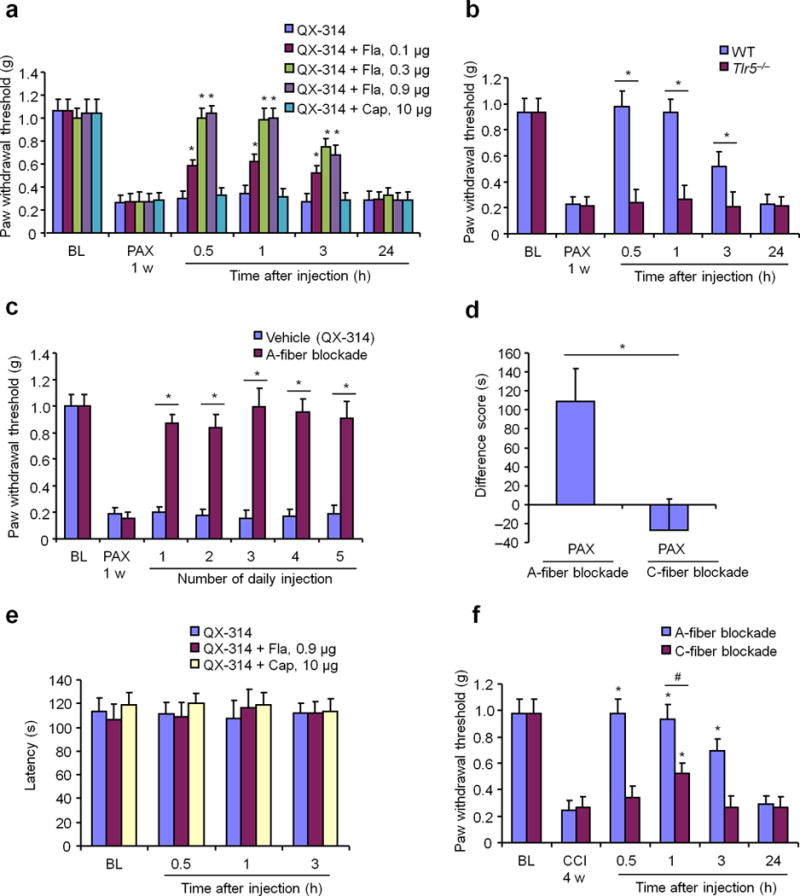
(a) Reversal of paclitaxel (PAX)-induced mechanical allodynia by intraplantar A-fiber blockade (0.1–0.9 μg flagellin (Fla)/6 mM QX-314) but not C-fiber blockade (10 μg capsaicin (Cap)/6 mM QX-314). *P<0.05, vs. vehicle, Two-Way ANOVA. n = 5 mice/group. (b) Intraplantar A-fiber blockade inhibits PAX-induced mechanical allodynia in WT but not Tlr5−/− mice. *P<0.05, Two-Way ANOVA. n = 5 mice/group. (c) Repeated applications of 0.3 μg flagellin/6 mM QX-314, once a day for 5 days, persistently inhibit PAX-elicited mechanical allodynia, assessed 1 h after each injection. QX-314 (6 mM) was used a vehicle. *P<0.05, Two-Way ANOVA. n = 5 mice/group. (d) Inhibition of ongoing pain by A-fiber (n = 7 mice) but not C-fiber (n = 5 mice) blockade, as revealed by CPP test showing difference score of time a mouse spent in nerve block-paired chamber (test time minus preconditioning time). *P<0.05, two-tailed student’s t-test. (e) Rota-rod test shows normal motor function following intraplantar A-fiber and C-fiber blockade. n = 5 mice/group. (f) Distinct effects of intraplantar A-fiber blockade vs. C-fiber blockade on CCI-induced mechanical allodynia. *P<0.05, Two-Way ANOVA. n = 5 mice/group. In b-d and f, A-fiber blockade was induced by 0.3 μg flagellin/6 mM QX-314 and C-fiber blockade was induced by 10 μg capsaicin/6 mM QX-314. All data were expressed as mean ± s.e.m.
Apart from evoked pain assessed by sensory stimuli-evoked paw withdrawal responses, chronic pain is also characterized by ongoing pain/spontaneous pain, which can be assessed by conditioned place preference (CPP)23, 24. In a two-chamber CPP test, A-fiber blockade by flagellin/QX-314 elicited significant CPP in PAX-treated mice, whereas C-fiber blockade by capsaicin/QX-314 had no effect (Fig. 4d, Supplementary Fig. 9d). Thus, A-fiber but not C-fiber blockade can suppress chemotherapy-induced ongoing pain. However, motor function, assessed by Rota-rod test, was not affected by either A-fiber or C-fiber blockade (Fig. 4e).
