

ABSTRACT
In Staphylococcus aureus, hundreds of small regulatory or small RNAs (sRNAs) have been identified, yet this class of molecule remains poorly understood and severely understudied. sRNA genes are typically absent from genome annotation files, and as a consequence, their existence is often overlooked, particularly in global transcriptomic studies. To facilitate improved detection and analysis of sRNAs in S. aureus, we generated updated GenBank files for three commonly used S. aureus strains (MRSA252, NCTC 8325, and USA300), in which we added annotations for >260 previously identified sRNAs. These files, the first to include genome-wide annotation of sRNAs in S. aureus, were then used as a foundation to identify novel sRNAs in the community-associated methicillin-resistant strain USA300. This analysis led to the discovery of 39 previously unidentified sRNAs. Investigating the genomic loci of the newly identified sRNAs revealed a surprising degree of inconsistency in genome annotation in S. aureus, which may be hindering the analysis and functional exploration of these elements. Finally, using our newly created annotation files as a reference, we perform a global analysis of sRNA gene expression in S. aureus and demonstrate that the newly identified tsr25 is the most highly upregulated sRNA in human serum. This study provides an invaluable resource to the S. aureus research community in the form of our newly generated annotation files, while at the same time presenting the first examination of differential sRNA expression in pathophysiologically relevant conditions.
IMPORTANCE
Despite a large number of studies identifying regulatory or small RNA (sRNA) genes in Staphylococcus aureus, their annotation is notably lacking in available genome files. In addition to this, there has been a considerable lack of cross-referencing in the wealth of studies identifying these elements, often leading to the same sRNA being identified multiple times and bearing multiple names. In this work, we have consolidated and curated known sRNA genes from the literature and mapped them to their position on the S. aureus genome, creating new genome annotation files. These files can now be used by the scientific community at large in experiments to search for previously undiscovered sRNA genes and to monitor sRNA gene expression by transcriptome sequencing (RNA-seq). We demonstrate this application, identifying 39 new sRNAs and studying their expression during S. aureus growth in human serum.
INTRODUCTION
In recent years, a number of studies have been carried out, employing both experimental and computational methods to identify regulatory or small RNAs (sRNAs) in Staphylococcus aureus (1–12). Hundreds of sRNAs have been identified, and many, in addition to the agr effector RNAIII, have been shown to play a role in gene regulation (while these molecules go by a variety of names such as regulatory RNAs or noncoding RNAs, we will use sRNAs to refer to them all, as previously recommended [13]). Despite advancements in sRNA identification, the roles of most of these molecules remain unknown, because in many cases, limited functional information can be gathered from analysis of their sequence alone.
One additional factor that has hampered the study of sRNAs in S. aureus has been the lack of a clear nomenclature and annotation system. This absence of a systematic identification and annotation process has led to the repeated discovery of the same sRNAs on multiple occasions, the reidentification of already known sRNAs (e.g., RNAIII), and even to important protein-coding genes being ascribed as sRNAs (e.g., the α-PSM transcript, which is not annotated in most S. aureus genome files). Recent work by Sassi et al. (14) established an online database for staphylococcal sRNAs; however, most sRNAs, including the well-studied RNAIII, are still not included in annotated S. aureus GenBank genome files. This is a marked oversight, as annotated genome files serve as the reference for global genomic and transcriptomic studies; thus, the absence of sRNAs from these files severely impedes their study and prevents us from gaining an overarching picture of regulatory circuits.
In S. aureus, most sRNA identification studies have been performed in a single background, the hospital-acquired methicillin-resistant S. aureus (MRSA) strain N315. The existence of many of these sRNAs has been demonstrated experimentally in strain N315 (1, 3, 4, 6, 7); however, very few of them have been investigated in other S. aureus strains, including the epidemic community-associated MRSA (CA-MRSA) strain USA300 (15). As such, their existence, location, and copy number in most S. aureus isolates is unknown, preventing us from gaining a sense of their role in the physiological and pathogenic differences between strains.
To better understand the sRNA content of multiple S. aureus strains, we explored the genomes of three well-studied S. aureus strains (USA300, MRSA252, and NCTC 8325). We identified the location(s) of previously discovered sRNAs and created new GenBank genome annotation files for each strain, inserting ~260 sRNAs. These newly annotated files serve as a valuable resource allowing us to do the following: (i) search the genome of each strain for as yet unidentified sRNAs without mistakenly reidentifying known species, and (ii) calculate expression values for these genes using transcriptome sequencing (RNA-seq) data.
To demonstrate the application of these new files, we performed RNA-seq on CA-MRSA strain USA300 growing in laboratory media and human serum and aligned the data to our newly created sRNA annotated genome files. Examining the data, we identified 39 novel putative sRNAs that had not been previously reported. These novel sRNAs were annotated, cross-referenced in the genomes of strains NCTC 8325 and MRSA252, and added to the newly created genome files as well. During the cross-referencing process, we observed numerous examples of inconsistent genome annotation in different S. aureus strains. We highlight examples clearly demonstrating genome misannotation and demonstrate how this phenomenon is adversely affecting the identification and characterization of sRNAs. Finally, we calculate expression values and examine the global sRNA expression profile of strain USA300, uncovering a wealth of molecules that display differential expression in human serum. The latter point is of significant importance, as it gives us a unique look into the sRNA transcriptome, not only during growth of S. aureus but also in a pathophysiologically relevant growth environment. The new genome annotation files described in this work have been deposited in the figshare depository and are freely available for download. We suggest that these newly reannotated genomes will be a valuable resource to the S. aureus research community for sRNA identification and analysis hereafter, as they can be incrementally added to as new sRNAs are discovered.
RESULTS
Annotation of sRNAs on the S. aureus genome.
Previous studies in our lab have utilized RNA-seq to determine the global transcriptomic profile of S. aureus in the community-associated MRSA strain USA300 (16, 17). Analysis of these data sets revealed a large number of S. aureus transcripts that map to intergenic regions where no protein-coding genes have been annotated. We hypothesized that many of these transcripts represent sRNAs because of the following. (i) Most S. aureus genome annotation files do not contain annotations for sRNA genes. (ii) Recent studies carried out in the S. aureus N315 background have demonstrated that there are several hundred sRNAs encoded in the S. aureus genome (12, 14). To facilitate improved global transcriptomic analysis of S. aureus by RNA-seq, we created new GenBank genome annotation files for three commonly used S. aureus strains, NCTC 8325, MRSA252, and USA300. To do this, we elected to expand the sRNA annotation and nomenclature system already present for 25 sRNAs in strain MRSA252 (see Table S1 in the supplemental material) and apply a similar annotation/nomenclature system to strains USA300 and NCTC 8325 (for details, see supplemental material). To include annotations for known sRNAs, we performed a literature search to identify studies in which sRNAs in S. aureus were reported. A total of 12 papers that employed a variety of methods to identify sRNAs, including computational approaches, microarray studies, cDNA cloning, and high-throughput sequencing, were investigated (1–12). Using the information provided in these publications, a list of 928 potential sRNAs was assembled (Table S2) (1–12). In order to ensure accurate annotation of each sRNA from this list, RNA-seq experiments were performed for each of the three strains growing under standard laboratory conditions. Reads generated from these works were aligned to the respective genomes and used as a guide to identify the specific location of each sRNA for each of the three strains (see Fig. S1 in the supplemental material).
This work condensed the 928 putative sRNAs to a total of 248 annotations for strain MRSA252, 254 annotations for strain NCTC 8325, and 264 annotations for strain USA300 (for a more-comprehensive explanation of how sRNAs were identified and annotated, see Text S1 in the supplemental material). Such a dramatic reduction in the number of sRNAs points to the scale of overlapping identification, reidentification, and duplicate naming that was extant in the literature for these elements and the poor state of sRNA curation in S. aureus genomes. Our newly generated GenBank files for all three strains represent the first comprehensive list of sRNAs annotated directly in the S. aureus genome, which will serve as a valuable reference point for future sRNA discovery, and more broadly, global transcriptomic analyses of S. aureus by RNA-seq.
Detection of novel sRNAs in strain USA300.
The creation of GenBank files containing annotations for previously identified sRNAs provided us with a unique opportunity to examine RNA-seq data for novel forms of these elements, without mistakenly reidentifying those that are already known. Accordingly, we set out to perform such an exploration using the community-associated MRSA isolate USA300, an undeniably relevant clinical strain for which no sRNA identification studies have yet been performed. To maximize the probability of identifying novel sRNAs, we performed RNA-seq with USA300 grown under both laboratory conditions (tryptic soy broth [TSB]), and in media that was more pathophysiologically relevant to its infectious lifestyle (human serum) (18).