Next, we compared the effects of A-fiber vs. C-fiber blockade in other neuropathic pain models: a diabetic neuropathy model induced by streptozotozin (STZ) injection, and a nerve injury model produced by chronic constriction injury (CCI). Notably, capsaicin-sensitive C-fiber nociceptors are not required for the development of STZ-induced mechanical allodynia25. Our results show that STZ-induced mechanical allodynia was reversed by intraplantar flagellin/QX-314 but not capsaicin/QX-314 treatment (Supplementary Fig. 9e). CCI elicits long-lasting mechanical allodynia for > 4 weeks26, and this allodynia was also reversed by flagellin/QX-314 in the maintenance phase between 2 and 4 weeks after CCI (Fig. 4f, Supplementary Fig. 11a–b). As expected, this reversal was compromised in Tlr5−/− mice (Supplementary Fig. 11a–b). In contrast, capsaicin/QX-314-induced C-fiber blockade only had mild inhibition on CCI-induced mechanical allodynia, although this blockade effectively suppressed heat hyperalgesia (Fig. 4f, Supplementary Fig. 11c,d).
Finally, we evaluated distinct role of A and C-fiber blockade in baseline pain. Of interest, flagellin/QX-314 treatment in naïve mice decreased withdrawal threshold and caused mechanical hypersensitivity, whereas capsaicin/QX-314 treatment increased withdraw threshold as previously reported16 (Supplementary Fig. 12a,b). The effects of A-fiber but not C-fiber blockade were abrogated by Tlr5 deletion (Supplementary Fig. 12a,b). This result suggests that Aβ-fibers might play distinct role in the normal and neuropathic pain conditions and supports the “gate control theory” that Aβ-fiber activation in the normal conditions inhibits pain transmission27.
In summary, we have identified TLR5 as a new marker for Aβ-fibers that co-express NF200 not only in primary sensory neuronal somata but also in their peripheral fibers in Meissner corpuscle-like structures of glabrous skin and central terminals in the deep dorsal horn. Consistently, a recent database from single-cell RNAseq analysis of mouse DRG neurons also shows Tlr5 mRNA expression primarily in Aβ-fibers28. Activation of TLR5 via flagellin led to selective QX-314 entry into TLR5-expressing DRG neurons and subsequent functional blockade of Aβ-fibers. This finding offers a highly-demanded research tool to study the function of Aβ-fibers. Neuropathic pain in preclinical models is driven by spontaneous discharges in primary sensory neurons, which mainly occur in Aβ-fibers3, 29, 30. Clinical neuropathic pain is also maintained by primary afferent activities31. Remarkably, flagellin/QX-314-elicited Aβ-fiber blockade but not capsaicin/QX-314-induced C-fiber blockade reversed chemotherapy- and nerve injury-evoked mechanical allodynia and chemotherapy-induced ongoing pain (Fig. 4). Future studies are needed to investigate how flagellin and TLR5 interaction can increase membrane permeability and whether this membrane change requires another partner (e.g., ion channel or transporter). Although our data support an important role of Aβ-fibers in sustaining neuropathic pain, we should not exclude the contributions of C-fibers and Aδ fibers, because (1) capsaicin-sensitive C-fibers were implicated in mechanical allodynia and ongoing pain23 (but also see1–3,32); (2) low-threshold C-mechanoceptors and Aδ-mechanoceptors may modulate mechanical hypersensitivity and allodynia14, 33–35; and (3) a small portion of Tlr5+ fibers could be TH+ low-threshold C-fibers and CGRP+ C/Aδ fibers. Furthermore, TLR5 could be expressed in CGRP-negative Aδ fibers. Future study is also required to characterize TLR5 expression in different afferent fibers when new tools such as Tlr5-GFP mice become available. However, our in vivo electrophysiology data suggested that the conduction of Aα and Aδ fibers is not affected by TLR5-mediated blockade (Fig. 3), indicating limited expression of TLR5 in these A-fibers. Notably, in neuropathic pain conditions the “gate” is already open, as a result of dis-inhibition in the spinal cord pain circuit. Therefore, Aβ-fiber stimulation could be sufficient to generate mechanical allodynia in neuropathic pain conditions36, 37.
Our findings suggest that development of TLR5-mediated Aβ-fiber blockade will lead to novel therapies for treating mechanical allodynia and ongoing pain, the cardinal symptoms of clinical neuropathic pain. In support of this notion, co-application of flagellin and QX-314 also effectively blocked sodium currents in large- but not small-diameter human DRG neurons. It is worthy to mention that unlike lipopolysaccharide, flagellin is well tolerated by animals38 and shows antitumor activity in immune therapy39. Given the high potency of flagellin (low nM range vs. μM range of capsaicin) to deliver QX-314 into Aβ-fibers, local treatment of flagellin together with QX-314 via intradermal or peri-neural route at low doses should be safe and feasible.
ONLINE METHODS
Reagents
We purchased capsaicin, QX-314, phallodin-rhodamine, lidocaine, streptozotozin (STZ), and paclitaxel from Sigma-Aldrich and flagellin from invivoGen.
Animals
Knockout Tlr5−/− and Myd88−/− mice and corresponding WT control mice (C57BL/6 background) were purchased from Jackson Laboratories and maintained at Duke animal facility. All the knockout mice were viable and showed no developmental defects. Young mice (4–6 weeks of both sexes) were used for electrophysiological studies in DRG neurons. Adult male mice (8–10 weeks) were used for behavioral and pharmacological studies. Male CD1 mice (8–10 weeks, Charles River) were also used for some behavioral and pharmacological studies. Mice were group-housed on a 12-hour light/12 hour dark cycle at 22±1 °C with free access to food and water. No statistical method was used to predetermine sample size. No randomization was applied to the animal experiments. Sample sizes were chosen based on our previous studies on similar tests11, 18, 20, 24. All the animal procedures were approved by the Institutional Animal Care & Use Committee of Duke University. Animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Animal models of pain and drug injection
Intraperitoneal (i.p.) injection of paclitaxel (PAX, 6 mg/kg for a single injection or 2 mg/kg for multiple injections at day 0, 2, 4, and 6) was given to generate chemotherapy-associated neuropathic pain20. Both CD1 mice (males, for dose-response experiment) and C57BL/6 mice (males, WT and Tlr5−/−) were used. To produce streptozotocin (STZ)-induced diabetic neuropathy, male CD1 mice received a single intraperitoneal injection of STZ (75 mg/kg), as previously reported40. To produce trauma-induced neuropathic pain model, we performed chronic constriction injury (CCI) in male C57BL/6 mice under isoflurane anesthesia24, 26. For intraplantar injection, drugs were injected using a Hamilton microsyringe (Hamilton) with a 30-gauge needle. Neuropathic pain behaviors were tested 1 week after STZ injection, 2 and 4 weeks after CCI, and 1 week after PAX injection.
Quantitative real-time RT-PCR
DRG tissues were rapidly isolated in RNAse free conditions. Total RNAs were extracted using RNeasy Plus Mini kit (Qiagen). Quantity and quality of the eluted RNA samples were verified by NanoDrop spectrophotometer (Thermo Fisher Scientific). Total RNAs (0.5 μg) were reverse-transcribed using the QuantiTect Reverse Transcription Kit according to the protocol of the manufacturer (Qiagen). Specific primers including were designed using IDT SciTools Real-Time PCR web tool and ordered from IDT (Integrated DNA Technologies). The sequences of murine TLR primers are described in Supplementary Table 1. We performed gene-specific mRNA analyses using the Bio-Rad CFX96 Real-Time RT-PCR system (BioRad). Quantitative PCR amplification reactions contained the same amount of reverse transcription (RT) product, including 7.5 μL of Kapa Sybr® Fast Bio-Rad iCycler 2X qPCR Master Mix (Kapa Biosystems) and 200 nM of forward and reverse primers in a final volume of 15 μL. The thermal cycling conditions comprised 3 min of polymerase activation at 95 °C, 45 cycles of 10 s denaturation at 95 °C, and 30 s annealing and extension at 60 °C, followed by a DNA melting curve for the determination of amplicon specificity. The expression level of the target mRNA was normalized to expression of GAPDH mRNA and analyze by the standard 2−ΔΔCT method41.
In situ hybridization
Digoxigenin (DIG)-labeled RNA probes were used for in situ hybridization. For TLR5 antisense probe, the mouse cDNA fragment was amplified by PCR with the antisense primer containing the T7 promoter sequence. The sequences of the primers for antisense probe were as follows: TLR5-F, 5′-TAACGAGTTCGTCTGCAACTGT-3′; TLR5-R, 5′-GCGTAATACGACTCACTATAGGGACCCTCTGATGGTCTCATGTCT-3′. Sense probe was included as control. For TLR5 sense probe, the same mouse cDNA fragment was amplified by PCR with the sense primer containing the T7 promoter sequence instead. In vitro transcription was then performed from the PCR-amplified template using T7 RNA polymerase (Roche) with Digoxigenin-UTP (Roche) for the synthesis of the antisense and sense probes. 20 μm DRG sections were used for in situ hybridization as we previously described42. Pre-hybridization, hybridization and washing were performed according to standard methods42. Sections were then incubated with alkaline phosphatase conjugated anti-Digoxigenin (1:3500; Roche) and mouse anti-NF200 (1:1000, mouse, N0142, Sigma), or CGRP antibody (1:1000, guinea pig, T-5027, Peninsula), or TH antibody (1:1000, Rabbit, AB152, EMD Millipore) overnight at 4 °C. After washing, the in situ signals were developed with Fast Red substrate and immunostaining signals were detected with Alexa Fluor 488 anti-mouse IgG (Molecular Probes, 1:1000) or FITC-conjugated secondary antibodies (1:400; Jackson ImmunoResearch) or Alexa Fluor 488 conjugated IB4 (Molecular Probes, I21411, 1:400). For quantification, 4–5 DRG sections from each mouse were selected and 3 mice were analyzed in each group. To determine the percentage of labeled neurons, the number of positive neurons was divided by the total number of neurons. Images were analyzed with NIH Image software or Adobe PhotoShop.