RNA-seq reads from bacteria grown under these conditions were mapped to the newly created USA300 GenBank file (containing 264 sRNA annotations), followed by a thorough examination for the presence of novel sRNA transcripts. A total of 39 potential sRNAs were identified from both conditions, which we named tsr1 to tsr39 for Tampa small RNA (Table 1) (for details regarding the criteria used for our determination of novel sRNAs, see Text S1 in the supplemental material). Although the majority of the tsr genes were located in intergenic regions, a number were identified as being antisense to annotated genes or as partially overlapping annotated coding DNA sequence (CDS) genes. The novel sRNAs were also added to our newly created USA300 genome file, using the nomenclature system described in Text S1, resulting in a GenBank file with a total of 303 sRNAs in the USA300 genome. This brings the total number of genes annotated on the USA300 genome from 2,850 to 3,153, representing an approximately 10% increase in annotated genes.
TABLE 1 .
Novel sRNAs identified in strain USA300
| sRNA designation | sRNA gene | Stranda | Location | Size (nt) | Chromosomal location of tsr gene in strain: |
||
|---|---|---|---|---|---|---|---|
| USA300b | MRSA252b | NCTC 8325b | |||||
| SAUSA300s265 | tsr1 | > | 52438−53094 | 656 | IG | − | − |
| SAUSA300s266 | tsr2 | < | 57712−57804 | 92 | IG | − | − |
| SAUSA300s267 | tsr3 | < | 61388−61550 | 162 | IG | − | − |
| SAUSA300s268 | tsr4 | > | 73511−74139 | 628 | AS | AS | AS |
| SAUSA300s269 | tsr5 | < | 79346−79425 | 79 | AS | − | − |
| SAUSA300s270 | tsr6 | > | 120785−120897 | 112 | IG | IG | IG |
| SAUSA300s271 | tsr7 | > | 169903−170079 | 176 | IG | − | IG |
| SAUSA300s272 | tsr8 | < | 170013−170214 | 201 | IG | − | IG |
| SAUSA300s273 | tsr9 | < | 228412−228796 | 384 | IG | CDS | CDS |
| SAUSA300s274 | tsr10 | > | 349895−350058 | 163 | IG | OL | OL |
| SAUSA300s275 | tsr11 | < | 356689−356782 | 93 | OL | OL | OL |
| SAUSA300s276 | tsr12 | > | 457272−457333 | 61 | IG | OL | AS |
| SAUSA300s277 | tsr13 | > | 484942−485025 | 83 | OL | OL | OL |
| SAUSA300s278 | tsr14 | > | 834340−834885 | 545 | IG | IG | AS |
| SAUSA300s279 | tsr15 | > | 896563−897379 | 816 | AS | AS | AS |
| SAUSA300s280 | tsr16 | < | 911246−911364 | 118 | AS | OL | OL |
| SAUSA300s281 | tsr17 | < | 973559−973971 | 412 | IG | CDS | CDS |
| SAUSA300s282 | tsr18 | < | 1074292−1074484 | 192 | IG | CDS | CDS |
| SAUSA300s283 | tsr19 | < | 1080302−1080394 | 92 | IG | IG | IG |
| SAUSA300s284 | tsr20 | < | 1154300−1154753 | 453 | IG | − | IG |
| SAUSA300s285 | tsr21 | < | 1154827−1156001 | 1174 | CDS | CDS3 | CDS3 |
| SAUSA300s286 | tsr22 | > | 1165484−1165865 | 381 | IG | CDS | CDS |
| SAUSA300s287 | tsr23 | > | 1256521−1256545 | 24 | IG | IG | IG |
| SAUSA300s288 | tsr24 | > | 1429517−1429754 | 237 | IG | − | IG |
| SAUSA300s289 | tsr25 | > | 1442862−1443042 | 180 | IG | AS | AS |
| SAUSA300s290 | tsr26 | > | 1641611−1641732 | 121 | IG | IG | − |
| SAUSA300s291 | tsr27 | < | 1642820−1642923 | 103 | IG | IG | − |
| SAUSA300s292 | tsr28 | < | 1715900−1715975 | 75 | OL | OL | OL |
| SAUSA300s293 | tsr29 | < | 1954961−1955091 | 130 | IG | − | IG |
| SAUSA300s294 | tsr30 | > | 2126434−2126545 | 111 | IG | − | − |
| SAUSA300s295 | tsr31 | < | 2244964−2245035 | 71 | IG | IG | IG |
| SAUSA300s296 | tsr32 | > | 2337922−2338072 | 150 | IG | IG | CDS |
| SAUSA300s297 | tsr33 | < | 2410564−2410648 | 84 | IG | IG | IG |
| SAUSA300s298 | tsr34 | > | 2591032−2591131 | 99 | IG | IG | IG |
| SAUSA300s299 | tsr35 | > | 2608047−2608594 | 547 | IG | CDS | CDS |
| SAUSA300s300 | tsr36 | < | 2608120−2608645 | 525 | IG | AS | AS |
| SAUSA300s301 | tsr37 | > | 2620285−2620621 | 336 | IG | CDS | CDS |
| SAUSA300s302 | tsr38 | < | 2664856−2664945 | 89 | IG | IG | IG |
| SAUSA300s303 | tsr39 | < | 2811278−2811330 | 52 | IG | IG | IG |
>, forward strand; <, reverse strand.
Characteristics of the chromosomal location of the tsr gene. IG, located in the intergenic region; −, absent, deleted, or no homologue; AS, antisense to the annotated gene; CDS, located within an existing annotated CDS; OL, partially overlaps CDS gene; CDS3, the corresponding locus contains three annotated CDSs.
To investigate the conservation of tsr genes in S. aureus, we analyzed their corresponding chromosomal loci on the S. aureus NCTC 8325 and MRSA252 genomes by performing a BLAST search (Table 1). Of the 39 tsr genes, 29 were found in strain MRSA252, while 32 were found in strain NCTC 8325. Annotations were again added to the MRSA252 and NCTC 8325 genome files for each of the tsr genes identified using the nomenclature outlined (see Text S1 in the supplemental material). Importantly, five of the tsr genes appear to be unique to USA300 (tsr1, tsr2, tsr3, tsr5, and tsr30), with no homologues found in either MRSA252 or NCTC 8325. In addition, five tsr genes were present in USA300 and NCTC 8325 but absent in MRSA252 (tsr7, tsr8, tsr20, tsr24, and tsr29), while two were present in USA300 and MRSA252 but absent in NCTC 8325 (tsr26 and tsr27) (see Fig. S2 in the supplemental material).
Northern blot analysis of tsr transcripts.
To validate our RNA-seq-based approach for sRNA discovery, five putative tsr transcripts were examined by Northern blot analysis. The transcripts investigated had different expression patterns in TSB compared to human serum. Three of the transcripts investigated (tsr8, tsr26, and tsr31) showed no alteration in expression between TSB and human serum, one (tsr25) demonstrated an increase in expression, while another (tsr33) showed a decrease (see Table S3 in the supplemental material). Northern blot analysis confirmed the predicted size and orientation of tsr25, tsr26, and tsr31 (Fig. 1). In the case of tsr8 and tsr33, bands were identified; however, they were considerably larger than those predicted by RNA-seq analysis (Fig. 1). For example, the predicted size of tsr33 was 85 nucleotides (nt); however, the size observed by Northern blotting was approximately 600 nt. When looking at the tsr33 transcript, one observes that it is located at the 3′ end of the sarR gene and is transcribed in the same orientation. The combined size of sarR and tsr33 would be around 600 nt, therefore suggesting that tsr33 represents a large 3′ untranslated region (UTR) for the sarR gene. However, RNA-seq alignment from strain USA300 growing in human serum demonstrates a much greater depth of coverage (and hence abundance) of tsr33 than of sarR, suggesting that a tsr33-specific RNA may exist under these conditions (see Fig. S3 in the supplemental material). On the basis of the above information, we predict that tsr33 is cotranscribed as a 3′ UTR of sarR, but under certain conditions (e.g., growth in human serum), it is possible that a tsr33 RNA may exist independently of sarR.
FIG 1 .

Northern blot analysis of tsr transcripts. Northern blotting was performed using oligonucleotide probes specific for the tsr transcripts (tsr8, tsr25, tsr26, tsr31, and tsr33). RNA-seq read alignments for each corresponding chromosomal location are shown, as are CDS genes (black arrows), and sRNAs (red arrows). The depth of read coverage on the genome is shown by the blue histograms.
The second tsr demonstrating a size difference was tsr8, which was predicted by RNA-seq to be 201 nt; however, two bands were observed by Northern blotting with approximate sizes between 300 and 400 nucleotides. A possible explanation for this may come from the fact that tsr8 and tsr7 are convergently transcribed, that the two transcripts are complementary at the 3′ ends, and that these regions overlap. The density of reads, in both directions, mapping to this region of complementarity make it difficult to predict the precise location of each transcript based on RNA-seq data, resulting in an underestimation of tsr8 size. Given our experimental findings, the size of tsr8 was amended to ~350 nt in the GenBank files. It is interesting to note that this type of genetic organization (convergent transcripts overlapping at the 3′ end) is common among toxin-antitoxin (TA) systems in S. aureus (19, 20), hence tsr7 and tsr8 could potentially represent a novel serum-induced TA system.