Immunohistochemistry
After appropriate survival times, animals were deeply anesthetized with isoflurane and perfused through the ascending aorta with PBS, followed by 4% paraformaldehyde. After the perfusion, the L4–L5 spinal cord segments and DRGs were removed and post-fixed in the same fixative overnight. Human DRGs from non-diseased donors were also post-fixed with 4% paraformaldehyde overnight. Spinal cord sections (15 μm or 30 μm for free-floating) and DRG sections (10 μm) were cut in a cryostat. Hind paw glabrous and hairy skins were also collected, postfixed in zamboni’s fixative solution, and cut in a cryostat (30 μm, free-floating). The sections were first blocked with 2% BSA for 1 h at room temperature. The sections were then incubated overnight at 4°C with the following primary antibodies: TLR5 antibody (1:300, rabbit, Santa Cruz), NF200 (1:1000, mouse, N0142, Sigma), Cytokeratin 20 (1:40, mouse, M701929-2, Dako), and CGRP antibody (1:1000, guinea pig, T-5027, Peninsula). The sections were then incubated for 2 h at room temperature with cyanine 3 (Cy3)- or FITC-conjugated secondary antibodies (1:400; Jackson ImmunoResearch). For double immunofluorescence, sections were incubated with a mixture of polyclonal and monoclonal primary antibodies, followed by a mixture of Cy3- and FITC- conjugated secondary antibodies. The stained and mounted sections were examined with a Nikon fluorescence microscope, and images were captured with a CCD Spot camera. Some sections were also examined under a Zeiss 510 inverted confocal microscope. To confirm the specificity of the TLR5 antibody, DRG, skin, and spinal cord sections from Tlr5−/− mice were also immunostained. For quantification, 4–5 sections from each DRG were selected and 4 animals were analyzed in each group. To determine the percentage of labeled neurons, the number of positive neurons was divided by the total number of neurons. Images were analyzed with NIH Image software or Adobe PhotoShop.
Primary cultures of mouse DRG neurons
We aseptically removed DRGs from 4–7 week-old mice and digested the tissues with collagenase (1.25 mg/ml, Roche) and dispase-II (2.4 units/ml, Roche) for 90 min, followed by 0.25% trypsin for 8 min at 37 °C43. We plated cells on glass cover slips coated with poly-D-lysine and laminin and grew them in a neurobasal defined medium (with 2% B27 supplement) in the presence of 5 μM AraC, at 37°C, with 5% CO2/95% air for 24 h before experiments.
Dye treatment in whole mount DRGs and dissociated DRG neurons of mice
As previously described44, The L4–L5 whole mount DRGs were carefully removed from the vertebral column and placed in cold oxygenated ACSF. The connective tissue was gently removed under a microscope and the ganglia were digested with a mixture of 1.0 mg/ml protease and 1.6 mg/ml collagenase (Sigma) for 30 min at 37°C. DRG was transferred into a holding chamber containing normal Mg2+-free ACSF bubbled with 95% O2 and 5% CO2 at room temperature for 3 hour. DRGs were incubated with 1 μM phallodin-rhodamine (Sigma) and 10 ng/ml flagellin for 15 min at 37°C after the transferred into a holding chamber at room temperature for 1 hour for removing nonspecific binding. DRGs were then fixed by 4% formalin and processed for immunohistochemistry as described above. In addition, dissociated DRG neurons were also treated with 10 μM phallodin-rhodamine or 10 μM phallodin-rhodamine + 0.3 nM flagellin for 5 min for simultaneous fluorescence imaging and action potential recordings.
Whole-cell patch clamp recordings in dissociated mouse DRG neurons
Whole-cell voltage and current clamp recordings were performed at room temperature (28°C) to measure transient sodium currents and action potentials, respectively, with Axopatch-200B or Multiclamp 700B amplifier (Axon Instruments) and Digidata 1440A data acquisition system (Axon Instruments)45. The patch pipettes were pulled from borosilicate capillaries (Chase Scientific Glass Inc.). When filled with the pipette solution, the resistance of the pipettes was 4–5 MΩ. The recording chamber (300 μl) was continuously superfused (3–4 ml/min). Series resistance was compensated for (> 80%), and leak subtraction was performed. Data were low-pass-filtered at 2 KHz, sampled at 10 KHz. The pClamp10 (Axon Instruments) software was used during experiments and analysis. For sodium current recording, pipette solution contained (in mM): CsCl 100, sodium L-glutamic acid 5, TEACl 30, CaCl2 0.1, MgCl2 2, EGTA 11, HEPES 10, adjusted to pH 7.4 with CsOH. The external solution was composed of (in mM): NaCl 60, choline chloride 60, TEACl 15, CaCl2 1, MgCl2 5, CoCl2 5, CdCl2 0.1, 4-aminopyridine 5, HEPES 10, glucose 10 adjusted to pH 7.4 with NaOH. In voltage-clamp experiments, the transient sodium current (INa) was evoked by a test pulse (200 ms, every 30 s) to 0 mV from the holding potential, −70 mV. For quantification, the peak sodium current was measured and INA was normalized for each cell by first sweep after drug application. Drugs were applied by bath perfusion for 3 min. The sweeps were obtained and included if we saw no run-down trace in pre-pulse testing. For current-clamp experiments, pipette solution contained (in mM): K-gluconate 140, CaCl2 1, MgCl2 2, EGTA 10, K2ATP 5, HEPES 10, adjusted to pH 7.4 with KOH. The external solution contained (in mM): NaCl 140, KCl 5, CaCl2 2, MgCl2 1, glucose 10, HEPES 10, adjusted to pH 7.4 with NaOH. Action potentials were evoked by 750–1000 pA current injection (3 ms, every 30s). The recording chamber (300 μl) was continuously perfused (3–4 ml/min) with focal perfusion system. All drugs were applied via the same focal perfusion system. Perfusion exchange time was 1–2 s from merging manifold from recording chamber. The data were recorded 2–3 min after whole-cell configuration. The baseline action potential was measured for 3 min before drug treatment. Capsaicin (30 mM) was dissolved in DMSO as 30,000 × stock solution. Flagellin (3 μM) was dissolved in distilled water as 1000 × stock solution. QX-314 (5 mM) was directly dissolved in external solution. Bath perfusion solution was used as vehicle/control.
Primary cultures and patch clamp recordings in human DRG neurons
Non-diseased human DRGs were obtained from donors through National Disease Research Interchange (NDRI) with permission of exemption from Duke IRB. Postmortem L3-L5 DRGs were dissected from 4 donors (67-year-old male, 52-year-old female, 50-year-old female, and 29-year-old male) and delivered in ice-cold culture medium to the laboratory at Duke University within 24–72 hours of death. Upon the delivery, DRGs were rapidly dissected from nerve roots and minced in a calcium-free HBSS (Gibco). Human DRG cultures were prepared as previously reported with some modifications46, 47. DRGs were digested at 37°C in humidified O2 incubator for 120 min with collagenase Type II (Worthington, 285 units/mg, 12 mg/ml final concentration) and dispase II (Roche, 1 unit/mg, 20 mg/ml) in PBS with 10 mM HEPES, pH adjusted to 7.4 with NaOH. DRGs were mechanically dissociated using fire-polished pipettes, filtered through a 100 μm nylon mesh and centrifuged (400 × g for 5 min). The pellet was resuspended, plated on 0.5 mg/ml poly-D-lysine-coated glass coverslips, and grown in Neurobasal medium supplemented with 10% FBS, 2% B-27 supplement, and 1% penicillin/streptomycin. Whole-cell patch clamp recordings in small (<50 μm) and large-diameter DRG neurons (55–80 μm) were conducted at room temperature using patch pipettes with resistances of 2–3 MΩ. The recording chamber was continuously superfused (3–4 ml/min). The data were acquired at a rate of 10 kHz and filtered at 3 kHz using an EPC-10 amplifier (HEKA, Germany) and Patchmaster software (HEKA, Germany). For sodium current recording, pipette solution contained (in mM): CsCl 130, NaCl 9, MgCl2 1, EGTA 10, HEPES 10, adjusted to pH 7.4 with CsOH. The external solution was composed of (in mM): NaCl 131, TEACl 10, CsCl 10, CaCl2 1, MgCl2 2, CdCl2 0.3, 4-aminopyridine 3, HEPES 10, glucose 10 adjusted to pH 7.4 with NaOH. In voltage-clamp experiments, the transient sodium current (INa) was evoked by a voltage pulse (50 ms, every 10 s) from −70 mV to 0 mV. The data were recorded for 5 min after whole-cell configuration. The baseline sodium currents were measured for at least 1 min before the drug treatments. The data analyses were performed without selection and exclusion after recordings.
Sciatic nerve compound action potential (CAP) recordings
Adult male mice (25–32g) were anaesthetized with urethane (1.5 g/kg, i.p.) and monitored for loss of hind paw pinch reflex with supplemental injections of urethane (0.2 g/kg). The animals were artificially ventilated with oxygen on a respirator. The left thigh was shaved and an incision made parallel to the femur. The muscle was parted by blunt forceps dissection to expose the sciatic nerve proximal to the trifurcation. A cuff electrode was placed loosely around the full circumference of the sciatic nerve. Skin flaps were raised to enclose a pool of mineral oil that covered the exposed regions of nerve. The hind paw was stimulated through a pair of shielded electrodes by a stimulus isolator (A365, World Precision Instruments) at 0.03 Hz (square-wave pulse of 100 μs duration). The evoked compound action potentials were amplified with a microelectrode AC amplifier (1800, AM systems), filtered (low cut-off 0.1Hz and hi cut-off 20 kHz) and digitized at 20–50 kHz (Digidata 1440A, Molecular Devices). Data were stored with a personal computer using pCLAMP 10 software and analyzed with Origin pro 8.0 (Origin Lab). The responses of different afferent fibers were characterized as previously reported48, 49.