For some of the tsr elements expressed at low levels, Northern blot detection proved unsuccessful (data not shown); therefore, we employed a reverse transcriptase PCR (RT-PCR)-based approach, which is inherently more sensitive. Using this methodology, we were able to validate the presence of an additional six transcripts, tsr1, tsr2, tsr18, tsr24, tsr29, and tsr32 (see Fig. S4 in the supplemental material), suggesting that our RNA-seq-based identification approach is effective at identifying legitimate sRNA molecules.
Inconsistent genome annotation in strains USA300, MRSA252, and NCTC 8325.
Twenty-seven tsr genes were found in the genomes of all three strains (USA300, MRSA252, and NCTC 8325). For 14 of these genes, the corresponding genomic loci were similarly annotated in all three strains, e.g., tsr6 is located in an intergenic region in all three strains, while tsr15 is located antisense to an annotated CDS. Interestingly, for 13 tsr genes, the genomic loci in the three strains studied are differentially annotated (Table 1). In many cases, the NCTC 8325 and MRSA252 genomes contain annotations for CDS genes, while the USA300 genome specifies these loci as being intergenic (e.g., tsr9, tsr17, tsr18, etc.). An open reading frame (ORF) search reveals that 11 tsr genes have the potential to encode proteins (of 30 amino acids or larger in size). Seven of these genes are annotated as CDS in strains MRSA252 and NCTC 8325. This raises the possibility that some tsr genes may in fact be protein-coding genes that were omitted from the USA300 genome annotation. Conversely, it is also possible that the NCTC 8325 and MRSA252 genomes may be incorrect and that these annotated genes do not encode proteins. Upon close examination, our data clearly demonstrate that incorrect genome annotation accounts for at least some of the discrepancies observed. For example, the tsr12 locus is annotated differently in all three strains (Fig. 2). In strain USA300, tsr12 is located in an intergenic region (between SAUSA300_0404 and hsdM). In strain MRSA252, tsr12 incompletely overlaps with a CDS gene annotated in the same orientation, while in strain NCTC 8325, it partially overlaps with a CDS gene annotated in the opposite direction. Bioinformatic analysis of these CDS genes reveals that they both specify very small proteins (43 amino acids in MRSA252 and 33 amino acids in NCTC 8325) that possess no known structural motifs and have no homology to any protein in the database beyond counterparts in a handful of other S. aureus strains. Furthermore, in USA300 and NCTC 8325, the tsr12 locus is 100% identical at the nucleotide level, making large-scale differences in coding sequences (e.g., inverse open reading frames) unlikely. Collectively, this demonstrates that there is likely misannotation in the genomes of MRSA252 and NCTC 8325 and that our suggested annotation of tsr12 as an sRNA is likely the correct one.
FIG 2 .

Variation in genome annotation of the tsr12 locus. RNA-seq read alignment data are shown for strains USA300 (A), MRSA252 (B), and NCTC 8325 (C). Annotations for CDS genes are shown by black arrows, and the depth of coverage is shown by the blue histograms. The location of tsr12 is shown by a red arrow. (A) There is no CDS annotation at the tsr12 locus in strain USA300. (B) In strain MRSA252, a gene (SAR0432) is annotated in the forward direction at the tsr12 locus. (C) In strain NCTC 8325, a gene (SAOUHSC_00396) is annotated in the reverse direction at the tsr12 locus.
tsr12 is an example where incorrect genome annotation is rather clear and raises important questions regarding how commonplace this type of genome misannotation may be. To investigate the issue of inconsistent genome annotation in S. aureus further, we selected another sRNA gene for additional study. Teg23 (SAUSA300s148, SARs145, SAOUHSCs144) was originally identified by Beaume et al. (7) as a potential 5′ UTR of the SAS083 gene in S. aureus strain N315. In strains NCTC 8325 and MRSA252, a gene is annotated in the position corresponding to SAS083 (SAOUHSC_02572 and SAR2384, respectively), but no gene is annotated in strain USA300 (Fig. 3A). Upon analysis, SAS083, SAOUHSC_02572, and SAR2384 once again encode small, hypothetical proteins with no known structural features, functional domains, or apparent homologues and are therefore likely misannotated genes (as noted above for the tsr12 locus).
FIG 3 .

Analysis of the Teg23 locus. (A) Teg23 locus in S. aureus strains N315, USA300, NCTC 8325, and MRSA252. Annotated CDS genes are shown in gray. The previously reported location of Teg23 is shown in white. (B) Northern blot analysis of Teg23. The Teg23 transcript was detected by Northern blot analysis with a strand-specific oligonucleotide probe. Two bands were detected and were designated Teg23.1 (approximately 215 nt) and Teg23.2 (approximately 310 nt). (C) Alignment of Teg23 DNA sequence in strains USA300, NCTC 8325, and MRSA252. Sequence corresponding to the SAOUHSC_02572 and SAR2384 CDS genes is highlighted in red and blue, respectively. Sequence potentially encoding an 80-amino-acid protein (Teg23P) in strain USA300 is highlighted in green. (D) Western blot to detect histidine-tagged Teg23P. No Teg23P protein was detected in S. aureus expressing the Teg23P-his plasmid (lane 1) or in a wild-type S. aureus strain without the plasmid (lane 2). Histidine-tagged RpoE was detected as a positive control (lane 3). (E) Quantitative RT-PCR analysis of Teg23 expression. Expression of Teg23 was analyzed in the wild-type S. aureus strain with or without the Teg23P-his plasmid. Approximately 3-fold-higher expression of Teg23 was detected in the strain expressing the Teg23P-his plasmid. Statistical significance was determined using Student's t-test. *, P < 0.05. (F) RNA-seq read alignment at the Teg23 locus in strain NCTC 8325. RNA-seq reads aligned to the NCTC 8325 genome are shown in blue. The locations of annotated genes are shown in gray. The locations of primers used for RT-PCR are shown in red. The predicted size and location of the Teg23.1 and Teg23.2 transcripts are shown by red arrows. (G) RT-PCR analysis of Teg23 transcript. PCR was performed using the primer pairs indicated and S. aureus genomic DNA (gDNA) (lane 1), cDNA from S. aureus containing the Teg23P-his plasmid (lane 2), cDNA from wild-type S. aureus (lane 3), or no DNA (lane 4). No PCR product was detected using primers 1 and 3 with S. aureus cDNA as the template.
Northern blot analysis using S. aureus USA300 grown in human serum (where Teg23 is strongly upregulated) detected a band of around 310 nt (as predicted by RNA-seq), alongside an additional band of approximately 215 nt (Fig. 3B). The smaller band (which we designated Teg23.1) was more abundant than the larger band (designated Teg23.2) and was repeatedly detected in multiple Northern blots (data not shown), suggesting that two forms of the transcript may exist within the cell. The CDS annotated in the NCTC 8325 genome at the Teg23 position (SAOUHSC_02572) potentially encodes a protein of 80 amino acids (Fig. 3C, red), while the corresponding locus in strain MRSA252 (SAR2384) potentially encodes a protein of 64 amino acids (Fig. 3C, blue). The difference is due to a 4-bp insertion in the MRSA252 genome that results in the creation of a stop codon (Fig. 3C). The nucleotide sequence of the 432-bp intergenic region from strain USA300 is almost identical with the corresponding locus in strain NCTC 8325 (431/432 identical), and it does not contain the 4-bp insertion found in MRSA252 (Fig. 3C). Therefore, an 80-amino-acid protein could potentially be generated from a transcript originating from this locus in USA300 (Fig. 3C, green), similar to NCTC 8325 (for simplicity, this potential protein will be referred to as Teg23P for Teg23 protein).
To test whether Teg23P is produced in strain USA300, we cloned a His6-tagged variant of its putative coding sequence, along with its native promoter, into an S. aureus shuttle vector. Western blots of USA300 strain bearing this construct revealed no detectable band using an antihistidine antibody, while the positive control (histidine-tagged RpoE) was detected (Fig. 3D). Quantitative RT-PCR (qRT-PCR) to confirm that the construct was being expressed demonstrated a 2.92-fold-higher expression of Teg23 in S. aureus containing the Teg23P-his6 plasmid (Fig. 3E). These data confirm that the plasmid gene-encoded Teg23 is expressed; however, no protein corresponding to Teg23P-his6 is generated. This strongly suggests that no protein corresponding to Teg23P is produced in strain USA300.