Behavioral testing
Animals were habituated to the environment for at least 2 days before the testing. All the behaviors were tested blindly. For testing mechanical sensitivity, we confined mice in boxes placed on an elevated metal mesh floor and stimulated their hindpaws with a series of von Frey hairs with logarithmically increasing stiffness (0.02–2.56g, Stoelting), presented perpendicularly to the central plantar surface. We determined the 50% paw withdrawal threshold by Dixon’s up-down method50. Thermal sensitivity was tested using hot plate and Hargreaves radiant heat apparatus51 (IITC Life Science). For the radiant heat test, the basal paw withdrawal latency was adjusted to 9–12 s, with a cutoff of 20 s to prevent tissue damage.
To test spontaneous/ongoing pain measurement, we used a single trial protocol for conditioned place preference (CPP)23,52. All mice underwent a 3-day pre-conditioning habituation and animal behavior was video-recorded. Analyses of the pre-conditioning (baseline) behavior showed no pre-existing chamber preference. On the conditioning day, mice received the vehicle control (QX-314 injection) paired with a randomly chosen chamber in the morning, and then the appropriate treatment of A-fiber block (QX-314 plus flagellin) or C-fiber block (QX-314 plus capsaicin) paired with the other chamber 4 h later. Chamber pairings were counterbalanced. On the test day, 20 h following the afternoon pairing, mice were placed in the CPP box with access to both chambers and the behavior was recorded for 15 min and analyzed by ANY-maze software for chamber preference.
To test motor function, A Rota-rod system (IITC Life Science Inc.) was used. Mice were tested for three trails separated by 10 min intervals. During the tests, the speed of rotation was accelerated from 4 to 40 rpm in 5 min. The falling latency was recorded.
To assess the inflammation after QX-314/flagellin treatment, paw volume was determined by water displacement plethysmometer (Ugo Basile, Italy). The Plethysmometer is a microcontrolled volume meter, specially designed for accurate measurement of the rodent paw swelling. It consists of a water filled Perspex cell into which the paw is dipped. A transducer of original design records small differences in water level, caused by volume displacement. The digital read-out shows the exact volume of the paw.
Statistical analyses
All the data were expressed as mean ± s.e.m, as indicated in the figure legends. The sample size is each experiment was based on our previous studies on such experiment24,41,44,53. Statistical analyses were completed with Prism GraphPad or Excel. Biochemical and behavioral data were analyzed using two-tailed student’s t-test (two groups) or Two-Way ANOVA followed by post-hoc Bonferroni test24. Electrophysiological data were tested using one-way ANOVA (for multiple comparisons) or Two-Way ANOVA (for multiple time points) followed by post-hoc Bonferroni test or student’s t-test (two groups)53. The criterion for statistical significance was P < 0.05.
Supplementary Material
Acknowledgments
This study is supported by US National Institutes of Health (NIH) R01 grants NS67686 (R.-R.J), NS87988 (R.-R.J), NS89479 (R.-R.J), DE17794 (R.-R.J), and DE22743 (R.-R.J), NIH R21 grants NS82985 (Z.-Z.X.) and NS91779 (Z.-Z.X.), NIH R01 grant DE19440 (F.W), Korea government grant 2012R1A3A2048834 (S.B.O.) and Korea National Research Foundation grant 2013R1A6A3A04065858 (Y.H.K).
Footnotes
All the authors have no financial interests in this study.
AUTHOR CONTRIBUTIONS
Z.-Z.X. developed the behavioral part of the project and designed and performed immunohistochemical and behavioral experiments, Y. H. K. recorded action potentials in mouse DRG neurons, compound potentials in intact mice, and sodium currents in human DRG neurons. S.B. initially tested the idea of flagellin/QX-314 blockade of sodium currents in mouse DRG neurons and examined dye (phallodin-rhodamine) uptake in DRGs. Y.Z performed in situ hybridization under the guidance of F.W., T.B. performed PCR experiment. R.-R. J. conceived and supervised the project. S.B.O and F.W. discussed the project, and R.-R. J., Z.-Z.X., S.B.O., and F.W. wrote the paper.