The RNA-seq read alignment data demonstrate that >99% of the Teg23 reads in strain NCTC 8325 terminate 88 nucleotides upstream of the annotated 3′ end of the gene (Fig. 3F). Consequently, transcript generated at this locus terminates before the end of the annotated gene, and therefore, an 80-amino-acid protein corresponding to the SAOUHSC_02572 gene is unlikely to be made. A similar pattern of transcript termination is observed in the S. aureus USA300 RNA-seq data set, further suggesting that the Teg23P protein could not be produced in this strain (data not shown). To test this further, we performed PCR using cDNA generated from total RNA from strain USA300. PCR performed using primers that bind within the Teg23 sequence (primers 2 and 3 [Fig. 4F]) generated products using template cDNA from USA300 and USA300 containing the Teg23P-his6 plasmid (Fig. 3G). In contrast, no product was generated in PCRs using one primer within the Teg23 sequence and a second primer located at the 3′ end of the Teg23P sequence (primers 1 and 3 [Fig. 3F]). These data show that the Teg23 RNA is being generated but that it terminates prior to the 3′ end of Teg23P, confirming the results from the Western blot analysis that Teg23 does not encode an 80-amino-acid protein.
FIG 4 .

Location and comparative expression analysis of the 303 S. aureus USA300 sRNAs. The black outer ring shows the chromosomal locations of 303 sRNAs in strain USA300 (the outer circle shows the forward strand, while the inner circle shows the reverse strand). The inner rings show sRNA gene expression profile in TSB (outside, blue), human serum (inside, red), and heat map (middle) showing differences in expression. The heights of the blue or red bars are proportional to the expression values (the maximum displayed expression value was 1,000).
While it is impossible to completely rule out the existence of a protein corresponding to Teg23P, these results strongly suggest that the CDS annotations for Teg23P in strains N315, MRSA252, and NCTC 8325 (i.e., SAS083, SAR2384, and SAOUHSC_02572, respectively) are incorrect. Teg23 was originally identified as a 5′ UTR for the SAS083 gene (i.e., the gene encoding Teg23P in N315) (7); however, the data presented herein demonstrates that this is incorrect and Teg23 likely represents a nontranslated sRNA. This comprehensively highlights how overannotation of genomes with CDS genes may be masking the identification of transcripts that encode sRNAs.
Global analysis of sRNA gene expression in strain USA300.
The inclusion of 303 sRNA gene annotations in the GenBank file of strain USA300 allowed us to calculate global gene expression values for sRNAs using our RNA-seq data sets. To examine variation in sRNA gene expression, we calculated and compared expression values for S. aureus USA300 growing in TSB and human serum, two conditions known to result in widespread changes in gene expression (18). The location, relative expression, and fold change for each of the 303 sRNA genes in strain USA300 are shown in Fig. 4 and Table S3 in the supplemental material. To identify sRNA genes with meaningful differences in expression, we applied cutoffs to eliminate genes expressed at low levels and those displaying fold changes that are less than 3-fold (see Text S1 in the supplemental material for details). This resulted in a total of 83 sRNAs displaying alterations in gene expression under the two conditions tested. Forty-two were upregulated in human serum (Table 2), while 41 were downregulated (Table 3). Of the newly identified tsr genes, 19 displayed differential regulation, with 16 downregulated in serum, while 3 (tsr18, tsr25, and tsr30) were upregulated. Interestingly, the newly identified tsr25 sRNA demonstrated the largest upregulation in serum of any sRNA (583-fold). To validate these findings, we performed Northern blot analysis of tsr25 and rsaOG (a previously identified sRNA which showed the second highest degree of upregulation in serum, 376-fold), confirming the predicted size and upregulation of both sRNAs in human serum (Fig. 5A). RNAIII was downregulated 13-fold in the RNA-seq data analysis, and we also confirmed this by Northern blotting (Fig. 5B). These results corroborate previously published data (18) and provide validation for the experimental techniques described herein.
TABLE 2 .
42 sRNAs that are upregulated in human serum versus TSB
| sRNA designation | sRNA gene name or feature | TSB expression value (RPKM)b | Serum expression value (RPKM) | Fold change |
|---|---|---|---|---|
| SAUSA300s289 | tsr25 | 60.67 | 35,374.06 | 583.02 |
| SAUSA300s046 | rsaOG | 90.99 | 34,267.77 | 376.61 |
| SAUSA300s053 | RsaG | 80.85 | 3,463.09 | 42.83 |
| SAUSA300s119 | ssr24 | 130.78 | 3,217.86 | 24.6 |
| SAUSA300s153 | Teg16 | 71.05 | 1,174.01 | 16.52 |
| SAUSA300s066 | Sau-63 | 104.99 | 1,690.95 | 16.11 |
| SAUSA300s030 | sprC | 149.58 | 2,144.32 | 14.34 |
| SAUSA300s026 | ssrS | 2,991.16 | 37,212.81 | 12.44 |
| SAUSA300s005 | ffs | 929.74 | 11,188.00 | 12.03 |
| SAUSA300s148 | Teg23 | 256.6 | 2,950.59 | 11.5 |
| SAUSA300s050 | RsaD | 2,737.91 | 30,473.59 | 11.13 |
| SAUSA300s226 | JKD6008sRNA173 | 50.05 | 539.79 | 10.78 |
| SAUSA300s013 | Lysine riboswitch | 659.93 | 6,917.84 | 10.48 |
| SAUSA300s260 | JKD6008sRNA396 | 16.13 | 150.26 | 9.31 |
| SAUSA300s294 | tsr30 | 470.64 | 4,383.00 | 9.31 |
| SAUSA300s027 | sprA1 | 1,097.34 | 9,306.35 | 8.48 |
| SAUSA300s233 | JKD6008sRNA205 | 96.96 | 717.53 | 7.4 |
| SAUSA300s003 | T-box riboswitch | 158.5 | 1,156.13 | 7.29 |
| SAUSA300s094 | Teg108 | 19.39 | 132.54 | 6.83 |
| SAUSA300s099 | Teg124 | 37.1 | 245.19 | 6.61 |
| SAUSA300s110 | sprA3 | 36.05 | 233.31 | 6.47 |
| SAUSA300s282 | tsr18 | 105.4 | 653.5 | 6.2 |
| SAUSA300s024 | GlmS ribozyme | 185.53 | 1,064.95 | 5.74 |
| SAUSA300s117 | ssr8 | 12.13 | 67.9 | 5.6 |
| SAUSA300s038 | sprG3 | 191.04 | 1,049.33 | 5.49 |
| SAUSA300s120 | ssr28 | 188.03 | 1,012.53 | 5.39 |
| SAUSA300s028 | sprA2 | 99.08 | 504.62 | 5.09 |
| SAUSA300s210 | JKD6008sRNA071 | 136.39 | 691.13 | 5.07 |
| SAUSA300s100 | Teg124 | 469.39 | 2,313.98 | 4.93 |
| SAUSA300s002 | SAMa riboswitch | 56.06 | 268.4 | 4.79 |
| SAUSA300s031 | sprD | 804.11 | 3,810.91 | 4.74 |
| SAUSA300s021 | SAM riboswitch | 239.09 | 941.21 | 3.94 |
| SAUSA300s075 | rsaOL | 273.5 | 1,010.42 | 3.69 |
| SAUSA300s091 | Teg60 | 95.9 | 350.09 | 3.65 |
| SAUSA300s034 | sprF3 | 1,227.97 | 4,261.30 | 3.47 |
| SAUSA300s016 | T-box riboswitch | 316.76 | 1,059.28 | 3.34 |
| SAUSA300s084 | Teg27 | 15,788.22 | 52,571.03 | 3.33 |
| SAUSA300s042 | rsaOC | 26.75 | 88.89 | 3.32 |
| SAUSA300s135 | ssr100 | 322.65 | 1,050.22 | 3.26 |
| SAUSA300s163 | Teg28as | 46.74 | 149.53 | 3.2 |
| SAUSA300s079 | rsaOU | 0 | 85.01 | ∞ |
| SAUSA300s151 | Teg134 | 0 | 204.13 | ∞ |
SAM, S-adenosylmethionine.
RPKM, reads per kilobase of transcript per million mapped reads.
TABLE 3 .
41 sRNAs that are downregulated in human serum versus TSB
| sRNA designation | sRNA gene name or feature | TSB expression value (RPKM)a | Serum expression value (RPKM) | Fold change |
|---|---|---|---|---|
| SAUSA300s277 | tsr13 | 509.65 | 0.43 | −1,187.67 |
| SAUSA300s113 | sbrC | 1,834.12 | 6.3 | −291.07 |
| SAUSA300s266 | tsr2 | 122.44 | 0.42 | −290.6 |
| SAUSA300s296 | tsr32 | 92.33 | 0.44 | −210.63 |
| SAUSA300s171 | Sau-6569 | 41,077.46 | 250.83 | −163.77 |
| SAUSA300s049 | RsaC | 8,079.72 | 77.42 | −104.37 |
| SAUSA300s004 | Purine riboswitch | 2,277.90 | 45.36 | −50.22 |
| SAUSA300s303 | tsr39 | 3,132.51 | 64.48 | −48.58 |
| SAUSA300s162 | Teg25as | 6,668.30 | 191.51 | −34.82 |
| SAUSA300s125 | ssr47 | 6,272.84 | 256.03 | −24.5 |
| SAUSA300s052 | RsaF | 35,374.06 | 1,713.79 | −20.64 |
| SAUSA300s275 | tsr11 | 398.93 | 20.01 | −19.94 |
| SAUSA300s301 | tsr37 | 390.39 | 23.36 | −16.72 |
| SAUSA300s292 | tsr28 | 513.06 | 31.91 | −16.08 |
| SAUSA300s297 | tsr33 | 8,987.32 | 592.2 | −15.18 |
| SAUSA300s062 | Sau-31 | 141.32 | 9.7 | −14.57 |
| SAUSA300s022 | RNAIII | 66,968.70 | 5,178.15 | −12.93 |
| SAUSA300s283 | tsr19 | 162.04 | 13.93 | −11.63 |
| SAUSA300s118 | ssr16 | 478.47 | 49.19 | −9.73 |
| SAUSA300s302 | tsr38 | 251.17 | 27.55 | −9.12 |
| SAUSA300s280 | tsr16 | 254.42 | 30.88 | −8.24 |
| SAUSA300s095 | Teg116 | 1,149.05 | 155.21 | −7.4 |
| SAUSA300s211 | JKD6008sRNA073 | 59.06 | 8 | −7.38 |
| SAUSA300s287 | tsr23 | 970.48 | 133.71 | −7.26 |
| SAUSA300s054 | RsaH | 4,110.36 | 629.99 | −6.52 |
| SAUSA300s087 | Teg41 | 481.01 | 74.55 | −6.45 |
| SAUSA300s078 | rsaOT | 17,890.11 | 2,790.22 | −6.41 |
| SAUSA300s127 | ssr54 | 3,245.19 | 584.15 | −5.56 |
| SAUSA300s051 | RsaE | 2,929.20 | 534.76 | −5.48 |
| SAUSA300s074 | rsaOI | 721.93 | 135.52 | −5.33 |
| SAUSA300s237 | JKD6008sRNA258 | 100.89 | 20.87 | −4.83 |
| SAUSA300s298 | tsr34 | 200.17 | 41.54 | −4.82 |
| SAUSA300s267 | tsr3 | 85.01 | 19.21 | −4.43 |
| SAUSA300s114 | sbrE | 424.58 | 102.21 | −4.15 |
| SAUSA300s073 | Sau-6072 | 265.82 | 64.27 | −4.14 |
| SAUSA300s276 | tsr12 | 295.29 | 73.2 | −4.03 |
| SAUSA300s291 | tsr27 | 135.22 | 34.48 | −3.92 |
| SAUSA300s204 | RsaX04 | 265.58 | 70.09 | −3.79 |
| SAUSA300s086 | Teg39 | 50.39 | 13.67 | −3.69 |
| SAUSA300s141 | ssr153 | 74.69 | 21.06 | −3.54 |
| SAUSA300s010 | T-box riboswitch | 487.82 | 149.37 | −3.27 |
RPKM, reads per kilobase of transcript per million mapped reads.
FIG 5 .
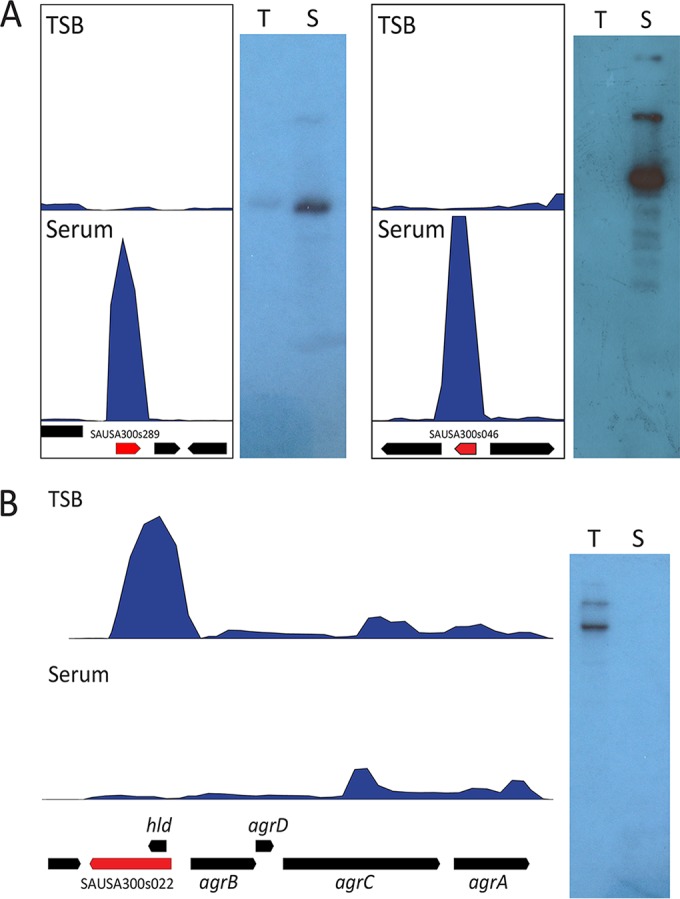
Northern blot analysis of serum-regulated sRNAs in strain USA300. (A) Analysis of sRNAs demonstrating upregulation in human serum. RNA-seq read alignment data and Northern blot analysis are shown for SAUSA300s289 (tsr25) and SAUSA300s046 (rsaOG) during growth in TSB (T) or human serum (S). (B) Analysis of RNAIII expression in human serum and TSB. RNA-seq read alignment data are shown for the entire agr locus. Northern blot analysis was performed using an oligonucleotide probe specific for the RNAIII transcript. Annotations for CDS genes are shown by black arrows, annotations for sRNAs are shown by red arrows, and depth of read coverage on the genome is shown by the blue histograms.
The data presented above represent the first global transcriptomic analysis of sRNA gene expression in the CA-MRSA strain USA300 and demonstrate the utility of the sRNA annotation files constructed in this study. The biological functions of most sRNAs are unknown, making it difficult to ascertain the impact on the bacterial cell of their differential regulation. Studying the variation in sRNA expression in response to changing environmental conditions may provide insight into which sRNAs play a role in the adaptive nature of S. aureus. tsr25 and rsaOG, for example, are upregulated >300-fold in human serum, suggesting a role for these molecules under these conditions. In addition, a number of conserved, well-studied sRNAs were among those differentially regulated in human serum. 4.5S RNA (SAUSA300s005) and ssrS (SAUSA300s026) demonstrated increased expression in human serum (12-fold increase for both). The increased expression of these RNA species may reflect physiological changes to the bacterial cell in this environmental niche (see Discussion).
DISCUSSION
In 1995, the release of the first fully sequenced bacterial genome heralded a new era of bacterial genomic research (21). Over the past 20 years, the number of sequenced bacterial genomes has risen exponentially, and new research strategies, techniques, and applications have emerged to exploit the opportunities that these resources provide. While raw genomic sequence data are valuable, the availability of fully annotated genome sequences, outlining the positions of known genes and genomic features, dramatically increases their utility. Global expression analysis techniques such as microarrays and RNA-seq depend heavily on annotated genome sequences as a reference source for genes in the bacterial cell. These techniques have proved extremely useful; however, recently, certain limitations to their application are becoming apparent. A major concern in this regard is that they do not provide expression data for genes that are not included in genome annotation files. Bacterial sRNAs represent a class of genes that are frequently absent from genome annotation files; consequently, their expression is rarely studied on a global level. In this work, we added annotations for 303 sRNA genes to the S. aureus USA300 genome, increasing the number of annotated genes from 2,850 to 3,153 (a 10% increase). Including sRNA gene annotations in S. aureus GenBank files facilitates global expression analysis of these understudied molecules. The 303 newly added annotations undoubtedly do not represent an exhaustive list of the complete S. aureus sRNA repertoire, as it is likely that subsequent studies, using different techniques and environmental conditions, will continue to add to this number. It is also likely that among the 303 annotations, there may be false positives as only 92 of the 303 sRNAs reported and annotated here (30%) have been confirmed by independent experimental methods other than high-throughput sequencing and microarray hybridization. Therefore, while the sRNA annotated GenBank files generated herein may not be definitive, they nonetheless represent a significant step forward and pave the way for future studies that can validate the existence, elucidate the role, and demonstrate the biological impact of these molecules on S. aureus physiology and virulence.
The 303 sRNA genes annotated in this study (representing 10% of all known genes on the S. aureus genome) were identified in S. aureus growing under a limited number of environmental conditions. How many sRNAs remain unidentified is unknown; however, on the basis of the data presented in this study and by others (14), it seems likely that sRNA genes may account for >15 to 20% of all transcripts in the S. aureus cell. Importantly, a benefit of the nomenclature system used in this study to annotate sRNAs (in addition to keeping original names intact) is that it can be expanded upon to accommodate new sRNAs as they are identified.