Reference List
- 1.Ossipov MH, Bian D, Malan TP, Jr, Lai J, Porreca F. Lack of involvement of capsaicin-sensitive primary afferents in nerve-ligation injury induced tactile allodynia in rats. Pain. 1999;79:127–133. doi: 10.1016/s0304-3959(98)00187-0. [DOI] [PubMed] [Google Scholar]
- 2.Minett MS, et al. Pain without nociceptors? Nav1.7-independent pain mechanisms. Cell Rep. 2014;6:301–312. doi: 10.1016/j.celrep.2013.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu CN, et al. Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain. 2000;85:503–521. doi: 10.1016/S0304-3959(00)00251-7. [DOI] [PubMed] [Google Scholar]
- 4.Brenneis C, et al. Phenotyping the function of TRPV1-expressing sensory neurons by targeted axonal silencing. J Neurosci. 2013;33:315–326. doi: 10.1523/JNEUROSCI.2804-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koltzenburg M, Lundberg LE, Torebjork HE. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1992;51:207–219. doi: 10.1016/0304-3959(92)90262-A. [DOI] [PubMed] [Google Scholar]
- 6.Ossipov MH, et al. Selective mediation of nerve injury-induced tactile hypersensitivity by neuropeptide Y. J Neurosci. 2002;22:9858–9867. doi: 10.1523/JNEUROSCI.22-22-09858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell JN, Raja SN, Meyer RA, Mackinnon SE. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32:89–94. doi: 10.1016/0304-3959(88)90027-9. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi F, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 9.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull. 2012;28:131–144. doi: 10.1007/s12264-012-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu T, et al. TLR3 deficiency impairs spinal cord synaptic transmission, central sensitization, and pruritus in mice. J Clin Invest. 2012;122:2195–2207. doi: 10.1172/JCI45414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawson SN, Waddell PJ. Soma neurofilament immunoreactivity is related to cell size and fibre conduction velocity in rat primary sensory neurons. J Physiol. 1991;435:41–63. doi: 10.1113/jphysiol.1991.sp018497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCoy ES, et al. Peptidergic CGRPalpha primary sensory neurons encode heat and itch and tonically suppress sensitivity to cold. Neuron. 2013;78:138–151. doi: 10.1016/j.neuron.2013.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li L, et al. The functional organization of cutaneous low-threshold mechanosensory neurons. Cell. 2011;147:1615–1627. doi: 10.1016/j.cell.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat Rev Neurosci. 2010;11:823–836. doi: 10.1038/nrn2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Binshtok AM, Bean BP, Woolf CJ. Inhibition of nociceptors by TRPV1-mediated entry of impermeant sodium channel blockers. Nature. 2007;449:607–610. doi: 10.1038/nature06191. [DOI] [PubMed] [Google Scholar]
- 17.Roberson DP, et al. Activity-dependent silencing reveals functionally distinct itch-generating sensory neurons. Nat Neurosci. 2013;16:910–918. doi: 10.1038/nn.3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park CK, et al. Extracellular MicroRNAs Activate Nociceptor Neurons to Elicit Pain via TLR7 and TRPA1. Neuron. 2014;82:47–54. doi: 10.1016/j.neuron.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith KD, et al. Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat Immunol. 2003;4:1247–1253. doi: 10.1038/ni1011. [DOI] [PubMed] [Google Scholar]
- 20.Liu XJ, et al. Nociceptive neurons regulate innate and adaptive immunity and neuropathic pain through MyD88 adapter. Cell Res. 2014 doi: 10.1038/cr.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavaletti G, Marmiroli P. Chemotherapy-induced peripheral neurotoxicity. Nat Rev Neurol. 2010;6:657–666. doi: 10.1038/nrneurol.2010.160. [DOI] [PubMed] [Google Scholar]
- 22.Binshtok AM, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28:14062–14073. doi: 10.1523/JNEUROSCI.3795-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King T, et al. Contribution of afferent pathways to nerve injury-induced spontaneous pain and evoked hypersensitivity. Pain. 2011 doi: 10.1016/j.pain.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu ZZ, et al. Neuroprotectin/Protectin D1 protects neuropathic pain in mice after nerve trauma. Ann Neurol. 2013 doi: 10.1002/ana.23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan GM, Chen SR, Pan HL. Role of primary afferent nerves in allodynia caused by diabetic neuropathy in rats. Neuroscience. 2002;114:291–299. doi: 10.1016/s0306-4522(02)00372-x. [DOI] [PubMed] [Google Scholar]
- 26.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 27.Melzack R, Wall PD. Pain mechanisms: a new theory. Science. 1965;150:971–979. doi: 10.1126/science.150.3699.971. [DOI] [PubMed] [Google Scholar]
- 28.Usoskin D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat Neurosci. 2015;18:145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 29.Devor M. Neuropathic pain and injured nerve: peripheral mechanisms. Br Med Bull. 1991;47:619–630. doi: 10.1093/oxfordjournals.bmb.a072496. [DOI] [PubMed] [Google Scholar]