An unexpected observation arising from this study is the lack of consistency in genome annotation across multiple strains of S. aureus. The examples highlighted in this study (specifically tsr12 and Teg23) demonstrate that this phenomenon occurs even at genomic loci with a high degree of homology (100% identical in the case of tsr12 in strains USA300 and NCTC 8325). Genome annotation is typically performed bioinformatically and is rarely validated and curated (22); therefore, this variation in annotation likely arises because different annotation pipelines have been used to annotate different genomes. This raises the important question of whether certain genomes have been under- and/or overannotated. It is likely that examples of both situations occur; however, on the basis of our data, it appears that the overannotation of genomes (i.e., the inclusion of CDS annotations that are not legitimate mRNA transcripts) is common and could have deleterious consequences for sRNA identification and study. sRNA transcripts that map to genomic regions containing CDS annotations will mistakenly be assumed to encode proteins. Highlighting this point is the data presented in Fig. 3, which strongly suggest that Teg23 is an sRNA and not a protein. Nonetheless, we acknowledge that this type of data, by its very nature, cannot exclude the possibility that such a gene/protein exists, and hence, once a CDS has been annotated, it is difficult or impossible to conclusively prove that it does not exist. Careful attention must be paid to the location of transcript start/stop sites, the existence of ribosome binding sites and the predicted function of annotated genes. We observed many instances of inconsistent genome annotation where a given gene encoded a protein of unknown function with no homologues and no functional domains. This lack of homology may indicate that while sequence analysis alone may suggest that a gene is possible at this locus, it is not certain to exist. The results presented herein are a timely reminder that, although genome annotation files are valuable resources that are increasingly relied upon by next-generation DNA sequencing technology, these annotations should be treated with a reasonable degree of caution and not seen as an infallible reference. Confirmation of the existence of a gene/transcript/protein by traditional biochemical methods (such as 5′ and 3′ rapid amplification of cDNA ends [RACE], primer extension, and in vivo translation) remains essential.
Including sRNA annotations in GenBank files allowed us to perform global sRNA expression analysis by RNA-seq for the first time in the CA-MRSA isolate USA300. The data generated demonstrated that 83 sRNAs are differentially expressed in TSB versus human serum (Tables 2 and 3). This represents 27% of the known sRNAs in strain USA300. It is not easy to interpret how their differential regulation affects the bacterial cell (because the biological functions of most of them are unknown); however, certain inferences can be made. The newly identified tsr25 sRNA demonstrated a 582-fold increase in expression in human serum. It is tempting to speculate that increased expression of tsr25 in serum suggests that it plays an important role during S. aureus bloodstream infections. A small number of conserved, well-studied sRNAs were also among the differentially regulated sRNAs in serum. 4.5S RNA, a component of the signal recognition particle, was upregulated 12-fold in human serum, perhaps reflecting altered protein secretion and/or protein composition in the cell membrane in this environment. Another important cellular RNA that has been well explored is 6S RNA (ssrS), which we demonstrate also has a 12-fold increase in expression during growth in serum. In Escherichia coli, 6S RNA binds to the housekeeping sigma factor σ70 and inhibits transcription from σ70-dependent promoters. It is thought that this diverts RNA polymerase to alternative sigma factors (such as the stress response sigma factor), resulting in increased expression of adaptive and stress-circumventing genes (23). The upregulation of ssrS in human serum suggests that a similar situation occurs in S. aureus, whereby σA-dependent genes are downregulated and σB-dependent genes are upregulated by the action of 6S RNA.
Examining the global transcriptome can provide valuable insights into bacterial physiology and adaptation to environmental conditions. In the past, global transcriptomic analysis has focused on protein-coding genes, but here, we conduct global transcriptomic analysis and include newly annotated sRNA genes. Like protein-coding genes, sRNA genes display differential regulation that allow bacteria to adapt to environmental changes. The annotation files presented herein, which facilitate this kind of global analysis will prove to be a valuable resource for the future study of sRNAs in S. aureus and will more generally broaden our understanding of regulatory circuits.
MATERIALS AND METHODS
Strains, plasmids, and primers.
Bacterial strains, plasmids, and primers used in this study are listed in Table 4. S. aureus and E. coli were grown routinely at 37°C with shaking in tryptic soy broth (TSB) and lysogeny broth (LB), respectively. Pooled human serum from anonymous donors, purchased from MP Biomedicals, was used for growth of S. aureus strain USA300. When necessary, antibiotics were added at the following concentrations: ampicillin, 100 µg ml−1; chloramphenicol, 5 µg ml−1.
TABLE 4 .
Bacterial strains, plasmids, and primers used in this study
| Bacterial strain, plasmid, or primer | Characteristic(s) or sequence | Reference or source | Commenta |
|---|---|---|---|
| S. aureus strains | |||
| USA300 Houston | Community-associated MRSA clinical isolate | 26 | |
| SH1000 | Laboratory strain; rsbU functional | 29 | |
| UAMS-1 | Osteomyelitis clinical isolate | 30 | |
| RN4220 | Restriction-deficient transformation recipient | 31 | |
| AW2192 | USA300 pMK4::teg23p-his6 | This study | |
| E. coli strain | |||
| DH5α | Routine cloning strain | Invitrogen | |
| Plasmids | |||
| pMK4 | Shuttle vector; Cmr | 32 | |
| pAW105 | pMK4::teg23P-his6 | This study | |
| Primers | |||
| OL1184 | 5′ TCTGGACCGTGTCTCAGTTCC 3′ | 27 | |
| OL1185 | 5′ AGCCGACCTGAGAGGGTGA 3′ | 27 | |
| OL2701 | 5′ CCAAATTTAGGCATGTCAAATCGGC 3′ | teg23 probe | |
| OL3201 | 5′ GGATTCCCAATTTCTACAGACAATGCA 3′ | tsr8 probe | |
| OL3208 | 5′ ACGGGCATATAAAAGGGGAATATTTGAAAA 3′ | tsr25 probe | |
| OU0121 | 5′ GTGTTAAAAAAATAACTGGGATGTG 3′ | tsr26 probe | |
| OL3216 | 5′ CTCACAAATTCTGTAAGGGGAGCGTAT 3′ | tsr31 probe | |
| OL3217 | 5′ TTATGTCCCAATGCTGAATAAATAACTTC 3′ | tsr33 probe | |
| OL3222 | 5′ ACGCGTCGACGCGCTTGTATTCGCTGCAGG 3′ | teg23P F | |
| OL3223 | 5′ CGGGATCCTTAGTGGTGGTGGTGGTGGTGCGCCAACAAGGTTTCAAGAGC 3′ | teg23P-his6 R | |
| OL3232 | 5′ CGCCAACAAGGTTTCAAGAGC 3′ | teg23 OL1 | |
| OL3281 | 5′ TAAACAACATACAGCCATTG 3′ | teg23 OL2 | |
| OL3282 | 5′ GAGAATTTGAAGGCAAGTAT 3′ | teg23 OL3 | |
| OL3880 | 5′ GCCAGGATAATGTAGTCTTAA 3′ | tsr1 F | |
| OL3882 | 5′ CCATTAATTTACTCAAACCG 3′ | tsr1 R | |
| OL3885 | 5′ GCTTCTGTTCGATCTC 3′ | tsr2 F | |
| OL3886 | 5′ CACGTCTTCTGATTAAAC 3′ | tsr2 R | |
| OL3916 | 5′ CATACCTCTTTAACAACAG 3′ | tsr18 F | |
| OL3917 | 5′ GGAGGAATTAATCATGTC 3′ | tsr18 R | |
| OL3935 | 5′ GAAGGGATCCAACACA 3′ | tsr24 F | |
| OL3937 | 5′ GTCTCGCCATTAATACTAC 3′ | tsr24 R | |
| OL3946 | 5′ GTCTTTTCACAACCAAAG 3′ | tsr29 F | |
| OL3948 | 5′ GGTTTTATCTTTGGAAAAAG 3′ | tsr29 R | |
| OL3956 | 5′ GATGCGGAAAATTTGG 3′ | tsr32 F | |
| OL3957 | 5′ GTGCGCAATGAATATTATG 3′ | tsr32 R |
F, forward; R, reverse.
RNA-seq.
Samples for transcriptome sequencing (RNA-seq) were prepared as follows for S. aureus USA300 and SH1000. Overnight cultures were diluted 1:100 in 100 ml of fresh TSB and grown at 37°C with shaking for 3 h. Exponentially growing cultures were then diluted and synchronized by inoculating fresh 100-ml flasks of TSB at an optical density at 600 nm (OD600) of 0.05, or in the case of strain USA300 growing in serum, 10 ml of human serum in a 50-ml tube. Synchronized cultures were grown for 3 h at 37°C with shaking, at which time bacteria were pelleted by centrifugation and stored at −80°C prior to RNA isolation. S. aureus UAMS-1 (a USA200 strain and close relative of MRSA252) overnight cultures were diluted to an optical density of 0.05 in 40 ml of TSB containing no dextrose (1:12.5 volume-to-flask volume ratio). Cultures were grown at 37°C with shaking for 4 h, before cells were harvested and stored as described above. For each RNA-seq sample, three replicate cultures were grown, and three biological replicate RNA isolations were performed (using the procedure outlined in reference 24). For each sample, equimolar amounts of the three biological replicate RNA preparations were mixed prior to rRNA reduction. The subsequent RNA-seq analysis therefore represents the average of three biological replicates. RNA-seq and data analysis were carried out using the protocol previously published by our research group (24).
Bioinformatics, sRNA identification, and genome annotation.
The CLC Genomics Workbench software platform (Qiagen) was used for all RNA-seq data analysis and for construction of new GenBank files. Annotated genome files for S. aureus N315, USA300, MRSA252, and NCTC 8325 (GenBank accession numbers NC_002745, CP000255, NC_002952, and NC_007795, respectively) were downloaded from NCBI. The location, sequence, and orientation of previously described sRNAs on the N315 genome were calculated based on the information provided in published manuscripts and supplemental information files of the relevant studies (1–12). Using the CLC Genomics Workbench built-in BLAST feature, the corresponding positions for sRNAs were identified in the genomes of strains USA300, MRSA252, and NCTC 8325. The location and orientation of each sRNA was then annotated.
Northern blots.
Northern blots to identify the size and abundance of sRNAs were performed by the method of Caswell et al. (25). Briefly, 10 µg of total RNA isolated from a 3 h USA300 culture in TSB were loaded on a 10% polyacrylamide gel (7 M urea, 1× Tris-borate-EDTA [TBE]) and separated by gel electrophoresis. The samples were then transferred via electroblotting to an Amersham Hybond N+ membrane (GE Healthcare). The membrane was exposed to UV light to cross-link samples to the membrane. Subsequently, membranes were prehybridized (1 h, 45°C) in ULTRAhyb-Oligo buffer (Ambion) and then incubated (16 h, 45°C) with sRNA-specific oligonucleotide probes end labeled with [γ-32P]ATP and T4 polynucleotide kinase (Thermo Scientific). After incubation, membranes were washed with 2×, 1×, and 0.5× SSC buffer (1× SSC is 300 mM sodium chloride and 30 mM sodium citrate) at 45°C for 30 min each to remove unspecific bound probes. Finally, X-ray film was exposed to membranes for sRNA detection.
Cloning of histidine-tagged Teg23P.
The genomic region containing Teg23P (including its native promoter) was amplified using USA300 genomic DNA and primers OL3222 and OL3223. The reverse primer OL3223, in addition to the gene-specific region, carries a sequence that encodes a hexahistidine (His6) tag, which allows the detection of a possible encoded protein via Western blotting. The amplified 761-bp product was cloned into shuttle vector pMK4, and the plasmid was transformed into chemically competent E. coli DH5α. The resulting colonies were screened for correct constructs employing a colony PCR approach using identical primers to those used for the amplification of the initial fragment. After identification of positive clones, the plasmid was verified via Sanger sequencing with the plasmid-specific standard primers M13Fw (Fw stands for forward) and M13Rv (Rv stands for reverse) (Eurofins MWG Operon). This construct was then transformed into S. aureus RN4220 and confirmed via PCR. Finally, the plasmid was transduced into S. aureus USA300 using a φ11 phage lysate, and after final confirmation of the construct using PCR, the strain was utilized for subsequent analysis.
qRT-PCR.
Quantitative reverse transcriptase PCR (qRT-PCR) was conducted as described by our research group previously (26), employing the teg23-specific primers OL3281, OL3282, OL3232, and OL3282 and RNA isolated from S. aureus USA300 cultures grown in TSB as described above for RNA-seq experiments. As a reference, 16S rRNA was amplified using primers OL1184 and OL1185 (27). All experiments were performed in triplicate.
Western blots.
The evaluation of Teg23 protein abundance was performed by Western immunoblotting as described previously (28). USA300 cells harboring the His6-tagged version of Teg23P were grown for 3 h in TSB, before they were pelleted and their cytoplasmic proteins were isolated. Following SDS-polyacrylamide gel electrophoresis and transfer of the separated proteins to a polyvinylidene difluoride (PVDF) membrane, detection was performed using anti-His monoclonal mouse antibody (Covance) and horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody. Subsequently, HRP activity was detected and visualized on X-ray film. Histidine-tagged RpoE, a protein previously described by our group, was included as a control for successful protein transfer and immunodetection (16).
Accession numbers.
The GenBank files generated have been deposited in figshare (https://dx.doi.org/10.6084/m9.figshare.2061132.v1). The files are provided in .gkb format and can be viewed using a variety of genome browser software (examples of freely available genome browsers include Artemis, Genome Compiler, and CLC Sequence Viewer). The RNA-seq data files have been deposited in GEO under accession number GSE74936. Newly identified sRNAs (i.e., the tsr sRNAs) have been deposited in GenBank under accession numbers KU639719-KU639757 for SAUSA300s265-SAUSA300s303, KU639758-KU639789 for SAOUHSCs255-SAOUHSCs286, and KU639790-KU639818 for SARs249-SARs277.
SUPPLEMENTAL MATERIAL
Supplemental methods. Download
Schematic outlining the procedure used to locate and annotate sRNAs in S. aureus. (A) Using information provided in previous studies, the approximate locations of reported sRNAs were identified. (B) RNA-seq was performed, and reads were aligned to the relevant S. aureus genome. (C) The locations of aligned reads were used to precisely locate and annotate sRNAs (blue arrow). Download
Distribution of tsr genes in strains USA300, NCTC 8325, and MRSA252. Five tsr genes are unique to strain USA300. An additional five are found in strains USA300 and NCTC 8325 (but not MRSA252), while two are found in USA300 and MRSA252 (but not NCTC 8325). Twenty-seven tsr genes are found in all three strains. Download
RNA-seq read alignment data for sarR and tsr33 during growth in human serum. Annotations for CDS genes are shown by black arrows, and the annotation for sRNA is shown by a red arrow. The depth of read coverage on the genome is shown by blue histogram. Greater depth of read coverage for tsr33 suggests that a sarR-independent tsr33 exists under these conditions. Download
Reverse transcriptase PCR-based detection of newly identified sRNAs in S. aureus. PCRs were performed with primers specific to newly identified sRNA genes. DNA-depleted RNA, DNA, and H2O were employed as the templates to verify the complete removal of DNA from RNA samples, the correct sizes of the respective amplicons, and the absence of DNA contaminations. Successful, size-specific amplification of a target from cDNA suggests the presence of transcript at the position where a novel sRNA annotation was added to the genome file. Download
25 miscellaneous RNAs from S. aureus MRSA252 genome.
Names and designations for 953 previously identified and 39 newly identified sRNAs in S. aureus strains USA300, MRSA252, and NCTC 8325.
Expression values for 303 S. aureus USA300 sRNA genes in TSB and human serum.
ACKNOWLEDGMENTS
We thank members of the Shaw and Rice labs for useful discussions. R.K.C. thanks Padraig Deighan for many years of advice and assistance.
Footnotes
Citation Carroll RK, Weiss A, Broach WH, Wiemels RE, Mogen AB, Rice KC, Shaw LN. 2016. Genome-wide annotation, identification, and global transcriptomic analysis of regulatory or small RNA gene expression in Staphylococcus aureus. mBio 7(1):e01990-15. doi:10.1128/mBio.01990-15.