- 30.Ma C, et al. Similar electrophysiological changes in axotomized and neighboring intact dorsal root ganglion neurons. J Neurophysiol. 2003;89:1588–1602. doi: 10.1152/jn.00855.2002. [DOI] [PubMed] [Google Scholar]
- 31.Haroutounian S, et al. Primary afferent input critical for maintaining spontaneous pain in peripheral neuropathy. Pain. 2014;155:1272–1279. doi: 10.1016/j.pain.2014.03.022. [DOI] [PubMed] [Google Scholar]
- 32.Kim YH, et al. TRPV1 in GABAergic interneurons mediates neuropathic mechanical allodynia and disinhibition of the nociceptive circuitry in the spinal cord. Neuron. 2012;74:640–647. doi: 10.1016/j.neuron.2012.02.039. [DOI] [PubMed] [Google Scholar]
- 33.Seal RP, et al. Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature. 2009;462:651–655. doi: 10.1038/nature08505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boada MD, et al. Fast-conducting mechanoreceptors contribute to withdrawal behavior in normal and nerve injured rats. Pain. 2014;155:2646–2655. doi: 10.1016/j.pain.2014.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith AK, O’Hara CL, Stucky CL. Mechanical sensitization of cutaneous sensory fibers in the spared nerve injury mouse model. Mol Pain. 2013;9:61. doi: 10.1186/1744-8069-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu Y, et al. A feed-forward spinal cord glycinergic neural circuit gates mechanical allodynia. J Clin Invest. 2013;123:4050–4062. doi: 10.1172/JCI70026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duan B, et al. Identification of spinal circuits transmitting and gating mechanical pain. Cell. 2014;159:1417–1432. doi: 10.1016/j.cell.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vijay-Kumar M, et al. Flagellin treatment protects against chemicals, bacteria, viruses, and radiation. J Immunol. 2008;180:8280–8285. doi: 10.4049/jimmunol.180.12.8280. [DOI] [PubMed] [Google Scholar]
- 39.Sfondrini L, et al. Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J Immunol. 2006;176:6624–6630. doi: 10.4049/jimmunol.176.11.6624. [DOI] [PubMed] [Google Scholar]
- 40.Alessandri-Haber N, Dina OA, Joseph EK, Reichling DB, Levine JD. Interaction of transient receptor potential vanilloid 4, integrin, and SRC tyrosine kinase in mechanical hyperalgesia. J Neurosci. 2008;28:1046–1057. doi: 10.1523/JNEUROSCI.4497-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berta T, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin Invest. 2014;124:1173–1186. doi: 10.1172/JCI72230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pagadala P, et al. Loss of NR1 Subunit of NMDARs in Primary Sensory Neurons Leads to Hyperexcitability and Pain Hypersensitivity: Involvement of Ca2+-Activated Small Conductance Potassium Channels. J Neurosci. 2013;33:13425–13430. doi: 10.1523/JNEUROSCI.0454-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus. Nat Neurosci. 2010;13:1460–1462. doi: 10.1038/nn.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie RG, et al. Blockade of persistent sodium currents contributes to the riluzole-induced inhibition of spontaneous activity and oscillations in injured DRG neurons. PLoS ONE. 2011;6:e18681. doi: 10.1371/journal.pone.0018681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee JH, et al. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell. 2014;157:1393–1404. doi: 10.1016/j.cell.2014.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 46.Baumann TK, Chaudhary P, Martenson ME. Background potassium channel block and TRPV1 activation contribute to proton depolarization of sensory neurons from humans with neuropathic pain. Eur J Neurosci. 2004;19(5):1343–51. doi: 10.1111/j.1460-9568.2004.03097.x. [DOI] [PubMed] [Google Scholar]
- 47.Steve D, et al. Human sensory neurons: Membrane properties and sensitization by inflammatory mediators. Pain. 2014;155(9):1861–70. doi: 10.1016/j.pain.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinto V, Derkach VA, Safronov BV. Role of TTX-sensitive and TTX-resistant sodium channels in Adelta- and C-fiber conduction and synaptic transmission. J Neurophysiol. 2008;99:617–628. doi: 10.1152/jn.00944.2007. [DOI] [PubMed] [Google Scholar]
- 49.Tsuchimochi H, McCord JL, Leal AK, Kaufman MP. Dorsal root tetrodotoxin-resistant sodium channels do not contribute to the augmented exercise pressor reflex in rats with chronic femoral artery occlusion. Am J Physiol Heart Circ Physiol. 2011;300:H652–H663. doi: 10.1152/ajpheart.00859.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- 51.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 52.King T, et al. Unmasking the tonic-aversive state in neuropathic pain. Nat Neurosci. 2009;12:1364–1366. doi: 10.1038/nn.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park CK, et al. Resolving TRPV1- and TNF-a-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1. J Neurosci. 2011;31:15072–15085. doi: 10.1523/JNEUROSCI.2443-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.