REFERENCES
- 1.Pichon C, Felden B. 2005. Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci U S A 102:14249–14254. doi: 10.1073/pnas.0503838102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM. 2006. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol 188:6739–6756. doi: 10.1128/JB.00609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geissmann T, Chevalier C, Cros MJ, Boisset S, Fechter P, Noirot C, Schrenzel J, François P, Vandenesch F, Gaspin C, Romby P. 2009. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res 37:7239–7257. doi: 10.1093/nar/gkp668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marchais A, Naville M, Bohn C, Bouloc P, Gautheret D. 2009. Single-pass classification of all noncoding sequences in a bacterial genome using phylogenetic profiles. Genome Res 19:1084–1092. doi: 10.1101/gr.089714.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abu-Qatouseh LF, Chinni SV, Seggewiss J, Proctor RA, Brosius J, Rozhdestvensky TS, Peters G, von Eiff C, Becker K. 2010. Identification of differentially expressed small non-protein-coding RNAs in Staphylococcus aureus displaying both the normal and the small-colony variant phenotype. J Mol Med (Berl) 88:565–575. doi: 10.1007/s00109-010-0597-2. [DOI] [PubMed] [Google Scholar]
- 6.Bohn C, Rigoulay C, Chabelskaya S, Sharma CM, Marchais A, Skorski P, Borezée-Durant E, Barbet R, Jacquet E, Jacq A, Gautheret D, Felden B, Vogel J, Bouloc P. 2010. Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res 38:6620–6636. doi: 10.1093/nar/gkq462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beaume M, Hernandez D, Farinelli L, Deluen C, Linder P, Gaspin C, Romby P, Schrenzel J, Francois P. 2010. Cartography of methicillin-resistant S. aureus transcripts: detection, orientation and temporal expression during growth phase and stress conditions. PLoS One 5:e10725. doi: 10.1371/journal.pone.0010725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Anderson KL, Roux CM, Olson MW, Luong TT, Lee CY, Olson R, Dunman PM. 2010. Characterizing the effects of inorganic acid and alkaline shock on the Staphylococcus aureus transcriptome and messenger RNA turnover. FEMS Immunol Med Microbiol 60:208–250. doi: 10.1111/j.1574-695X.2010.00736.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olson PD, Kuechenmeister LJ, Anderson KL, Daily S, Beenken KE, Roux CM, Reniere ML, Lewis TL, Weiss WJ, Pulse M, Nguyen P, Simecka JW, Morrison JM, Sayood K, Asojo OA, Smeltzer MS, Skaar EP, Dunman PM. 2011. Small molecule inhibitors of Staphylococcus aureus RnpA alter cellular mRNA turnover, exhibit antimicrobial activity, and attenuate pathogenesis. PLoS Pathog 7:e1001287. doi: 10.1371/journal.ppat.1001287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nielsen JS, Christiansen MH, Bonde M, Gottschalk S, Frees D, Thomsen LE, Kallipolitis BH. 2011. Searching for small sigmaB-regulated genes in Staphylococcus aureus. Arch Microbiol 193:23–34. doi: 10.1007/s00203-010-0641-1. [DOI] [PubMed] [Google Scholar]
- 11.Xue T, Zhang X, Sun H, Sun B. 2014. ArtR, a novel sRNA of Staphylococcus aureus, regulates alpha-toxin expression by targeting the 5′ UTR of sarT mRNA. Med Microbiol Immunol 203:1–12. doi: 10.1007/s00430-013-0307-0. [DOI] [PubMed] [Google Scholar]
- 12.Howden BP, Beaume M, Harrison PF, Hernandez D, Schrenzel J, Seemann T, Francois P, Stinear TP. 2013. Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob Agents Chemother 57:3864–3874. doi: 10.1128/AAC.00263-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sassi M, Augagneur Y, Mauro T, Ivain L, Chabelskaya S, Hallier M, Sallou O, Felden B. 2015. SRD: a Staphylococcus regulatory RNA database. RNA 21:1005–1017 doi: 10.1261/rna.049346.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tenover FC, Goering RV. 2009. Methicillin-resistant Staphylococcus aureus strain USA300: origin and epidemiology. J Antimicrob Chemother 64:441–446. doi: 10.1093/jac/dkp241. [DOI] [PubMed] [Google Scholar]
- 16.Weiss A, Ibarra JA, Paoletti J, Carroll RK, Shaw LN. 2014. The delta subunit of RNA polymerase guides promoter selectivity and virulence in Staphylococcus aureus. Infect Immun 82:1424–1435. doi: 10.1128/IAI.01508-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krute CN, Carroll RK, Rivera FE, Weiss A, Young RM, Shilling A, Botlani M, Varma S, Baker BJ, Shaw LN. 2015. The disruption of prenylation leads to pleiotropic rearrangements in cellular behavior in Staphylococcus aureus. Mol Microbiol 95:819–832 doi: 10.1111/mmi.12900. [DOI] [PubMed] [Google Scholar]
- 18.Malachowa N, Whitney AR, Kobayashi SD, Sturdevant DE, Kennedy AD, Braughton KR, Shabb DW, Diep BA, Chambers HF, Otto M, DeLeo FR. 2011. Global changes in Staphylococcus aureus gene expression in human blood. PLoS One 6:e18617. doi: 10.1371/journal.pone.0018617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sayed N, Jousselin A, Felden B. 2012. A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat Struct Mol Biol 19:105–112. doi: 10.1038/nsmb.2193. [DOI] [PubMed] [Google Scholar]
- 20.Pinel-Marie ML, Brielle R, Felden B. 2014. Dual toxic-peptide-coding Staphylococcus aureus RNA under antisense regulation targets host cells and bacterial rivals unequally. Cell Rep 7:424–435. doi: 10.1016/j.celrep.2014.03.012. [DOI] [PubMed] [Google Scholar]
- 21.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, Bult CJ, Tomb JF, Dougherty BA, Merrick JM, McKenney K, Sutton G, FitzHugh W, Fields C, Gocayne JD, Scott J, Shirley R, Liu L-I, Glodek A, Kelley JM, Weidman JF, Phillips CA, Spriggs T, Hedblom E, Cotton MD, Utterback TR, Hanna MC, Nguyen DT, Saudek DM, Brandon RC, Fine LD, Fritchman JL, Fuhrmann JL, Geoghagen NSM, Gnehm CL, McDonald LA, Small KV, Fraser CM, Smith HO, Venter JC. 1995. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 22.Richardson EJ, Watson M. 2013. The automatic annotation of bacterial genomes. Brief Bioinform 14:1–12. doi: 10.1093/bib/bbs007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wassarman KM. 2007. 6S RNA: a small RNA regulator of transcription. Curr Opin Microbiol 10:164–168. doi: 10.1016/j.mib.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 24.Carroll RK, Weiss A, Shaw LN. 2016. RNA-sequencing of Staphylococcus aureus messenger RNA. Methods Mol Biol 1373:131–141 doi: 10.1007/7651_2014_192. [DOI] [PubMed] [Google Scholar]
- 25.Caswell CC, Gaines JM, Ciborowski P, Smith D, Borchers CH, Roux CM, Sayood K, Dunman PM, Roop RM II. 2012. Identification of two small regulatory RNAs linked to virulence in Brucella abortus 2308. Mol Microbiol 85:345–360. doi: 10.1111/j.1365-2958.2012.08117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kolar SL, Nagarajan V, Oszmiana A, Rivera FE, Miller HK, Davenport JE, Riordan JT, Potempa J, Barber DS, Koziel J, Elasri MO, Shaw LN. 2011. NsaRS is a cell-envelope-stress-sensing two-component system of Staphylococcus aureus. Microbiology 157:2206–2219. doi: 10.1099/mic.0.049692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koprivnjak T, Mlakar V, Swanson L, Fournier B, Peschel A, Weiss JP. 2006. Cation-induced transcriptional regulation of the dlt operon of Staphylococcus aureus. J Bacteriol 188:3622–3630. doi: 10.1128/JB.188.10.3622-3630.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carroll RK, Robison TM, Rivera FE, Davenport JE, Jonsson IM, Florczyk D, Tarkowski A, Potempa J, Koziel J, Shaw LN. 2012. Identification of an intracellular M17 family leucine aminopeptidase that is required for virulence in Staphylococcus aureus. Microbes Infect 14:989–999. doi: 10.1016/j.micinf.2012.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ. 2002. sigmaB modulates virulence determinant expression and stress resistance: characterization of a functional rsbU strain derived from Staphylococcus aureus 8325-4. J Bacteriol 184:5457–5467. doi: 10.1128/JB.184.19.5457-5467.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gillaspy AF, Hickmon SG, Skinner RA, Thomas JR, Nelson CL, Smeltzer MS. 1995. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun 63:3373–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kreiswirth BN, Löfdahl S, Betley MJ, O’Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 32.Sullivan MA, Yasbin RE, Young FE. 1984. New shuttle vectors for Bacillus subtilis and Escherichia coli which allow rapid detection of inserted fragments. Gene 29:21–26. doi: 10.1016/0378-1119(84)90161-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download
Schematic outlining the procedure used to locate and annotate sRNAs in S. aureus. (A) Using information provided in previous studies, the approximate locations of reported sRNAs were identified. (B) RNA-seq was performed, and reads were aligned to the relevant S. aureus genome. (C) The locations of aligned reads were used to precisely locate and annotate sRNAs (blue arrow). Download
Distribution of tsr genes in strains USA300, NCTC 8325, and MRSA252. Five tsr genes are unique to strain USA300. An additional five are found in strains USA300 and NCTC 8325 (but not MRSA252), while two are found in USA300 and MRSA252 (but not NCTC 8325). Twenty-seven tsr genes are found in all three strains. Download
RNA-seq read alignment data for sarR and tsr33 during growth in human serum. Annotations for CDS genes are shown by black arrows, and the annotation for sRNA is shown by a red arrow. The depth of read coverage on the genome is shown by blue histogram. Greater depth of read coverage for tsr33 suggests that a sarR-independent tsr33 exists under these conditions. Download
Reverse transcriptase PCR-based detection of newly identified sRNAs in S. aureus. PCRs were performed with primers specific to newly identified sRNA genes. DNA-depleted RNA, DNA, and H2O were employed as the templates to verify the complete removal of DNA from RNA samples, the correct sizes of the respective amplicons, and the absence of DNA contaminations. Successful, size-specific amplification of a target from cDNA suggests the presence of transcript at the position where a novel sRNA annotation was added to the genome file. Download
25 miscellaneous RNAs from S. aureus MRSA252 genome.
Names and designations for 953 previously identified and 39 newly identified sRNAs in S. aureus strains USA300, MRSA252, and NCTC 8325.
Expression values for 303 S. aureus USA300 sRNA genes in TSB and human serum.